
ABSTRACT
Alternative splicing (AS) in response to changing external conditions often requires alterations in the ability of sequence-specific RNA-binding proteins to bind to cis-acting sequences in their target pre-mRNA. While daily oscillations in AS events have been described in several organisms, cis-acting sequences that control time of the day-dependent AS remain largely elusive. Here we define cis-regulatory RNA elements that control body-temperature driven rhythmic AS using the mouse U2af26 gene as a model system. We identify a complex network of cis-regulatory sequences that regulate AS of U2af26, and show that the activity of two enhancer elements is necessary for oscillating AS. A minigene comprising these U2af26 regions recapitulates rhythmic splicing of the endogenous gene, which is controlled through temperature-regulated SR protein phosphorylation. Mutagenesis of the minigene delineates the cis-acting enhancer element for SRSF2 within exon 6 to single nucleotide resolution and reveals that the combined activity of SRSF2 and SRSF7 is required for oscillating U2af26 AS. By combining RNA-Seq with an siRNA screen and individual-nucleotide resolution cross-linking and immunoprecipitation (iCLIP), we identify a complex network of SR proteins that globally controls temperature-dependent rhythmic AS, with the direction of splicing depending on the position of the cis-acting elements. Together, we provide detailed insights into the sequence requirements that allow trans-acting factors to generate daily rhythms in AS.
1. Introduction
Alternative splicing (AS) is a regulated process that dramatically increases the coding potential of the genome. Through variable combinations of exonic sequences, multiple different mRNAs can be generated from a single gene [Citation1]. While it is estimated that ~95% of human multi-exon genes generate at least two mRNA isoforms [Citation2,Citation3], the biological function of most of these isoforms remains enigmatic [Citation4,Citation5]. However, there are several studies that characterise the functional difference of AS isoforms in detail (e.g. [Citation6–Citation9]), associate certain splice isoforms with diseases [Citation10] or globally demonstrate differences of AS isoforms [Citation11,Citation12], highlighting the broad biological importance of this process. Accumulating evidence shows that sequence-specific RNA-binding proteins (RBPs) (trans-acting factors) binding to specific sites within the pre-mRNA (cis-acting elements) determine the outcome of AS [Citation13,Citation14]. Sequence specific RBPs are often expressed in a development- or tissue-specific manner, enabling the differential regulation of splicing patterns of co-regulated exons in different cellular backgrounds [Citation14]. However, AS is rarely regulated by a single pair of cis- and trans-acting elements; it is rather the interplay of several trans-acting factors binding to different cis-acting sequences that together control the splicing decision [Citation15–Citation18]. Indeed, it was recently shown that virtually each exonic nucleotide can contribute to the splicing outcome [Citation19], emphasising the importance of different cis-regulatory RNA sequences. Consequently, changes in cis-acting sequences have been suggested to be the main determinant of species-specific AS [Citation20,Citation21]. Additionally, mutations affecting cis-regulatory elements are associated with a variety of diseases in human [Citation22–Citation24], further highlighting the requirement to understand the sequence characteristics determining splicing outcome.
We have previously characterised an AS event in the mouse U2af26 gene, which is regulated in a time of the day-dependent manner in peripheral clocks [Citation25]. Here, skipping of two variable exons (6 and 7) generates a novel protein variant of U2af26 that directly controls circadian clock resetting in vivo. This and several other oscillating AS events are driven by rhythmic body temperature changes, which directly controls SR protein phosphorylation [Citation26]. While we have demonstrated an involvement of trans-acting factors, particularly SRSF2 and SRSF7, the cis-regulatory elements required for oscillating AS remain unknown. For oscillating transcription, it was previously demonstrated that the presence of an 18 bp E-box element within the promoter region is sufficient [Citation27]. This E-box is recognised by the trans-acting core circadian transcription factors CLOCK and BMAL1 and the presence of an E-box is predictive for circadian gene expression [Citation28,Citation29]. Here, we set out to identify sequence elements within pre-mRNAs that, in analogy to E-boxes, mediate oscillating AS. We have used U2af26 variable exons 6 and 7 as model system and square wave temperature cycles in cell culture, which mimic 24-hour rhythms of AS in vivo [Citation26]. Using systematic mutational analysis of a minigene, we define the cis-regulatory elements required for the recruitment of SRSF2 and SRSF7 to the U2af26 pre-mRNA. We further demonstrate that not a single cis-regulatory element, but the presence of several elements is essential for oscillating AS. Using an siRNA screen, RNA-Seq and iCLIP data we show that most rhythmic exons are co-regulated by several SR proteins, suggesting this to be a general principle to obtain oscillating AS.
2. Results
2.1. A U2af26 minigene recapitulates endogenous temperature-controlled AS
AS of U2af26 variable exons 6 and 7 is regulated in a time of the day dependent manner in mouse peripheral clocks [Citation25]. Mechanistically, this depends on body temperature mediated changes in the phosphorylation level of two SR proteins, namely SRSF2 and SRSF7 [Citation26]. To identify the cis-regulatory elements mediating rhythmic U2af26 AS, we cloned a minigene containing U2af26 exons 5 to 8 and the complete intervening introns (wt-minigene, ), see also supplementary materials and methods for minigene sequences). After transfection into N2A cells, the wt-minigene was processed into the expected fl (joining of exons 5, 6, 7 and 8), ∆7 and Δ67 isoforms and an additional isoform still containing intron 5 (called IR for intron retention, )). We noted that the variable exons are less efficiently included in the minigene context (compare with endogenous splicing, )), suggesting that splicing activators become limiting due to overexpression of the minigene.
Figure 1. A U2af26-minigene recapitulates endogenous splicing regulation.
(a) Comparison of endogenous and minigene U2af26 splicing. Top: exon/intron structure of the wt-minigene including sizes of exons and introns. Highlighted in grey is the part of U2af26 genomic region directly cloned to generate the minigene reporter. Bottom: Shown are representative gels for endogenous (left) and minigene (right) specific RT-PCRs from N2A cells.(b) The U2af26 wt-minigene recapitulates splicing regulation by overexpression of SRSF2/7. N2A cells were co-transfected with the U2af26 wt-minigene and SRSF2 or 7. AS was analysed using minigene-specific primers (left, representative gel). %∆67- and %∆7-isoform were quantified relative to the GFP control (right, n = 9, mean ± SD, ***p < 0.001). See also Figure S1(a) for a complete quantification and an explanation how the fold-change %∆67 is calculated and Figure S1(b) for overexpression validation. (c) Knockdown of SRSF2 or SRSF7. N2A cells were transfected with siRNAs against SRSF2 or SRSF7 and with the wt-minigene. AS was analysed using minigene-specific primers (left, representative gel). %∆67-isoform was quantified relative to the siRNA control (n = 6–8, mean +/- SD, **p < 0.01, ***p < 0.001). See also Figure (S1(c), (d)). (d) The U2af26 wt-minigene recapitulates splicing regulation by heat shock. 3T3 cells were transfected with the wt-minigene and heat shocked (42°C, red) for 2 hours. AS was analysed using minigene-specific primers (left, representative gel). %IR-isoform was quantified relative to 37°C (n = 3, mean +/- SD, **p < 0.01). See also Figure S1(e). (e) The U2af26 wt-minigene recapitulates splicing regulation by cold shock. N2A cells were transfected with the wt-minigene and cold shocked (32°C, blue) for 2 hours. AS was analysed using minigene-specific primers (left, representative gel). %∆67-isoform was quantified relative to 37°C (n = 3, mean +/- SD, *p < 0.05). (f) Temperature-regulated minigene splicing. N2A cells were transfected with the wt-minigene and treated with square-wave temperature cycles (12h 34°C (blue)/12h 38°C (red)) for 3 days (see Figure S1(f) for a temperature scheme). AS was analysed on the third day using minigene-specific primers (left, representative gel, all lanes come from a single gel but are rearranged for clarity). %∆67-isoform was quantified relative to 72 hours (n = 3, mean +/- SD, *p < 0.05, ***p < 0.001).
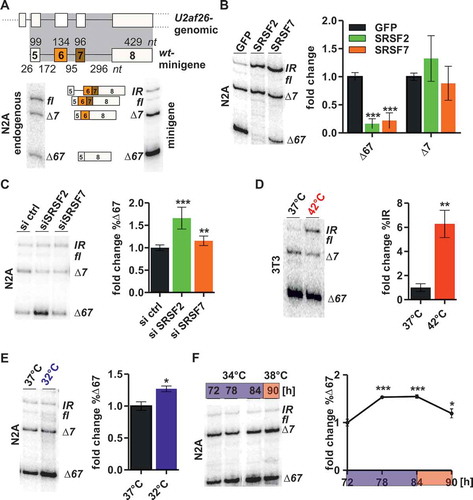
We have previously shown that endogenous U2af26 alternative exon inclusion is promoted by SRSF2 and 7, whose activity is regulated by temperature-mediated changes in their phosphorylation level [Citation26]. Consistently, overexpression of SRSF2 or SRSF7 led to a strong increase in exon inclusion (the IR isoform seems to replace the fl isoform in the minigene context), whereas knockdown showed an antagonistic but only moderate effect, especially for SRSF7 (,)). Importantly, overexpression of SRSF2 or SRSF7 only promoted inclusion of exons 6 and 7 together, while the ∆7 isoform was not affected (), see Figure S1(a) for a complete quantification and an explanation how the fold-change %∆67 is calculated, see Figure S1(b), S1(c) and S1(d) for validation of overexpression and knockdown). This is in line with the observation that only the ∆67-isoform is regulated in a circadian manner in mice and by temperature changes in cell culture [Citation25,Citation26]. Furthermore, heat shock increases the longest isoform in the minigene background ()) similar to the effect on endogenous U2af26 alternative exon inclusion [Citation26]. For heat shock we used 3T3 cells, since for endogenous splicing the heat shock response was slightly stronger in this cell type [Citation26], but obtained similar results in N2A cells (Figure S1(e)). Antagonistically, cold shock promotes exon skipping ()) and consequently minigene AS oscillated under an external square wave temperature rhythm (12-hours 34°C/12-hours 38°C; ), S1(f). In summary, these data establish a minigene system that recapitulates oscillating AS and confirm the presence of all cis-regulatory elements required for temperature-mediated splicing regulation through SRSF2 and SRSF7.
2.2. An enhancer element in U2af26 exon 6 mediates SRSF2 dependent splicing regulation
To identify cis-regulatory elements involved in rhythmic U2af26 splicing, we conducted a systematic mutational analysis. First, exonic and intronic sequences were individually replaced by sequences from the β-globin gene to identify silencer and enhancer elements that control U2af26 splicing (), left, see also Figure S2(a) for further details and supplementary materials and methods for minigene sequences and Figure S2(b) for the same analysis in 3T3 cells). This analysis identified three splicing silencers located in introns 5 and 6, and in exon 7. All of them repress exon 6 inclusion, as the respective mutants show a strong increase in exon 6 inclusion with ∆7 becoming the main isoform. In addition, we identified one enhancer element in exon 6 that is necessary for the inclusion of exons 6/7, as its replacement led to complete exon skipping (main isoform: ∆67, )). To confirm silencers and enhancers mediating exon 6 inclusion we generated a 2-exon reporter, containing exons 5 and 6 only. In this minimal reporter mutating intron 5 promoted splicing while mutating exon 6 prevented splicing (Figure S2(c)), consistent with the identification of a silencer in intron 5 and an enhancer in exon 6. Since the ∆7 isoform is neither regulated by temperature [Citation26] nor by SRSF2/7 ()) we focused further analyses on the enhancer element in exon 6. Moreover, this was the only sequence required for the inclusion of both exons 6 and 7, suggesting an involvement in regulating oscillating AS of U2af26. To address this, we first tested splicing of the various mutant minigenes in response to knockdown of SRSF2 (we focused here on SRSF2, as SRSF7 knockdown had only a very moderate effect on wt minigene splicing): Consistent with the presence of a cis-regulatory sequence within exon 6, splicing of all mutant minigenes except the one lacking exon 6 (E6mut) remained responsive to SRSF2 knockdown ()). Next, we investigated splicing of the E6mut and I5mut minigenes in response to SRSF2 and SRSF7 overexpression. Mutation of intron 5 did not impair the SRSF2/7 response ()). In contrast, after mutation of exon 6, overexpression of SRSF2 or SRSF7 resulted only in a moderate decrease of the ∆67 isoform ()), confirming the presence of an SRSF2/7-responsive exonic splicing enhancer (ESE) in exon 6. Interestingly SRSF7 overexpression still promoted some mild formation of the longest isoform, suggesting that SRSF7 binds independently of exon 6, but alone is not able to promote full exon inclusion (see also below).
Figure 2. Mapping cis-regulatory elements in U2af26.
(a) Exon/intron structures of U2af26 minigenes (left). From the wt-minigene (containing genomic exon 5 to 8 sequence) exons and introns were individually replaced by β-globin sequence (marked in violet; exons were replaced by β-globin exon 2 sequence of the same length, introns were replaced by complete β-globin intron 1 including the branch point adenosine). Mutations were designed not to affect the direct splice sites (see also Figure S2(a)). N2A cells were transfected with the minigenes and AS was analysed using minigene-specific primers (middle, representative gel, all bands come from a single gel but are rearranged for clarity). Here, and in the following panels wt data from are shown and marked in light grey. Right: Summary of identified cis-regulatory elements. See also Figure S2(b) for analysis of the same minigenes in 3T3 cells.(b) SRSF2-knockdown response of mutated minigenes. N2A cells were transfected with the indicated siRNAs and indicated minigenes. %∆67-isoform was quantified relative to control siRNA (n = 5–6, mean +/- SD, ***p < 0.001).(c) SRSF2/7-overexpression response of mutated minigenes. N2A cells were transfected as in , comparing the I5mut (left) and the E6mut minigene (right). %∆67-isoform was quantified relative to GFP control (n = 6, mean +/- SD, ***p < 0.001).(d) Heat shock response of mutated minigenes. 3T3 cells were transfected with the indicated minigenes and exposed to heat shock for 2 hours (red). For each minigene fold change of % of the longest isoform was quantified relative to 37°C (n = 3, mean +/- SD, *p < 0.05, **p < 0.01).(e) Cold shock response of the E6 mutant. N2A cells were transfected and exposed to cold shock for 2 hours (blue). %∆67-isoform was quantified relative to 37°C (n = 3, mean +/- SD).(F) Temperature-regulated E6mut minigene splicing. N2A cells were transfected and exposed to oscillating temperature changes as in . %∆67-isoform was quantified relative to 72 hours (n = 3, mean +/- SD).
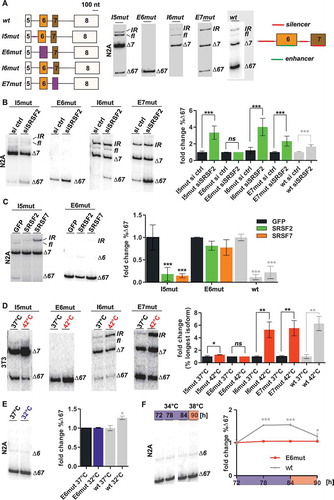
We then addressed the splicing response of the different minigenes to a 2-hour heat-shock. While mutations of intron 6 or exon 7 (I6mut, E7mut) did not exhibit changes in exon6/7 inclusion compared to the wt, the heat-shock response was strongly abrogated in I5mut and completely abolished when exon 6 was absent (E6mut; )). Furthermore, the E6mut minigene was not responsive to cold-shock or square wave temperature cycles anymore (,)). In summary, this mutational analysis identified an enhancer element in exon 6 necessary for temperature-controlled AS by SRSF2, which forms the regulatory basis for rhythmic AS in vivo.
2.3. The exon 6 enhancer is sufficient for SRSF2 dependent splicing regulation
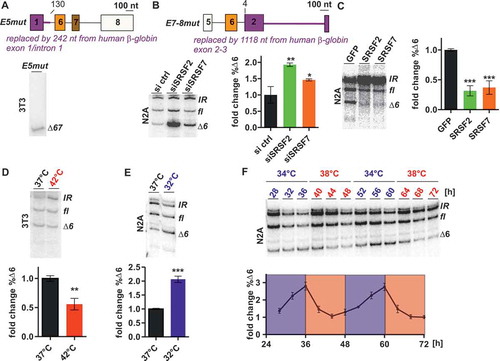
To address whether U2af26 exon 6 is also sufficient for the regulation by SRSF2/7 and temperature we investigated its splicing in a heterologous context. To this end, exon 6 together with the surrounding 3ʹ and 5ʹ splice sites, was cloned in between exon 10 and 14 of the unrelated CamKII gene [Citation30] (E6het, )), see also supplementary materials and methods for minigene sequences). After transfection into N2A cells, the E6het minigene was processed into the expected fl and ∆6 isoforms and an additional isoform still containing the intron upstream of exon 6 (IR, ), first lane). We noted that exon 6 inclusion is more efficient in the heterologous context compared to wt-minigene splicing (see )), consistent with the presence of silencer elements within introns 5 and 6, and exon 7 ()). To test whether the enhancer in exon 6 is responsive to SRSF2 and/or SRSF7 we compared splicing of the E6het-minigene in response to SRSF2/7 overexpression and knockdown (,b)). Interestingly, exon 6 inclusion was moderately promoted by SRSF2 overexpression, whereas SRSF7 had no effect ()). Similarly, exon 6 skipping was promoted only by knockdown of SRSF2 but not SRSF7 ()). In contrast, skipping of exons 6/7 in endogenous U2af26 was promoted by depletion of either SRSF2 or SRSF7 (data not shown). These data show that the enhancer element located within U2af26 exon 6 is specific for SRSF2, which is consistent with our previously published in vivo binding data. Using iCLIP data from P19, we identified a cluster of SRSF2 crosslinks (iCLIP-tags) in the second half of exon 6; in contrast SRSF7 crosslinks were not detectable [Citation26,Citation31]. The SRSF2 iCLIP-tags in exon 6 overlap with five predicted SRSF2 binding motifs ()), which were deduced from binding studies and functional SELEX experiments [Citation32,Citation33]. To functionally validate the identified SRSF2-binding sites we introduced five CC to AA point mutations into the E6het-minigene, which should prevent SRSF2 binding to exon 6 (mutCC, )). Indeed, these mutations completely abolished exon 6 inclusion (compare lanes 1 and 3 in ,)) confirming the functional importance of the SRSF2 binding site identified by iCLIP. Next, we tested AS regulation by temperature: The presence of exon 6 was indeed sufficient for its regulation by heat ()), as the E6het-minigene showed increased exon inclusion at 42°C, which was completely abolished in the mutCC construct ()). However, the E6het construct did not show a response to cold shock or square wave temperature cycles (,f)), suggesting that additional sequences are required for rhythmic AS regulation. Since exon 6 is solely responsive to SRSF2 this indicates a more prominent role of SRSF2 in controlling splicing upon heat shock and a more prominent role of SRSF7 and/or other factors in the cold shock response. In summary, these data define the SRSF2-responsive element in exon 6 at single nucleotide resolution and show that the combined activity of more than one cis-regulatory element and more than one SR protein, in the case of U2af26 SRSF2 and 7, is required for oscillating, temperature-controlled splicing regulation.
Figure 3. An enhancer in U2af26 exon 6 mediates regulation through SRSF2.
(a, b) SRSF2/7 response of a minigene with exon 6 in a heterologous context (top, exon/intron structure; black: heterologous sequence from midge CamKII exons 10 and 14 with adjacent introns; the numbers above the introns indicate the start and end positions in U2af26 introns 5 and 6). The E6het-minigene was either co-transfected with SRSF2 or 7 (a) or transfected after knockdown of SRSF2 or 7 (b). Splicing was analysed and %∆6-isoform was quantified and normalised to control (n ≥ 3, mean +/- SD, **p < 0.01, ***p < 0.001).(c) Sequence of U2af26 exon 6 nucleotides 68 to 121. Below is number of SRSF2 iCLIP-tags [Citation31], overlapping and neighboring SRSF2 consensus binding sites ([Citation43], small letters indicate small differences of the U2af26 sequence to the consensus binding site), and mutations introduced (mutCC) highlighted in red.(d) Heat shock response after SRSF2-binding site mutation. N2A cells were transfected with the indicated minigenes and splicing was analysed after 2-hours heat shock. %∆6-isoform was quantified and normalised to E6het at 37°C (n = 3, mean +/- SD, **p < 0.01, ***p < 0.001).(e, f) Temperature-regulated minigene splicing. N2A cells were transfected with the wt or the E6mut minigene and exposed to cold shock (e) or temperature cycles of 34 and 38°C (f). %∆6-isoform was quantified (n = 2–3, mean +/- SD, ***p < 0.001).
![Figure 3. An enhancer in U2af26 exon 6 mediates regulation through SRSF2.(a, b) SRSF2/7 response of a minigene with exon 6 in a heterologous context (top, exon/intron structure; black: heterologous sequence from midge CamKII exons 10 and 14 with adjacent introns; the numbers above the introns indicate the start and end positions in U2af26 introns 5 and 6). The E6het-minigene was either co-transfected with SRSF2 or 7 (a) or transfected after knockdown of SRSF2 or 7 (b). Splicing was analysed and %∆6-isoform was quantified and normalised to control (n ≥ 3, mean +/- SD, **p < 0.01, ***p < 0.001).(c) Sequence of U2af26 exon 6 nucleotides 68 to 121. Below is number of SRSF2 iCLIP-tags [Citation31], overlapping and neighboring SRSF2 consensus binding sites ([Citation43], small letters indicate small differences of the U2af26 sequence to the consensus binding site), and mutations introduced (mutCC) highlighted in red.(d) Heat shock response after SRSF2-binding site mutation. N2A cells were transfected with the indicated minigenes and splicing was analysed after 2-hours heat shock. %∆6-isoform was quantified and normalised to E6het at 37°C (n = 3, mean +/- SD, **p < 0.01, ***p < 0.001).(e, f) Temperature-regulated minigene splicing. N2A cells were transfected with the wt or the E6mut minigene and exposed to cold shock (e) or temperature cycles of 34 and 38°C (f). %∆6-isoform was quantified (n = 2–3, mean +/- SD, ***p < 0.001).](/cms/asset/dd418258-14d0-4d44-9bc3-08002a564bb9/krnb_a_1502587_f0003_c.jpg)
2.4. A network of cis-regulatory elements allows oscillating U2af26 AS
To investigate whether regulation by SRSF2 and SRSF7 is sufficient for oscillating AS, we generated a minigene containing potential binding sites for both SR proteins. Our iCLIP data indicated additional binding of SRSF2 and SRSF7 to exon 5 and intron 5 sequences [Citation26]. To address the functional importance of these sequence elements, we used the wt-minigene and replaced exon 5 and parts of intron 5 by β-globin sequence (E5mut, ), see also supplementary materials and methods for minigene sequences). This resulted in the exclusive formation of the ∆67-isoform, suggesting that exon 5 (or the 5’ss in intron 5 not mutated in I5mut but mutated in E5mut) contains additional enhancer elements for exon 6/7 inclusion that are regulated by SRSF2/7 ()). Next, we generated a minigene that contains only the enhancer elements within exon/intron 5 and exon 6, whereas exon 7 and 8 were replaced by sequences from β-globin exon 2 to 3 (E7-8mut, ), see also supplementary materials and methods or minigene sequences). After transfection into N2A cells, the E7-8mut-minigene was processed into the expected fl and ∆6 and an IR isoform (), first lane). Exon 6 processing in this minigene context was responsive to both, SRSF2 and SRSF7, as knockdown of either SR protein increased the ∆6-isoform strongly ()). Antagonistically to the knockdown, overexpression of SRSF2 or SRSF7 promoted exon 6 inclusion ()). Furthermore, we now found AS of exon 6 to be responsive to both heat shock and cold shock (,)), suggesting that the SRSF7 binding sites (absent in the E6het construct) mediate the cold response. Consistent with our hypothesis that the combined activity of SRSF2 and SRSF7 is required for rhythmic AS, this minigene showed oscillating AS with a period of 24-hours over 48 hours under square wave temperature cycles ()).
Figure 4. Enhancers in U2af26 exons 5 and 6 are sufficient for oscillating AS.
(a) Identification of an enhancer element in U2af26 exon 5. Top: From the wt-minigene exon 1 and parts of intron 1 (up to nt 130, top) were replaced by β-globin sequence (marked in violet). 3T3 cells were transfected with the minigenes and AS was analysed using minigene-specific primers (bottom: representative gel).(b, c) SRSF2/7 response of the E7-8mut-minigene. The E7-8mut-minigene (top; violet sequence E2-E3 from β-globin) was transfected after knockdown (b) or overexpression (c) of either SRSF2 or 7 in N2A cells. Splicing was analysed and %∆6-isoform was quantified and normalised to the control (n = 3, mean +/- SD, *p < 0.05, **p < 0.01, ***p < 0.001).(d, e, f) Temperature response of the E7-8mut-minigene. Cells were transfected and splicing was analysed after heat shock (d), cold shock (e), or two consecutive days of temperature cycles (f). %∆6-isoform was quantified and normalised to 37°C (d, e) or time point 72-hours (f; n = 3–4, mean +/- SD, **p < 0.01, ***p < 0.001).
2.5. Oscillating AS is generally co-regulated by several SR proteins
To investigate, whether temperature-responsive rhythmic AS is globally co-regulated by several SR proteins we performed RNA-Seq of P19 cells synchronised with square-wave temperature cycles. P19 cells were used, since we have previously obtained binding data for SR proteins using iCLIP in this cell line [Citation31] and P19 cells reproduce oscillating AS of U2af26 and our E7-8mut-minigene (), S3A and [Citation26]). Using MISO, we identified 97 splicing events (of different splicing types), which are alternatively spliced at 34°C vs. 38°C () and Table S1). These 97 events show a significant overlap with previously identified cold-responsive splicing events from 12-hour cold-shocked MEFs (), p = 1.731*10−33, data from [Citation26,Citation34]), including previously validated splicing events in Tor1aip2, Bat2d, Azi2, Eif4h, Tbp, Fam115a, and Cd151 (see [Citation26]), and show similar GO term enrichments such as ‘RNA binding’ (2.37*10−13) or ‘RNA splicing’ (1.56*10−5). We were able to validate temperature-responsive oscillating AS for 6 out of 6 tested splicing events using N2A cells () and S3(b)), thus confirming that temperature-responsive splicing is not a cell-type specific effect but appears to be a general phenomenon. Additionally, 24 of the 25 splicing events identified by both RNA sequencing analyses were regulated in the same direction (Table S1), suggesting that the list of identified splicing events represents a reliable source of temperature-oscillating splicing events. Of note, 20 out of 61 exon skipping events (SE) showed cold-induced exon skipping, as for U2af26 (e.g. Mettl3, ), cold-skipped), while 41 events showed more inclusion (e.g. Eif4h, Figure S3(b), heat-skipped).
Figure 5. Temperature-dependent rhythmic AS regulation by SR proteins.
(a) P19 cells reproduce endogenous oscillation of U2af26 AS. P19 cells were treated with square-wave temperature cycles and AS was analysed at the indicated time-points using U2af26-specific primers (top, representative gel). %∆67-isoform was quantified (bottom, n = 4, mean +/- SD); red arrowheads indicate samples used for RNA-Seq. (b) Identification of temperature-responsive oscillating splicing events by RNA-Seq. Identified events are categorised into alternative 3’ss (alt 3’ss), alternative 5’ss (alt 5’ss), mutually exclusive exons (Mxe), retained intron (RI), and skipped exon (SE).(c) Comparison of the 97 oscillating splicing events (yellow) with our previously identified 202 cold-responsive splicing events (grey). See also Table S1. (d) Square-wave temperature rhythms. In N2A cells 6 exemplary targets were analysed by splicing sensitive radioactive RT-PCR and splicing isoforms were quantified. For each the percentage of one isoform (for SE events always the skipping isoform) was calculated (n = 3, mean +/- SD). See Figure S3B for additional 5 targets.(e, f) Temperature-controlled exons are co-regulated by several SR proteins. AS of cold-skipped (e) and heat-skipped (f) exons was analysed after SR protein knockdown. N2A cells were transfected with siRNA pools against the depicted SR proteins and splicing was analysed (as in [Citation26]). For each event skipping was quantified. Mean of 2 independent experiments +/- SD. The dotted lines represent 2.5 SD of the ctrl siRNA. siRNAs showing an effect of more than 2.5 SD are highlighted, in blue if the knockdown resembles the effect of cold, in red if the knockdown resembles the effect of heat.
![Figure 5. Temperature-dependent rhythmic AS regulation by SR proteins.(a) P19 cells reproduce endogenous oscillation of U2af26 AS. P19 cells were treated with square-wave temperature cycles and AS was analysed at the indicated time-points using U2af26-specific primers (top, representative gel). %∆67-isoform was quantified (bottom, n = 4, mean +/- SD); red arrowheads indicate samples used for RNA-Seq. (b) Identification of temperature-responsive oscillating splicing events by RNA-Seq. Identified events are categorised into alternative 3’ss (alt 3’ss), alternative 5’ss (alt 5’ss), mutually exclusive exons (Mxe), retained intron (RI), and skipped exon (SE).(c) Comparison of the 97 oscillating splicing events (yellow) with our previously identified 202 cold-responsive splicing events (grey). See also Table S1. (d) Square-wave temperature rhythms. In N2A cells 6 exemplary targets were analysed by splicing sensitive radioactive RT-PCR and splicing isoforms were quantified. For each the percentage of one isoform (for SE events always the skipping isoform) was calculated (n = 3, mean +/- SD). See Figure S3B for additional 5 targets.(e, f) Temperature-controlled exons are co-regulated by several SR proteins. AS of cold-skipped (e) and heat-skipped (f) exons was analysed after SR protein knockdown. N2A cells were transfected with siRNA pools against the depicted SR proteins and splicing was analysed (as in [Citation26]). For each event skipping was quantified. Mean of 2 independent experiments +/- SD. The dotted lines represent 2.5 SD of the ctrl siRNA. siRNAs showing an effect of more than 2.5 SD are highlighted, in blue if the knockdown resembles the effect of cold, in red if the knockdown resembles the effect of heat.](/cms/asset/553de577-264b-4fae-93a3-273b1abf317d/krnb_a_1502587_f0005_c.jpg)
Next, we used an established siRNA screen targeting SR proteins [Citation26] to investigate whether rhythmic AS is generally co-regulated by several SR proteins. Investigating six targets, we find that both cold-induced ()) and heat-induced ()) skipping events are always co-regulated by at least 2 different SR proteins. Knockdown of all SR proteins affects AS of at least one target, indicating that most/all SR proteins could be involved in temperature-regulated splicing regulation. SRSF2, SRSF7 and SRSF11 regulated four out of six targets, suggesting that these proteins play a more dominant role in temperature-dependent splicing regulation (,f)). Interestingly, SRSF2 knockdown mimics the effect of colder temperature for all four SRSF2-responsive targets, consistent with our previous observation that cold-induced hyperphosphorylation inhibits SRSF2 [Citation26]. In contrast, SRSF11 knockdown resembles the effect of heat for all responsive targets, indicating that for SRSF11 the cold-induced isoform (likely hyperphosphorylated) might be more active, whereas the protein is inactivated by heat. While SRSF2 knockdown always recapitulates the effect of colder temperature, it can result both in more exon skipping (Bat2d; U2af26 and hnRNPA2B1 in [Citation26]) or in more exon inclusion (Atp13a3, Ppp3cb, Mff). To gain mechanistic insights into these antagonistic outcomes, we compared SRSF2 binding to cold-skipped (potentially SRSF2 activated) and heat-skipped (potentially SRSF2 repressed) variable exons and the corresponding flanking exons using our iCLIP data [Citation31]. For cold-skipped exons we detected more SRSF2 iCLIP tags within the alternative exon and the downstream constitutive exon, while heat-skipped exons showed more binding to the upstream constitutive exon ()). These data indicate that binding of SRSF2 to the alternative exon enhances its inclusion, whereas binding to the upstream exon enhances its skipping as proposed previously [Citation35,Citation36]. In line with this, cold-skipped exons are enriched in a sequence motif containing CAG or CTG (26 occurrences; 13 of 20 exons; E-value 1.14e-03; Figure S4(a)). This motif resembles the SRSF2 consensus binding motif derived from all iCLIP data (Figure S4(b)), or from SRSF2 crosslink sites within cold-skipped exons (Figure S4(c)). The CAG/CTG motif is not significantly enriched in heat-skipped exons (8 occurrences; 8 of 40 exons; E-value 1.5e-01). Interestingly, the position of the SRSF2 binding site in the flanking exons seems to play a crucial role in determining the direction of the temperature effect ()). In summary, these data reveal extensive co-regulation of temperature-responsive rhythmic AS by several SR proteins, and highlight the importance of the position of the respective cis-regulatory binding sites.
Figure 6. RNA maps show distinct Srsf2-binding patterns for cold- and heat-induced exons.
(a) Srsf2-binding to the 3’ss (−100 to + 100 nucleotides) of 21 cold-skipped (blue) and 41 heat-skipped (red) exon skipping events using our previously published iCLIP data from P19 cells stably expressing GFP-tagged Srsf2 near endogenous level [Citation31]. Binding was investigated for the upstream constitutive exon, the variable exon itself and for the downstream constitutive exon. The red and blue line represent the sum of iCLIP tags for each position on the pre-mRNA. (b) Model for temperature-regulated (top: cold/bottom: hot) AS mediated by SR proteins. (I) Only dephosphorylated SRSF2 binding to the variable and/or downstream exon promotes exon inclusion. (II) Only dephosphorylated SRSF2 binding to the upstream exon promotes exon skipping. (III) Co-regulation by SRSF2 and a second SR protein (SRSFX) allows oscillating AS, indicated by the clock.
![Figure 6. RNA maps show distinct Srsf2-binding patterns for cold- and heat-induced exons. (a) Srsf2-binding to the 3’ss (−100 to + 100 nucleotides) of 21 cold-skipped (blue) and 41 heat-skipped (red) exon skipping events using our previously published iCLIP data from P19 cells stably expressing GFP-tagged Srsf2 near endogenous level [Citation31]. Binding was investigated for the upstream constitutive exon, the variable exon itself and for the downstream constitutive exon. The red and blue line represent the sum of iCLIP tags for each position on the pre-mRNA. (b) Model for temperature-regulated (top: cold/bottom: hot) AS mediated by SR proteins. (I) Only dephosphorylated SRSF2 binding to the variable and/or downstream exon promotes exon inclusion. (II) Only dephosphorylated SRSF2 binding to the upstream exon promotes exon skipping. (III) Co-regulation by SRSF2 and a second SR protein (SRSFX) allows oscillating AS, indicated by the clock.](/cms/asset/2b4d50f7-7caf-4973-a028-4d05a6dcf5ba/krnb_a_1502587_f0006_c.jpg)
3. Discussion
Here, we have combined an in detail minigene analysis of a single target – U2af26 – and a global RNA sequencing based approach to understand how SR proteins and their respective cis-regulatory elements shape rhythmic AS in response to oscillating temperature changes. In summary, these data show that rhythmic AS of U2af26, as observed under daily changes in body temperature, is not mediated by a single cis-regulatory element but requires the combined activity of at least two enhancer motifs. We have performed a systematic mutational analysis of a minigene and identified three silencer and two enhancer elements mediating U2af26 variable exon inclusion. In addition, we defined the binding site for the trans-acting factor SRSF2 in exon 6, and provide further data indicating binding of SRSF7 to an enhancer within exon/intron 5. The combined activity of both activators appears to be essential for exon 6/7 inclusion, as removing one enhancer element (this study) or one activator [Citation26] almost completely abolished their inclusion. Additionally, U2af26 variable exon inclusion is influenced by three silencer elements in introns 5 and 6, and in exon 7, which interestingly all prevent exon 6 inclusion. We therefore hypothesise that they impact on U2af26 AS by a common mechanism. Interestingly, RNA secondary structure predictions suggest the formation of a stable fold by the U2af26 variable region from intron 5 to exon 7 (data not shown), which could regulate variable exon inclusion. Mutating either intron 5 or 6, or exon 7 could disturb this secondary structure, thereby allowing exon 6 inclusion leading to the formation of the ∆7 isoform. However, exon 7 is not repressed by the silencer motifs, suggesting that two mechanisms control formation of the ∆7 and ∆67 isoforms and act independently of one another. It is further interesting to note that the fraction of RNAs skipping only exon 7 is not further regulated by SRSF2/7 (e.g. )) which could be explained by a certain amount of pre-mRNAs acquiring a secondary structure that only allows the formation of the ∆7-isoform, with the fraction of these pre-mRNAs being determined by the silencer elements. The ∆67 isoform is controlled in a rhythmic manner whereas the ∆7 isoform is induced upon T cell activation (data not shown) and in both cases the other isoform does not change at all, providing further evidence for independent regulatory mechanisms.
Temperature-mediated changes in U2af26 splicing can be largely explained by changes in the phosphorylation level of SRS2/7 [Citation26], but it is an intriguing hypothesis that temperature also alters the accessibility of cis-regulatory elements by changes in the RNA secondary structure [Citation37]. This idea is in line with a recent report showing that in vivo binding of RBPs is mainly determined by local RNA secondary structures [Citation38]. In case of U2af26, we find that exon inclusion is inhibited by colder temperature. Colder temperature could stabilise local RNA secondary structures including the enhancer elements and thereby prevent binding of SRSF2/7. In contrast, higher temperature could disturb this local secondary structures thereby allowing recognition of the enhancer elements. Therefore, temperature-responsive splicing could be a combined result of altered SR protein activity and altered availability of cis-regulatory elements.
In a global RNA sequencing approach, we were able to identify temperature-oscillating AS events. We further find that all tested splicing events are responsive to knockdown of several SR proteins (at least 2) indicating that oscillating AS is generally co-regulated by several SR proteins. We speculate that temperature changes effect the activity of several/all SR proteins, which would be consistent with our previously published Western blots against phosphorylated SR proteins indicating temperature-dependent SR protein phosphorylation for all SR repeat containing proteins [Citation26]. It seems possible that the phosphorylation differences resulting from moderate temperature changes (as under square wave temperature cycles) are mostly not sufficient to result in oscillating AS, if an event is regulated by a single SR protein. In contrast, if a splicing event is regulated by several SR proteins acting in concert – which are all modulated by temperature – it becomes sensitive to moderate temperature changes. Our iCLIP data additionally indicate that the position of the cis-regulatory elements is crucial for the direction of the splicing regulation: for SRSF2 we found elevated binding to the cold skipped variable exons (and the downstream exon), while heat skipped exons showed more binding to the upstream constitutive exon. These data are consistent with the following model ()): Binding of SRSF2 to the alternative exon (or the downstream exon) activates variable exon inclusion. However, upon cold induced hyper-phosphorylation SRSF2 becomes deactivated resulting in more exon skipping (I). In contrast, binding to the upstream exon promotes exon skipping. Here, cold mediated deactivation of SRSF2 results in more variable exon inclusion (II). This is consistent with the RNA binding pattern and the direction of splicing regulation observed for several D. melanogaster SR proteins [Citation39] and for SRSF2 in an MS2-tethering approach [Citation36]. If SRSF2 (or a different SR protein) co-regulates a splicing event together with a second SR protein (e.g. SRSF7 for U2af26) this can be sufficient for oscillating AS to occur (III). In summary, rhythmic AS is the result of at least two cis-acting elements bound by two different SR proteins that together use body temperature cycles as input to produce time of the day dependent AS patterns as output.
4. Materials and methods
4.1. Minigene constructs
Cloning was performed using 3-step PCRs, restriction digest (enzymes are indicated in the supplement), and ligation into pcDNA3.1(+). 2-exon constructs were cloned using a reverse primer in intron 6 and the 4-exon constructs as template. Primer sequences are available upon request. All minigene sequences were confirmed by sequencing. See supplementary materials and methods for minigene sequences.
4.2. Cell culture and transfections
We used DMEM medium for NIH3T3 and P19 cells (for P19 cells, plates were coated with 0.1% gelatin), N2A cells were maintained in 50% DMEM/50% OptiDMEM + GlutaMAX (life technologies). All media were supplemented with 10% FBS and Pen/Strep (Invitrogen). For temperature shocks, 12-well plates were sealed with parafilm and incubated swimming in a water bath with the indicated temperatures for 2 hours. For square-wave temperature cycles we used two incubators set to 34°C and 38°C and shifted the cells every 12 hours. Transfections of N2A and NIH3T3 using Rotifect (Roth) were performed according to the manufacturer’s instructions. For the siRNA screen, pools of four siRNAs targeting SR proteins were obtained from Dharmacon; these and other siRNA sequences are available upon request. All transfections were performed in 12-well format. Cells transfected with 0.8 µg minigenes (or co-transfected with overexpression vectors; 0.4 µg each) were harvested 48-hours after transfection. For minigene knockdown experiments N2A cells were first transfected with a single siRNA against Srsf2 or Srsf7 (final concentration: 0.08 µM), 24-hours later the minigenes were transfected and harvested 48-hours later for RNA extraction. Knockdown and overexpression of Srsf2 and Srsf7 were confirmed as previously described using Western blot and quantitative RT-PCRs (see [Citation26], also for cloning of Srsf2 and Srsf7 overexpression constructs). Additionally, SRSF7 levels upon overexpression and knockdown were investigated using an antibody against endogenous SRSF7 (C18943, AssayBioTech).
4.3. RNA extraction and RT-PCR
RNA extraction and radioactive RT-PCRs were done as previously described [Citation25]. Briefly, RNA was extracted using RNATri (Bio&Sell) and 1µg RNA was used in a gene-specific RT-reaction. For analysis of minigene splicing the RNA was additionally digested with DNase I and re-purified. Low-cycle PCR with a 32P-labeled forward primer was performed, products were separated by denaturing PAGE and quantified using a Phosphoimager and ImageQuantTL software. Quantifications are given as mean values of several independent biological replicates, error bars represent standard deviation, p-values were calculated using Student’s unpaired t-test. Significance is indicated by asterisks (*p < 0.05; **p < 0.01; ***p < 0.001). See supplementary material and methods for primer sequences (Table S2).
4.4. RNA-Seq and iCLIP analysis
For RNA-Seq P19 cells were synchronised using square wave temperature cycles (34°C 12-hours, 38°C 12-hours) for two days. The cells were harvested after 48 and 60 hours and total RNA was isolated from biological duplicates. Sequencing libraries were generated after removal of ribosomal RNA (RiboZero®), and the inserts were sequenced on a HiSeq2000 instrument yielding on average 65 million paired-end 75-bp reads. Reads were aligned to the mm10 reference genome using TopHat (version 2.0.13) with default parameters [Citation40]. Mixture of Isoforms (MISO) [Citation41] Bayesian Inference model (version 0.5.3) was then used to estimate splicing patterns. Events with a minimum PSI difference of 0.1 and a Bayes factor greater than 5 between all 34°C and all 38°C samples were included in the list of temperature responsive exons (Table S1).
Normalised significant iCLIP crosslink sites (peaks) mapped to mm9 were used to generate RNA maps [31]. Bedgraph files were imported into R using import.bedGraph and corrected for score and strand. The MISO output table including the genomic coordinates of the regulated and flanking exons were loaded into R and converted into a Granges object. The region around regulated exons (and upstream and downstream constitutive exons) was extended by 300 nt in both directions using flank. Crosslink sites were counted in the extended regions using countOverlaps and attributed to the 5’ss or 3’ss. RNA maps were plotted with R plot.
For in vivo binding motifs of SRSF2, a z-score analysis for enriched 8-mers around significant binding sites was performed as described previously [Citation31]. All occurrences of an 8-mer within the evaluated interval (−30, −5), (5, 30) were counted. The top enriched 30 k-mers were used to calculate the in vivo binding consensus motif. Ten nucleotides around the binding sites were excluded from the analysis to avoid crosslink bias. Search for enriched sequence motifs in cold-shock regulated exons was performed by MEME using default options [Citation42].
Supplemental Material
Download MS Word (5.9 MB)Acknowledgments
The authors would like to thank the HPC Service of ZEDAT, Freie Universität Berlin, for computing time, the ‘Forschungsförderung’ of the Freie Universität Berlin for ‘Initiativmittel’ to MP and members of the Heyd lab for constructive comments on this work. This work was funded through DFG grants HE5398/3 and HE5398/4 to FH. Additional support from the DFG came from the SFB958/A21 to FH and SFB902 to MMM. MP is founded by a post-doc stipend of the Peter and Traudl Engelhorn Foundation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
References
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. PubMed PMID: 12626338; eng.
- Pan Q, Shai O, Lee LJ, et al. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet. 2008 Dec;40(12):1413–1415. PubMed PMID: 18978789; eng.
- Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008 Nov 27;456(7221):470–476. PubMed PMID: 18978772; PubMed Central PMCID: PMCPmc2593745. eng.
- Tress ML, Abascal F, Valencia A. Alternative splicing may not be the key to proteome complexity. Trends Biochem Sci. 2017 Feb;42(2):98–110. PubMed PMID: 27712956; eng.
- Blencowe BJ. The relationship between alternative splicing and proteomic complexity. Trends Biochem Sci. 2017 Jun;42(6):407–408. PubMed PMID: 28483376; eng.
- Gabut M, Samavarchi-Tehrani P, Wang X, et al. An alternative splicing switch regulates embryonic stem cell pluripotency and reprogramming. Cell. 2011 Sep 30;147(1):132–146. PubMed PMID: 21924763; eng.
- Wollscheid HP, Biancospino M, He F, et al. Diverse functions of myosin VI elucidated by an isoform-specific alpha-helix domain. Nat Struct Mol Biol. 2016 Apr;23(4):300–308. PubMed PMID: 26950368; PubMed Central PMCID: PMCPMC4964928. eng.
- Kanemori Y, Koga Y, Sudo M, et al. Biogenesis of sperm acrosome is regulated by pre-mRNA alternative splicing of Acrbp in the mouse. Proc Natl Acad Sci U S A. 2016 Jun 28;113(26):E3696–E705. PubMed PMID: 27303034; PubMed Central PMCID: PMCPMC4932935. eng.
- Wilhelmi I, Kanski R, Neumann A, et al. Sec16 alternative splicing dynamically controls COPII transport efficiency. Nat Commun. 2016 Aug;05(7):12347. PubMed PMID: 27492621; PubMed Central PMCID: PMCPMC4980449. eng.
- Galarza-Munoz G, Briggs FBS, Evsyukova I, et al. Human epistatic interaction controls IL7R splicing and increases multiple sclerosis risk. Cell. 2017 Mar 23;169(1):72–84.e13. PubMed PMID: 28340352; PubMed Central PMCID: PMCPMC5456452. eng.
- Ellis JD, Barrios-Rodiles M, Colak R, et al. Tissue-specific alternative splicing remodels protein-protein interaction networks. Mol Cell. 2012 Jun 29;46(6):884–892. PubMed PMID: 22749401; eng
- Yang X, Coulombe-Huntington J, Kang S, et al. Widespread expansion of protein interaction capabilities by alternative splicing. Cell. 2016 Feb 11;164(4):805–817. PubMed PMID: 26871637; PubMed Central PMCID: PMCPmc4882190. eng.
- Barash Y, Calarco JA, Gao W, et al. Deciphering the splicing code. Nature. 2010 May 6;465(7294):53–59. PubMed PMID: 20445623; eng.
- Xiong HY, Alipanahi B, Lee LJ, et al. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science (New York, NY). 2015 Jan 09;347(6218):1254806. PubMed PMID: 25525159; PubMed Central PMCID: PMCPMC4362528. eng.
- Schultz AS, Preussner M, Bunse M, et al. Activation-dependent TRAF3 exon 8 alternative splicing is controlled by CELF2 and hnRNP C binding to an upstream intronic element. Mol Cell Biol. 2017 Apr 1;37(7). DOI:10.1128/mcb.00488-16. PubMed PMID: 28031331; PubMed Central PMCID: PMCPMC5359431. eng.
- Heyd F, Lynch KW. Degrade, move, regroup: signaling control of splicing proteins. Trends Biochem Sci. 2011 Aug;36(8):397–404. PubMed PMID: 21596569; PubMed Central PMCID: PMCPMC3155649. eng.
- Fu XD, Ares M Jr. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet. 2014 Oct;15(10):689–701. PubMed PMID: 25112293; PubMed Central PMCID: PMCPMC4440546. eng.
- Preussner M, Schreiner S, Hung LH, et al. HnRNP L and L-like cooperate in multiple-exon regulation of CD45 alternative splicing. Nucleic Acids Res. 2012 Jul;40(12):5666–5678. PubMed PMID: 22402488; PubMed Central PMCID: PMCPMC3384337. eng.
- Julien P, Minana B, Baeza-Centurion P, et al. The complete local genotype-phenotype landscape for the alternative splicing of a human exon. Nat Commun. 2016 May;10(7):11558. PubMed PMID: 27161764; PubMed Central PMCID: PMCPMC4866304. eng.
- Gao Q, Sun W, Ballegeer M, et al. Predominant contribution of cis-regulatory divergence in the evolution of mouse alternative splicing. Mol Syst Biol. 2015 Jul 01;11(7):816. PubMed PMID: 26134616; PubMed Central PMCID: PMCPMC4547845. eng.
- Barbosa-Morais NL, Irimia M, Pan Q, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science (New York, NY). 2012 Dec 21;338(6114):1587–1593. PubMed PMID: 23258890; eng.
- Lorson CL, Hahnen E, Androphy EJ, et al. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999 May 25;96(11):6307–6311. PubMed PMID: 10339583; PubMed Central PMCID: PMCPMC26877. eng..
- Herdt O, Neumann A, Timmermann B, et al. The cancer-associated U2AF35 470A>G (Q157R) mutation creates an in-frame alternative 5ʹ splice site that impacts splicing regulation in Q157R patients. RNA (New York, NY). 2017 Dec;23(12):1796–1806. PubMed PMID: 28893951; PubMed Central PMCID: PMCPMC5689001. eng.
- Scotti MM, Swanson MS. RNA mis-splicing in disease. Nat Rev Genet. 2016 Jan;17(1):19–32. PubMed PMID: 26593421; eng.
- Preussner M, Wilhelmi I, Schultz AS, et al. Rhythmic U2af26 alternative splicing controls PERIOD1 stability and the circadian clock in mice. Mol Cell. 2014 May 22;54(4):651–662. PubMed PMID: 24837677; eng.
- Preussner M, Goldammer G, Neumann A, et al. Body temperature cycles control rhythmic alternative splicing in mammals. Mol Cell. 2017 Aug 03;67(3):433–446.e4. PubMed PMID: 28689656; eng.
- Darlington TK, Lyons LC, Hardin PE, et al. The period E-box is sufficient to drive circadian oscillation of transcription in vivo. J Biol Rhythms. 2000 Dec;15(6):462–471. PubMed PMID: 11106063; eng.
- Bunger MK, Wilsbacher LD, Moran SM, et al. Mop3 is an essential component of the master circadian pacemaker in mammals. Cell. 2000 Dec 22;103(7):1009–1017. PubMed PMID: 11163178; PubMed Central PMCID: PMCPMC3779439. eng.
- Gekakis N, Staknis D, Nguyen HB, et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science (New York, NY). 1998 Jun 05;280(5369):1564–1569. PubMed PMID: 9616112; eng.
- Kaiser TS, Poehn B, Szkiba D, et al. The genomic basis of circadian and circalunar timing adaptations in a midge. Nature. 2016 Dec 01;540(7631):69–73. PubMed PMID: 27871090; PubMed Central PMCID: PMCPMC5133387. eng.
- Muller-McNicoll M, Botti V, de Jesus Domingues AM, et al. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev. 2016 Mar 1;30(5):553–566. PubMed PMID: 26944680; PubMed Central PMCID: PMCPmc4782049. eng.
- Cavaloc Y, Bourgeois CF, Kister L, et al. The splicing factors 9G8 and SRp20 transactivate splicing through different and specific enhancers. RNA (New York, NY). 1999 Mar;5(3):468–483. PubMed PMID: 10094314; PubMed Central PMCID: PMCPMC1369774. eng.
- Liu HX, Chew SL, Cartegni L, et al. Exonic splicing enhancer motif recognized by human SC35 under splicing conditions. Mol Cell Biol. 2000 Feb;20(3):1063–1071. PubMed PMID: 10629063; PubMed Central PMCID: PMCPMC85223. eng.
- Liu Y, Hu W, Murakawa Y, et al. Cold-induced RNA-binding proteins regulate circadian gene expression by controlling alternative polyadenylation. Sci Rep. 2013;3:2054. . PubMed PMID: 23792593; PubMed Central PMCID: PMCPmc3690385. eng.
- Han J, Ding JH, Byeon CW, et al. SR proteins induce alternative exon skipping through their activities on the flanking constitutive exons. Mol Cell Biol. 2011 Feb;31(4):793–802. PubMed PMID: 21135118; PubMed Central PMCID: PMCPMC3028638. eng.
- Erkelenz S, Mueller WF, Evans MS, et al. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA (New York, NY). 2013 Jan;19(1):96–102. PubMed PMID: 23175589; PubMed Central PMCID: PMCPMC3527730. eng.
- Narberhaus F, Waldminghaus T, Chowdhury S. RNA thermometers. FEMS Microbiol Rev. 2006 Jan;30(1):3–16. PubMed PMID: 16438677; Eng.
- Taliaferro JM, Lambert NJ, Sudmant PH, et al. RNA sequence context effects measured in vitro predict in vivo protein binding and regulation. Mol Cell. 2016 Oct 20;64(2):294–306. PubMed PMID: 27720642; PubMed Central PMCID: PMCPMC5107313.
- Bradley T, Cook ME, Blanchette M. SR proteins control a complex network of RNA-processing events. RNA (New York, NY). 2015 Jan;21(1):75–92. PubMed PMID: 25414008; PubMed Central PMCID: PMCPMC4274639. eng.
- Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012 Mar;7(3):562–578. PubMed PMID: 22383036; PubMed Central PMCID: PMCPmc3334321. eng.
- Katz Y, Wang ET, Airoldi EM, et al. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010 Dec;7(12):1009–1015. PubMed PMID: 21057496; PubMed Central PMCID: PMCPmc3037023. Eng.
- Bailey TL, Elkan C The value of prior knowledge in discovering motifs with MEME. Proc Int Conf on Intell Systems for Mol Biol. 1995;3:21–29. PubMed PMID: 7584439; eng.
- Pandit S, Zhou Y, Shiue L, et al. Genome-wide analysis reveals SR protein cooperation and competition in regulated splicing. Mol Cell. 2013 Apr 25;50(2):223–235. PubMed PMID: 23562324; PubMed Central PMCID: PMCPmc3640356. eng.