
ABSTRACT
Non-coding RNAs (ncRNAs) are powerful regulators of gene expression but medium-sized (50–300 nts in length) ncRNAs (msRNAs) are barely picked-up precisely by RNA-sequencing. Here we describe msRNA-sequencing (msRNAseq), a modified protocol that associated with a computational analyses pipeline identified about ~1800 msRNA loci, including over 300 putatively novel msRNAs, in human and murine cells. We focused on the identification and initial characterization of three POLIII-derived transcripts. The validation of these uncharacterized msRNAs identified an ncRNA in antisense orientation from the POLR3E locus transcribed by POLIII. This msRNA, termed POLAR (POLR3E Antisense RNA), has a strikingly short half-life, localizes to paraspeckles (PSPs) and associates with PSP-associated proteins indicating that msRNAseq identifies functional msRNAs. Thus, our analyses will pave the way for analysing the roles of msRNAs in cells, development and diseases.
Introduction
Current ncRNA sequencing strategies mainly focus on either long ncRNAs (e.g. lincRNA) with more than 200–300 nucleotides (nts) in length or short ncRNAs (e.g. miRNA) with a size below 30 nts. Due to library preparation protocols these analyses, however, barely cover medium-sized ncRNAs with a length between 50 and 300 nts. An essential limitation of small RNA library protocols is that 5ʹ-end modifications in msRNAs, e.g. cap structures or triphosphates, are not removed routinely. The analysis of lncRNAs usually involves RNA fragmentation to achieve coverage of the complete RNA body. This essentially limits the coverage of msRNAs which are fragmented as well and thus fall below size limits of following library preparation steps or yield read peaks not reflecting the full-length transcript. Detailed protocols have been established for short ncRNAs as well as long ncRNAs highlighting the importance of, e.g., RNA fragmentation, size selection and 5ʹ/3ʹ-end modifications [Citation1,Citation2]. Finally, annotation analyses of msRNAs are complicated by the fact that these are frequently encoded by multiple gene or pseudogene loci. Despite these limitations, hundreds of msRNAs had been identified over recent decades. They are transcribed by all three RNA polymerases, but POLIII solely synthesizes msRNAs [Citation3]. Like other RNAs, msRNAs are processed and/or modified, for instance the splicing and pseudouridylation of tRNAs [Citation4]. Functional analyses revealed that msRNAs serve essential roles in splicing (e.g. snRNAs) [Citation5], RNA processing (e.g. RNAse P) [Citation6], transcription (e.g. 7SK) [Citation7] and mRNA translation (e.g. tRNAs) [Citation8]. In support of this, msRNAs have been implicated in human diseases like cancer (e.g. tRNAs) [Citation9] or genetic disorders like the Prader-Willi-Syndrome (snoRNAs) [Citation10]. Despite their multiple roles and potential pathophysiological relevance, a genome-wide experimental survey of msRNA-properties and -expression had not been performed systematically to the best of our knowledge.
Here we describe a protocol for the isolation, sequencing and computational analysis of msRNAs. In addition to the sole identification of msRNAs, we demonstrate that msRNAseq allows analysing the biogenesis, processing, editing and modification of msRNAs. Among the identified msRNAs we characterized POLAR, the first known msRNA recruited to paraspeckles.
Materials and methods
Cell culture and transfection
HEK293T/17 and B16-F10 cells were purchased from the ATCC and cultured in DMEM supplemented with GlutaMAX and 10% FBS (Life Technologies). For RNAi HEK293 cells were seeded at a density of 500.000 cells per well (6-well plate) and transfected with Lipofectamine RNAiMAX (Life Technologies) according to manufacturer’s protocols. For La-directed siRNAs, cells were harvested after 72 hours post-transfection. Due to strong proliferation effects cells with BDP1-directed knockdowns were terminated after 36 hours. The POLAR gene was amplified from HEK293/T17 cells and cloned into the ZeroBlunt TOPO Vector (Life Technologies). For plasmid transfections HEK293 cells were treated with GeneIn (amsbio) or Lipofectamine 2000 (Life Technologies) according to manufacturer’s protocols. For overexpression analysis, cells were harvested two days after transfection and subjected to RNA extraction/RT-qPCR or Western Blotting.
Preparation of msRNAs, library preparation and msRNAseq
Total RNA was prepared by TRIZOL extraction from HEK293 and B16-F10 cells. 15 µg of total RNA was then treated with RppH (NEB) according to manufacturer’s protocols to remove 5ʹ-Pyrophosphates and Caps for 1 h at 37°C. De-phosphorylated total RNA was separated on a TBE-Urea cushion gel (bottom 22%, top 7% acryl amide) together with an RNA ladder. Afterwards the gels were stained with EtBr and RNAs ranging from 50 to 300 nts were excised. Gel slices were now spun through perforated tubes resulting in crushed gel pieces. These slices were now supplemented with extraction buffer (20 mM Tris 7.4, 250 mM Na-acetate, 1 mM EDTA, 0,25% SDS) and incubated at 65°C for 1 h. The slurry was centrifuged through spin filters to separate gel pieces from RNA solution. The RNA was precipitated with isopropanol and dissolved in water. RNA integrity was checked by TBE-Urea gel electrophoresis. Library preparation and RNA sequencing were performed by Vertis Biotechnologie AG (Freising, Germany). For library preparation, the Illumina TruSeq protocol was used. The RNA sequencing was performed on a NextSeq 500 system resulting in single reads from the 5ʹ-end (150 bp read length).
Bioinformatic analyses
The resulting unpaired reads were adapter-trimmed using cutadapt (version 1.6) [Citation11]. Trimming was performed with a short adapter (AGATCGGAAG) and a minimum overlap of seven nucleotides. Final read length was restricted to a minimum of 40 nts. No quality trimming was performed to retain a maximum number of msRNA reads. Next, adapter-trimmed reads were aligned with bowtie2 (version 2.2.4) to the respective genomes (human: hg38, mouse: mm10) [Citation12]. Only uniquely mapped reads were used, allowing one mismatch in the seed region of the read. Subsequently, genome coverages were calculated in a strand-specific manner using genomeCoverageBed within bedtools (v2.25.0) [Citation13]. For each strand, adjacent nucleotides with coverages of at least 50 reads were combined to coherent regions with a minimum length of 40 nucleotides. Furthermore, these coverage regions were checked for an integrated core region to allow detection of precursor vs mature msRNAs. These core regions were identified by comparing the fold change in a nucleotide wise manner for the outer nucleotides at both ends of the coverage region. If the fold change was at least three times higher compared to the previous nucleotide, coverage region was narrowed by nucleotide trimming until a minimum length of 40 nucleotides was reached, generating the final core region. Smaller regions were discarded and the initial coverage region was reported. For each core region, consensus sequences were calculated compared to the respective genomes by variant calling mpileup within SAMtools (version 1.1) [Citation14]. Resulting consensus sequences were converted to FASTA format and the reverse complement was generated for the minus strand. Consensus sequences of the reported core regions were aligned to a generated msRNA database by NCBI-Blast (version 2.6.0). This database consists of known msRNAs from different sources like NCBI, ENSEMBL, GtRNAdb, snOPY and miRBase. Blast results were processed to gather hit information about known and unknown sequences from the coverage regions. Sequences retrieving no database hits were crosschecked for obvious contamination sources like mitochondrial RNA, ribosomal RNA and histone mRNAs. Identified msRNA loci can be found in Supp. Table S2.
POLAR deletion by CRISPR/Cas9
HEK293T/17 cells were transfected with a Cas9-coding plasmid (pcDNA_Cas9_T2A_GFP) as well as sgRNA-Plasmids (sgControl, sgPOLAR 1, sgPOLAR3 or sgPOLAR4 and 5) using Lipofectamine 2000 (Life Technologies). GFP-positive cells were sorted by FACS and the diluted cell suspension was plated on 15-cm plates. After 7 days single colonies were picked and cultured. After successful expansion total RNA was isolated and POLAR levels were determined by RT-qPCR. Genomic sequencing (Sanger) was applied to verify the genomic modification.
Western and Northern blotting
Blotting techniques were essentially performed as previously described using the materials provided in Supp. Table S1Citation15.
Cell fractionation, RT-qPCR and RNA pulldown
Subcellular fractionation of HEK293/T17 was performed by re-suspending 2 million cells in 200 µl of Cyto-Buffer (10 mm HEPES-KOH pH 7.2, 5 mm MgCl2, 150 mm KCl, 80 µg/ml Digitonin), incubating for 7 min at room temperature and centrifugation for 3 min at 4.000 rpm. The supernatant was removed and served as cytoplasmic fraction. The pellet was re-suspended again with the Cyto-Buffer, cells were centrifuged and the supernatant was discarded. The remaining pellet served as nuclear fraction. Both fractions were subjected to RNA-isolation using TRIZOL (Sigma) and RT-qPCR. RT-qPCR and RNA pulldowns were conducted as previously described using indicated materials (Supp. Table S1) [Citation15,Citation16].
Determination of RNA half life
HEK293 cells were seeded at a density of 500.000/6-well 24 h before treatment. On the next day, cells were treated for indicated time points with Actinomycin D at a final concentration of 3.1 µg/ml. Treatment was stopped by addition of TRIZOL. Subsequently, RNA was isolated and subjected to RT-qPCR.
Fluorescence in situ hybridization (FISH)
For FISH OVCAR-3 cells were seeded on coverslips. Cells were washed with PBS and fixed with 1% paraformaldehyde for 10 min at RT. After washing with PBS, cells were permeabilized with 70% Ethanol for 1 h at 4°C. After two washing steps with PBS, cells were incubated with PerfectHyb Plus (Sigma) for 5 min at RT. Probes were now added to a final concentration of 2 ng/µl and incubated for 4 h at RT. Subsequently, the buffer was removed and coverslips were washed twice with 2xSSC/0.1% SDS. Protein staining was now performed as previously described [Citation15]. Coverslips were stained with DAPI, mounted and imaged using Leica TCS SP5 X confocal microscope.
Results
Analysing msRNA by msRNA-sequencing (msRNAseq)
Aiming to establish a protocol allowing the genome-wide analysis of expressed msRNAs, we developed an unbiased RNA-sequencing approach emanating from a modified RNA preparation protocol (). Total RNA isolated from human (HEK293) or mouse (B16-F10) cells was treated with RNA 5ʹ-Pyrophosphohydrolase (RppH) to remove 5ʹ-modifications and generate RNA species with a 5ʹ-monophosphate suitable for the ligation of RNA-linkers. To enrich msRNAs ranging from 50 to 300 nts, total RNA was gel-purified and enrichment was monitored by TBE-Urea-gel electrophoresis (, B). Finally, a novel computational analysis pipeline was required to extract msRNA sequence information derived by RNA-sequencing (). In available human and mouse genome versions msRNA annotations are incomplete due to outlined reasons. Therefore, we applied an unbiased approach solely relying on the mapping of reads to the respective genomes. Only genomic position that showed coverage of at least 50 reads with a minimum length of 40 nts were considered. Within these msRNA candidate regions, we applied a coverage trimming algorithm distinguishing low abundant trailer sequences from the mature msRNA region, e.g. pre-snoRNAs vs mature snoRNAs. To check if these genomic regions were already reported, we applied a BLAST-based alignment to our msRNA-database including known msRNAs.
Figure 1. MsRNA Sequencing. A The purification scheme for msRNAs used in this study. After De-capping of total RNA, a gel-based RNA size selection ranging from 50–300 nts was performed. C – Cap; P – Phosphate. B A representative example for an msRNA-purification performed from HEK293 (human) and B16-F10 cells (mouse) cells is shown (15% TBE-Urea-Gel stained with Syto60). T – Total RNA; MS – Purified msRNAs. C The bioinformatic pipeline for the analysis of msRNAs is depicted. D All identified coverage regions from HEK293 cells were classified by their blast hits. The fraction of each msRNA family was calculated according to their obtained amount of reads. E The coverage regions were identified as in D. The fraction of each msRNA family was calculated according to the number of genes they were mapped to. Note that contaminants mainly contain fragments of large ribosomal RNAs, mitochondrial RNAs and histone mRNAs
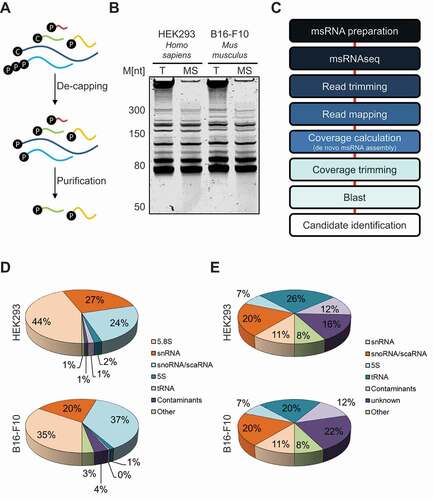
To evaluate msRNAseq and the computational analyses pipeline we aimed for an overview of identified msRNAs. To this end, mapped reads and respective Blast-hits were summed and categorized (). 5.8S rRNA was by far most frequently sequenced individual msRNA (HEK293: 44%, B16-F10: 35%). This was expected since rRNA was deliberately not depleted before library preparation to reduce biases by rRNA depletion methods. Other major msRNA species included snRNAs and snoRNAs. The most abundant contaminant sequences were derived from larger rRNAs, mitochondrial RNAs and histone mRNAs. The latter was expected since histone mRNAs are the smallest family of mRNAs (starting at ~300nts). However, these contaminants just made up a small fraction of reads (HEK293: 1%, B16-F10: 4%). Finally, we analysed the number of msRNA genes mapped to the respective genomes (). This was of particular interest, since most msRNAs are encoded by multiple genes. Notably, the major classes of msRNAs (snRNAs, snoRNAs, 5S rRNA and tRNAs) each were present on multiple genomic positions in human and mouse reflecting the manifold genes producing these transcripts. Interestingly, a large portion of genes (HEK293: 16%, B16-F10: 22%) could not be mapped to known msRNA genes suggesting the identification of previously unannotated msRNAs.
Characterization of msRNA 3ʹ-end processing
With extensive msRNA sequencing at hand we aimed to analyse the biogenesis of msRNAs in more detail. Accordingly, the poorly understood 3ʹ-end formation of Y RNAs, another abundant msRNA family comprising four members (Y1, Y3, Y4 and Y5), was analysed () [Citation17]. Coverage sharply decreased before the reported 3ʹ-end of Y1, Y3 and Y4. This, however, was not observed for Y5 containing an Oligo-U stretch at its 3ʹ-end indicating that Y1, Y3 and Y4 are 3ʹ-trimmed leading to the release of the last 5–6 nts, as suggested earlier [Citation18]. Intriguingly, trimmed Y RNAs are mainly cytoplasmic whereas Y5 is enriched in the nucleus suggesting a link of trimming and subcellular sorting [Citation19]. The mainly nuclear La protein associates with the 3ʹ-located Oligo-U tract of Y RNAs and was proposed to modulate their nuclear export and turnover [Citation20]. In agreement, the depletion of La by two distinct siRNAs led to severely diminished levels of Y5 but not the trimmed Y RNAs in HEK293 cells (). Together this indicated that the poly-U stretch of the Y5 RNA is essential to retain this ncRNA in the nucleus and prevent rapid Y5 decay. In contrast, the other Y RNAs apparently act largely independent of La and accumulate in the cytoplasm where they are stabilized by the Ro60 (TROVE2) protein [Citation21].
Figure 2. MsRNA modifications and editing. A The normalized coverage at the 3ʹ-end of human Y RNAs was determined. The Oligo-U tract at the end of the Y RNAs is indicated and just significantly retained within Y5. B Northern and Western Blot analyses were performed in HEK293 cells transfected with siRNAs targeting La (siLa#1, siLa#2). Mock and control siRNA (siC) transfections served as controls. Northern Blots (NB) were probed for Y RNAs and U2 as loading control. Western Blots (WB) were probed with anti-La and VCL-antibodies. C Three different murine msRNAs (Pre-let7b, SNORA4 and SNORD86) were analysed for uridylation/adenylation at their 3ʹ-ends. The sequence of the region is depicted on top of the bars and the actual 3ʹ-end is highlighted. Not encoded U’s (Pre-let7b) or A’s (SNORA4 and SNORD86) are indicated as fraction of mismatched nucleotides in red. Whenever mismatched nucleotides cannot be discriminated from authentic nucleotides light red and grey were used for the bars. D Three different human tRNAs (tRNAAla(AGC), tRNAAla(UGC) and tRNAIle(AAU)) were analysed for A-I editing within their anticodon region. The fraction of mismatching nucleotides (A-to-G conversion) is indicated in red. The sequence of the region is depicted on top of the bars and the anticodon loop is highlighted. E Two different human tRNAs (tRNAArg(UCU) and tRNATyr(GUA)) were analysed for their splicing pattern. Coverage plots of each tRNA are shown on the left (total coverage in black and split reads in grey). The intron region is highlighted in blue
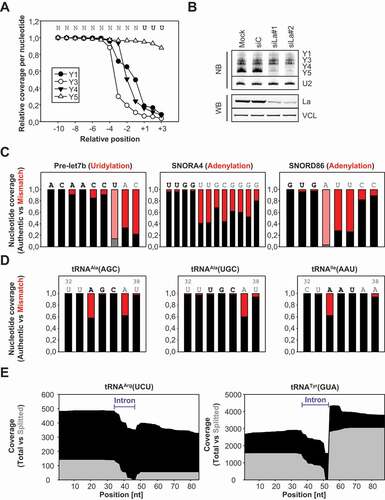
Next to Y RNAs, various msRNAs are modified at the 3ʹ-end modulating their turnover or biogenesis [Citation22]. For instance, the cytoplasmic uridylation of de-adenylated RNAs by TUTases is implicated in their degradation by the exonuclease DIS3L2 [Citation23]. One prominent example for this is the oligo-uridylation of the pre-let7 family [Citation24]. In agreement, we observed a significant uridylation with one to two added uridines in the pre-let7b miRNA in B16-F10 cells expressing this pre-miRNA at sufficient levels for the presented analyses (). For some snoRNAs oligo-adenylation has been described and was reported to enhance the PARN-dependent 3ʹ-end maturation of these msRNAs [Citation25]. In support of this, msRNAseq revealed oligo-adenylation of a variety of snoRNAs including murine SNORA4 and SNORD86 (). Interestingly, these snoRNAs showed adenylation to varying degrees resulting in the addition of at least eight (SNORA4) or four adenines (SNORD86).
Analysis of tRNA modification and processing
TRNAs are subject to a variety of nucleotide modifications. To test if some of these modifications can be detected by msRNAseq we searched for mismatches in human tRNA genes. First, we focused on ADAT-catalysed (adenosine deaminase acting on tRNA) A-to-I editing [Citation26]. This is detectable by RNA-sequencing due to the mis-incorporation of nucleotides by reverse transcriptases frequently reading inosine as guanine resulting in a characteristic A-to-G conversion [Citation27]. For these studies, three different loci encoding for tRNAAla(AGC), tRNAAla(UGC) or tRNAIle(AAU) were picked. These tRNAs show high degrees of modification within their anticodon regions [Citation28]. Consistently, msRNAseq revealed a prominent A-to-G conversion in the anticodon region: a) positions 34 and 37, tRNAAla(AGC); b) position 37, tRNAAla(UGC); c) position 34, tRNAIle(AAU) ().
To test if msRNAseq also indicates tRNA splicing intermediates we evaluated two transcripts: tRNAArg(UCU) and tRNATyr(GUA) (Supp. ) [Citation29]. Since we did not perform a de-acylation of the RNA used for msRNAseq, we expected that a significant amount of the sequenced tRNAs should represent nascent tRNAs and processing intermediates. So the here presented data just give limited information on the abundant matured tRNAs but allows the analysis of tRNA biogenesis. In HEK293 cells, msRNAseq revealed a substantial amount of nascent, unspliced tRNAs with decreased total coverage within the respective intronic regions of both tRNAs (black coverage, ). This indicated that distinct intermediates of tRNA processing could be detected. This included nascent pre-tRNAs, tRNA fragments splitted during splicing (indicated in grey coverage) and matured, spliced tRNAs.
Identification of POLIII-transcripts
RNA polymerase III is responsible for synthesizing the majority of msRNAs. Three different promoter types and pre-initiation complexes for this polymerase had been described (Type I, II and III) [Citation30]. The genes encoding for tRNAs are usually Type II genes, harbouring specific sequences (A- and B-Boxes) to initiate their transcription by TFIIIC and thereby POLIII [Citation31]. In order to identify putative novel Type II POLIII-genes, we applied a motif-based search (using the FIMO package) [Citation32] on the 333 putative candidate msRNAs which we obtained by msRNAseq (). We could clearly identify 14 msRNAs that contained A- and B-Boxes and thus resembled putative Type II genes. Next we crosschecked these 14 RNAs for occurrence in a tRNA database (tRNAdb) [Citation33,Citation34]. Here it emerged that six of these genes were listed as tRNAs and excluded from further analyses. Additionally, we investigated if the remaining eight msRNAs showed major peaks in publically available ChIP data sets (derived from ENCODE). We could clearly detect BDP1/BRF1/POLR3G peaks in close proximity to three of these msRNA genes, indicating that these genes contain respective transcription factor recognition motifs. Indeed, we found A- and B-Boxes matching the consensus sequence in all three genes (). These three msRNAs were used for further characterization and named after their genomic location PAMR1 interspersed RNA (PAMIR), C1QTNF6 interspersed RNA (COIR) and POLR3E antisense RNA (POLAR) (Supp. ).
Figure 3. Identification of POLIII-derived msRNAs. A The pipeline for the identification of putatively novel msRNAs is depicted. B Comparison of the consensus sequences of A- and B-Boxes compared to the three high confidence msRNA candidates obtained by msRNAseq (POLAR, PAMIR and COIR). C Total RNA from transient siRNA-mediated depletions of BDP1 in HEK293 cells was subjected to RT-qPCR analyses. EEF2 mRNA served as negative control, whereas POLAR-, PAMIR- and COIR-levels significantly decreased upon BDP1 knockdown. D Nucleo-cytoplasmic fractionation of HEK293 was performed and resulting RNAs were subjected to RT-qPCR analyses. For the depicted RNAs the ratio between nuclear and cytoplasmic fractions was determined. PPIA mRNA served as cytoplasmic and U1 snRNAs as nuclear control
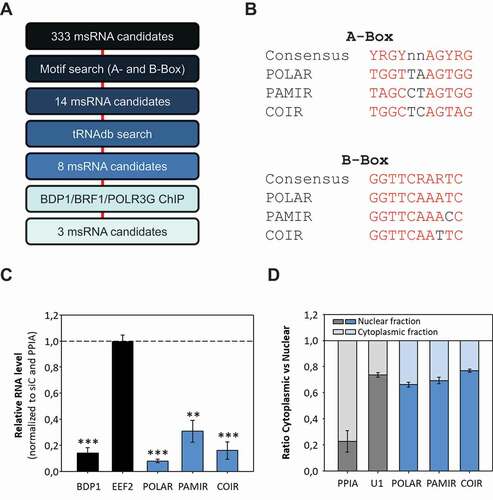
POLIII-driven transcription of POLAR, PAMIR and COIR was supported by the depletion of BDP1 using siRNAs (). RT-qPCR showed that msRNA-levels were strongly decreased upon BDP1 depletion whereas EEF2 mRNA levels (POLII-driven) were unchanged. The subcellular localization of these msRNAs was analysed by RT-qPCR upon fractionation (cytoplasmic vs nuclear) of HEK293 cells (). PPIA mRNA was mainly observed in the cytoplasm whereas U1 snRNA as well as all three msRNAs were enriched in the nuclear fraction. These results showed that msRNAseq experimentally identified three previously unannotated msRNAs, which are transcribed by a Type II POLIII-driven mechanism and localize to the nucleus.
Characterization of the msRNA POLAR
The msRNAseq analyses suggested various unannotated msRNAs which could not be assigned to any of the well-characterized msRNA families like snoRNAs or tRNAs. The locus encoding one of these msRNA candidates was mapped to chr16:22,298,494–22,298,627 (hg38) in the first intron of the human POLR3E gene, notably in antisense orientation ( upper panel). Therefore, this msRNA was named POLR3E Antisense RNA (POLAR). ChIP-seq data for POLIII-associated proteins (BDP1, BRF1 and POLR3G) in K562 cells (derived from ENCODE) suggested POLIII-driven synthesis of POLAR ( bottom panel) [Citation35]. The POLIII-driven tRNALeu locus, located upstream of the POLR3E locus, served as positive control. BDP1 and BRF1 ChIP-signals were observed immediately upstream of both loci, tRNALeu and POLAR. POLR3G ChIP signals were found within both genes. Since POLIII-transcripts lack poly-A-tails and usually end in an Oligo-U terminator we tested if POLAR can be reverse transcribed by Oligo-dT or random hexamers (R6) only (Supp. ). RT-PCR analyses confirmed that mRNAs like EEF2 are primed by dT and R6 whereas POLAR was only amplified by R6-priming suggesting that POLAR lacks a poly(A) tail.
Figure 4. Identification and characterization of POLAR. A The msRNA coverage at the human POLR3E is shown (top panel). The POLR3E antisense RNA (POLAR) was identified as msRNA. The genes encoding for POLR3E and tRNALeu are indicated. ENCODE ChIP-seq data of three POLIII-associated proteins was included for the same genomic region (bottom panel). B The La protein was overexpressed as SBP-tagged version or depleted by two independent siRNAs (siLa#1, siLa#2) in HEK293 cells. Total RNA from these experiments was subjected to RT-qPCR analyses and POLAR levels were determined. RNA levels were normalized to the empty vector control or control siRNA transfections. C Stability of indicated RNAs was determined by Actinomycin D treatment in HEK293 cells. D RNA pulldowns were performed from OVCAR3 cells and subjected to Western Blot analyses. Beads only and Y4 pulldowns were used as negative controls for PSP-associated proteins (PSF, NONO and RBM14). VCL and EIF3A served as negative controls to test for unspecific binding. La was used as control, associating with both POLIII-transcripts. E OVCAR3 cells were subjected to fluorescence in situ hybridizations (FISH). POLAR-FISH (left column) was combined with immunostainings against paraspeckle-associated proteins (NONO, PSF and PSP1) or the splicing speckle-associated protein SC35. The second column shows the staining for the individual RBP. The cells were counterstained with DAPI to label the nuclei (third column). A representative intensity profile of FISH and RBP signals is shown on the right, with areas indicated in the DAPI panel (dotted line). Statistical significance was determined by student’s t-test (ns – not significant, * – p < 0,05, ** – p < 0,01). Bar 5 µm
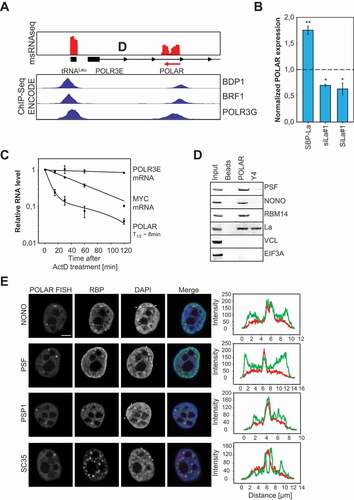
A key regulator of POLIII-transcript homoeostasis and turnover is the La protein typically binding at an oligo-U tract at the 3ʹ-end of nascent POLIII-transcripts [Citation36]. To check if POLAR, like Y5, is regulated by La, the protein was depleted or overexpressed in HEK293 cells and RNA abundance was analysed by RT-qPCR (; Supp. ). Similar to Y5, the depletion of La resulted in significantly decreased POLAR levels whereas abundance was markedly increased by the overexpression of La. If the La-directed stabilization of POLAR was associated with the binding of the protein to the msRNA was analysed by La-pulldowns in HEK293 cells (Supp. ). In contrast to the PPIA and EEF2 mRNAs, POLAR like nascent Y4 (positive control) was significantly enriched by La precipitation. Together this provided strong evidence that POLAR is a POLIII-driven transcript bound by the La protein.
Most POLIII-driven ncRNAs are characterized by long half-lifes, e.g. 5S rRNA or tRNAs. To test if this also holds for POLAR, we performed RNA decay analyses using Actinomycin D to impair global RNA synthesis (). POLR3E and MYC mRNAs served as controls. While POLR3E mRNA remained stable over the 2 h time course, MYC mRNA was readily degraded with a half-life of approximately 45 minutes as previously reported [Citation37]. Surprisingly, POLAR was rapidly degraded with an estimated half-life of only 8 minutes.
Aiming to identify the cellular role of POLAR CRISPR/Cas9-directed knockout clones were established using four distinct sgRNAs and one sgRNA combination (Supp. ). In view of the antisense orientation of POLAR and POLR3E, it was tempting to speculate that POLAR modulates POLR3E synthesis. However, neither POLR3E mRNA nor protein levels were substantially affected in nine analysed clones showing severely reduced POLAR expression (Supp. , F). This was further validated by the transient overexpression of POLAR from a plasmid. However, although overexpressed at least 100-fold, POLR3E mRNA abundance remained essentially unchanged indicating that POLAR-expression in trans has no significant impact on POLR3E mRNA expression in HEK293 cells (Supp. ). It has to be noted that in a recent report the mouse homolog of POLAR was identified [Citation38]. The authors suggest that the transcription of POLAR by POLIII interferes with the POLII-driven transcription of the POLR3E gene and thereby decreases transcriptional output from this protein-coding gene. In contrast to these findings, we did not observe significant changes of POLR3E mRNA or protein upon POLAR deletion (Supp. ). Future studies have to reveal if this discrepancy can be for instance explained by species-specific effects or cell-type-specific regulation.
Various POLIII-driven ncRNAs are essential co-regulators of mRNA processing modulating the splicing and/or 3ʹ-end processing of mRNAs, e.g. snRNAs or Y RNAs [Citation15,Citation39]. Associated with these cellular functions, some of these msRNAs are enriched in discrete nuclear foci like splicing speckles and Cajal bodies [Citation40]. If this is also observed for POLAR was analysed by fluorescence in situ hybridization (FISH) (). Although POLAR was expressed in all tested cell lines, OVCAR-3 and HEK293 showed the highest expression of POLAR indicating OVCAR-3 as the preferred cell model for FISH (Supp. ). Consistent with the fractionation analyses (see ), POLAR was localized to the nucleoplasm but absent from nucleoli. However, POLAR was strongly enriched in discrete nuclear foci suggesting an association with nuclear RNA speckles. However, counter-staining for the SC35 protein, a widely used marker of splicing speckles, revealed no significant co-localization. In view of their irregular shape and varying number, we speculated that POLAR associates with paraspeckles (PSPs) that are observed in the interchromatin space of most human cells [Citation41]. This was confirmed by the counterstaining with the PSP-marker proteins NONO, PSF and PSP1 (). In contrast to SC35, all three PSP-markers showed a significant co-localization with POLAR in discrete nuclear foci suggesting that the msRNA is enriched in PSPs. If POLAR also associates with PSP-proteins was analysed by RNA-pulldown studies using biotinylated in vitro transcribed POLAR and OVCAR-3 cell lysates (). The Y4 ncRNA served as negative control since although transcribed by POLIII the transcript is enriched in the cytoplasm and has not been reported in PSPs [Citation42]. Consistent with its enrichment in PSPs, POLAR co-purified the PSP components NONO, PSF and RBM14 as well as the La protein that was confirmed to bind POLAR in protein pulldowns (see Supp. ). Notably, VCL and EIF3A, were neither co-purified with Y4 nor POLAR. However, nascent Y4 comprising a poly-U stretch at its 3ʹ-end effectively co-purified La, as previously reported [Citation42].
The NEAT1 lncRNA is an essential scaffold of PSPs and it was proposed that PSPs and NEAT1 evolved in the lineage of placental mammals [Citation43,Citation44]. To investigate the evolutional conservation of POLAR we analysed available genomes for the POLAR locus. POLAR genes were identified in several species of placental mammals, including mouse and human (Supp. ). In contrast, we could not find any evidence of POLAR conservation in other mammals like marsupials. Taken together these results suggest that both PSPs/NEAT1 and POLAR evolved in the lineage of placental mammals supporting the view that POLAR serves an essential role in the poorly understood function of paraspeckles.
Discussion
MsRNAs, with a size range of 50–300 nts, are inadequately represented by currently used RNA-sequencing protocols due to insufficient removal of 5ʹ-modifications hindering adapter ligation and RNA fragmentation degrading msRNAs beyond a limiting size threshold. In addition, most currently used analysis pipelines of RNA-sequencing data discard reads not uniquely mapped to the genome, as observed for many msRNAs transcribed from multiple genomic loci (e.g. Y RNAs, snRNAs). The here presented msRNAseq protocol in combination with modified annotation and analyses pipelines largely overcomes these limitations. In addition to the identification of msRNAs, the presented protocol allows the quantitative and qualitative analysis of msRNA modification, processing and splicing. Importantly, a continuous coverage of 50 reads over a length of at least 40 nucleotides was used as a threshold for the identification of msRNA. Accordingly, we expect to have missed a substantial fraction of msRNAs yet to be discovered. Presumably due to the size selection included in the msRNAseq protocol, contamination by larger, even highly abundant RNAs was negligible. Contaminations with large rRNAs were minor and could be reduced further by applying rRNA depletion. Consistent with their small size (starting from ~300 nts) histone mRNAs presented the only significant contamination by protein-coding transcripts.
Like observed for other ligation-based RNA-sequencing methods, msRNAseq is biased in respect to the absolute quantification of RNA species since the removal of 5ʹ- or 3ʹ-end modifications is incomplete. Therefore, it is likely that read coverage in msRNAseq is still somewhat biased towards transcripts that already naturally can be ligated to RNA adapters (e.g. snoRNAs). Despite these limitations, msRNAseq proved suitable for the relative quantification of msRNAs and identified major previously known msRNAs including 5.8S rRNA, snoRNAs and snRNAs. Matured tRNAs were barely observed due to the lack of de-acylation. If applied, we expect that msRNAseq also identifies large portions of matured tRNAs. However, here we intentionally focused on pre-tRNAs since this allows precise genomic mapping due to flanking sequences and enabled us to analyse tRNA biogenesis including tRNA splicing intermediates readily identified by msRNAseq.
In addition to the sole identification of msRNAs, msRNAseq allowed the analysis of msRNA modifications and processing. For pre-miRNAs and snoRNAs, the addition of non-encoded nucleotides was observed at the cost of sufficiently flexible alignments. Associated with this decreased alignment accuracy and the low abundance of such modified RNA molecules, the number of added nucleotides cannot be determined more precisely. This may be improved by further depleting rRNAs and thereby allowing analyses at a greater sequencing depth. Another modification observed in msRNAs is the A-to-I editing of tRNAs. This is readily observed due to error-prone reverse transcriptase and thus does not involve chemical modification. The robust identification of A-to-I editing in ~40% of respective tRNA reads suggests a substantial degree of modification within the analysed tRNAs. Analyses of Y RNAs by msRNAseq revealed that only nuclear Y5 retains a significant Oligo-U tract at its 3ʹend allowing association of the La protein and stabilization of this ncRNA. Finally, the characterization of tRNA reads identified nascent next to splicing intermediates of specific tRNAs indicating that msRNAseq is a versatile technique suitable for the characterization of msRNA processing, biogenesis and modification.
Aiming to evaluate if previously unannotated msRNAs identified by msRNAseq simply indicate transcriptional background or functional ncRNAs we characterized the POLIII-driven transcripts POLAR, PAMIR and COIR. In depth analyses showed that POLAR is the first (to the best of our knowledge) msRNA localized to paraspeckles (PSP). POLAR also interacts with PSP-associated proteins like PSF, NONO or RBM14 in RNA pulldowns. Intriguingly, the evolutionary conservation of POLAR suggests that it co-evolved with NEAT1 and thus PSPs in placental mammals. Thus, future studies will now have to reveal the role of POLAR in PSPs. In view of its binding to PSP-associated proteins and short half-life, one could speculate that this ncRNAs serves scaffolding roles for PSP-associated proteins and/or their nuclear sorting as already suggested for other msRNAs [Citation45]. The association of POLAR with PSPs could also indicate a pathophysiological role since NEAT1 and thus PSPs are upregulated in a variety of cancers [Citation46–48].
In summary, we present a powerful technique that is suitable not only for the identification of msRNAs but also allows characterizing their processing, biogenesis and modification. This will pave the way for unravelling the plethora of yet uncharacterized ncRNAs.
Supplemental Material
Download Zip (385.6 KB)Acknowledgments
The authors acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation), project number 391498659, RTG 2467 “Intrinsically Disordered Proteins – Molecular Principles, Cellular Functions, and Diseases”.
Disclosure statement
The authors report no conflict of interest.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Podnar J, Deiderick H, Huerta G, et al. Next-generation sequencing RNA-seq library construction. Curr Protoc Mol Biol. 2014;106(4):21 1–19.
- McGinn J, Small CB. RNA library construction for high-throughput sequencing. Methods Mol Biol. 2014;1093:195–208.
- Orioli A, Pascali C, Pagano A, et al. RNA polymerase III transcription control elements: themes and variations. Gene. 2012;493:185–194.
- Kessler AC, Silveira d’Almeida G, Alfonzo JD. The role of intracellular compartmentalization on tRNA processing and modification. RNA Biol. 2018;15:554–566.
- Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15:108–121.
- Jarrous N. Roles of RNase P and its subunits. Trends Genet. 2017;33:594–603.
- AJ CQ, Bugai A, Barboric M. Cracking the control of RNA polymerase II elongation by 7SK snRNP and P-TEFb. Nucleic Acids Res. 2016;44:7527–7539.
- Rak R, Dahan O, Pilpel Y. Repertoires of tRNAs: the couplers of genomics and proteomics. Annu Rev Cell Dev Biol. 2018;34:239–264.
- Goodarzi H, Nguyen HCB, Zhang S, et al. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell. 2016;165:1416–1427.
- Cavaille J, Buiting K, Kiefmann M, et al. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc Natl Acad Sci U S A. 2000;97:14311–14316.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. Vol. 17. 2011.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359.
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842.
- Li H, Handsaker B, Wysoker A, et al. The sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079.
- Kohn M, Ihling C, Sinz A, et al. The Y3** ncRNA promotes the 3ʹ end processing of histone mRNAs. Genes Dev. 2015;29:1998–2003.
- Wachter K, Kohn M, Stohr N, et al. Subcellular localization and RNP formation of IGF2BPs (IGF2 mRNA-binding proteins) is modulated by distinct RNA-binding domains. Biol Chem. 2013;394:1077–1090.
- Kohn M, Pazaitis N, Why HS. YRNAs? About versatile RNAs and their functions. Biomolecules. 2013;3:143–156.
- Wolin SL, Belair C, Boccitto M, et al. Non-coding Y RNAs as tethers and gates: insights from bacteria. RNA Biol. 2013;10:1602–1608.
- Gendron M, Roberge D, Boire G. Heterogeneity of human Ro ribonucleoproteins (RNPS): nuclear retention of Ro RNPS containing the human hY5 RNA in human and mouse cells. Clin Exp Immunol. 2001;125:162–168.
- Simons FH, Rutjes SA, van Venrooij WJ, et al. The interactions with Ro60 and La differentially affect nuclear export of hY1 RNA. RNA. 1996;2:264–273.
- Xue D, Shi H, Smith JD, et al. A lupus-like syndrome develops in mice lacking the Ro 60-kDa protein, a major lupus autoantigen. Proc Natl Acad Sci U S A. 2003;100:7503–7508.
- Song J, Mo B, Chen X. Uridylation and adenylation of RNAs. Sci China Life Sci. 2015;58:1057–1066.
- Viegas SC, Silva IJ, Apura P, et al. Surprises in the 3ʹ-end: ‘U’ can decide too! Febs J. 2015;282:3489–3499.
- Heo I, Joo C, Kim Y-K, et al. TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell. 2009;138:696–708.
- Berndt H, Harnisch C, Rammelt C, et al. Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. RNA. 2012;18(5):958–972.
- Schaub M, Keller W. RNA editing by adenosine deaminases generates RNA and protein diversity. Biochimie. 2002;84:791–803.
- Oakes E, Vadlamani P, Hundley HA. Methods for the Detection of Adenosine-to-Inosine Editing Events in Cellular RNA. Methods Mol Biol. 2017;1648:103–127.
- Su AA, Randau L. A-to-I and C-to-U editing within transfer RNAs. Biochemistry (Mosc). 2011;76:932–937.
- Hopper AK. Transfer RNA post-transcriptional processing, turnover, and subcellular dynamics in the yeast Saccharomyces cerevisiae. Genetics. 2013;194:43–67.
- Schramm L, Hernandez N. Recruitment of RNA polymerase III to its target promoters. Genes Dev. 2002;16:2593–2620.
- Ramsay EP, Vannini A. Structural rearrangements of the RNA polymerase III machinery during tRNA transcription initiation. Biochim Biophys Acta Gene Regul Mech. 2018;1861:285–294.
- Cuellar-Partida G, Buske FA, McLeay RC, et al. Epigenetic priors for identifying active transcription factor binding sites. Bioinformatics. 2012;28:56–62.
- Chan PP, Lowe TM. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016;44:D184–9.
- Juhling F, Morl M, Hartmann RK, et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–62.
- An integrated encyclopedia of DNA elements in the human genome. Nature. ENCODE Project Consortium. 2012;489:57–74.
- Maraia RJ, Mattijssen S, Cruz-Gallardo I, et al. The La and related RNA-binding proteins (LARPs): structures, functions, and evolving perspectives. Wiley Interdiscip Rev RNA. 2017;8(6):10.1002/wrna.1430.
- Weidensdorfer D, Stohr N, Baude A, et al. Control of c-myc mRNA stability by IGF2BP1-associated cytoplasmic RNPs. RNA. 2009;15:104–115.
- Yeganeh M, Praz V, Cousin P, et al. Transcriptional interference by RNA polymerase III affects expression of the Polr3e gene. Genes Dev. 2017;31:413–421.
- Will CL, Luhrmann R. Spliceosome structure and function. Cold Spring Harb Perspect Biol. 2011;3:a003707-a003707.
- Machyna M, Heyn P, Neugebauer KM. Cajal bodies: where form meets function. Wiley Interdiscip Rev RNA. 2013;4:17–34.
- Fox AH, Lam YW, Leung AKL, et al. Paraspeckles: a novel nuclear domain. Curr Biol. 2002;12:13–25.
- Fabini G, Rutjes SA, Zimmermann C, et al. Analysis of the molecular composition of Ro ribonucleoprotein complexes. Identification of novel Y RNA-binding proteins. Eur J Biochem. 2000;267:2778–2789.
- Clemson CM, Hutchinson JN, Sara SA, et al. An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol Cell. 2009;33:717–726.
- Hutchinson JN, Ensminger AW, Clemson CM, et al. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39.
- Täuber H, Hüttelmaier S, Köhn M. POLIII-derived non-coding RNAs acting as scaffolds and decoys. J Mol Cell Biol. 2019;11:880–885.
- Choudhry H, Albukhari A, Morotti M, et al. Tumor hypoxia induces nuclear paraspeckle formation through HIF-2alpha dependent transcriptional activation of NEAT1 leading to cancer cell survival. Oncogene. 2015;34:4482–4490.
- Pan LJ, Zhong T-F, Tang R-X, et al. Upregulation and clinicopathological significance of long non-coding NEAT1 RNA in NSCLC tissues. Asian Pac J Cancer Prev. 2015;16:2851–2855.
- Guo S, Chen W, Luo Y, et al. Clinical implication of long non-coding RNA NEAT1 expression in hepatocellular carcinoma patients. Int J Clin Exp Pathol. 2015;8:5395–5402.