
ABSTRACT
Inhibition of tRNA aminoacylation has proven to be an effective antimicrobial strategy, impeding an essential step of protein synthesis. Mupirocin, the well-known selective inhibitor of bacterial isoleucyl-tRNA synthetase, is one of three aminoacylation inhibitors now approved for human or animal use. However, design of novel aminoacylation inhibitors is complicated by the steadfast requirement to avoid off-target inhibition of protein synthesis in human cells. Here we review available data regarding known aminoacylation inhibitors as well as key amino-acid residues in aminoacyl-tRNA synthetases (aaRSs) and nucleotides in tRNA that determine the specificity and strength of the aaRS-tRNA interaction. Unlike most ligand-protein interactions, the aaRS-tRNA recognition interaction represents coevolution of both the tRNA and aaRS structures to conserve the specificity of aminoacylation. This property means that many determinants of tRNA recognition in pathogens have diverged from those of humans—a phenomenon that provides a valuable source of data for antimicrobial drug development.
Introduction
All living organisms encode information in the form of deoxyribonucleic acid (DNA), a template that is transcribed into messenger ribonucleic acid (mRNA) and subsequently translated into a corresponding sequence of amino acids during protein synthesis [Citation1]. The physiochemical basis of translational fidelity hinges on correct coupling of each amino acid to its cognate tRNA molecule [Citation2] and accurate base pairing between a codon in mRNA and an anticodon of an aminoacyl-tRNA [Citation3]. Fidelity is thus ensured by the specificity of aaRSs, which couple each amino acid to their cognate tRNA molecule and recognize both substrates with exquisite precision. For some aaRSs, proofreading mechanisms further ensure high fidelity by reversing incorrect aminoacylation reactions, e.g. leucyl-tRNA synthetase (LeuRS) [Citation4]. Aminoacyl-tRNAs are synthesized by the 3′-esterification of tRNAs with the appropriate amino acids. The majority of aminoacyl-tRNAs accomplish direct aminoacylation of a particular tRNA with the corresponding amino acid via a two-step reaction:
In the first step of this reaction scheme, ATP is hydrolyzed to form an aminoacyl-adenylate (AA-AMP) intermediate with the concomitant release of inorganic pyrophosphate (PPi). This AA-AMP intermediate is subsequently ligated to the tRNA in the second step, releasing AMP. The high-fidelity conversion of genetic information requires precise operation of this network of aaRSs [Citation5] with the rules governing interactions between aaRSs and tRNAs termed “the second genetic code” [Citation6]. The reaction shown in holds true for all known aaRSs, indicating a high degree of functional conservation during molecular evolution.
Scheme 1.

To date, biochemical assays have been extensively used to elucidate genetic determinants of aminoacylation within both aaRS enzymes and tRNA, as well as to identify numerous aminoacylation inhibitors. Available assays are capable of discriminating between perturbative effects on either step of the reaction shown in . Step (1) of this reaction has largely been studied using the ATP–PPi exchange assay, which measures the rate of conversion of radiolabeled [32P]-PPi into ATP. In this assay, purified aaRS is added to a reaction mixture containing its cognate amino acid, Mg, and [32P]-ATP. Aliquots are taken and quenched at different time points, ATP is filtered from the solution, and [32P]-PPi quantified through liquid scintillation counting [Citation7,Citation8]. More recently a spectrophotometric assay to detect release of PPi has been developed, applicable for high-throughput screening [Citation9]. In this assay, inorganic pyrophosphatase (PPiase) and malachite green are added to the reaction mixture; PPiase converts PPi into inorganic phosphate (Pi), which reacts with malachite green and is detected by monitoring absorbance at 620 nm9. While PPi exchange assays are typically conducted in the absence of tRNA [Citation8], authors of the spectrophometric protocol note that the addition of tRNA enables additional rounds of catalysis, thereby producing a larger signal [Citation9].
Aminoacylation assays are typically used to measure activity of both steps shown in , assaying formation of AA-tRNA. Early aminoacylation assays used radiolabeled amino acid (either 3H or 14C) in a reaction mixture containing aaRS, tRNA, ATP, and corresponding aaRS buffer. Aliquots of the reaction mix are removed at incremental time points, spotted on filter paper to quench the reaction, and quantified through liquid scintillation counting [Citation10,Citation11]. An improved assay was later devised enabling use of saturating concentrations of amino acid by instead radiolabeling tRNA is at its 3′ end with 32P using CCA-adding enzyme [Citation12]. After quenching, aminoacylated tRNA is then digested with P1 nuclease and the radioactive products are separated and quantified by thin layer chromatography (TLC) [Citation12]. Together with ATP-PPi exchange, aminoacylation assays comprise the most common biochemical method used to characterize aaRS enzymes.
As aminoacylation is an essential step in protein synthesis, in vitro translation assays have also been used for early characterization of aaRS inhibitors. When the mechanism of an aaRS inhibitor antimicrobial is unknown, in vitro translation assays can be used to identify the pathway affected, as was the case for the antimicrobial pentamidine [Citation13]. In another recently described assay, termed Selective Toeprinting in PURE System (SToPS) [Citation14], an artificially constructed gene containing codons specifying all 20 amino acids is translated in the PURE [Citation15] in vitro translation system. Primer extension inhibition (toeprinting) is then used to identify the site of translational arrest [Citation14]. This method is able to both detect aaRS inhibitor activity, and identify which aaRS enzyme is affected within a mixture [Citation14].
Developing inhibitors of aminoacylation has proven to be an effective strategy for development of antimicrobial drugs [Citation16]. While most aminoacylation inhibitors identified to date target the amino acid or adenylate binding pockets within aaRS enzymes, a growing literature on experimental inhibitors of aminoacylation includes drugs targeting both aaRS tRNA binding sites, and those which target the tRNA itself directly [Citation16,Citation17]. Notably, the aaRS-tRNA interaction represents the coevolution of both genes, which have had the opportunity to diverge across different domains of life. Thus, the atomic determinants mediating every point of intermolecular contact across each the aaRS-tRNA interaction contain valuable structure-activity information for the structure-guided design of novel aminoacylation inhibitors. Recent advancements in structure-guided drug design can greatly accelerate the process of drug development [Citation18], however atomic-level characterization of target sites is critical for these approaches [Citation19,Citation20].
Here we review available data regarding known aminoacylation inhibitors, as well as key determinants in both the aaRS and tRNA macromolecules that determine the specificity and strength of the aaRS-tRNA interaction. We focus on the molecular mechanisms of binding, turnover, and specificity in the aaRS-tRNA interaction with respect to drug design, utilizing the increasing body of available of genomic data, in vitro mutational analysis, and structural information [Citation21,Citation22].
Inhibition of aaRS as an antimicrobial strategy
Antimicrobial drugs must fulfill the fundamental requirement of exhibiting toxicity against the targeted organism while leaving human physiology unaffected. Although all living organisms perform protein translation, selective inhibition of microbial protein synthesis has been shown to be one of the most common and effective approaches in antibiotic development [Citation23,Citation24]. Due to their central role in protein biosynthesis, aaRS proteins are excellent targets, particularly since many microbial aaRSs have significantly diverged from their human homologs. Phylogenic sequence analyses across the three domains of life have revealed ancient divergence between bacterial and archaeal variants of nine aaRS genes (PheRS, TyrRS, LeuRS, IleRS, GluRS, TrpRS, HisRS, ProRS, and AspRS), followed by subsequent divergence of eukaryotic sequences from the archaeal lineage— a phenomenon termed a ‘full canonical pattern’ [Citation25]. Similarly, a systematic comparison of over 4,000 tRNA sequences across the three domains of life identified numerous kingdom-specific characteristics for certain tRNA isoacceptors [Citation26]. However, active site residues within their corresponding aaRS catalytic domains show a higher degree of structural conservation, likely reflecting the identical biochemical requirements for amino acid and adenylate recognition across different organisms [Citation25,Citation27,Citation28].
All known aaRSs are multi-domain proteins, and are subdivided into two distinct classes based on the structure of their catalytic domain [Citation25]. Within the active site of each aaRS catalytic domain, there exist three distinct binding pockets responsible for recognition of amino acid, adenylate, and tRNA moieties ()[Citation22, Citation27]. While some aaRS inhibitors target the separate editing domain, the majority bind one or more pockets within the catalytic domain [Citation16, Citation22]. Structurally, most aaRS inhibitor antibiotics act as non-cleavable mimics of AA-AMP, which is the natural intermediate produced during aminoacylation (see ) [Citation16, Citation22]. The best described example of such an inhibitor is mupirocin, which is one of only three approved aaRS inhibitor antibiotics prescribed for human or veterinary use (alongside halofuginone and tavaborole) [Citation29-Citation31]. Mupirocin (marketed as Bactroban) is a broad-spectrum topical antibiotic that mimics isoleucyl-AMP binding to inhibit IleRS across several species of bacteria. This antibiotic exhibits a strong preference for bacterial IleRS, and has been shown to inhibit Escherichia coli (E. coli) IleRS protein ∼8,000-fold more strongly than the corresponding protein variant isolated from rat liver [Citation32]. Mupirocin exhibits this high degree of specificity despite binding to the highly structurally conserved region of IleRS; structural analysis of a susceptible Thermus thermophilus (T. thermophilus) protein revealed its binding site to differ by only two amino acids relative to the resistant eukaryotic homolog [Citation33]. Apart from mupirocin, numerous experimental aaRS inhibitors have been developed that mimic amino acid and/or AMP binding, which have previously been the subject of extensive review [Citation16,Citation22] (For antimicrobials discussed here and their corresponding cellular targets and binding sites, see Table S3).
Figure 1. Druggable regions for inhibition of aminoacylation. Examples of drugs targeting each region of either the aaRS enzyme of tRNA substrate are shown. Acceptor stem and anticodon tRNA regions (highlighted in red) are frequently strong determinants for aaRS recognition. Notable residues comprising the anticodon (N34, N35, N36), CCA-tail (A76, C75, C74) and discriminator base (N73) are also shown. AaRS regions involved in tRNA recognition are also depicted, including the tRNA, ATP, and amino acid sites in the catalytic domain, as well as the anticodon recognition region (shown in red) of the anticodon binding domain.
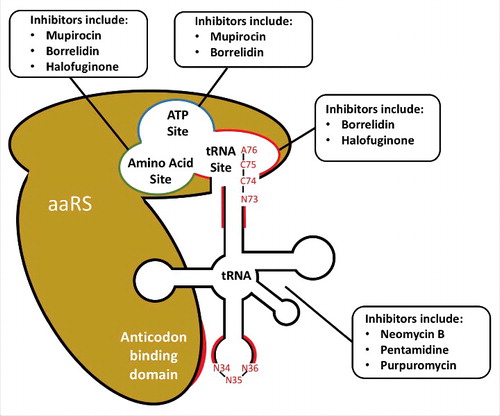
The aaRS tRNA recognition site is a target for antimicrobial inhibition
As mupirocin illustrates, a mere two amino-acid difference between prokaryotic and eukaryotic aaRS binding sites can alone confer inhibitor selectivity. However, divergent substrate binding determinants within amino acid and AMP binding sites are rare considering the universal biochemical requirements for recognition of these substrates. Conversely, tRNAs have coevolved alongside each aaRS; therefore, the binding criterion for tRNA substrates exhibits a higher degree of divergence between distantly related organisms. Thus, it is surprising that among the many characterized aaRS inhibitors relatively few occupy the tRNA binding pocket [Citation16,Citation22]. Nonetheless, the existing aaRS inhibitors that target the tRNA binding pocket demonstrate the efficacy of this mechanism as a therapeutic strategy.
Halofuginone, an antiprotozoal agent commonly used in livestock with multiple potential clinical indications in humans [Citation30, has been shown to inhibit ProRS by mimicking both proline and tRNAPro within the binding site [Citation34]. Similarly, phenyl-thiazolylurea-sulfonamides have been shown to exhibit broad-spectrum antibiotic activity by inhibiting PheRS, and a recent crystal structure revealed that member compound 1-(3-((4-pyridin-2-ylpiperazin-1-yl)sulfonyl)phenyl)-3-(1,3-thiazol-2-yl)urea occupies the tRNA binding pocket of Phe [Citation22,Citation35].
Co-crystal structures of borrelidin—an experimental macrolide antimicrobial and anticancer drug—with human and E. coli threonyl-tRNA synthetase (ThrRS) recently revealed this compound to utilize a ‘four-site’ inhibitory mechanism [Citation36]. Borrelidin interacts with adenylate, threonine, and tRNAThr binding sites in both ThrRS protein variants, thereby inhibiting interaction for all three enzyme substrates [Citation36]. Unusually, borrelidin was found to occupy a fourth pocket within the aaRS active site that is uninvolved in substrate binding but essential for the inhibitory action of this antibiotic [Citation36].
The mechanisms of halofuginone, phenyl-thiazolylurea-sulfonamides, and borrelidin indicate that targeting the tRNA binding pocket may represent an efficacious strategy for antimicrobial drugs. As each of these compounds were discovered without knowledge of their mechanism of action [Citation22], development of novel compounds targeting this binding site would require detailed information on the divergence of human and microbial tRNA binding. Recognition of each tRNA by its cognate aaRS is determined by a relatively small number of nucleotides termed “identity elements”, while misacylation by non-cognate synthetases is prevented by anti-determinant elements [Citation37,Citation38]. Although the coevolution of mutual recognition between cognate aaRS and tRNA molecules is essential for protein synthesis, the molecular shapes involved in each interaction are fundamentally arbitrary [Citation39].
tRNA identity elements determine aaRS specificity
Identity elements for all tRNAs were first biochemically elucidated for E. coli, and available data remains the most comprehensive for this organism [Citation5,Citation37,Citation40]. Individual identity elements vary in their importance; following mutation, nucleotides resulting in a large decrease in aminoacylation efficiency (k cat/K m) are considered ‘strong’, while those leading to a smaller decrease are considered ‘weak’ [Citation37]. Across all tRNAs, strong identity elements are typically concentrated in the distal ends of the tRNA, corresponding to the acceptor stem and anticodon stem-loop regions [Citation37,Citation41,Citation42]. Within the acceptor stem position N73 is commonly found as a strong identity element, termed the ‘discriminator base’ [Citation37,Citation43]. Located adjacent to the ubiquitously conserved CCA tail, the discriminator base was the first identity element identified [Citation43], and has been found to play a role in CCA addition [Citation44]. The anticodon is itself frequently an identity determinant, with recognition governed by a separate anticodon-binding aaRS domain; however, there are many notable instances wherein recognition is anticodon-independent [Citation37,Citation45].
While the tRNA regions containing identity elements are strongly conserved, numerous differences in identity element nucleotides have been observed across different domains of life [Citation27]. These differences have been shown to prevent cross-complementation of incompatible aaRS-tRNA pairs, a characteristic used to explain the evolutionary patterns observed for many aaRS genes [Citation27,Citation46]. Given the critical importance of aminoacylation to protein synthesis, identifying tRNA identity elements that diverge between humans and pathogens can highlight pathogen-specific interactions that can be targeted for antimicrobial drug design.
Across different organisms containing tRNA of prokaryotic origin, tRNA regions confirmed as identity elements in E. coli can be readily compared. Such prokaryotic tRNAs encompass not only bacterial tRNAs but also other endosymbiotic organelles of prokaryotic origin, including mitochondrial and apicoplastic tRNAs [Citation47]. These organelles encode many of their own tRNA•aaRS pairs that exhibit more bacteria-like characteristics [Citation48], and further show low levels of redundancy for tRNA genes enabling ready identification of consensus sequences. Since many eukaryotic tRNA and aaRS genes are highly divergent from their bacterial counterparts, developing antimicrobials that inhibit aaRS interactions with bacterial or apicoplastic tRNA identity elements is a feasible strategy. Notably, such antimicrobials must not inhibit mitochondrial tRNA•aaRS interactions in order to limit off-target effects in the eukaryotic host.
Eukaryotic genomes show substantially higher levels of tRNA sequence redundancy compared to prokaryotes, complicating bioinformatic analysis. Notably, the human genome has been found to be unexpectedly enriched for tRNA isodecoder tRNAs, which contain the same anticodon sequence but differing tRNA bodies with respect to one another [Citation49]. In a study using tRNASer, different isodecoders were found to vary in their suppression efficiencies by as much as 20-fold, corresponding to differing levels of aminoacylation [Citation49]. This variability has complicated identification of critical tRNA identity elements for each isoacceptor in human cytosolic tRNA. Nonetheless, bioinformatic analysis has been used to identify consensus sequences for a number of isoacceptors [Citation47, while biochemical analysis was applied to determine identity elements for another subset of tRNA variants [Citation37,Citation50–Citation54].
As a result, identifying divergence in identity elements regions across tRNAs of eukaryotic origin presents a greater challenge. Nevertheless, multiple aaRS inhibitors of natural origin have been shown to be efficacious against eukaryotic pathogens (recently reviewed at length [Citation55), including halofuginone, which has been approved for veterinary use against coccidia [Citation55]. A recent bioinformatics survey examined aaRS genes in Leishmania major (L. major), the pathogen responsible for leishmaniasis. In this study, 11 new or extended aaRS domain structures were identified, as well as a putative novel AspRS paralog [Citation56]. Following additional structural studies, these atypical aaRS proteins could likely serve as drug design targets. Indeed, existing crystal structures of L. major TyrRS and MetRS complexes have revealed multiple structural features within the catalytic domain that diverge from human homologs [Citation57,Citation58]. This observation suggests that the parsing of metagenomic aaRS sequences could be further applied to identify potential target aaRS genes within other pathogens in the order Trypanosomatida, which includes Trypanosoma brucei (T. brucei, responsible for trypanosomiasis) and Trypanasoma cruzi (T. cruzi, responsible for Chagas disease), as these organisms are likely to share many features with L. major.
Post-transcriptional modifications in tRNA contribute to tRNA identity in a domain-specific manner [Citation59]. To date, over 100 different types of post-transcriptional modifications of RNAs have been identified across all three domains of life. Among the three major species of RNA (mRNA, rRNA, and tRNA), post-transcriptional modifications are most commonly found in tRNA [Citation60-Citation63]. Modified nucleosides in several E. coli, yeast, and archaeal tRNA variants have been biochemically shown to constitute significant identity element features, with replacement by unmodified nucleosides leading to significant loss of aminoacylation activity [Citation64]. Modifications affecting identity are most commonly found at position 34 (within the anticodon) and position 37 (adjacent to the anticodon) [Citation65]. Modifications at these positions are thought to have evolved to further stabilize AU-rich codon-anticodon pairs, as well as to prevent mistranslation and frameshifting [Citation65]. At these positions, modifications have been found to diverge across the three domains of life (see ref [Citation65], Table 5), and are thought to represent evolution of differing mRNA decoding strategies [Citation65]. Modified bases in E. coli tRNAIle and yeast tRNAAsp have also been shown to act as identity anti-determinants, preventing misacylation by E. coli MetRS and yeast ArgRS, respectively [Citation66,Citation67]. Notably, modified nucleosides cannot be identified through DNA sequencing data alone, and it is thought that many cryptic nucleotide modifications remain unidentified [Citation68]. While confirmation of a modified nucleoside requires detailed structural characterization, modifications can be predicted through phylogenetic analysis of known modifications [Citation63]. Recently, the tRNAmodviz web server (http://genesilico.pl/trnamodviz) has been made available as an online tool to facilitate predictive analysis of tRNA modifications [Citation63].
Identification of aaRS-tRNA interactions for antimicrobial development
To ascertain whether a given aaRS-tRNA recognition interaction may constitute a suitable drug target, it is necessary to compare critical atoms involved in both human and pathogenic organisms. While critical residues can be identified within a given aaRS, this analysis requires detailed structural and/ or biochemical characterization of a given enzyme. Fortunately, identification of such critical residues is more facile in tRNA. As detailed above, the high degree of conservation of tRNA identity element positions and the trove of experimentally confirmed identity elements in E. coli together enable identification of divergent prokaryotic tRNA recognition interactions within metagenomic sequencing data.
In selecting a specific interaction to target for drug design, off-target activity is best avoided by initially focusing on prokaryotic tRNA identity elements that are divergent from their mitochondrial counterparts. As proof of concept, we have chosen E. coli as our target bacteria species in consideration of the extensive available data regarding this organism. We have compared identity elements previously determined biochemically in E. coli to their corresponding residue in Homo sapiens (H. sapiens) mitochondria, shown in Table S1. This analysis identified divergent identity elements in seventeen tRNA sequences.
Before selecting a target for drug development, it is also essential to further validate each potential position by assessing possible interactions with cytosolic human aaRSs. However, there is substantially less available data regarding identity elements for human cytosolic tRNAs (see above discussion on eukaryotic consensus tRNA sequences). Thus, identity elements that diverge between E. coli and human mitochondrial tRNAs were compared against available biochemical data for six H. sapiens cytosolic tRNAs ().
Table 1. Identity elements in E. coli tRNA vs human mitochondrial tRNA vs. human cytosolic tRNA.
This analysis identified three E. coli tRNA genes containing identity elements divergent from both human cytosolic and mitochondrial genes: tRNASer, containing divergent residues G2:C71; tRNAGly, containing divergent residue U73; and tRNAPhe, containing divergent residues G27:C43, G28:C42, G44, and U45. Thus, drugs disrupting aaRS recognition of these regions would be expected to inhibit E. coli growth while remaining nontoxic to human cells.
Each of the nucleotides detailed above were evaluated for potential tRNA modifications using the tRNAmodviz database [Citation63]. Although modification data was unavailable for mitochondrial tRNAGly and mitochondrial tRNAPhe, available E. coli and mitochondrial data were not predicted to show any modifications at these positions. While human cytosolic tRNAGly and tRNASer were also not predicted to have modifications at the sites listed above, cytosolic tRNAPhe was predicted to contain pseudouridine at position 27 and position 28, with no modifications predicted at positions 42–45. Despite potential nucleotide modification, these identity element residues remain useful guides for drug development as they are divergent from the homologous positions in E. coli tRNA. However, to identify drugs inhibiting recognition at these positions, exact knowledge of the nucleotides governing tRNA recognition interactions is essential to guide virtual screening of drug candidates.
Although E. coli provides an attractive test case for tRNA target identification given the extensive biochemical data known for this organism, there exist numerous pathogenic microbes of equal or greater clinical importance. As regions containing identity elements tend to be conserved [Citation41], bioinformatic analysis can predict identity elements in other prokaryotic species by identifying tRNA residues homologous to confirmed identity elements in E. coli. We extended meta-analysis of identity element conservation between bacteria and human mitochondria to two clinically relevant pathogens: Gram-positive bacteria Staphylococcus aureus (S. aureus) and Gram-negative bacteria Pseudomonas aeruginosa (P. aeruginosa) (Table S2). These two species were selected on the basis of their differing Gram-types and their classification as ‘ESKAPE’ pathogens, which encompass a broad-spectrum group of bacteria against which new therapies are urgently needed [Citation69,Citation70].
Susceptibility profiles across bacterial species can vary widely for different antibiotics. While drugs with a broader spectrum of activity are essential for treatment when the infective agent is unknown, narrow-spectrum antibiotics are typically preferred to reduce side effects of therapy and stall the emergence of antibiotic resistance [Citation71]. As shown in Table S2, many tRNA identity elements are not only divergent between mitochondria and bacteria, but also between different bacterial species. Consequently, the spectrum breadth of aaRS inhibitor antibiotics that function by inhibiting specific recognition interactions can be readily predicted.
Using data shown in Table S2, we have predicted activity spectra of putative antibiotics that target the specific identity elements identified in . A compound disrupting recognition of either E. coli tRNASer residues G2:C71, E. coli tRNAGly residue U73, or E. coli tRNAPhe residues G44 or U45 would also be expected to be efficacious against both P. aeruginosa and S. aureus, acting as a broad-spectrum antibiotic against both Gram-negative and Gram-positive bacteria. In contrast, a compound disrupting E. coli tRNAPhe residues G27:C43 or G28:C42 would be expected to be efficacious against P. aeruginosa, but not against S. aureus, thereby acting as a more narrow-spectrum antibiotic efficacious against Gram-negative bacteria.
This approach can readily be extended to predict the spectrum of drugs that recognize identity elements in other microbial species where sequencing data is available. However, in the absence of biochemical data, an important caveat to this cross-species prediction lies in the variable strengths of predicted identity elements which have been previously observed to vary across taxons [Citation37,Citation72,Citation73].
Direct targeting of tRNAs by small-molecule inhibitors
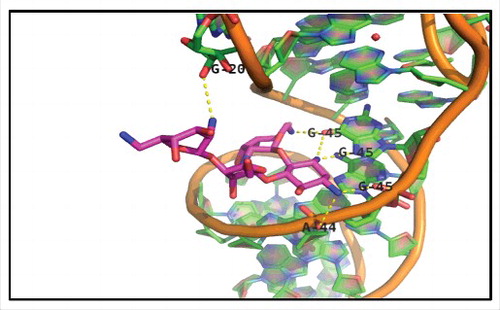
To disrupt a key aaRS-tRNA interaction, it is viable to target either the aaRS or tRNA macromolecule (see ). Generally, RNA molecules pose a more challenging drug target compared to proteins. Nonetheless, direct drug binding to the tRNA has also been shown to be a viable strategy for inhibiting aminoacylation. Targeting the anticodon stem loop region of tRNA may prove a promising approach, as tRNA identity elements are frequently concentrated in this region. While aminoglycoside antibiotics are best known for their role in inhibiting translation through direct interaction with the 30S ribosomal subunit, several have been shown to also bind tRNA [Citation74,Citation75]. Notably, aminoglycoside antibiotic Neomycin B was shown to inhibit the in vitro aminoacylation of E. coli tRNAPhe 76. Co-crystallization of yeast tRNAPhe with Neomycin B showed that the inhibitor directly interacts with residues G20, A44 and G45 in the anticodon stem loop region, occupying a known metal ion binding site (). Drug binding thus inhibits aminoacylation by blocking aaRS interaction with these major identity element determinants [Citation37,Citation76,Citation77]. Notably, Trana Discovery Inc. has developed a strategy for identifying novel drug candidates binding the anticodon stem loop region. Their approach utilizes fluorescent probes mimicking the anticodon stem loop to assay for drug candidate binding and has been applied to develop a high-throughput screen for antibiotics against S. aureus [Citation78].
Figure 2. X-ray crystal structure of Neomycin B bound to tRNAPhe. Shown at 2.6A° resolution (PDB file 1I9V), Neomycin B occupies a binding cleft on tRNAPhe one full helical turn above the anticodon stem-loop. Interactions within 3.5 Å include nucleotides 20, 44, and 45, which are represented by dashed yellow lines.
Other compounds have been shown to bind the T or D-loop regions of tRNA. In one example, the aminoglycoside tobramycin was shown to competitively inhibit in vitro aminoacylation of yeast tRNAAsp, yet did not affect formation of aspartate adenylate as measured by ATP–PPi exchange [Citation79]. Fluorescence anisotropy experiments using fluorescein-labeled tobramycin revealed a direct binding interaction with tRNAAsp (KD = 267 nM) [Citation79], and selective 2′-hydroxyl acylation analyzed by primer extension (SHAPE) analysis subsequently revealed this compound disrupted interactions between the T- and D-loop regions [Citation80].
Multiple RNase P inhibitors have also been shown to bind the T- and D-loops of tRNA. While these compounds do not affect aminoacylation, they demonstrate the viability of targeting identity elements within these regions. Bis-benzimidazole compounds have been shown by UV-visible and fluorescence spectrophotometry to directly bind different tRNA species, with hydroxyl radical footprinting showing binding in the D- and T- loops [Citation81]. Porphyrin and porphine compounds have also been shown by fluorescence spectroscopy to bind tRNA and inhibit RNase P via putative displacement of a metal ion in the D-loop/T-loop region [Citation82].
Another strategy is to bind the acceptor stem of tRNA to prevent tRNA exit from the aaRS active site. AN2690 (also known as Tavaborole), a boron-containing antimicrobial, was shown in a crystal structure to bind yeast tRNALeu at the 3′ terminal adenosine, causing the tRNA to become trapped inside the aaRS editing site [Citation83]. Similarly, plant tumor control agent Agrocin 84 has been shown to be processed inside the cell to produce TM84, a Leu-AMP analog that crystal structures show to also bind to the 3′ terminus of tRNALeu 84. This binding interaction traps the aaRS-tRNA complex in an ‘aminoacylation-like’ conformation, preventing further enzymatic turnover [Citation84].
Antimicrobial agents pentamidine and purpuromycin have been shown to inhibit aminoacylation through nonspecific tRNA binding mechanisms. These compounds have both been shown to inhibit in vitro aminoacylation of different synthetases while leaving formation of adenylate unaffected, as measured by ATP–PPi exchange [Citation13,Citation85]. Unusually, both compounds were also shown to exhibit a nonspecific tRNA binding mechanism. For pentamidine, free hydroxyl radical footprinting found that pentamidine protected many regions of tRNALeu, encompassing nearly the entire RNA phosphate backbone. Isothermal titration calorimetry (ITC) further revealed a large number of pentamidine binding sites, with binding driven largely through hydrophobic interactions [Citation13]. Pentamidine has also been shown to inhibit self-splicing of the nuclear group I introns in the rRNA of multiple fungal species [Citation86–Citation88], consistent with a mechanism of nonspecific targeting of structured rRNA species in addition to tRNA [Citation13]. Purpuromycin was shown by aminoacylation assay to inhibit the aminoacylation of several bacterial tRNA variants, and was further found to bind the acceptor stem region [Citation85]. Authors further noted that the addition of a mixture of total tRNA to the in vitro mixture restored aminoacylation activity, concluding that purpuromycin directly interacts with many tRNA species in a nonspecific manner [Citation85].
Taken together, these findings demonstrate multiple strategies for directly targeting regions of interest in tRNA. While nonspecific mechanisms such as those utilized by pentamidine and purpuromycin may be difficult to recapitulate through rational design, identity elements in the anticodon stem loop and D-loop/T-loop regions provide more feasible drug targets. Targeting metal ion binding pockets also appears to be a viable strategy to both occlude recognition of nearby identity element residues and further destabilize tRNA through displacement of metal ions. Metal ions can readily be identified within available structural data [Citation89], with the importance of metal ions to RNA folding and function having previously been the topic of review [Citation90]. However, efforts to target RNA for drug binding need to take into account the polyanionic charge of the backbone as well as the abundance of hydrogen bond donors and acceptors [Citation91]. Despite recent advances, structure-guided drug development tools for targeting RNA remain less developed than those for proteins [Citation92].
Inhibiting aaRS enzymes through tRNA mimics
Development of a compound designed to mimic tRNA recognition would be expected to competitively inhibit the cognate aaRS. Early experiments found that synthetic oligonucleotides mimicking recognition elements of tRNA competitively inhibit the formation of aminoacyl-tRNA, while leaving the formation of aminoacyl-adenylate unaffected [Citation93,Citation94]. While these oligonucleotides themselves are not viable as drugs due to their inability to traverse cellular membranes, they do serve to validate the strategy of tRNA mimics as aaRS inhibitors. The existence of aaRS inhibitors targeting the tRNA binding site (described above) further demonstrate the capacity of small molecules to mimic tRNA binding.
As mentioned above, previous reviews of the aaRS literature have long established three distinct activities within the catalytic domain mediating aminoacylation activity: (1) the amino-acid binding site, (2) the ATP binding site and (3) the tRNA binding site [Citation28,Citation95]. To elucidate structure-activity insights within the scope of this review, we focus on key aaRS motifs that interact with tRNA identity elements. Within the catalytic domain of all aaRS enzymes, the tRNA binding site interacts with the acceptor stem of the tRNA, comprising the canonical tRNA 3′-CCA end as well as N72 and the N73 ‘discriminator base’ important for tRNA specificity recognition. This site buries the 3′ terminal nucleotide A76 and makes contacts with other acceptor stem nucleotides between N72–76, with these nucleobases often found in flipped conformation in known x-ray crystal structures of the aaRS-tRNA complexes. The tRNA acceptor stem binding site is directly adjacent to the ATP site, suggesting that this orientation is critical for proper aminoacyl adenylate formation. In the absence of ATP, A76 has been observed to occupy the ATP site [Citation96]. This structural homology between the A76 base and ATP may explain the existence of multiple dual-site inhibitors of aaRS (eg., borrelidin, halofuginone) and the absence of any known inhibitors that only bind the tRNA acceptor stem site [Citation22].
All aaRS enzymes excluding SerRS, LeuRS, and AlaRS also contain a separate anticodon binding domain, recognizing anticodon nucleotides distal from the aforementioned catalytic site [Citation97]. These anticodon residues (found at tRNA positions 34–36) mediate the interaction between tRNA and the genetic code, and consequently vary only under extremely rare circumstances [Citation98,Citation99]. Consequently, interactions of anticodon residues do not provide viable drug targets considering their near universal conservation between pathogenic species and humans. However, nucleotides adjacent to the anticodon (e.g., positions 31–39) have been observed to act as tRNA identity elements (see Table S1). These residues comprise what has been termed the proximal ‘extended anticodon’, with each nucleotide in this region having evolved to mediate uniform mRNA decoding by modulating the stability of codon-anticodon pairs and the strength of the interaction between tRNA and the ribosomal A-site [Citation65]. Divergent interactions at these positions may present viable drug targets, however care would need to be taken to avoid inhibition of anticodon recognition by human aaRS proteins.
Many aaRSs also contain an editing site, responsible for hydrolyzing amino acids which have either been incorrectly adenylated (termed pre-transfer editing) or aminoacylated (termed post-transfer editing. Although editing sites predominantly recognize amino acid moieties, post-transfer editing often involves binding to 3′ terminal regions of tRNA [Citation29,Citation100]. As compounds AN2690 and TM84 (described above) demonstrate, it is possible to inhibit of aaRS activity through interactions with the aaRS editing site [Citation83,Citation84]. However, targeting interactions between tRNA and the aaRS editing site would also necessitate consideration of the catalytic site during drug development, as both the editing and catalytic aaRS sites bind the tRNA acceptor stem.
Analysis of available crystal structures of aaRS-tRNA complexes make it clear that many tRNA identity elements mediate interactions outside of previously described binding pockets. In these cases, the aaRS-tRNA interface bears more similarity to a protein-protein interaction rather than a ligand interface. While approaches for drugging such comparatively featureless interactions continue to improve, such regions within aaRS structures nonetheless present more challenging targets for small molecule drug binding [Citation101,Citation102].
Towards identification of selective aaRS inhibitors targeting the tRNA binding site, eukaryotic GlyRS genes have been identified as particularly phylogenetically divergent in comparison to their prokaryotic homologs [Citation103]. Prokaryotic GlyRS protein variants are known to form form α2β2 heterotetramers, belonging to aaRS subclass IIC; eukaryotic GlyRS protein variants conversely form α2 homodimers, belonging to aaRS subclass IIA [Citation104,Citation105]. Functionally, this is reflected by the inability of eukaryotic and prokaryotic GlyRS enzymes to cross-aminoacylate tRNAGly species originating from the other domain [Citation104]. Intriguingly, this selectivity is almost entirely explained by a single difference at the N73 discriminator base, with eukaryotic and prokaryotic tRNAGly containing A73 and U73, respectively (). While recent co-crystal structures were unable to provide ‘smoking gun’ evidence of the exact structural basis of tRNA acceptor stem recognition by GlyRS, they have provided strong evidence that the acceptor site of GlyRS is an appropriate target for the rational design of antimicrobials [Citation106].
Prokaryotic PheRS represents another high-priority target aaRS for rational design of tRNA binding site inhibitors. Analogous to GlyRS, PheRS proteins of different domain origin also exhibit different quaternary structures, and based on sequence analysis it has been proposed that prokaryotic and human enzymes exhibit different tRNA binding modes [Citation107,Citation108]. As mentioned above, phenyl-thiazolylurea-sulfonamides are a novel antibiotics class highly specific for bacterial PheRS, exhibiting nanomolar inhibitory IC50 values in aminoacylation assays using both Gram-negative and Gram-positive bacterial PheRS protein variants [Citation109]. X-ray crystal structures of 1-(3-((4-pyridin-2-ylpiperazin-1-yl)sulfonyl)phenyl)-3-(1,3-thiazol-2-yl)urea bound to P. aeruginosa PheRS revealed that selectivity is determined by a single amino acid Met99, which is present in prokaryotic PheRS but mutated to leucine in both cytoplasmic and mitochondrial versions of mammalian PheRS [Citation109].
Structural dynamics dictate functionality in the aaRS-tRNA complex
A growing body of evidence supports the idea that aaRSs are a highly dynamic enzyme class that undergo induced fit upon binding tRNA and during aminoacylation, with major conformational changes occurring for both macromolecules. Structural dynamics analyses during aaRS activity have been performed through co-crystallization at various steps of tRNA binding, aminoacylation and turnover. For example, a series of high-resolution structures of E. coli leucyl-tRNA synthetase ternary complexes probed both the aminoacylation and editing conformations of the complex [Citation110]. Furthermore, a co-crystallized inactive complex of E. coli AspRS and yeast tRNAAsp supports a distinct tRNA-initiated control mechanism that initiates loop movement in the AspRS catalytic domain, opening the acceptor stem site of AspRS [Citation96]. A serendipitous high-resolution structure of human GlyRS demonstrated two distinct conformational states of GlyRS recognition during tRNA complexation within a single crystal lattice, supporting conformational change in GlyRS loop regions to allow tRNA acceptor stem entry for the first step of aminoacylation [Citation111]. A high degree of flexibility in this region during both tRNA binding and aminoacylation was further supported by limited molecular dynamics studies [Citation111].
These critical structural studies have provided excellent snapshots of aaRS dynamics steps, and should further enable high-performance molecular dynamics studies. However, it remains a priority in the field to solve additional human cytosolic aaRS-tRNA complexes, as relatively few have been co-crystallized successfully with their cognate tRNAs [Citation111–Citation113]. Modern advancements in graphics processing computing over the past five years have further enabled microsecond-scale simulations of even large multi-chain systems including aaRS-tRNA complexes. A complete set of high-resolution mammalian aaRS-tRNA complex structures will be an invaluable tool towards the preparation of accurate dynamical models of aminoacylation for the rational design of species-specific therapeutic inhibitors.
Conclusion
Following its discovery, the genetic code has famously been described as a “frozen accident”, as it has remained largely unchanged throughout nature [Citation114]. By contrast, the so-called “second genetic code” has proven far more malleable, with the network of interactions between aaRSs and tRNAs exhibiting substantial variation between distant phylogenies. Targeting such interactions in pathogens where they diverge from those in humans is likely to become an increasingly viable antimicrobial development strategy as structure-guided drug design tools continue to improve. Considering the efficacy of current antimicrobial drugs that target aaRS activity, as well as the abundant and growing body of data surrounding these enzymes, it is clear that this class of protein will remain valuable for future drug development.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed
Financial disclosures statement
Authors JMH, EB, DS, and CM are stakeholders in DS Therapeutics, a for-profit company with commercial interest in developing pharmaceuticals.
supp_data_1429879.docx
Download MS Word (40.7 KB)Additional information
Funding
References
- Ibba M, Soll D. Quality control mechanisms during translation. Science. 1999;286:1893–7. doi:10.1126/science.286.5446.1893
- Ibba M, Curnow AW, Soll D. Aminoacyl-tRNA synthesis: divergent routes to a common goal. Trends Biochem Sci. 1997;22:39–42. doi:10.1016/S0968-0004(96)20033-7
- Smith YA. tRNA on the ribosome: A waggle theory. tRNA: Structure, biosynthesis and function. Washington: DC: ASM Press; 1995.pp. 443–69
- Lincecum TL, Jr., Tukalo M, Yaremchuk A, et al. Structural and mechanistic basis of pre- and posttransfer editing by leucyl-tRNA synthetase. Mol Cell. 2003;11:951–63. doi:10.1016/S1097-2765(03)00098-4
- Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. 2000;69:617–50. doi:10.1146/annurev.biochem.69.1.617
- de Duve C Transfer RNAs: The second genetic code. Nature. 1988;333:117–8. doi:10.1038/333117a0
- Fersht AR, Ashford JS, Bruton CJ, et al. Active site titration and aminoacyl adenylate binding stoichiometry of aminoacyl-tRNA synthetases. Biochemistry. 1975;14:1–4. doi:10.1021/bi00672a001
- Francklyn CS, First EA, Perona JJ, et al. Methods for kinetic and thermodynamic analysis of aminoacyl-tRNA synthetases. Methods. 2008;44:100–18. doi:10.1016/j.ymeth.2007.09.007
- Cestari I, Stuart K. A spectrophotometric assay for quantitative measurement of aminoacyl-tRNA synthetase activity. J Biomol Screen. 2013;18:490–7. doi:10.1177/1087057112465980
- Loftfield RB. The mechanism of aminoacylation of transfer RNA. Prog Nucleic Acid Res Mol Biol. 1972;12:87–128. doi:10.1016/S0079-6603(08)60660-1
- Eigner EA, Loftfield RB. Kinetic techniques for the investigation of amino acid: tRNA ligases (aminoacyl-tRNA synthetases, amino acid activating enzymes). Methods Enzymol. 1974;29:601–19. doi:10.1016/0076-6879(74)29053-0
- Wolfson AD, Pleiss JA, Uhlenbeck OC. A new assay for tRNA aminoacylation kinetics. RNA. 1998;4:1019–23. doi:10.1017/S1355838298980700
- Sun T, Zhang Y. Pentamidine binds to tRNA through non-specific hydrophobic interactions and inhibits aminoacylation and translation. Nucleic Acids Res. 2008;36:1654–64. doi:10.1093/nar/gkm1180
- Orelle C, Szal T, Klepacki D, et al. Identifying the targets of aminoacyl-tRNA synthetase inhibitors by primer extension inhibition. Nucleic Acids Res. 2013;41:e144. doi:10.1093/nar/gkt526
- Tuckey C, Asahara H, Zhou Y, et al. Protein synthesis using a reconstituted cell-free system. Curr Protoc Mol Biol. 2014;108: 16 31 1–22
- Vondenhoff GH, Van Aerschot A. Aminoacyl-tRNA synthetase inhibitors as potential antibiotics. Eur J Med Chem. 2011;46:5227–36. doi:10.1016/j.ejmech.2011.08.049
- Chopra S, Reader J. tRNAs as antibiotic targets. Int J Mol Sci. 2014;16:321–49. doi:10.3390/ijms16010321
- Lionta E, Spyrou G, Vassilatis DK, et al. Structure-based virtual screening for drug discovery: principles, applications and recent advances. Curr Top Med Chem. 2014;14:1923–38. doi:10.2174/1568026614666140929124445
- Coates AR, Halls G, Hu Y. Novel classes of antibiotics or more of the same? Br J Pharmacol. 2011;163:184–94. doi:10.1111/j.1476-5381.2011.01250.x
- Becker D, Selbach M, Rollenhagen C, et al. Robust Salmonella metabolism limits possibilities for new antimicrobials. Nature. 2006;440:303–7. doi:10.1038/nature04616
- Loman NJ, Pallen MJ. Twenty years of bacterial genome sequencing. Nature reviews Microbiology. 2015;13:787–94. doi:10.1038/nrmicro3565
- Fang P, Guo M. Evolutionary limitation and opportunities for developing tRNA synthetase inhibitors with 5-binding-mode classification. Life (Basel). 2015;5:1703–25
- Wilson DN. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol. 2014;12:35–48. doi:10.1038/nrmicro3155
- Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nat Rev Microbiol. 2010;8:423–35. doi:10.1038/nrmicro2333
- O'Donoghue P, Luthey-Schulten Z. On the evolution of structure in aminoacyl-tRNA synthetases. Microbiol Mol Biol Rev. 2003;67:550–73. doi:10.1128/MMBR.67.4.550-573.2003
- Marck C, Grosjean H. tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea, and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA. 2002;8:1189–232. doi:10.1017/S1355838202022021
- Woese CR, Olsen GJ, Ibba M, et al. Aminoacyl-tRNA synthetases, the genetic code, and the evolutionary process. Microbiol Mol Biol Rev. 2000;64:202–36. doi:10.1128/MMBR.64.1.202-236.2000
- Schimmel P, Tao J, Hill J. Aminoacyl tRNA synthetases as targets for new anti-infectives. Faseb J. 1998;12:1599–609
- Silvian LF, Wang J, Steitz TA. Insights into editing from an ile-tRNA synthetase structure with tRNAile and mupirocin. Science. 1999;285:1074–7. doi:10.1126/science.285.5430.1074
- Pines M, Spector I. Halofuginone – the multifaceted molecule. Molecules. 2015;20:573–94. doi:10.3390/molecules20010573
- Tavaborole topical solution (Kerydin) for onychomycosis. Med Lett Drugs Ther. 2015;57:35–6
- Hughes J, Mellows G. Interaction of pseudomonic acid A with Escherichia coli B isoleucyl-tRNA synthetase. Biochem J. 1980;191:209–19. doi:10.1042/bj1910209
- Nakama T, Nureki O, Yokoyama S. Structural basis for the recognition of isoleucyl-adenylate and an antibiotic, mupirocin, by isoleucyl-tRNA synthetase. J Biol Chem. 2001;276:47387–93. doi:10.1074/jbc.M109089200
- Zhou H, Sun L, Yang XL, et al. ATP-directed capture of bioactive herbal-based medicine on human tRNA synthetase. Nature. 2013;494:121–4. doi:10.1038/nature11774
- Abibi A, Ferguson AD, Fleming PR, et al. The role of a novel auxiliary pocket in bacterial phenylalanyl-tRNA synthetase druggability. J Biol Chem. 2014;289:21651–62. doi:10.1074/jbc.M114.574061
- Fang P, Yu X, Jeong SJ, et al. Structural basis for full-spectrum inhibition of translational functions on a tRNA synthetase. Nat Commun. 2015;6:6402. doi:10.1038/ncomms7402
- Giege R, Sissler M, Florentz C. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 1998;26:5017–35. doi:10.1093/nar/26.22.5017
- Freyhult E, Moulton V, Ardell DH. Visualizing bacterial tRNA identity determinants and antideterminants using function logos and inverse function logos. Nucleic Acids Res. 2006;34:905–16. doi:10.1093/nar/gkj478
- Ardell DH. Computational analysis of tRNA identity. FEBS letters. 2010;584:325–33. doi:10.1016/j.febslet.2009.11.084
- Saks ME, Sampson JR, Abelson JN. The transfer RNA identity problem: A search for rules. Science. 1994;263:191–7. doi:10.1126/science.7506844
- Giege R. Toward a more complete view of tRNA biology. Nat Struct Mol Biol. 2008;15:1007–14. doi:10.1038/nsmb.1498
- Carter CW, Jr., Wolfenden R. tRNA acceptor stem and anticodon bases form independent codes related to protein folding. Proc Natl Acad Sci U S A. 2015;112:7489–94. doi:10.1073/pnas.1507569112
- Crothers DM, Seno T, Soll G. Is there a discriminator site in transfer RNA? Proc Natl Acad Sci U S A. 1972;69:3063–7. doi:10.1073/pnas.69.10.3063
- Wende S, Bonin S, Gotze O, et al. The identity of the discriminator base has an impact on CCA addition. Nucleic Acids Res. 2015;43:5617–29. doi:10.1093/nar/gkv471
- Ambrogelly A, Gundllapalli S, Herring S, et al. Pyrrolysine is not hardwired for cotranslational insertion at UAG codons. Proc Natl Acad Sci U S A. 2007;104:3141–6. doi:10.1073/pnas.0611634104
- Shiba K, Motegi H, Schimmel P. Maintaining genetic code through adaptations of tRNA synthetases to taxonomic domains. Trends Biochem Sci. 1997;22:453–7. doi:10.1016/S0968-0004(97)01135-3
- Putz J, Giege R, Florentz C. Diversity and similarity in the tRNA world: overall view and case study on malaria-related tRNAs. FEBS letters. 2010;584:350–8. doi:10.1016/j.febslet.2009.11.050
- Salinas-Giege T, Giege R, Giege P. tRNA biology in mitochondria. Int J Mol Sci. 2015;16:4518–59. doi:10.3390/ijms16034518
- Geslain R, Pan T. Functional analysis of human tRNA isodecoders. J Mol Biol. 2010;396:821–31. doi:10.1016/j.jmb.2009.12.018
- Breitschopf K, Gross HJ. The exchange of the discriminator base A73 for G is alone sufficient to convert human tRNA(Leu) into a serine-acceptor in vitro. EMBO J. 1994;13:3166–9
- Achsel T, Gross HJ. Identity determinants of human tRNA(Ser): sequence elements necessary for serylation and maturation of a tRNA with a long extra arm. EMBO J. 1993;12:3333–8
- Stehlin C, Burke B, Yang F, et al. Species-specific differences in the operational RNA code for aminoacylation of tRNAPro. Biochemistry. 1998;37:8605–13. doi:10.1021/bi980364s
- Hipps D, Shiba K, Henderson B, et al. Operational RNA code for amino acids: species-specific aminoacylation of minihelices switched by a single nucleotide. Proc Natl Acad Sci U S A. 1995;92:5550–2. doi:10.1073/pnas.92.12.5550
- Nazarenko IA, Peterson ET, Zakharova OD, et al. Recognition nucleotides for human phenylalanyl-tRNA synthetase. Nucleic Acids Res. 1992;20:475–8. doi:10.1093/nar/20.3.475
- Pham JS, Dawson KL, Jackson KE, et al. Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. Int J Parasitol Drugs Drug Resist. 2014;4:1–13. doi:10.1016/j.ijpddr.2013.10.001
- Foth BJ, McFadden GI. The apicoplast: a plastid in Plasmodium falciparum and other Apicomplexan parasites. Int Rev Cytol. 2003;224:57–110. doi:10.1016/S0074-7696(05)24003-2
- Larson ET, Kim JE, Zucker FH, et al. Structure of Leishmania major methionyl-tRNA synthetase in complex with intermediate products methionyladenylate and pyrophosphate. Biochimie. 2011;93:570–82. doi:10.1016/j.biochi.2010.11.015
- Larson ET, Kim JE, Castaneda LJ, et al. The double-length tyrosyl-tRNA synthetase from the eukaryote Leishmania major forms an intrinsically asymmetric pseudo-dimer. J Mol Biol. 2011;409:159–76. doi:10.1016/j.jmb.2011.03.026
- Grosjean H, de Crecy-Lagard V, Marck C. Deciphering synonymous codons in the three domains of life: Co-evolution with specific tRNA modification enzymes. FEBS letters. 2010;584:252–64. doi:10.1016/j.febslet.2009.11.052
- Weigele P, Raleigh EA. Biosynthesis and Function of Modified Bases in Bacteria and Their Viruses. Chem Rev. 2016;116:12655–87. doi:10.1021/acs.chemrev.6b00114
- Cantara WA, Crain PF, Rozenski J, et al. The RNA Modification Database, RNAMDB: 2011 update. Nucleic Acids Res. 2011;39:D195–201. doi:10.1093/nar/gkq1028
- Machnicka MA, Milanowska K, Osman Oglou O, et al. MODOMICS: A database of RNA modification pathways–2013 update. Nucleic Acids Res. 2013;41:D262–7. doi:10.1093/nar/gks1007
- Machnicka MA, Olchowik A, Grosjean H, et al. Distribution and frequencies of post-transcriptional modifications in tRNAs. RNA Biol. 2014;11:1619–29. doi:10.4161/15476286.2014.992273
- Giegé R LJ. Transfer RNA aminoacylation and modified nucleosides. In: H G, ed. DNA and RNA modification enzymes: Structure, mechanism, function and evolution. Austin, Texas, USA: Landes Bioscience; 2009.p. 476–92
- Grosjean H, Westhof E. An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Res. 2016;44:8020–40. doi:10.1093/nar/gkw608
- Perret V, Garcia A, Grosjean H, et al. Relaxation of a transfer RNA specificity by removal of modified nucleotides. Nature. 1990;344:787–9. doi:10.1038/344787a0
- Putz J, Florentz C, Benseler F, et al. A single methyl group prevents the mischarging of a tRNA. Nat Struct Biol. 1994;1:580–2. doi:10.1038/nsb0994-580
- Limbach PA, Crain PF, McCloskey JA. Summary: the modified nucleosides of RNA. Nucleic Acids Res. 1994;22:2183–96. doi:10.1093/nar/22.12.2183
- Boucher HW, Talbot GH, Bradley JS, et al. Bad bugs, no drugs: No ESKAPE! An update from the infectious diseases society of America. Clin Infect Dis. 2009;48:1–12. doi:10.1086/595011
- Pendleton JN, Gorman SP, Gilmore BF. Clinical relevance of the ESKAPE pathogens. Expert Rev Anti Infect Ther. 2013;11:297–308. doi:10.1586/eri.13.12
- Leekha S, Terrell CL, Edson RS. General principles of antimicrobial therapy. Mayo Clin Proc. 2011;86:156–67. doi:10.4065/mcp.2010.0639
- Bonnefond L, Giege R, Rudinger-Thirion J. Evolution of the tRNA(Tyr)/TyrRS aminoacylation systems. Biochimie. 2005;87:873–83. doi:10.1016/j.biochi.2005.03.008
- Becker HD, Giege R, Kern D. Identity of prokaryotic and eukaryotic tRNA(Asp) for aminoacylation by aspartyl-tRNA synthetase from Thermus thermophilus. Biochemistry. 1996;35:7447–58. doi:10.1021/bi9601058
- Kotra LP, Haddad J, Mobashery S. Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob Agents Chemother. 2000;44:3249–56. doi:10.1128/AAC.44.12.3249-3256.2000
- Davis BD. Mechanism of bactericidal action of aminoglycosides. Microbiol Rev. 1987;51:341–50
- Mikkelsen NE, Johansson K, Virtanen A, et al. Aminoglycoside binding displaces a divalent metal ion in a tRNA-neomycin B complex. Nat Struct Biol. 2001;8:510–4. doi:10.1038/88569
- Kirk SR, Tor Y. tRNA(Phe) binds aminoglycoside antibiotics. Bioorg Med Chem. 1999;7:1979–91. doi:10.1016/S0968-0896(99)00170-4
- Discovery T. Staphylococcus aureus 201 HTS-assay white paper. 2011
- Walter F, Putz J, Giege R, et al. Binding of tobramycin leads to conformational changes in yeast tRNA(Asp) and inhibition of aminoacylation. EMBO J. 2002;21:760–8. doi:10.1093/emboj/21.4.760
- Wang B, Wilkinson KA, Weeks KM. Complex ligand-induced conformational changes in tRNA(Asp) revealed by single-nucleotide resolution SHAPE chemistry. Biochemistry. 2008;47:3454–61. doi:10.1021/bi702372x
- Hori Y, Bichenkova EV, Wilton AN, et al. Synthetic inhibitors of the processing of pretransfer RNA by the ribonuclease P ribozyme: enzyme inhibitors which act by binding to substrate. Biochemistry. 2001;40:603–8. doi:10.1021/bi002378f
- Hori Y, Rogert MC, Tanaka T, et al. Porphyrins and porphines bind strongly and specifically to tRNA, precursor tRNA and to M1 RNA and inhibit the ribonuclease P ribozyme reaction. Biochim Biophys Acta. 2005;1730:47–55. doi:10.1016/j.bbaexp.2005.06.003
- Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759–61. doi:10.1126/science.1142189
- Chopra S, Palencia A, Virus C, et al. Plant tumour biocontrol agent employs a tRNA-dependent mechanism to inhibit leucyl-tRNA synthetase. Nat Commun. 2013;4:1417. doi:10.1038/ncomms2421
- Kirillov S, Vitali LA, Goldstein BP, et al. Purpuromycin: an antibiotic inhibiting tRNA aminoacylation. Rna. 1997;3:905–13
- Liu Y, Tidwell RR, Leibowitz MJ. Inhibition of in vitro splicing of a group I intron of Pneumocystis carinii. J Eukaryot Microbiol. 1994;41:31–8. doi:10.1111/j.1550-7408.1994.tb05931.x
- Miletti KE, Leibowitz MJ. Pentamidine inhibition of group I intron splicing in Candida albicans correlates with growth inhibition. Antimicrob Agents Chemother. 2000;44:958–66. doi:10.1128/AAC.44.4.958-966.2000
- Zhang Y, Bell A, Perlman PS, et al. Pentamidine inhibits mitochondrial intron splicing and translation in Saccharomyces cerevisiae. RNA. 2000;6:937–51. doi:10.1017/S1355838200991726
- Putignano V, Rosato A, Banci L, et al. MetalPDB in 2018: A database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2018;46(D1):D459–D464. doi:10.1093/nar/gkx989
- Draper DE. A guide to ions and RNA structure. RNA. 2004;10:335–43. doi:10.1261/rna.5205404
- Hermann T. Strategies for the Design of Drugs Targeting RNA and RNA-Protein Complexes. Angewandte Chemie. 2000;39:1890–904. doi:10.1002/1521-3773(20000602)39:11%3c1890::AID-ANIE1890%3e3.0.CO;2-D
- Chen L, Calin GA, Zhang S. Novel insights of structure-based modeling for RNA-targeted drug discovery. J Chem Inf Model. 2012;52:2741–53. doi:10.1021/ci300320t
- Deutscher M. The Effect of Polynucleotides on Aminoacyl-Rna Synthetases. 1. Inhibition by Synthetic Polynucleotides Biochem Biophys Res Commun. 1965;19:283–8. doi:10.1016/0006-291X(65)90455-9
- Beltchev B, Grunberg-Manago M. Competitive inhibition of the acceptor activity of tRNA(Tyr II)(E. coli) by a combination of oligo G and a CCA terminated nineteen residue oligonucleotide of tRNA(Tyr II)(E. coli). FEBS letters. 1970;12:27–9. doi:10.1016/0014-5793(70)80586-5
- Schimmel P. Aminoacyl tRNA synthetases: general scheme of structure-function relationships in the polypeptides and recognition of transfer RNAs. Annu Rev Biochem. 1987;56:125–58. doi:10.1146/annurev.bi.56.070187.001013
- Moulinier L, Eiler S, Eriani G, et al. The structure of an AspRS-tRNA(Asp) complex reveals a tRNA-dependent control mechanism. EMBO J. 2001;20:5290–301. doi:10.1093/emboj/20.18.5290
- Wolf YI, Aravind L, Grishin NV, et al. Evolution of aminoacyl-tRNA synthetases–analysis of unique domain architectures and phylogenetic trees reveals a complex history of horizontal gene transfer events. Genome Res. 1999;9:689–710
- Keeling PJ. Genomics: Evolution of the genetic code. Curr Biol. 2016;26:R851–R3. doi:10.1016/j.cub.2016.08.005
- Mukai T, Englert M, Tripp HJ, et al. Facile Recoding of Selenocysteine in Nature. Angewandte Chemie. 2016;55:5337–41. doi:10.1002/anie.201511657
- Fukai S, Nureki O, Sekine S, et al. Structural basis for double-sieve discrimination of L-valine from L-isoleucine and L-threonine by the complex of tRNA(Val) and valyl-tRNA synthetase. Cell. 2000;103:793–803. doi:10.1016/S0092-8674(00)00182-3
- Bai F, Morcos F, Cheng RR, et al. Elucidating the druggable interface of protein-protein interactions using fragment docking and coevolutionary analysis. Proc Natl Acad Sci U S A. 2016;113:E8051–E8. doi:10.1073/pnas.1615932113
- Watkins AM, Arora PS. Structure-based inhibition of protein-protein interactions. Eur J Med Chem. 2015;94:480–8. doi:10.1016/j.ejmech.2014.09.047
- Forster C, Szkaradkiewicz K, Perbandt M, et al. Human tRNA(Gly) acceptor-stem microhelix: crystallization and preliminary X-ray diffraction analysis at 1.2 A resolution. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:858–61. doi:10.1107/S1744309107041528
- Shiba K, Schimmel P, Motegi H, et al. Human glycyl-tRNA synthetase. Wide divergence of primary structure from bacterial counterpart and species-specific aminoacylation J Biol Chem. 1994;269:30049–55
- Mazauric MH, Reinbolt J, Lorber B, et al. An example of non-conservation of oligomeric structure in prokaryotic aminoacyl-tRNA synthetases. Biochemical and structural properties of glycyl-tRNA synthetase from Thermus thermophilus. Eur J Biochem/FEBS. 1996;241:814–26. doi:10.1111/j.1432-1033.1996.00814.x
- Qin X, Hao Z, Tian Q, et al. Cocrystal structures of glycyl-tRNA synthetase in complex with tRNA suggest multiple conformational states in glycylation. J Biol Chem. 2014;289:20359–69. doi:10.1074/jbc.M114.557249
- Rodova M, Ankilova V, Safro MG. Human phenylalanyl-tRNA synthetase: cloning, characterization of the deduced amino acid sequences in terms of the structural domains and coordinately regulated expression of the alpha and beta subunits in chronic myeloid leukemia cells. Biochem Biophys Res Commun. 1999;255:765–73. doi:10.1006/bbrc.1999.0141
- Finarov I, Moor N, Kessler N, et al. Structure of human cytosolic phenylalanyl-tRNA synthetase: evidence for kingdom-specific design of the active sites and tRNA binding patterns. Structure. 2010;18:343–53. doi:10.1016/j.str.2010.01.002
- Abibi A, Ferguson AD, Fleming PR, et al. The role of a novel auxiliary pocket in bacterial phenylalanyl-tRNA synthetase druggability. J Biol Chem. 2014;289:21651–62. doi:10.1074/jbc.M114.574061
- Palencia A, Crepin T, Vu MT, et al. Structural dynamics of the aminoacylation and proofreading functional cycle of bacterial leucyl-tRNA synthetase. Nat Struct Mol Biol. 2012;19:677–84. doi:10.1038/nsmb.2317
- Deng X, Qin X, Chen L, et al. Large Conformational Changes of Insertion 3 in Human Glycyl-tRNA Synthetase (hGlyRS) during Catalysis. J Biol Chem. 2016;291:5740–52. doi:10.1074/jbc.M115.679126
- Wang C, Guo Y, Tian Q, et al. SerRS-tRNASec complex structures reveal mechanism of the first step in selenocysteine biosynthesis. Nucleic Acids Res. 2015;43:10534–45
- Shen N, Guo L, Yang B, et al. Structure of human tryptophanyl-tRNA synthetase in complex with tRNATrp reveals the molecular basis of tRNA recognition and specificity. Nucleic Acids Res. 2006;34:3246–58. doi:10.1093/nar/gkl441
- Crick FH. The origin of the genetic code. J Mol Biol. 1968;38:367–79. doi:10.1016/0022-2836(68)90392-6
- Breitschopf K, Achsel T, Busch K, et al. Identity elements of human tRNA(Leu): structural requirements for converting human tRNA(Ser) into a leucine acceptor in vitro. Nucleic Acids Res. 1995;23:3633–7. doi:10.1093/nar/23.18.3633
- Hou YM, Schimmel P. Evidence that a major determinant for the identity of a transfer RNA is conserved in evolution. Biochemistry. 1989;28:6800–4. doi:10.1021/bi00443a003
- Juhling F, Morl M, Hartmann RK, et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–62. doi:10.1093/nar/gkn772