
ABSTRACT
Function of bacterial small non-coding RNAs (sRNAs) and overall RNA metabolism is largely shaped by a vast diversity of RNA-protein interactions. However, in non-model bacteria with defined non-coding transcriptomes the sRNA interactome remains almost unexplored. We used affinity chromatography to capture proteins associated in vivo with MS2-tagged trans-sRNAs that regulate nutrient uptake (AbcR2 and NfeR1) and cell cycle (EcpR1) mRNAs by antisense-based translational inhibition in the nitrogen-fixing α-rhizobia Sinorhizobium meliloti. The three proteomes were rather distinct, with that of EcpR1 particularly enriched in cell cycle-related enzymes, whilst sharing several transcription/translation-related proteins recurrently identified associated with sRNAs. Strikingly, MetK, the synthetase of the major methyl donor S-adenosylmethionine, was reliably recovered as a binding partner of the three sRNAs, which reciprocally co-immunoprecipitated with a FLAG-tagged MetK variant. Induced (over)expression of the trans-sRNAs and MetK depletion did not influence canonical riboregulatory traits, `for example, protein titration or sRNA stability, respectively. An in vitro filter assay confirmed binding of AbcR2, NfeR1 and EcpR1 to MetK and further revealed interaction of the protein with other non-coding and coding transcripts but not with the 5S rRNA. These findings uncover a broad specificity for RNA binding as an unprecedented feature of this housekeeping prokaryotic enzyme.
Introduction
Post-transcriptional regulation of gene expression by small non-coding RNAs (sRNAs) underlies virtually any physiological adaptation of bacteria to external changes [Citation1,Citation2]. A large class of sRNAs act by base-pairing with usually multiple trans-encoded mRNAs, thereby influencing message translation and/or decay [Citation3–6]. Trans-acting sRNAs and their target mRNAs typically exhibit short and discontinuous stretches of sequence complementarity, and thus their base-pairing interactions are inefficiently established without the assistance of proteins [Citation7]. The bacterial Sm-like protein Hfq has been long regarded as a major node in post-transcriptional RNA networks, promoting sRNA stability and facilitating base-pairing of trans-sRNAs with their targets [Citation6,Citation8]. Hfq substrates extend beyond sRNA transcripts to include other RNA species and even DNA [Citation9]. Nevertheless, Hfq is not ubiquitous (ie almost half of species lack a recognizable homolog) and in many bacteria expressing a canonical Hfq, particularly gram-positive, this protein has limited or even null contribution to riboregulation [Citation6,Citation10]. More recently, gradient profiling by sequencing (Grad-seq) of Salmonella ribonucleoprotein complexes identified ProQ as a novel binding partner of a large set of highly structured sRNAs, which envisages a global Hfq-like role of this protein in trans-encoded mRNA regulation and virulence [Citation11–13]. Indeed, both Hfq and ProQ share several RNA partners, indicating overlapping or competing functions of these two RNA chaperones [Citation14]. However, ProQ is not as widespread as Hfq in bacteria, which suggests that other yet undiscovered RNA-binding proteins (RBPs) with constrained phylogenetic distribution or unique to particular species may fulfil a chaperone function in riboregulation [Citation15]. Affinity chromatography using aptamer-tagged RNAs as baits has been one of the experimental approaches of choice to capture the proteomes associated with a number of trans-sRNAs in enterobacteria and in the Hfq-less ε-proteobacteria Helicobacter pylori [Citation16–19]. This procedure consistently recovered three major proteins as common interacting partners of E. coli trans-sRNAs controlling target mRNA translation; Hfq, the small ribosomal subunit protein S1 and the β–subunit of the RNA polymerase (RNAP) [Citation19]. Reciprocally, stoichiometric amounts of Hfq and ribosomal protein S1 have been found in RNAP preparations from E. coli cultures, further suggesting that these three proteins are in vivo assembled into a complex of a yet unknown role in riboregulation [Citation20]. A functionally relevant Hfq-interacting protein is the major single-strand endoribonuclease RNase E, which is a catalytic component of the enterobacterial RNA degradosome that also includes the exoribonuclease polynucleotide phosphorylase (PNPase), RNA helicase RhlB and the glycolytic enzyme enolase [Citation21]. Through interaction with RNase E, Hfq recruits the degradosome complex to the sRNA-mRNA interplay, thus promoting irreversible target mRNA degradation subsequent to primary sRNA-mediated translational repression [Citation22].
Instead of acting by base-pairing interactions with mRNAs, several sRNAs bind to and antagonize the activity of certain proteins. This minor class of RNA regulators typically mimic DNA or RNA motifs that are specifically recognized by certain proteins. The widely conserved 6S RNA and the CsrB family of sRNAs are well-characterized examples of riboregulators that act by target mimicry, outcompeting the σ70 RNAP holoenzyme and the carbon storage regulator CsrA at their cognate targets in gene promoters and mRNAs, respectively [Citation23,Citation24]. Base-pairing and protein titration have long been considered mutually exclusive mechanisms of sRNA activity. A remarkable exception is McaS, an E. coli trans-RNA that relies on a dual mechanism involving both Hfq-dependent antisense interaction with a target mRNA and CsrA titration for controlling biofilm formation [Citation25]. More recently, a new role has been uncovered for CsrA as RNA matchmaker in Bacillus subtillis, which further highlights the functional plasticity and diversity of the RBPs for the regulation of transcription, translation and RNA turnover [Citation26,Citation27].
Sinorhizobium meliloti is a genetically tractable soil-dwelling α-proteobacterium that is well known for its ability to establish nitrogen-fixing endosymbiosis with legume plants [Citation28]. Previous high-throughput surveys revealed that S. meliloti expresses hundreds of trans-sRNAs with regulatory potential, but scarcely six of those have assigned functions [Citation29–34]. Here we focus on three characterized stress-induced S. meliloti trans-sRNAs that are widely conserved in α–proteobacteria; AbcR2 (ABC transporter RNA2) belonging to the so-called αr15 family [Citation29], NfeR1 (nodule formation efficiency RNA1; αr14 family), with symbiotic functions [Citation33], and EcpR1 (elongated cell phenotype RNA1; SmelC291 family), which is involved in cell-cycle progression [Citation30]. These sRNAs primarily act by canonical antisense base-pairing to downregulate nutrient uptake (AbcR2 and NfeR1) and cell-cycle master regulators (EcpR1) mRNAs [Citation30,Citation33,Citation35]. The S. meliloti genome encodes a functional Hfq chaperone but only 14% of the annotated trans-sRNAs co-immunoprecipitate with this protein [Citation35]. This set includes AbcR2 but not NfeR1 and EcpR1. In this work, we have used affinity chromatography to pull-down proteins associated with MS2-tagged versions of these sRNAs. Besides several proteins functionally related to the flow of genetic information, we identified the enzyme S-adenosylmethionine (SAM) synthetase (MetK) as a binding partner of AbcR2, NfeR1 and EcpR1. In vitro assays not only confirmed binding of MetK to these sRNA transcripts but also uncovered the ability of this metabolic enzyme to interact with other RNA species.
Results
Aptamer-tagged trans-sRNAs are stably expressed in S. meliloti
S. meliloti sRNAs were tagged at their primary (AbcR2, NfeR1) or prevalent processed 5ʹ-end (EcpR1) with two different MS2 tandem repeats of either 43 nt (MS2) or 50 nt (MS2*). For that, engineered plasmid pSRK-C was used as a backbone for cloning and constitutive (over)expression of the full-length aptamer-tagged sRNAs [Citation29] (). As predicted by the RNAfold algorithm, fusion of the sRNAs to either of the aptamer variants preserved the predicted functional stem loops of the wild-type transcripts [Citation29,Citation30,Citation33,Citation35,Citation36] (Supplementary Fig. S1). As control vectors, we used pSRKMS2-Term or pSRKMS2*-Term, which express the corresponding tag followed by a canonical Rho-independent transcriptional terminator (T1). All plasmid constructs were first conjugated individually into an S. meliloti strain expressing a 3× FLAG-tagged Hfq from its chromosomal locus (SmhfqFLAG) [Citation35].
Figure 1. Expression of aptamer-tagged trans-sRNAs in S. meliloti. (A) Schematics (drawn to scale) of the genetic constructs to express the MS2/MS2*-tagged AbcR2, NfeR1 and EcpR1 sRNAs, and the Term control from an engineered constitutive Plac’ promoter. The expected length (nt) of the RNA species derived from each construct is indicated. The arrowhead indicates the processing site of the full-length EcpR1 wild-type transcript. Sites used for cloning were BamHI (B), XbaI (X) and HindIII. (B, C, and D) Northern blot detection of aptamer-tagged sRNAs. Total RNA from strain SmhfqFLAG transformed with either pSRK-MS2-Term, pSRKMS2/MS2*-AbcR2, pSRKMS2/MS2*-NfeR1 or pSRKMS2/MS2*-EcpR1, was probed with specific oligonucleotides targeting AbcR2 (A), NfeR1 (B) and EcpR1 (C), respectively. The detected RNA species and their length (nt) are indicated to the left of each panel. 5S rRNA was probed as RNA loading control. Lanes: 1, MS2-Term-expressing cells; 2, bacteria expressing the corresponding MS2-tagged sRNA; 3, bacteria expressing MS2*-tagged sRNA. Expression of wild-type endogenous sRNAs was induced by growth of bacteria in MM to log phase (NfeR1) or by an osmotic upshift (AbcR2 and EcpR1)
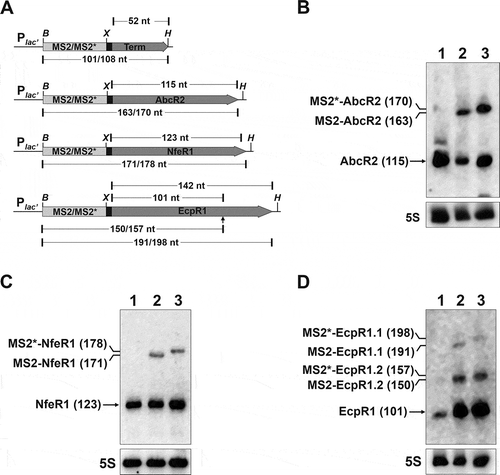
To assess whether the aptamers affected sRNA expression and stability we subjected RNA from all transconjugants to Northern analysis. Bacteria were previously grown in conditions promoting sRNA expression from the respective chromosomal loci, that is, salt stress for AbcR2 and EcpR1 and minimal medium (log phase) for NfeR1. Membranes were subsequently probed with radiolabelled oligonucleotides targeting the corresponding sRNA, the 5S rRNA (), and the MS2/MS2* tag (Supplementary Fig. S2A). The 101-nt and 108-nt RNA species expressed from pSRKMS2-Term and pSRKMS2*-Term, respectively, were reliably detected in control bacteria, indicating that MS2/MS2* aptamer transcription ends efficiently when followed by the T1 terminator (Supplementary Fig. S2A). Aptamer-tagged 163-nt and 170-nt AbcR2 sRNAs exhibited comparable expression levels (). The NfeR1 variants (171 and 178 nt-long transcripts), although reliably detected, did not reach the high levels of the endogenous wild-type molecule (123-nt) ( and Supplementary Fig. S2A). Considering that NfeR1 is among the S. meliloti transcripts with highest coverage in reported RNAseq datasets [Citation33,Citation37,Citation38], this is more likely due to the different strength of the endogenous and plasmid promoters rather than to a lower stability of the tagged sRNA. MS2/MS2*-EcpR1 versions are processed at the 3ʹ-end similar to the chromosomally encoded EcpR1 [Citation30], promoting the accumulation of major 150-nt (MS2-EcpR1.2) and 157-nt (MS2*-EcpR1.2) long transcripts along with the less-abundant full-length 191-nt (MS2-EcpR1.1) and 198-nt (MS2*-EcpR1.1) variants ().
We used the experimentally confirmed AbcR2-prbA interaction [Citation35] to test if the tagged sRNAs retained their ability for target regulation (Supplementary Fig. S2B). A S. meliloti AbcR2 deletion mutant (Rm1021ΔR2) [Citation29] harbouring the reporter plasmid pRprba::egfp was independently transformed with the control plasmids pSRK-C or pSRKMS2/MS2*-Term, or with constructs constitutively expressing the wild-type AbcR2 transcript (pSRK-R2) and its tagged variants (pSRKMS2/MS2*-AbcR2). Fluorescence of double transconjugants with the different plasmids combinations confirmed that MS2-AbcR2, but not MS2*-AbcR2, significantly reduced prbA expression to almost the same extent as wild-type AbcR2, indicating that the MS2 aptamer version did not affect the regulatory properties of the sRNA. Therefore, we used the shorter MS2 version as the tag of all three sRNAs for further assays.
AbcR2 is a Hfq-dependent sRNA [Citation29,Citation35]. Hence, to optimize the affinity purification procedure for S. meliloti, we reasoned that Hfq should be enriched in the elution fraction obtained from cells transformed with pSRKMS2-AbcR2, as compared to the controls [Citation39]. The SmhfqFLAG strain enabled Western-blot tracking of Hfq during the affinity purification process. Successful-specific Hfq recovery in the MS2-AbcR2 eluate was taken as the reference for setting the appropriate affinity chromatography variables, that is, starting number of cells, salt concentration in buffer and incubation conditions. Following the optimized protocol described in Materials and Methods, Hfq was found enriched in the MS2-AbcR2 elution fraction (Supplementary Fig. S3A, top panel), thus supporting the reliability of the method. A silver-stained polyacrylamide gel showed enrichment of MS2-MBP (Maltose Binding Protein) in the eluted fractions, also anticipating distinct proteome profiles associated with MS2-AbcR2 and control samples (Supplementary Fig. S3A, bottom panel).
Identification of sRNA-protein partners
Plasmids expressing the MS2-Term control, the 163-nt MS2-AbcR2, the 171-nt MS2-NfeR1, or the 191-nt MS2-EcpR1 RNA species were next mobilized to the corresponding sRNA deletion mutant strain. The available plasmids pSRK-R2 [Citation29] and pSRK-NfeR1 [Citation33], constitutively expressing the wild-type untagged AbcR2 and NfeR1 sRNAs, respectively, were also used as negative controls to assess unspecific protein binding in affinity chromatography. As a similar control for EcpR1, we generated pSRK-EcpR1, expressing its stable and functional 101-nt processed form [Citation30]. Specific recovery of tagged sRNAs in the eluted fractions was systematically verified by RT-PCR (Supplementary Fig. S3B). Proteins bound to controls and tagged sRNAs were first resolved by SDS-PAGE, which revealed distinct protein patterns across samples (Supplementary Fig. S3C) that were further analysed by liquid chromatography-mass spectrometry (LC-MS/MS) (Supplementary Table S1, ‘RAW’ sheet). Confirming the Western-blot data, Hfq was exclusively detected as a partner of the tagged AbcR2 sRNA.
MS analysis identified a total of 198 proteins with a false discovery rate (FDR) ≤2% ( and B). Data obtained from MS2-sRNA samples presented a poor correlation (r = 0.26–0.34, Supplementary Fig. S3D), indicating a strong specificity of protein partners for each sRNA. Moreover, this weak correlation was also observed when compared the control samples (r = 0.03–0.43, Supplementary Fig. S3D). Hence, given that control samples that were used to demarcate non-specific binders showed different protein complexity, we decided to exclude the 92 proteins identified in the controls for further analysis. Thus, we analysed the distribution of the 106 proteins exclusively present in the MS2-sRNA samples (Supplementary Table S1, ‘Putative sRBPs’ sheet). Fifteen proteins were identified associated with MS2-AbcR2 (40% identified with more than 1 peptide), 11 with MS2-NfeR1 (36%) and 95 with MS2-EcpR1 (60%). Of these, seven proteins were exclusive of MS2-AbcR2, four of MS2-NfeR1 and 81 of MS2-EcpR1, which showed a considerable higher number of associated proteins. MS2-EcpR1 dataset shared seven proteins with MS2-AbcR2 and six with MS2-NfeR1 but there is no overlap between the last two samples. Interestingly, MetK (a conserved SAM synthetase) was the only protein recovered with all the MS2-tagged sRNAs but not with any of the controls.
Figure 2. Identification of proteins associated with the sRNAs. (A) Heatmap representation of protein abundance as revealed by MS. Columns refer to the different pSRK-MS2-sRNAs used for these experiments and the respective controls, rows represent individual proteins. For visualization purposes, the white-blue colour bar represents the number of peptides identified for the corresponding proteins. (B) Venn diagram showing the overlap of all identified proteins within tagged-sRNAs and all control samples. (C) Heatmap representation of enriched GO terms (rows) from comparative GO [Citation72] among proteins exclusively present in the MS2-sRNA samples indicated in the columns. The red-blue colour bar represents the degree of enrichment (p-value) for the described GO terms in the samples obtained by hypergeometric test when considering the 6,177 genes of S. meliloti 1021 strain annotated in the GO database as the background set
![Figure 2. Identification of proteins associated with the sRNAs. (A) Heatmap representation of protein abundance as revealed by MS. Columns refer to the different pSRK-MS2-sRNAs used for these experiments and the respective controls, rows represent individual proteins. For visualization purposes, the white-blue colour bar represents the number of peptides identified for the corresponding proteins. (B) Venn diagram showing the overlap of all identified proteins within tagged-sRNAs and all control samples. (C) Heatmap representation of enriched GO terms (rows) from comparative GO [Citation72] among proteins exclusively present in the MS2-sRNA samples indicated in the columns. The red-blue colour bar represents the degree of enrichment (p-value) for the described GO terms in the samples obtained by hypergeometric test when considering the 6,177 genes of S. meliloti 1021 strain annotated in the GO database as the background set](/cms/asset/57f13dc4-fb16-42aa-a54f-d9a9ac11f33f/krnb_a_1829365_f0002_oc.jpg)
Proteins specifically identified in the MS2-EcpR1 sample were enriched for metabolism, such as the biosynthetic process of different amino acids (isoleucine P < 3 × 10−4, valine P < 9 × 10−4), glycolysis (P < 0.02) and the pentose phosphate shunt (P < 0.02); as well as for cellular functions such as DNA replication (P ≤ 0.05) and DNA binding (P < 4 × 10−5) (), both related to cell-cycle regulation, which is the role uncovered previously for EcpR1 [Citation30]. The proteomes associated with the three sRNAs were enriched for terms related to translation, for example, ‘translation’ (P < 0.05, P < 7 × 10−7 and P < 0.0001 for AbcR2, NfeR1 and EcpR1, respectively), ‘structural constituent of ribosome’ (P < 0.01, P < 3 × 10−7 and P < 0.0002) and ‘rRNA binding’ (P < 4 × 10−5 and P < 0.0006 for NfeR1 and EcpR1, respectively) ().
Some of these proteins are suspected to be assembled into higher-order complexes with Hfq [Citation21]. Therefore, to discriminate between proteins directly bound to the Hfq-dependent AbcR2 sRNA and those putatively complexed with Hfq, we subjected the HfqFLAG associated proteome (ie proteins co-immunoprecipitated with HfqFLAG) to MS analysis and compared it with that of the MS2-AbcR2 eluate (Supplemental Table S2). Only Hfq was common to both protein sets, whereas none of the known enterobacterial Hfq interacting proteins (eg RNase E, ribosomal protein S1 or RNAP) [Citation19,Citation21,Citation40] were identified in our study as S. meliloti Hfq partner. However, PNPase, some ribosomal proteins (including S1) and RpoA (RNA polymerase) co-purified with either one or two of the sRNAs under study. These findings anticipate major differences in content and assembly mechanisms of the protein complexes assisting riboregulation in distantly related bacteria.
In vivo validation of novel sRNA-protein interactions
Because our experimental approach was conceived as an initial screen for sRNA-binding proteins, we next sought to validate some of the putative sRNA–protein interactions to set a stringent threshold and further select the bonafide sRNA-protein partners among the candidates identified by LC-MS/MS. For this, we performed a complementary reverse approach in which FLAG-tagged proteins were expressed from an IPTG-inducible promoter in pSRKKm derivatives. CoIP-RNA samples were subsequently screened for the presence of the expected sRNA partners by RT-PCR (). As a control, we used cells transformed with the pSK-FLAG empty vector. We tagged a set of protein candidates that fulfilled the following criteria: (i) proteins exclusively present in one, two or all the MS2-tagged samples – Hfq, MinD (cell cycle-related protein), DnaN (DNA polymerase III), Rrf (ribosome recycling factor), RpoA and MetK.; (ii) proteins associated with sRNAs but also present at least in one control sample – DnaK (heat shock protein 70) and HspC2 (heat shock protein). All the tagged proteins were detectable in the cell lysates upon IPTG-induction, stably produced and efficiently recovered by IP (Supplementary Fig. S4A and B).
Figure 3. In vivo confirmation of specific sRNA-protein complexes. Agarose gel showing RT-PCR products for detection of sRNAs (right) in CoIP-RNA eluates of the indicated FLAG-tagged proteins or FLAG negative control (top). cDNA synthesized from total RNA isolated from stationary cells grew in TY or MM was used as positive control of sRNA expression. Length (bp) of the PCR products is indicated to the left
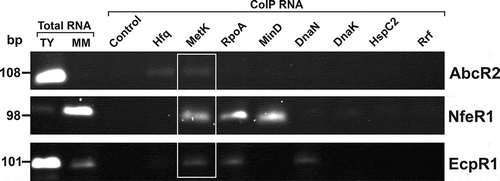
Before reverse transcription, all CoIP-RNA samples were systematically checked for the absence of contaminant DNA by PCR amplification with primer pairs specific for each sRNA (Supplementary Fig. S4C). As expected, RT-PCR revealed that AbcR2 was specifically enriched in the Hfq CoIP-RNA () whereas none of the sRNAs were detectable in the FLAG control. This analysis did not confirm the presence of any of the sRNAs in CoIP-RNA from tagged-proteins that appeared in at least one of the initial control samples, irrespective the number of peptides (). These results further support the removal of all crossed controls from our final list of putative RBPs (Supplementary Table S1, ‘Putative sRBPs’ sheet). We further confirmed specific enrichment of NfeR1 and EcpR1 in MinD-FLAG and DnaN-FLAG CoIP-RNAs, respectively, whereas both sRNAs were identified in RpoA-FLAG CoIP-RNA. Conversely, we were not able to detect EcpR1 upon IP in the strain accumulating Rrf-FLAG (). Confirming the interaction with MetK, the three sRNAs were recovered in CoIP-RNA from MetK-FLAG expressing bacteria. According to these results, we only considered as bonafide partners of the sRNA under study proteins identified with at least two peptides or one peptide but sequence coverage ≥10% and not present in any of the controls (Supplementary Table S1, ‘Putative sRBPs’ sheet, highlighted in grey). Applying these criteria, we have finally kept 66 proteins from our starting catalogue as the most probable sRNA partners: 10 for AbcR2 (6 exclusives), 4 for NfeR1 (2 exclusives) and 58 for EcpR1 (53 exclusives). This curated list of candidates kept the functional enrichment described above.
MetK-sRNA interaction does not influence canonical riboregulatory traits
In bacteria, sRNA–protein interactions typically result in either interference with protein activity or sRNA protection from degradation. Since MetK is not a recognizable RNA chaperone, we first assessed whether binding to NfeR1, AbcR2 and EcpR1 influenced SAM homoeostasis. We reasoned that SAM accumulation might parallel increased transcription of metK, which occurs in a single copy and is essential in S. meliloti. We, therefore, constructed a metK conditional deletion strain (SmΔmetK) that was complemented with plasmid pSKmetKFLAG expressing a FLAG-tagged MetK protein upon IPTG induction, as verified by Western-blot (Supplementary Fig. S5A). Leaky transcription of metKFLAG from the inducible lacZ promoter was enough to render cells viable (data not shown). We next used this strain as recipient of a translational fusion of the S. meliloti metZ gene to the eGFP reporter, that includes a predicted SAM-II riboswitch (pRmetZ::eGFP) (Supplementary Fig. S5B). MetZ is involved in methionine biosynthesis, which is inhibited by the MetK-dependent intracellular SAM accumulation presumably sensed by the riboswitch fused to eGFP. As predicted, the fluorescence of SmΔmetK bacteria co-transformed with plasmids pSKmetKFLAG and pRmetZ::eGFP decreased as MetK accumulated with increased concentrations of the inducer (Supplementary Fig. S5B). These data validate the metZ SAM-II riboswitch as reporter of intracellular SAM levels in S. meliloti. Therefore, we next mobilized pRmetZ::eGFP to NfeR1/AbcR2 and EcpR1 deletion mutants (Sm2B2020 and Sm4011ecpR1) and co-transformed the resulting strains with plasmids expressing each sRNA upon IPTG induction, ie Sm2B2020 with pSKNfeR1+ or pSKAbcR2+, and Sm4011ecpR1 with pSKEcpR1 +. Double transconjugants were grown in TY broth to exponential phase and then induced for sRNA expression with 0.5 mM IPTG for a further 24 h. Induced expression of NfeR1 and EcpR1 in these conditions has been shown to result in productive target regulation and gain-of-function phenotypes [Citation30,Citation33]. In our assays, the fluorescence of the reporter strains was not altered upon IPTG addition to cultures (Supplementary Fig. S5C), suggesting that overexpression of the sRNAs did not influence SAM levels. On the other hand, Western-blot probing of lysates from an S. meliloti derivative strain expressing MetKFLAG from the chromosome (SmmetKFLAG) cultured in conditions that promote endogenous expression of the three sRNAs (ie stationary phase and salt shock) did not reveal obvious alterations of MetK accumulation (Supplementary Fig. S5D; left panel), consistent with the housekeeping function of the enzyme. In line with this observation, similar experiments with Sm2B2020 harbouring either pSKNfeR1+, pSKAbcR2+ or pSKEcpR1+, revealed that accumulation of MetK was not altered either by sRNA-induced expression (Supplementary Fig. S5D; right panel).
Finally, we tested whether MetK depletion influenced sRNA stability using NfeR1 as a proof of principle. This sRNA was reliably detected by Northern blot probing of MetKFLAG CoIP-RNA from SmmetKFLAG (Supplementary Fig. S6A). However, its accumulation pattern in rifampicin-treated SmΔmetK bacteria complemented with pSKmetKFLAG was not altered regardless IPTG-induced MetK levels (Supplementary Fig. S6B). Together these results hint at a non-canonical novel function of MetK as sRNA-binding protein in bacteria.
In vitro assays confirmed ability of MetK to bind trans-sRNAs and other RNA species
To further confirm MetK-sRNA interactions we performed binding assays with the purified protein and radiolabelled in vitro transcribed full-length AbcR2, NfeR1 and EcpR1 RNA species. Mass spectrometry discarded contamination of our MetK preparation with Hfq, as a possibility reported previously for other Ni-affinity purifications of His-tagged proteins from E. coli [Citation41]. Binding reactions were analysed by dot-blot on nitrocellulose and polyvinylidene difluoride (PVDF) membranes that collect RNA-protein complexes and free RNA, respectively (). Increased protein concentrations in the reaction mixtures resulted in reliable detection of increased amounts of bound sRNA for all three transcripts. KD values derived from data of three independent experiments were 45.6, 21.2 and 6.7 nM, for AbcR2, NfeR1 and EcpR1, respectively, indicating different affinities of each sRNA for association with MetK (). Addition of a 100-fold molar excess of cold sRNA outcompeted binding of the radiolabelled sRNAs to MetK, whereas each sRNA was exclusively recovered in the PVDF membrane upon incubation with 2 µM bovine serum albumin (BSA), which has no recognized RNA-binding ability (Supplementary Fig. S7). These two negative binding controls validate all three sRNA–MetK interactions.
Figure 4. In vitro analysis of MetK-RNA interactions. Dot-blot assays to test binding of MetK to AbcR2, NfeR1, EcpR1, SmelC812, tRNAMet, RmInt1, SMb20420 and 5S RNA species. Radiolabelled transcripts (*) alone (1 nM) or with a molar excess of the corresponding cold RNA (cRNA; 100 nM) were incubated with increased concentrations of purified MetK as indicated on top of the panel. RNA-protein complexes and free RNA were collected by blotting of the reaction mixtures on nitrocellulose (left) and PVDF (right) membranes, respectively. Signal intensities were plotted in the graphs shown to the right. Binding affinities (KD values) quoted in the text were calculated from data of three (in some cases four) independent experiments
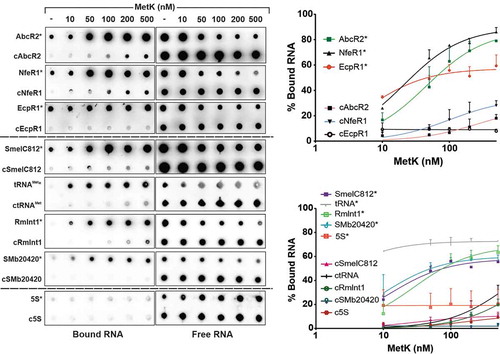
To assess specificity of MetK interaction with RNA, in a new series of experiments, we similarly probed MetK binding to a set of different Hfq-independent RNA species expressed by S. meliloti (): the antisense sRNA SmelC812 (165 nt) [Citation38], tRNAMet (76 nt), the group II ribozyme RmInt1 (748 nt) [Citation42], the mRNA annotated as SMb20420 (627 nt) and the 5S rRNA (120 nt). The assays revealed binding of MetK to all these in vitro synthesized transcripts except for the 5S rRNA. In this case, KD values calculated from 3 to 4 independent assays were 12.4, 1.3, 28.4 and 10.31 nM for SmelC812, tRNAMet, RmInt1 and SMb20420, respectively, predicting a remarkable strong binding affinity of MetK for tRNA substrates. Together, our data thus uncover an unexpected promiscuity of the metabolic enzyme MetK for RNA binding.
Discussion
The non-coding transcriptome of the legume symbiont S. meliloti is one of the best characterized among those of its α-proteobacterial counterparts [Citation34,Citation37,Citation38]. However, besides the well-known RNA chaperone Hfq, the repertoire of RBPs and their role in riboregulation remains unexplored in this bacterium [Citation35]. The reported profiling of glycerol gradient-sorted ribonucleoprotein complexes in Salmonella identified ProQ as a novel bacterial RNA chaperone with expected widespread Hfq-like functions [Citation11,Citation12,Citation14]. ProQ orthologues are identifiable by a domain of the FinO protein that mediates antisense RNA regulation of F plasmid transfer [Citation15]. However, in the large α–subgroup of proteobacteria, the occurrence of ProQ/FinO-domain proteins is likely restricted to the species Rhizobium leguminosarum and Caulobacter cresecentus [Citation15]. Similarly, rhizobial genomes lack genes putatively encoding members of the well-characterized CsrA translational inhibitors [Citation43]. The larger sets of sRNAs uncovered in these environmentally relevant α-proteobacteria as compared to enterobacteria, and the known functional protein redundancy (eg RNases) anticipate an even larger number of RBPs encoded by the multipartite rhizobial genomes. Therefore, specific screens are required to identify RBPs with putative roles in riboregulation and overall RNA metabolism within this group of bacteria. Here, we describe S. meliloti sRNA-associated proteomes as revealed by affinity chromatography-based capture. Remarkably, this screening and its further validation by independent methodologies identified the synthetase of the major methyl donor MetK as unexpected non-canonical bacterial sRNA-binding protein. Notably, our results also provide evidence of a striking ability of MetK for binding functionally diverse RNA species.
We selected as baits three characterized S. meliloti trans-sRNAs, which are widely conserved in α-proteobacterial species, namely AbcR2, NfeR1 and EcpR1 [Citation44,Citation45]. These sRNAs down-regulate their corresponding sets of target mRNAs via short and imperfect antisense interactions at the translation initiation region [Citation29,Citation30,Citation33]. This canonical RNA silencing mechanism is known to be assisted by RNA-chaperones and ribonucleases, but only Hfq, and the RNases YbeY and RNase E have been related to the activity of functionally characterized S. meliloti sRNAs [Citation29–31,Citation46]. To screen for proteins associated with AbcR2, NfeR1 and EcpR1, we have adapted an in vivo affinity chromatography approach previously described to identify sRNA-protein partners in E. coli [Citation18]. This method is based on the tagging of the sRNA of interest with the MS2 aptamer that specifically binds to the fusion protein MS2-MBP, which enables column immobilization of the in vivo assembled sRNA-protein complexes [Citation39]. Due to the variability of the phage MS2 coat protein binding motif [Citation47,Citation48], we tested two MS2 variants of different length (MS2 and MS2*) fused to the 5ʹ-end of the sRNAs. The rationale of choosing 5ʹ-end tagging is based on transcription homogeneity and higher efficiency for target regulation already reported for other bacterial sRNAs modified in this position [Citation18]. Indeed, Northern blot probing reliably detected accumulation of all the tagged transcripts as discrete full-length RNA species. However, we noticed that only tagging with the shorter MS2 version preserved the regulatory ability of the sRNAs, as verified with the AbcR2-prbA regulatory pair. Likewise, MS2*-tagged E.coli sRNAs of similar length have been also observed impaired for target regulation [Citation18].
Affinity chromatography captured rather different protein sets as putative binding partners of AbcR2, NfeR1 and EcpR1 sRNAs. Despite the stringent criteria used to filter out the most probable unspecific binders the final list of candidates was unexpectedly large, particularly for EcpR1. Nonetheless, further supporting the accuracy of the approach and the selection thresholds, Hfq was specifically recovered associated with AbcR2. Intriguingly, no promising candidates to fulfil an Hfq-like chaperone role (ie with recognizable RNA-binding domains; RBDs) were envisaged among the protein partners of the Hfq-independent NfeR1 and EcpR1 sRNAs. However, all the three proteomes shared components related to transcription, translation and RNA turnover. Similarly, this procedure has repeatedly captured proteins functionally related to these processes (eg RNA polymerase, ribosomal protein S1, or RNases) as trans-sRNA partners in enterobacteria and H. pylori [Citation11,Citation17,Citation19]. Regardless of their involvement in the control of different biological processes, most bacterial trans-sRNAs used as baits for the identification of RBPs share translation inhibition of the target mRNAs as regulatory mechanism. The recurrent co-purification of the core components of the RNA polymerase and ribosomal proteins with these sRNAs suggest coupling of transcription with sRNA-mediated translational control by a hitherto uncharacterized universal mechanism in bacteria. Our data do not predict the arrangement of the identified S. meliloti sRNA-binding proteins into higher order protein complexes with similar content to those involved in riboregulation in model enterobacteria, for example, the Hfq-containing degradosome. Rather, the findings suggest that these proteins are either direct partners of the tested sRNAs or associate to a yet unidentified novel protein scaffold.
Besides the proteins related to the flow of genetic information, our curated lists of putative RBPs contain a striking number of metabolic enzymes, particularly associated to EcpR1, which is unprecedented among bacterial proteins linked to riboregulation. Nonetheless, it has been already reported that binding of the small alarmone synthetase RelQ from the Gram-positive pathogen Enterococcus faecalis to single-stranded RNA allosterically antagonizes the enzyme activity [Citation49,Citation50], thus anticipating the ability of metabolic enzymes to bind RNA in bacteria. In addition, recent screenings for RBPs in eukaryotic organisms have revealed that a large fraction of metabolic enzymes indeed bind to polyadenylated RNAs via non-classical RBDs [Citation51–53]. Moreover, comprehensive studies of two conserved enzymes across all animal kingdoms, glyceraldehyde-3-phosphate and aconitase, demonstrated that these proteins have dual functions where RNA binding is incompatible with the enzymatic activity [Citation54–56]. Interestingly, previous transcriptomic data revealed that the induction of the S. meliloti EcpR1 sRNA via the alarmone nucleotide guanosine tetraphosphate (ppGpp) as second messenger results in the differential expression of metabolic-related genes [Citation30]. These findings add further evidence supporting the anticipated occurrence of different regulatory layers interconnecting metabolism and cell cycle in bacteria and eukaryotic cells [Citation57,Citation58]. In this scenario, EcpR1 could be a key hub of the S. meliloti cell cycle regulatory network modulating both accumulation of mRNAs and activity of metabolic enzymes involved in the process.
Affinity chromatography, CoIP with the FLAG-tagged protein and in vitro assays unambiguously confirmed that one of such metabolic enzymes, MetK, is a common interacting partner of AbcR2, NfeR1 and EcpR1 sRNAs. Massive binding to sRNAs may hint at a chaperone-like role of MetK in promoting RNA stability. However, probing of NfeR1 sRNA upon rifampicin treatment of MetK-depleted cells argued against this possibility. As documented for other prokaryotic and eukaryotic RNA–protein interactions, we, therefore, considered protein titration as a plausible consequence of MetK binding to sRNAs. To test this hypothesis, we set up a SAM-II riboswitch-based assay that reliably sensed increased SAM accumulation upon MetK synthesis, thus becoming a novel genetic tool to investigate the biology of macromolecule methylation in rhizobia. However, our assays did not reveal either significative alterations of intracellular SAM levels or MetK accumulation upon endogenous or induced (over)expression of either of the three sRNAs. These findings predict that the function of MetK in riboregulation, if any, is not canonical.
Of note, our in vitro assays reliably revealed that MetK is also able to bind other non-coding and protein-coding RNA species with diverse cellular functions, thus anticipating an unexpected promiscuity of a protein lacking recognizable RBDs for RNA binding in bacteria. All the tested transcripts are highly divergent in length (76 to 748 nt) and primary nucleotide sequence, which suggests that interaction with MetK most likely relies on the recognition of structural modules with widespread occurrence among bacterial transcripts, as reported for ProQ [Citation59]. In this regard, MetK evidenced a particular strong affinity for binding to tRNA. This broad RNA-binding specificity is also a feature of well-characterized RNA-chaperones such as Hfq. The plethora of RNA ligands and molecular interactions explain the diversity of newly discovered Hfq functions in protein synthesis beyond its recognized role in sRNA-mediated regulation [Citation9]. To date, MetK has been solely viewed as a key enzyme involved in macromolecule metabolism and epigenetic control of gene expression. Thus, our data add this protein to the emerging group of moonlighting enzymes with likely novel functions in riboregulation and other RNA-dependent cellular processes that merit further investigation in the near future.
Materials and methods
Bacterial strains and cultivation conditions
All bacterial strains and plasmids used in this work are listed in Supplementary Table S3. LB medium was used to routinely grown E. coli at 37°C and complex tryptone yeast (TY) [Citation60] or minimal medium (MM) [Citation61] for rhizobia at 30°C. The following antibiotics were added when required: streptomycin (600 µg/ml), tetracycline (10 µg/ml), kanamycin (50 µg/ml for E. coli and 180 µg/ml for Sinorhizobium strains) and gentamycin (8 µg/ml for E. coli and 30 µg/ml for rhizobia) [Citation62–64]. For growth in liquid media, the antibiotic concentration was reduced to 50%. For induction of FLAG-tagged protein and sRNA expression, IPTG was added to a final concentration of 0.5 mM to exponential phase cultures, unless otherwise indicated.
Cell pellets for RNA isolation, affinity chromatography and immunoprecipitation experiments were obtained as follows. Bacterial strains were streaked onto TY-agar and incubated at 30°C for up to 72 h. Bacteria in single colonies were grown at OD600 ~ 2.0 in TY or in MM at OD600 ~ 0.6. Endogenous AbcR2 and EcpR1 expression was induced by an osmotic upshift (400 mM NaCl during 1 h) in MM broth. Cells equivalent to 240 OD600 (eg 120 ml of a culture with OD600 ~ 2.0) were harvested by centrifugation 10 min at 5,500 × g and 4°C. Pelleted cells were then washed once with 0.1% sarcosyl in Tris-EDTA pH 8.0 (TE) buffer and once in buffer A (20 mM Tris–HCl pH 8.0, 150 mM KCl, 1 mM MgCl2, 1 mM DTT), centrifuged for 5 min at 6,000 r.p.m. (4°C) and stored at −80°C.
Oligonucleotides, plasmids construction and generation of S. meliloti derivative strains
DNA oligonucleotides used in this study are listed in Supplementary Table S4. For systematic 5´-end aptamer-tagging of the sRNAs of interest, MS2 and MS2* aptamers were generated by annealing of two complementary oligonucleotides that leave BamHI and XbaI compatible overhangs and then inserted between these sites into the mid-copy pSRKKm derivative pSRK-C [Citation29,Citation65], yielding vectors pSRK-MS2 and pSRK-MS2*. The full-length AbcR1, NfeR1 and EcpR1 coding sequences (from its primary or prevalent processed 5ʹ-ends) were amplified by PCR from their corresponding transcription start sites using Sm2B3001 genomic DNA as template and a pair of primers that incorporate XbaI and HindIII sites to the 5ʹ- and 3ʹ-ends of the amplicons, respectively. These PCR products were digested and inserted between the corresponding sites in pSRK-MS2 and pSRK-MS2* to yield pSRK-MS2/MS2*-AbcR2, pSRK-MS2/MS2*-EcpR1 and pSRK-MS2/MS2*-NfeR1 that express the MS2/MS2* 5´-tagged sRNAs constitutively. A Rho-independent transcriptional terminator (T1) generated by oligo hybridization was also cloned downstream of MS2 and MS2* to generate the control plasmids pSRK-MS2/MS2*-Term. Control plasmid pSRK-EcpR1 constitutively overexpressing the untagged wild-type EcpR1 sRNA from its transcription starting site 2 (TSS2) [Citation30] was similarly obtained by PCR amplification and cloning into pSRK-C as a BamHI- SacI fragment.
To endow several S. meliloti proteins with a FLAG-tag at their C-terminus, the tag sequence was cloned into plasmid pSRKKm to yield pSK-FLAG, which carries an inducible Lac promoter closely upstream the NdeI restriction site enabling IPTG-induced expression of the tagged protein. To construct vector pSK-FLAG, the complementary oligos XBTagF and BXTagR, carrying the 3× FLAG sequence followed by a TGA stop codon, were annealed and cloned as XbaI-BamHI fragments into pSRKKm. The coding regions of selected proteins, devoid of the respective stop codon, were amplified by PCR using primers that introduce NdeI and XbaI sequences to be inserted into the corresponding restriction sites of pSK-FLAG. Plasmids expressing tagged sRNAs and proteins were conjugated into the appropriate S. meliloti strains (wild-type or sRNA deletion mutants) by biparental mating involving E. coli S17-1 [Citation66]. At least three different transconjugants were selected for further independent determinations. Plasmids pSKEcpR1+, pSKNfeR1+ [Citation33], and pSKAbcR2+, were similarly mobilized as required to strains Sm4011ecpR1 [Citation30], SmmetKFLAG or Sm2B2020 for IPTG-induced transcription of the untagged wild-type EcpR1, NfeR1 and AbcR2 sRNAs, respectively. Plasmids pSKEcpR1+ and pSKAbcR2+ were generated by PCR amplification of EcpR1 from its prevalent processed 5ʹ-end and full-length AbcR2, respectively, as described [Citation29,Citation30].
Strain SmmetKFLAG, the Sm2B2019 derivative chromosomally encoding a MetK variant tagged with the FLAG epitope at the C-terminus, was generated by replacement of the wild-type allele via pK18metKFLAG-mediated double recombination as described [Citation67]. Plasmid pK18metKFLAG was generated by PCR amplification of the MetK-FLAG coding sequence from pSK_MetKFLAG and a 1,300-bp fragment of its downstream region from Sm2B3001 genomic DNA with primer pairs EcoNdemetkF/BamTGAflagR and 3metKOBamHI_F/4metKOPstII_R, respectively. The first fragment was restricted with EcoRI/BamHI and the second with BamHI/XbaI. All restriction sites were incorporated into the PCR primers except XbaI, which is encoded in the genome. In the same ligation reaction, both digestion products were inserted between the EcoRI and XbaI sites of the suicide vector pK18mobsacB [Citation68] to yield pK18metKFLAG. Proper expression of metKFLAG in strain SmmetKFLAG was confirmed by Western blot.
To achieve MetK depletion in strain Sm2011, its coding sequence was deleted by pK18ΔmetK-mediated double recombination as described [Citation29] in the presence of plasmid pSKmetK, which expresses the wild-type metK gene upon IPTG induction. Plasmid pK18ΔmetK was generated by PCR amplification of Sm2011 DNA with the pair of primers 1metKOEcoRI_F/2metKKOBamHI_R and 3metKOBamHI_F/4metKOPstII_R, ligation of the resulting fragments at cohesive BamHI overhangs and insertion of the tandem between the EcoRI and PstII sites of pK18mobsacB.
Finally, as reporter of intracellular SAM accumulation we constructed plasmid pRmetZ::eGFP, which expresses a translational fusion of the SAM-II riboswitch, preceding the metZ gene, to eGFP. For that, a metZ-derived fragment extending from its transcription start site [Citation37] to the 25th codon was amplified with primers TSS_metZ_Fw and metZ_75_Rv, and inserted between the BamHI and NheI sites of pR_eGFP [Citation29]. Fluorescence of S. meliloti single or double transconjugants carrying the reporter construct pR_eGFP was measured on adjusted stationary TY cultures in a Tecan Infinite M200 reader.
Northern blot hybridization
Total RNA was isolated from bacterial pellets by acid phenol/chloroform extraction as previously described [Citation69]. For Northern analysis, RNA samples (typically 10–20 μg) were subjected to electrophoresis on 6% polyacrylamide/7 M urea gels in TBE Tris-Borate-EDTA at ~30 mA and electro-transferred to nylon membranes, which were subsequently probed with 5ʹ-end radiolabeled 25mer oligonucleotides specific for the AbcR2, NfeR1 and EcpR1 sRNAs following a previously described protocols [Citation30,Citation69].
Affinity chromatography of S. meliloti tagged sRNAs
The MS2 coat protein, carrying a double mutation to avoid oligomerization [Citation70], is N-terminally fused to the MBP yielding the recombinant MS2-MBP. Production of the fusion protein was IPTG-induced in E. coli DH5α and purified by FPLC over an amylose column (GE Healthcare, Cat#: 28–9187-79) in 20 mM HEPES pH 7.9, 200 mM KCl and 1 mM EDTA. The sample was subsequently purified over a heparin column (GE Healthcare) using the same buffer with a 20–400 mM KCl gradient to remove bound nucleic acids. Affinity chromatography was performed following a recently published protocol [Citation39]. Briefly, S. meliloti bacteria expressing MS2-tagged sRNAs were harvested as described above, resuspended in 8 ml buffer A and disrupted using a Branson Sonifier sonicator in three cycles of 10 s bursts at 32 W with a microprobe. The lysate was then centrifuged (15 min, 16,000 × g, 4°C) and the supernatant was incubated with 200 pmol of MS2-MBP bait protein for 5 min. The column was prepared for affinity purification by 3 washes with 800 µl buffer A, loading of 100 µl amylose resin (NEB) and immobilization of 200 pmol of MS2-MBP diluted in buffer A. The mixture of cell lysate and half of the bait protein was then loaded into the amylose column, which interacts non-covalently with the MBP moiety. Unspecific-binding was removed by three column washes and then 800 µl buffer A containing 12 mM maltose were added to elute aptamer-tagged sRNA-protein complexes.
RNA and proteins contained in the eluted fractions were separated by phenol–chloroform–isoamylalcohol [25:24:1 (v/v)] extraction followed by EtOH precipitation of the aqueous phase. The organic lower phase containing the proteins was precipitated with acetone (3× vol.) overnight at −20°C. Protein pellets were washed with acetone, resuspended in 50 µl of 1× protein loading buffer, and stored at −20°C. Analytical samples from lysates, supernatant, wash and eluate fractions were kept as reference.
LC-MS/MS and protein identification
MS analysis was performed at the Proteomics Service from Instituto de Parasitología y Biomedicina ‘López-Neyra’ (CSIC, Granada). Protein samples equivalent to 120 OD600 were run 10 min in a 4% SDS-PAGE. The gel lane was cut into 10 slices and subjected to in-gel tryptic manual digestion. The resulting peptides were fractionated using an Easy n-LC II chromatography system (Proxeon) in line with an Amazon Speed ETD mass spectrometer (Bruker Daltonics).
The raw data files were processed using DataAnalysis software v4.3 (Bruker-Daltonics) and searched against the UniProtTrembl database using Mascot 2.4 (Matrix Science) integrated together with ProteinScape v4.0 (Bruker-Daltonics). Peptide precursor mass tolerance was set at 0.5 Da, and MS/MS tolerance was set at 0.5 Da. Search criteria included carbamidomethylation of cysteine (+57.02 Da) as a fixed modification and oxidation of methionine (+15.99 Da) as a variable modification. Searches were performed with a maximum of two missed cleavage for tryptic digestion. The reverse database search option was enabled, and all peptide data were filtered to satisfy the false discovery rate (FDR) of ≤2%. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [Citation71] partner repository with the dataset identifier PXD006410.
Significantly enriched Gene Ontology (GO) terms (P < 0.05, hypergeometric test) for the proteins associated with the different sRNAs were identified with the GO Term Finder tool http://www.comparativego.com [Citation72]. S. meliloti 1021 strain, containing 6,177 genes annotated in this database was considered as the background set. Proteins analysed include those identified associated with the different sRNAs but did not appear in any control sample. Therefore, we analysed 15, 11 and 95 proteins included in the subsets of putative partners of AbcR2, NfeR1 and EcpR1, respectively. Relationship between samples was assessed using the number of peptides to calculate the Pearson correlation coefficient in excel (r).
Protein immunoblot and silver staining
For Western blot, aliquots equivalent to 0.2 OD of cell lysates, flow-through or wash fractions were resuspended in 2× protein loading buffer (0.05 OD for silver staining). For both procedures, half of the elution fractions (25 µl) were loaded (120 OD). Cell protein fractions were denatured by heating at 95°C for 5 min, resolved by SDS-PAGE and blotted onto a polyvinylidene difluoride membrane (P 0.45, Amersham). Membranes were probed with a monoclonal anti-FLAG antibody (Sigma #F7425; 1:5,000) as reported [Citation67]. Blots were developed by incubation for 5 min in blotting detection reagent (ECL, Amersham) and signals were detected with a ChemiDoc system (BioRad). Polyacrylamide gels were alternatively stained as appropriate either with Coomassie blue or the Bio-Rad silver staining kit according to the manufacturer’s instructions.
CoIP assays with 3× FLAG-tagged proteins
For co-immunoprecipitation (CoIP), S. meliloti carrying the plasmids encoding FLAG-tagged proteins were induced with IPTG and harvested in the conditions resembling cognate sRNA expression described before [Citation29,Citation30,Citation33]. CoIP-RNA was obtained from frozen pellets equivalent to 240 OD600 using the ANTI-FLAG M2 affinity gel (Sigma) as described [Citation67]. RNA-protein complexes were digested with DNAseI (QIAGEN) prior to phenol-chloroform-isoamylalcohol extraction as described for the affinity purification. Protein complexity of the cell culture, total cell lysate, supernatant, wash and eluate (5 μl) was analysed by Western blotting. The RNA from the aqueous phase was reverse transcribed into cDNA for PCR analysis. Similar CoIP experiments were conducted with strain SmmetKFLAG. In this case, the protein fraction was analysed by LC-MS/MS as described above, and total and CoIP-RNA were subjected to Northern hybridization with a specific NfeR1 probe.
RT-PCR analysis of RNA samples
To detect the sRNAs under study, input, affinity purified and CoIP RNA from 24 OD600 equivalents (1 μl) (at least two independent eluates per sample) were subjected to First-Strand cDNA Synthesis Using 100 U SuperScript™ II RT (Invitrogen). RT was performed with 100 ng random hexamers and RNAse OUT (Invitrogen) according to the manufactures´ instructions. PCR was performed with 1 μl of cDNA samples using Phusion® High-Fidelity DNA Polymerase (NEB) with the sRNA-specific primers listed in Supplemental Table S2. Specific amplification of the target transcripts was confirmed by DNA sequencing.
MetK purification
The MetK coding sequence was PCR amplified from S. meliloti Rm1021 genomic DNA using primers metK3ʹXhoI and NdeImetKF2. The fragment was checked by sequencing and inserted between the NdeI and XhoI sites of pET-16b (Novagen), yielding p16metK, which encodes a MetK variant tagged with 10 His residues at its N-terminus. For purification of recombinant MetK, plasmid p16metK was transformed into Rosetta-gami (DE3) pLysS (Novagen). The cells were grown at 37°C in 1 l of LB medium supplemented with 100 µg/ml ampicillin and 50 µg/ml chloramphenicol to an OD600 of 0.5. Protein production was induced at 20ºC by addition of IPTG to a final concentration of 0.3 mM. After 18–20 h of growth, cells were harvested by centrifugation at 5,000 xg for 15 min at 4°C. Cells were resuspended in 25 ml buffer B (50 mM Tris-HCl buffer pH 8, 0.5 M NaCl) and lysed by three consecutive passes through a French’s Press 1000 PSIG. The lysate was centrifuged at 38,000 xg for 1 h at 4ºC. The supernatant was loaded onto a HisTrapTM HP 5 ml column (GE Healthcare) equilibrated with buffer B, which was then washed with three column volumes of the same buffer. Then, column was washed with three column volumes of 5% buffer C (50 mM Tris-HCl buffer pH 8, 0.5 M NaCl and 1 M imidazole). Protein was eluted in a 5%-100% gradient of buffer C during 15 min. Fractions were subjected to SDS-PAGE and those containing MetK were pooled and dialysed overnight against 50 mM Tris-ClH pH 8, 0.5 M NaCl, glycerol 10% at 4°C. Protein concentration was measured using Bradford assay. The purified protein was stored at −80°C.
In vitro synthesis and labelling of RNA
The AbcR2, NfeR1, EcpR1, SmelC812 [Citation38], tRNAMet, SMb20420 and 5S rRNA transcripts were amplified for in vitro transcription from Sm2B3001 genomic DNA and the group II ribozyme RmInt1 from plasmid pKGEMA4 [Citation42] with the primer pairs listed in Supplementary Table S4. All forward primers incorporate the T7 promoter sequence. PCR templates (250 ng) were transcribed with T7 RNA polymerase. Synthetized transcripts were DNase I-treated, dephosphorylated with Antartic Phosphatase (New England Biolabs), purified on 6% polyacrylamide-7 M urea gel and quantified on a Qubit3 Fluorometer (Thermo Fisher Scientific). For binding assays, transcripts were radiolabelled at the 5ʹ-end with ɣ-32P-ATP using T4 Polinucleotide Kinase (New England Biolabs).
Filter binding assays
Radiolabeled transcripts (1 nM) were incubated for 30 min at 30°C with the indicated concentrations of purified recombinant MetK (0–500 nM) in a 10-µl reaction containing 1x Binding Buffer (50 mM Tris-HCl, 0.5 mM NaCl, 1 mM DTT). Binding specificity was assessed by competition experiments in the presence of a molar excess (100 nM) of the corresponding unlabelled sRNA. BSA (2 µM) was also used instead of MetK as negative protein control for binding to the trans-sRNAs. Reactions were loaded into a dot-blot device (Bio-Dot; Bio-Rad) provided with nitrocellulose and PVDF membranes (Amersham). Before and after sample blotting, wells were rinsed three times with 50 µl of DTT-less Binding Buffer. Membranes were finally exposed to a Phosphor Imager screen (Bio-Rand) and quantified with the Quantity One software (Rio-Rad).
Supplemental Material
Download Zip (6.8 MB)Acknowledgments
We thank Prof. Henning Urlaub (Institute for Clinical Chemistry, University Medical Center Göttingen) for kindly providing the plasmid for MS2-MBP expression. The proteomic analyses were performed in the Proteomic Unit of IPBLN-CSIC that belongs to ProteoRed, PRB2-ISCIII, supported by grant PT13/0001.
Disclosure statement
Authors declare no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Wagner EGH, Romby P. Small RNAs in bacteria and archaea: who they are, what they do, and how they do it. In: Theodore Friedmann JCD, Stephen FG, editors. Adv Genet. Vol. 90. Academic Press, Amsterdam; 2015. p. 133–208.
- Waters LS, Storz G. Regulatory RNAs in bacteria. Cell. 2009 Feb 20;136(4):615–628.
- Storz G. An expanding universe of noncoding RNAs. Science. 2002 May;296(5571):1260–1263.
- Sharma CM, Darfeuille F, Plantinga TH, et al. A small RNA regulates multiple ABC transporter mRNAs by targeting C/A-rich elements inside and upstream of ribosome-binding sites. Genes Dev. 2007 Nov;21(21):2804–2817.
- Desnoyers G, Bouchard MP, Massé E. New insights into small RNA-dependent translational regulation in prokaryotes. Trends Genet. 2013 Feb;29(2):92–98.
- Sun X, Zhulin I, Wartell RM. Predicted structure and phyletic distribution of the RNA-binding protein Hfq. Nucleic Acids Res. 2002 Sep;30(17):3662–3671.
- Vogel J, Wagner EG. Target identification of small noncoding RNAs in bacteria. Curr Opin Microbiol. 2007 Jun;10(3):262–270.
- Valentin-Hansen P, Eriksen M, Udesen C. The bacterial Sm-like protein Hfq: a key player in RNA transactions. Mol Microbiol. 2004 Mar;51(6):1525–1533.
- Dos Santos RF, Arraiano CM, Andrade JM. New molecular interactions broaden the functions of the RNA chaperone Hfq. Curr Genet. 2019;65(6):1313–1319.
- Romby P, Charpentier E. An overview of RNAs with regulatory functions in gram-positive bacteria. Cell Mol Life Sci. 2010 Jan;67(2):217–237.
- Smirnov A, Forstner KU, Holmqvist E, et al. Grad-seq guides the discovery of ProQ as a major small RNA-binding protein. Proc Natl Acad Sci U S A. 2016 Oct 11;113(41):11591–11596.
- Smirnov A, Wang C, Drewry LL, et al. Molecular mechanism of mRNA repression in trans by a ProQ‐dependent small RNA. Embo J. 2017;36(8):1029–1045.
- Westermann AJ, Venturini E, Sellin ME, et al. The major RNA-binding protein proq impacts virulence gene expression in salmonella enterica serovar typhimurium. mBio. 2019;10(1).
- Melamed S, Adams PP, Zhang A, et al. RNA-RNA Interactomes of ProQ and Hfq reveal overlapping and competing roles. Mol Cell. 2020;77(2):411–425.e7.
- Olejniczak M, Storz G. ProQ/FinO-domain proteins: another ubiquitous family of RNA matchmakers? Mol Microbiol. 2017;104(6):905–915.
- Windbichler N, Schroeder R. Isolation of specific RNA-binding proteins using the streptomycin-binding RNA aptamer. Nat Protoc. 2006;1(2):637–640.
- Rieder R, Reinhardt R, Sharma C, et al. Experimental tools to identify RNA-protein interactions in Helicobacter pylori. RNA Biol. 2012;9(4):520–531.
- Said N, Rieder R, Hurwitz R, et al. In vivo expression and purification of aptamer-tagged small RNA regulators. Nucleic Acids Res. 2009 November 1;37(20):e133.
- Windbichler N, von Pelchrzim F, Mayer O, et al. Isolation of small RNA-binding proteins from E. coli: evidence for frequent interaction of RNAs with RNA polymerase. RNA Biol. 2008 Jan-Mar;5(1):30–40.
- Sukhodolets MV, Garges S. Interaction of Escherichia coli RNA polymerase with the ribosomal protein S1 and the Sm-like ATPase Hfq. Biochemistry. 2003 Jul 8;42(26):8022–8034.
- Sobrero P, Valverde C. The bacterial protein Hfq: much more than a mere RNA-binding factor. Crit Rev Microbiol. 2012 Nov;38(4):276–299.
- De Lay N, Schu DJ, Gottesman S. Bacterial small RNA-based negative regulation: hfq and its accomplices. J Biol Chem. 2013;288(12):7996–8003.
- Babitzke P, Romeo T. CsrB sRNA family: sequestration of RNA-binding regulatory proteins. Curr Opin Microbiol. 2007 Apr;10(2):156–163.
- Wassarman KM. 6S RNA: a small RNA regulator of transcription. Curr Opin Microbiol. 2007 Apr;10(2):164–168.
- Jørgensen MG, Thomason MK, Havelund J, et al. Dual function of the McaS small RNA in controlling biofilm formation. Genes Dev. 2013 May 15;27(10):1132–1145.
- Müller P, Gimpel M, Wildenhain T, et al. A new role for CsrA: promotion of complex formation between an sRNA and its mRNA target in Bacillus subtilis. RNA Biol. 2019;16(7):972–987.
- Holmqvist E, Vogel J. RNA-binding proteins in bacteria. Nature Rev Microbiol. 2018;6(10):601–615.
- Jones KM, Kobayashi H, Davies BW, et al. How rhizobial symbionts invade plants: the Sinorhizobium-Medicago model. Nat Rev Microbiol. 2007 Aug;5(8):619–633.
- Torres-Quesada O, Millan V, Nisa-Martinez R, et al. Independent activity of the homologous small regulatory RNAs AbcR1 and AbcR2 in the legume symbiont Sinorhizobium meliloti. PLoS One. 2013;8(7):e68147.
- Robledo M, Frage B, Wright PR, et al. A stress-induced small RNA modulates alpha-rhizobial cell cycle progression. PLoS Genet. 2015;11(4):e1005153.
- Baumgardt K, Šmídová K, Rahn H, et al. The stress-related, rhizobial small RNA RcsR1 destabilizes the autoinducer synthase encoding mRNA sinI in Sinorhizobium meliloti. RNA Biol. 2015;13(5):486–499.
- Lagares A Jr., Ceizel Borella G, Linne U, et al. Regulation of polyhydroxybutyrate accumulation in Sinorhizobium meliloti by the trans-encoded small RNA MmgR. J Bacteriol. 2017 Feb 06;199(8):e00776–16.
- Robledo M, Peregrina A, Millán V, et al. A conserved α-proteobacterial small RNA contributes to osmoadaptation and symbiotic efficiency of rhizobia on legume roots. Environ Microbiol. 2017;19(7):2661–2680.
- Robledo M, García-Tomsig NI, Jiménez-Zurdo JI. Riboregulation in Nitrogen-Fixing Endosymbiotic Bacteria. Microorganisms. 2020;8(3):384.
- Torres-Quesada O, Reinkensmeier J, Schluter JP, et al. Genome-wide profiling of Hfq-binding RNAs uncovers extensive post-transcriptional rewiring of major stress response and symbiotic regulons in Sinorhizobium meliloti. RNA Biol. 2014;11(5):563–579.
- Gruber AR, Lorenz R, Bernhart SH, et al. The Vienna RNA websuite [Research Support, Non-U.S. Gov’t]. Nucleic Acids Res. 2008 Jul 1;36:70–74.
- Schlüter JP, Reinkensmeier J, Barnett MJ, et al. Global mapping of transcription start sites and promoter motifs in the symbiotic α-proteobacterium Sinorhizobium meliloti 1021. BMC Genomics. 2013;14:156.
- Schlüter JP, Reinkensmeier J, Daschkey S, et al. A genome-wide survey of sRNAs in the symbiotic nitrogen-fixing alpha-proteobacterium Sinorhizobium meliloti. BMC Genomics. 2010;11:245.
- Robledo M, Matia-Gonzalez AM, Garcia-Tomsig NI, et al. Identification of small RNA-protein partners in plant symbiotic bacteria. Methods Mol Biol. 2018;1737:351–370.
- Arraiano CM, Andrade JM, Domingues S, et al. The critical role of RNA processing and degradation in the control of gene expression. FEMS Microbiol Rev. 2010 Sep;34(5):883–923.
- Milojevic T, Sonnleitner E, Romeo A, et al. False positive RNA binding activities after Ni-affinity purification from Escherichia coli. RNA Biol. 2013;10(6):1066–1069.
- Nisa-Martinez R, Jimenez-Zurdo JI, Martinez-Abarca F, et al. Dispersion of the RmInt1 group II intron in the Sinorhizobium meliloti genome upon acquisition by conjugative transfer. Nucleic Acids Res. 2007 JAN;35(1):214–222.
- Jiménez-Zurdo JI, Valverde C, Becker A. Insights into the noncoding RNome of nitrogen-fixing endosymbiotic α-proteobacteria. Mol Plant-Microbe Interact. 2013;26(2):160–167.
- Del Val C, Romero-Zaliz R, Torres-Quesada O, et al. A survey of sRNA families in alpha-proteobacteria [Research Support, Non-U.S. Gov’t]. RNA Biol. 2012 Feb;9(2):119–129.
- Reinkensmeier J, Schlüter J-P, Giegerich R, et al. Conservation and occurrence of trans-encoded sRNAs in the rhizobiales. Genes (Basel). 2011;2(4):925–956.
- Saramago M, Peregrina A, Robledo M, et al. Sinorhizobium meliloti YbeY is an endoribonuclease with unprecedented catalytic features, acting as silencing enzyme in riboregulation. Nucleic Acids Res. 2017 December 6;45(3):1371–1391.
- Batey RT, Kieft JS. Improved native affinity purification of RNA. RNA. 2007 Aug;13(8):1384–1389.
- Deckert J, Hartmuth K, Boehringer D, et al. Protein composition and electron microscopy structure of affinity-purified human spliceosomal B complexes isolated under physiological conditions. Mol Cell Biol. 2006 Jul;26(14):5528–5543.
- Beljantseva J, Kudrin P, Andresen L, et al. Negative allosteric regulation of Enterococcus faecalis small alarmone synthetase RelQ by single-stranded RNA. Proc Natl Acad Sci U S A. 2017 Apr 04;114(14):3726–3731.
- Hauryliuk V, Atkinson GC. Small Alarmone Synthetases as novel bacterial RNA-binding proteins. RNA Biol. 2017 Aug;18. DOI:10.1080/15476286.2017.1367889
- Baltz AG, Munschauer M, Schwanhausser B, et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell. 2012 Jun 8;46(5):674–690.
- Castello A, Fischer B, Eichelbaum K, et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell. 2012 Jun 8;149(6):1393–1406.
- Matia-Gonzalez AM, Laing EE, Gerber AP. Conserved mRNA-binding proteomes in eukaryotic organisms. Nat Struct Mol Biol. 2015 Dec;22(12):1027–1033.
- Nagy E, Rigby WF. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold). J Biol Chem. 1995 Feb 10;270(6):2755–2763.
- Nagy E, Henics T, Eckert M, et al. Identification of the NAD(+)-binding fold of glyceraldehyde-3-phosphate dehydrogenase as a novel RNA-binding domain. Biochem Biophys Res Commun. 2000 Aug 28;275(2):253–260.
- Walden WE, Selezneva AI, Dupuy J, et al. Structure of dual function iron regulatory protein 1 complexed with ferritin IRE-RNA. Science. 2006 Dec 22;314(5807):1903–1908.
- Beaufay F, Coppine J, Mayard A, et al. A NAD-dependent glutamate dehydrogenase coordinates metabolism with cell division in Caulobacter crescentus. Embo J. 2015 Jul;34(13):1786–1800.
- Krall AS, Christofk HR. Cell cycle: division enzyme regulates metabolism. Nature. 2017 Jun;546(7658):357–358.
- Holmqvist E, Li L, Bischler T, et al. Global Maps of ProQ Binding in vivo Reveal Target Recognition via RNA Structure and Stability Control at mRNA 3ʹ Ends. Mol Cell. 2018;70(5):971–982.e6.
- Beringer JE. R factor transfer in Rhizobium leguminosarum. J Gen Microbiol. 1974 Sep;84(1):188–198.
- Robertsen BK, Aman P, Darvill AG, et al. Host-symbiont interactions: V. the structure of acidic extracellular polysaccharides secreted by Rhizobium leguminosarum and Rhizobium trifolii. Plant Physiol. 1981 Mar;67(3):389–400.
- Casse F, Boucher C, Julliot JS, et al. Identification and Characterization of Large Plasmids in Rhizobium meliloti using Agarose Gel Electrophoresis. J Gen Microbiol. 1979 August 1;113(2):229–242.
- Meade HM, Long SR, Ruvkun GB, et al. Physical and genetic characterization of symbiotic and auxotrophic mutants of Rhizobium meliloti induced by transposon Tn5 mutagenesis. J Bacteriol. 1982 Jan;149(1):114–122.
- Bahlawane C, McIntosh M, Krol E, et al. Sinorhizobium meliloti regulator MucR couples exopolysaccharide synthesis and motility [Research Support, Non-U.S. Gov’t]. Mol Plant Microbe Interact. 2008 Nov;21(11):1498–1509.
- Khan SR, Gaines J, Roop RM, et al. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol. 2008;74(16):5053–5062.
- Simon R, Priefer U, Puhler A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria [10.1038/nbt1183-784]. Nat Biotech. 1983;1(9):784–791.
- Torres-Quesada O, Oruezabal RI, Peregrina A, et al. The Sinorhizobium meliloti RNA chaperone Hfq influences central carbon metabolism and the symbiotic interaction with alfalfa. BMC Microbiol. 2010;10:71.
- Schafer A, Tauch A, Jager W, et al. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum [Research Support, Non-U.S. Gov’t]. Gene. 1994 Jul 22;145(1):69–73.
- Del Val C, Rivas E, Torres-Quesada O, et al. Identification of differentially expressed small non-coding RNAs in the legume endosymbiont Sinorhizobium meliloti by comparative genomics. Mol Microbiol. 2007 Dec;66(5):1080–1091.
- LeCuyer KA, Behlen LS, Uhlenbeck OC. Mutants of the bacteriophage MS2 coat protein that alter its cooperative binding to RNA. Biochemistry. 1995;34(33):10600–10606.
- Vizcaíno JA, Côté RG, Csordas A, et al. The Proteomics Identifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 2012;41(D1):D1063–D1069.
- Fruzangohar M, Ebrahimie E, Ogunniyi AD, et al. Comparative GO: a web application for comparative gene ontology and gene ontology-based gene selection in bacteria. PLoS One. 2013;8(3):e58759.