
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
All species transcribe ribosomal RNA in an immature form that requires several enzymes for processing into mature rRNA. The number and types of enzymes utilized for these processes vary greatly between different species. In low G + C Gram-positive bacteria including Bacillus subtilis and Geobacillus stearothermophilus, the endoribonuclease (RNase) M5 performs the final step in 5S rRNA maturation, by removing the 3ʹ- and 5ʹ-extensions from precursor (pre) 5S rRNA. This cleavage activity requires initial complex formation between the pre-rRNA and a ribosomal protein, uL18, making the full M5 substrate a ribonucleoprotein particle (RNP). M5 contains a catalytic N-terminal Toprim domain and an RNA-binding C-terminal domain, respectively, shown to assist in processing and binding of the RNP. Here, we present structural data that show how two Mg2+ ions are accommodated in the active site pocket of the catalytic Toprim domain and investigate the importance of these ions for catalysis. We further perform solution studies that support the previously proposed 3ʹ-before-5ʹ order of removal of the pre-5S rRNA extensions and map the corresponding M5 structural rearrangements during catalysis.
Introduction
All species utilize 3 to 4 ribosomal RNAs (rRNAs) and ribosomal proteins (r-proteins) to assemble a functional ribosomal particle. The rRNAs are initially transcribed as pre-rRNAs that require trimming at the 5ʹ- and 3ʹ-end to become mature rRNAs within ribosomes. The rRNA maturation process diverges greatly between species, and, even within the same kingdom, the proteins used for a specific processing reaction can vary significantly. As such, the Gram-negative model organism Escherichia coli and the low G + C Gram-positive model organism Bacillus subtilis share only two processing pathways i.e. the RNase III-dependent pathway used for the release of pre-rRNAs (16S, 23S, 5S) from an initial long pre-rRNA transcript (30S) [Citation1–3] and the 16S specific YbeY/YqfG pathway used for processing of the 3ʹ-end of 16S rRNA. All remaining steps towards mature rRNAs occur via entirely different sets of proteins where E. coli utilizes RNase E, G, AM, T, PH, R and II, and PNPase and B. subtilis uses RNases J1, M5 and mini-RNase III to perform the same reactions (summarized in [Citation4]). The RNase M5 enzyme (M5) is responsible for processing of the pre-5S rRNA at both the 5ʹ- and 3ʹ-end to produce the mature 5S rRNA. The substrate-cleavage occurs in a double-stranded (ds) RNA segment of pre-5S rRNA by staggered cuts [Citation5–10]. M5 activity depends on the initial association of pre-5S rRNA with the r-protein uL18 [Citation6,Citation11], as uL18 maintains the substrate fold for optimal enzyme recognition and permits cleavage [Citation10,Citation12,Citation13]. This initial association makes the actual substrate for M5 a ribonucleoprotein (RNP) particle of pre-5S rRNA and uL18.
M5 is an endoribonuclease belonging to the Toprim family of proteins also encompassing DnaG-like primases, DNA topoisomerases (bacterial type IA, II, and VI), OLD family nucleases, and RecR family of DNA repair proteins [Citation14]. Toprim proteins cleave and/or join single- or double-stranded DNA or RNA strands, and are catalytically dependent on, or stimulated by, Mg2+ ions [Citation15–17]. M5 is the only known Toprim domain-containing protein that cleaves both strands of its dsRNA substrate. Toprim domains are most often found in combination with other domains, reflecting the differences in substrate specificity and activity of proteins across this family [Citation14]. This is also the case for M5 that contains both an N-terminal catalytic Toprim domain (NTD) and a C-terminal domain (CTD) with an RNA-binding domain (RBD) fold, that has not been observed in any other structurally characterized proteins [Citation10]. The CTD fold covers four α-helices most closely resembling the death domain fold [Citation10]. Proteins of the death domain family, however, operate in distinctively different pathways to that of M5, adding to the novelty of the CTD fold.
The structure of M5 is not known, but we recently solved the crystal structures of the M5 NTD and CTD and the cryoEM structure of inactive M5D58A bound to its RNP substrate in the full 50S ribosomal particle [Citation10]. M5 binds to the 50S particle as a monomer, with the catalytic NTD located at the pre-5S rRNA 3ʹ-cleavage site. Additional biochemical studies showed that the M5 CTD, that exhibits a novel RNA-binding fold, drives the binding to the RNP substrate via conserved residues from two of its four α-helices, α6 and α7. Initial attachment of the CTD, in turn, positions the catalytic NTD at the 3ʹ-cleavage site, thus also playing a role in direct facilitation of catalysis. uL18 acts as an RNA chaperone that moulds the pre-5S rRNA into an optimal fold for initial recognition by the CTD, and also positions the rRNA-precursor extensions by the NTD for cleavage. In addition to revealing the roles of each M5 domain and the uL18 cofactor r-protein, the cryoEM data also provided mechanistic details about the rRNA cleavage reaction. The 3ʹ- and 5ʹ-strand together form a dsRNA helix, and since M5 is bound to the 3ʹ-strand, it is predicted to initially cleave this before removing the extension from the opposite 5ʹ-strand. This is consistent with a previous observation that a substrate lacking the 5ʹ-strand was a poorer substrate for M5 than a substrate lacking the 3ʹ-strand that was as good a substrate as the intact RNA [Citation8]. We previously proposed that structural rearrangements of M5 domains and the 5ʹ-strand – subsequent to 3ʹ-extension removal – would allow M5 to cleave the 5ʹ-strand [Citation10], and that this cleavage reaction would proceed via a mechanism involving two Mg2+ ions coordinated by acidic residues.
Here, we present the crystal structure of the catalytic domain of M5 bound to two Mg2+ ions, which reveals the role of each catalytic site acidic residue in metal coordination. We further use small-angle X-ray scattering (SAXS) to study the solution structure of M5, and hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS) to investigate the structural rearrangements that would allow cleavage of the 5ʹ-strand subsequent to removal of the 3ʹ-extension. Together, this study provides new details on the mechanism used by M5 for cleavage of the dsRNA substrate.
Results
The NTD of M5 is an extended Toprim domain
The NTD of M5 presents a Toprim domain fold, which is classically comprised of three α-helices flanking a central β-sheet of three parallel β-strands [Citation10,Citation14]. Compared to this core fold, the M5 NTD contains an additional α-helix (α4, residues 100–109) at its C-terminus. We investigated the effect of this helix on protein integrity, by mapping the boundaries for a minimal, well-folded NTD protein. Three protein constructs were prepared, spanning the conserved core of the Toprim domain (residues 1–80), the core and the additional helix (residues 1–110), or the latter plus three residues assigned to the linker (residues 1–113). The folding of each protein was evaluated by 1D NMR, revealing that only the longest construct, including the entire additional helix and part of the linker, yielded well-folded protein (). Other Toprim domains are well folded in the absence of such an additional element, and it is therefore surprising that M5 requires this segment for stabilization of the fold. Analysis of the NTD crystal structure (PDB 6TG6) reveals that residues R104 and Y111 – located at the beginning and end, respectively, of the additional helix – form hydrogen bonds to residues in other elements of the NTD i.e. R104 to A21, and Y111 to E5 (which in turn hydrogen bonds to K76) (), which could have a stabilizing effect on the entire fold.
Figure 1. Structural organization of M5. (A) 1D 1H NMR spectra for three constructs of M5 NTD: residues 1–80 (left), 1–110 (middle), and 1–113 (right) showing that only the construct spanning residues 1–113 is folded. (B) Structure of the M5 NTD zoomed on the additional α-helix (α4: residues 100–109). A residue within (R104), and just after the helix (Y111) form hydrogen bonds to residues in other structural elements (A21 and E5-to-K76, respectively), stabilizing the Toprim domain structure. These hydrogen bonds are shown in black dots. (C) Schematic and structural outline of M5 domains comprising an extended N-terminal Toprim domain (NTD), a linker region and a C-terminal RNA-binding domain (CTD). Secondary structure elements are indicated on the M5 structure taken from the cryoEM structure of M5 bound to the 50S (pre5S) particle (PDB 6TPQ). (D) Native mass spectrum of full-length M5 showing that the protein is a monomer with a molecular mass of 20,989 ± 1 Da corresponding to its precise theoretical molecular mass
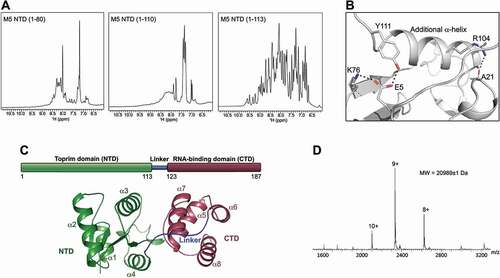
The M5 NTD thus spans residues 1–113 covering the entire catalytic Toprim domain. The NTD shows a central β-sheet surrounded by four α-helices (α1-α4) and a long loop with two small α-helical segments (αS1 and αS2), connecting β4 to β4 (). The NTD is followed by a flexible linker (residues 114–122), and an RNA-binding C-terminal domain of four α-helices (α5-α8) (CTD, residues 123–187) (). Toprim domains usually function as monomers but can be part of multimeric proteins formed by interaction via other domains. Previous cryoEM structural data revealed that M5 binds to the 50S ribosomal subunit as a monomer [Citation10]. Native mass spectrometry (Native MS) () for M5 and its individual CTD and NTD (Supplementary , B) confirmed that this protein is a monomer in solution.
Although the structure of its individual domains (NTD and CTD) is known, the overall structural organization of isolated full-length M5 is not. During crystallization trials to obtain a full-length M5 structure, the flexible linker connecting the NTD and CTD of M5 was highly prone to proteolysis, which did not change with extensive mutations and/or deletions in this linker region. All further structural studies were therefore performed using the catalytic NTD of M5.
The catalytic NTD of M5 binds two Mg2+ ions
The activity of Toprim domain-containing proteins depends on the presence of Mg2+ ions. In classical Toprim domains, these ions are coordinated directly or via water to one, two, or all three of the highly conserved acidic residues; E10, D56, D58 (B. subtilis M5 numbering) [Citation14], and sometimes to additional acidic residues outside this triad [Citation18]. To investigate whether M5 binds Mg2+ ions, the thermostability of the catalytic NTD was evaluated by recording the protein melting temperature (Tm) by differential scanning fluorimetry (DSF) in the absence and presence of Mg2+ ions (). The difference in Tm (ΔTm) between the apo- and metal-bound protein (ΔTm of 7.6°C) indicated, as expected, binding of the NTD to Mg2+ ions.
Figure 2. Metal binding in the M5 N-terminal domain. (A) Plot of melting temperature (Tm) of the N-terminal domain (NTD) of M5 in the absence or presence of Mg2+ ions, measured in degrees Celsius (C) from a DSF experiment. Significant difference is indicated by asterisks according to a standard P-test. (B) Electrostatic surface potential view of the M5 NTD co-crystallized with two Mg2+ ions (green spheres) bound in the active site. Water molecules that coordinate the ions are shown as red spheres. Red and blue surfaces indicate negative and positive electrostatic potentials, respectively, while white surfaces represent neutral residues. (C) Electron density maps of the active site in the Mg2+-bound M5 (Mg2+ as green spheres, water molecules as red spheres). The composite difference 2 Fo-Fc density omit map (blue) is contoured at 1.5σ. The difference Fo-Fc omit map (green) for the M5 model refined without the Mg2+ ions in the active site and inner-sphere water molecules is contoured at 5σ. (D) The acidic residues (E10, D14, D56, D58 and E96, stick representation) and water molecules (W, red spheres) in the active site of M5 that participate in binding of the two Mg2+ ions (A and B, green spheres). The octahedral arrangement around metal A and square-planar arrangement around metal B are indicated by outlining the bonds (black dots) to the inner-sphere O-atoms. (E) Superimposition of the Mg2+-bound and apo NTD structures. Residues (D14, D56 D58, E96) that coordinate Mg2+ ions (A, B) are shown as sticks. (F) Superimposition of the Mg2+-bound NTD structure onto the NTD from the RNA-bound M5 model. An O-atom from A117 in the RNA cleavage site is positioned such that it could coordinate a Mg2+ ion (ion B, in Fig. 2B). Substrate pre-5S rRNA is shown in orange with the extensions in yellow. The 3ʹ-cleavage site is indicated
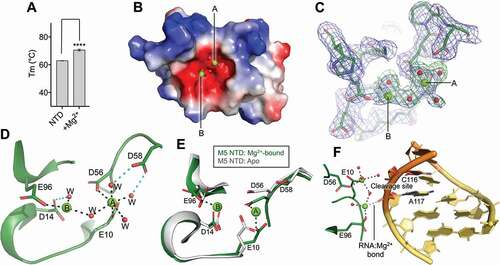
In our previous structure of the M5 NTD (PDB: 6TG6) co-crystallization with ion(s) in the active site was prohibited due to steric blockage by residues from symmetry-related molecules [Citation10]. To determine the binding site(s) of the catalytic ion(s) in the NTD, we, therefore, searched for alternative crystal packings that would allow protein:ion co-crystallization. Crystals were obtained with such alternative packing with a protein:Mg2+ ratio of 1:12.5 in the crystallization conditions (). This yielded a 2.14 Å resolution structure with two Mg2+ ions bound in the highly charged active site pocket (, C, Supplementary ). In this structure, the first metal ion (A) shows an octahedral coordination sphere with the inner-sphere oxygen ligands donated by two O-atoms from two conserved residues, E10 and D56, from the catalytic triad, with bonding distances of 2.05 and 2.21 Å, respectively (), and four water molecules with bonding distances between 1.94 and 2.68 Å. Two of these water molecules are further kept in place by coordination to the third residue from the conserved catalytic triad, D58. The second ion (B) is coordinated in a square-planar arrangement involving O-atoms from the acidic residue E96 (2.02 Å) and two water molecules (1.97 and 2.35 Å, respectively), one of which is kept in place by further coordination to the acidic residue D14 (). One water molecule is shared between the two Mg2+ ions. The average coordination distance observed for the six (A, octahedrally coordinated Mg2+) and three (B, square planar coordinated Mg2+) inner-sphere oxygen ligands of each metal ion is consistent with Mg2+ ions occupying these sites [Citation19]. The two Mg2+ ions are positioned 4.2 Å apart, in agreement with distances found between Mg2+ in two-ion-dependent RNases when no substrate is bound [Citation20]. The binding of Mg2+ causes a slight tightening of the active site pocket compared to the apo-structure [Citation10] that relocates residue D56 1.3 Å towards the metal-binding site and rotated residue E10 at the Cβ-atom and residue E96 at the Cδ-atom (), to allow the coordination of one Mg2+ ion in the octahedral arrangement and one in the square planar arrangement. In the Mg2+-bound structure, all five acidic residues in the active site that contribute to metal ion binding are fixed in position by an elaborate network of hydrogen bonds, often involving coordination of the side-chain O-atom to the backbone N-atom of the same residue, or to strictly conserved residues such as G11 and G61 (Supplementary ). The three free coordination sites on the square-planar coordinated Mg2+ ion (B) could be taken up by atoms from the pre-5S rRNA substrate upon binding. Indeed, superimposition of the Mg2+-bound NTD with the NTD of RNA-bound M5 reveals that a phosphate group O-atom from nucleotide (nt) (A117) within the 3ʹ-cleavage site (C116-A117) is well positioned to take up a free coordination-position in the square-planar arrangement () with a coordination distance of 2.31 Å. The two Mg2+ ions could move closer in a substrate-bound complex, compared to their positions in the unbound structure, as previously observed for RNase H [Citation21], which could, in turn, induce additional RNA:ion interactions. The positioning of two Mg2+ ions in the active site, coordinated directly or through water to all conserved acidic residues of the active site, suggests that M5 acts via a two-ion cleavage mechanism.
Table 1. Crystallographic data. Processing and refinement statistics for the crystallized RNase M5 NTD
Two Mg2+ ions are required for RNase M5 activity
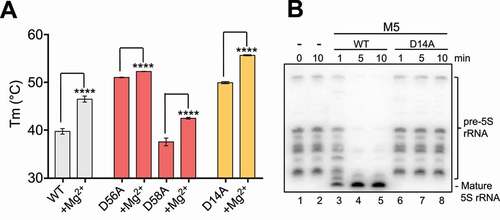
The Mg2+-bound NTD structure revealed two sites that each coordinate one Mg2+ ion. Residues from the first site (E10, D56 and D58) were previously shown to be indispensable for activity [Citation22]. Tm studies using full-length proteins mutated at either one of the aspartate residues in this site (M5D56A or M5D58A) showed that the mutant proteins are less thermostabilised in the presence of Mg2+ ions compared to wild-type (WT) enzyme, but still able to bind Mg2+ (ΔTmWT = 7.8°C, ΔTmD56A = 1.3°C, ΔTmD58A = 5.0°C) (). This suggests that an Mg2+ ion is still accommodated by M5 in the second ion binding site. Residues from the second site (D14, E96) were never previously investigated. We, therefore, set out to map their individual contribution to M5 Mg2+-binding and activity. Alanine-mutation of E96 persistently yielded insoluble protein when recombinantly expressed in E. coli, showing that E96 not only coordinates Mg2+, but also stabilizes the M5 fold. Tm studies of soluble M5D14A protein showed an overall more stable protein than M5WT, that could still coordinate Mg2+ with a ΔTmD14A = 5.7°C (), likely in the first ion binding site. For activity studies, we isolated 50S ribosomal subunits with pre-5S rRNAs (50S(pre-5S)) from a B. subtilis strain lacking the M5 gene (∆rnmV) [Citation5]. The pre-5S rRNAs produced from the 10 rrn operons in the ∆rnmV strain vary in length from 125 to 165 nucleotides (nts) and no mature 5S rRNA (116 nts) is produced (). Addition of recombinant M5D14A to 50S(pre-5S) substrate particles showed no maturation of pre-5S rRNA, while the WT enzyme efficiently generated mature 5S rRNA (). The complete loss of activity upon M5D14A mutation shown here, and M5E10A, -D56A, or −D58A shown previously [Citation22], together confirms that both Mg2+ ions are required for activity, establishing a two-metal ion catalytic mechanism for M5.
Figure 3. Key residues in M5 active site. (A) Plot of melting temperature (Tm) of the full-length M5 wild-type (WT) or mutant D56A, D58A and D14A protein in the absence or presence of Mg2+ ions, measured in degree Celsius (C) from a DSF experiment. Significantly different samples are indicated by asterisks according to a standard P-test. (B) Northern blot showing removal of 5ʹ- and 3ʹ-extensions from 50S(pre-5S) particles by M5 wild-type (M5WT) (lanes 3–5), and lack thereof by M5D14A mutant protein (lanes 6–8) at different time points. Lanes 1–2 show pre-5S rRNA at the first and last time point in the absence of M5 protein
The M5 domains interact via α7 in the apo form and when bound to the 3ʹ-cleavage site of the 50S(pre-5S) particle
The cryoEM structure of M5 bound to the 50S(pre-5S) particle (PDB: 6TPQ, EMD-10,543) shows a protein primed for cleavage of the 3ʹ-strand but without contacts to the 5ʹ-cleavage site [Citation10]. Despite the cryoEM structure being obtained at 200x excess of M5 to ribosome, no density for M5 was observed at the 5ʹ-strand, indicating that the 3ʹ-extension is removed prior to the 5ʹ-extension. We hypothesized that 5ʹ-strand cleavage would occur by relocation of the transiently bound NTD to the 5ʹ-strand, subsequent to removal of the 3ʹ-extension [Citation10]. The CTD was previously shown to anchor the protein to the 50S(pre-5S) particle [Citation10], and the presence of free CTD mixed in with full-length M5 for cryoEM structure determination did not reveal binding of the single domain at alternative sites near the pre-5S rRNA. Therefore, binding of the NTD to the 5ʹ-cleavage site must occur without co-relocation of the CTD, but instead via stretching of the flexible linker and/or conformational change of the pre-5S rRNA structure after initial removal of the 3ʹ-extension. To understand the domain flexibility of M5 through the linker, we collected SAXS data for M5 (Supplementary ). The Kratky plot derived from the SAXS scattering data shows a well-folded protein in solution. To study the proximity of the two M5 domains in solution, two models were generated in which the NTD and CTD were separated by the maximum distance allowed by the linker (Supplementary ). The theoretical SAXS curve for both of these models shows a poor curve fit at the lower scattering angles (Supplementary ), supporting the idea that the M5 domains are associated in solution. This agrees with NMR spectra recorded on full-length M5 and its CTD fragment, showing the interaction between the M5 CTD and NTD (). Taken together, these data show that the M5 NTD and CTD domains are associated in solution.
Figure 4. M5 domain interactions. (A) NMR HSQC spectra of full-length M5 (in black) and M5 CTD (in red). The chemical shift variations observed between the two spectra, seen for several resonances, shows interaction between the NTD and CTD. (B) The experimental scattering curve for full-length M5 (black) plotted against the theoretical scattering curve (red) for a representative M5 solution model from each subclass and the RNA-bound M5 structure. The goodness-of-fit (χ2) is indicated. The plot was created using the FoXS server [Citation36]. (C) Five models of full-length M5 in solution calculated from the SAXS data compared to the RNA-bound cryoEM structure (PDB 6TPQ). CTD: dark red, NTD: dark green, Linker: dark blue. The RNA-bound model is taken from the cryoEM model of M5 bound to the 50S ribosomal particle
![Figure 4. M5 domain interactions. (A) NMR HSQC spectra of full-length M5 (in black) and M5 CTD (in red). The chemical shift variations observed between the two spectra, seen for several resonances, shows interaction between the NTD and CTD. (B) The experimental scattering curve for full-length M5 (black) plotted against the theoretical scattering curve (red) for a representative M5 solution model from each subclass and the RNA-bound M5 structure. The goodness-of-fit (χ2) is indicated. The plot was created using the FoXS server [Citation36]. (C) Five models of full-length M5 in solution calculated from the SAXS data compared to the RNA-bound cryoEM structure (PDB 6TPQ). CTD: dark red, NTD: dark green, Linker: dark blue. The RNA-bound model is taken from the cryoEM model of M5 bound to the 50S ribosomal particle](/cms/asset/84b3ba71-a224-4c94-9b08-68bddc11cbb3/krnb_a_1885896_f0004_c.jpg)
The scattering data were next used for calculation of M5 solution structures, which yielded five models with a goodness-of-fit value (χ2) between 1.307 and 1.348 for the theoretical vs. the experimental SAXS curves (). A poor χ2 of 25.78 is seen for the theoretical SAXS curve vs. the 50S(pre-5S)-bound M5 structure, highlighting that there are structural differences between substrate-bound and unbound M5. To compare the model structures of M5 in solution, we grouped them into three structural subclasses using a dendrogram derived by average linkage clustering of the structural similarity matrix (DALI Z-score) [Citation23] (, C, Supplementary ), with each class defined by the location of the linker and the position of the NTD relative to the CTD. The NTD in subclass 1 interacts with α7 of the CTD via the NTD loop and αS1 (). The NTD in subclass 2 interacts with the same part of the CTD as subclass 1 (CTD α7), but via a different part of the NTD (NTD α2 and the end of NTD α3) (). In subclass 3, CTD α7 interacts with NTD α2 and the end of NTD α3, like subclass 2, but also includes the CTD loop connecting α7 with α8 (). In the dendrogram, 50S(pre-5S)-bound M5 groups with subclasses 2 and 3 reflecting some similarities in domain organization (Supplementary ). In the 50S(pre-5S)/M5 complex – as for subclass 2 and 3 – the M5 NTD interface is created primarily by NTD α3, but the CTD interaction is primarily via CTD α6 rather than α7. The interface between M5 domains in the 50S(pre-5S)/M5 complex also involves regions in NTD αS1, NTD α4, CTD α7 and the CTD loop connecting α7 with α8 (). These multiple elements together provide an overall tighter structure of M5, which sets the structure of M5 in complex with 50S(pre-5S) apart from the solution M5 models. Analysis with the InterProSurf server [Citation24] shows that the five solution models bury fewer atoms in the NTD:CTD interface, compared to the 50S(pre-5S)-bound M5 protein, consistent with the idea that M5 is less compact in solution. Taken together, these data show that M5 bound at the 3ʹ-strand of substrate rRNA differs from unbound M5, but indicate that CTD α7 is involved in interaction with the M5 NTD in both forms.
Docking of the NTD on the 5ʹ-strand via superimposition of the 3ʹ- (nts G115 to A118) and 5ʹ- (nts U-1 to U2) cleavage sites revealed that the linker would have to stretch to around 48 Å (from V110 αC to T122 αC) for the NTD to reach the 5ʹ-strand while keeping the CTD stationed. The five docking models displayed 22.7–29.6 Å linker lengths, indicating that M5 does not adapt a fold in solution that is compatible with the positioning of the NTD at the 5ʹ-cleavage site, while the CTD stays anchored to the 50S(pre-5S) substrate. The cryoEM structure with M5 bound at the 3ʹ-strand displays a linker length 24 Å, similar in range to the solution structures. The linker length thus does not allow the NTD to reach the 5ʹ-cleavage site, and M5, therefore, cannot cleave the 5ʹ-strand by a simple relocation of the NTD to the 5ʹ-cleavage site. This suggests that the RNA substrate must undergo a conformational change after cleavage of the 3ʹ-extension such that the 5ʹ-strand can be bound by the NTD.
M5 interdomain interactions change when binding to the 5ʹ-strand of pre-5S rRNA
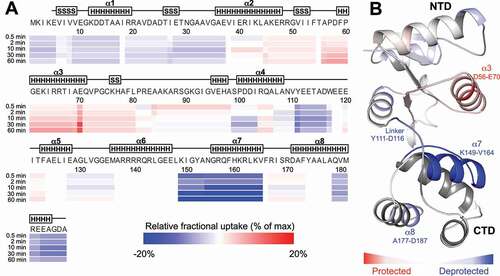
After initial removal of the 3ʹ-extension, cleavage of the 5ʹ-strand requires a movement of i) the NTD to the 5ʹ-strand, ii) the 5ʹ-strand to the NTD, or iii) an induced fit in which both components move towards each other. The lack of alternative CTD binding sites on the pre-5S rRNA substrate, and the limited length of the NTD-CTD linker, eliminates the possibility that the NTD can reach the 5ʹ-cleavage site without movement of the substrate strand, ultimately pointing at an induced-fit mechanism. To examine the structural and dynamic changes associated with M5 binding to the 5ʹ-cleavage site, hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS) was performed using catalytically inactive M5D56A and an RNP substrate of pre-5S rRNA and uL18. In these studies, an in vitro produced RNP substrate was used in which the 3ʹ-extension was removed from the pre-5S rRNA to enforce binding of the NTD to the 5ʹ-strand. Native MS of this complex showed a 1:1 complex stoichiometry between M5 and the RNP substrate (Native MS = 67,184 ± 2 Da, theoretical molecular mass = 67,181 Da) (Supplementary Figure S4A). HDX-MS indicated that when M5 binds to the 5ʹ-cleavage site, the largest structural alterations are a protection of α3 in the NTD and a deprotection of α7 in the CTD and to a lesser extent α8 at the C-terminus of the CTD (, Supplementary Figure S4B,C). NTD α3 contains the catalytic residues, D56 and D58. The clear protection of M5 at this site thus highlights the interaction between the NTD and the 5ʹ-cleavage site as this enters the active site pocket. The CTD α7 helix forms an interdomain interaction with the NTD in all M5 solution models. The deprotection of this element is thus likely to be a consequence of broken interdomain interactions in M5 being replaced by weaker interactions with the pre-5S rRNA in the RNP-bound M5 and/or with the NTD. The first part of the linker (Y111-W117) was also identified as deprotected upon M5/RNP complex formation, showing a potential requirement for the flexibility of this element in the binding of M5 NTD to the 5ʹ-strand. The C-terminal end of α8 interacts with α6 in the CTD structure, and is not involved in RNA-binding in the cryoEM structure. Its deprotection could correspond to a difference in compaction between free and RNA-bound CTD structures. Together, these data show that M5 binds to the RNP substrate through local M5 domain rearrangements, resonating with that the dsRNA-cleavage by M5 proceeds via an induced-fit mechanism.
Figure 5. Local rearrangements of M5 for 5ʹ-strand cleavage. (A) Heat map of the relative fractional uptake (RFU) differences between free inactive M5D56A and M5D56A bound to the RNP substrate (uL18:pre-5S rRNA) lacking the 3ʹ-extension. The differences are given for the shortest identified peptide for each residue and for all deuteration time-points (0.5 min, 2 min, 10 min, 30 min and 60 min). Each residue is colour-scaled from −20% to +20% of relative fractional uptake (RFU) difference (from blue to red, respectively). (B) Data obtained at the 30 min deuteration time-point shown on the structure of M5 (cartoon representation) extracted from the ribosome-bound M5D58A cryoEM structure (PDB: 6TPQ, EMD-10,543). Residues are coloured in accordance with the HDX-MS data shown in (A) where zones that are significantly protected or deprotected are shown in red or blue, respectively
Discussion
Here, we present new structural data for the Mg2+ cofactor-bound RNase M5 that matures pre-5S rRNA in Gram-positive bacteria. Based on this structure, we performed biochemical studies to elucidate how the protein cleaves its double-stranded rRNA substrate. We confirm that M5 functions as a monomer and show that it coordinates two Mg2+ ions via two clusters of acidic residues, i.e, the conserved Toprim triad (E10, D56 and D58) and conserved residues E96 and D14. Residues in the first cluster were previously shown to be indispensable for activity [Citation22]. For the second cluster, we establish that residue D14 is likewise essential for activity, and that residue E96 is crucial for proper M5 folding. M5 cleaves its dsRNA substrate one strand at a time using a Toprim domain, similarly to type1A RNA topoisomerases [Citation25–27] that cleave one strand of a dsRNA. The Toprim domain in topoisomerases coordinates catalytic Mg2+ ion(s), but uses a tyrosine – not present in M5 – to form a covalent intermediate with RNA [Citation28]. M5, therefore, acts via an entirely different mechanism to that of classical Toprim domain-containing proteins. Instead, M5 operates similarly to a recently characterized Toprim domain-containing OLD protein (PDB: 6NK8) [Citation18], in which acidic residues and water molecules, coordinated to two Mg2+, facilitate cleavage of one strand of the RNA in the dsRNA substrate.
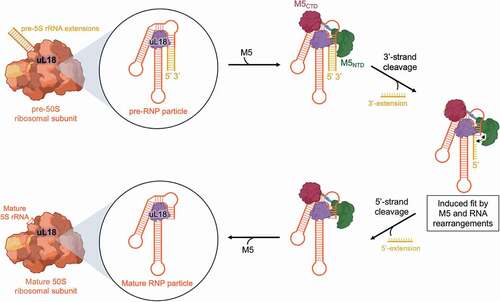
In our published cryoEM structure of M5 [Citation10], the enzyme is bound at the 3ʹ-cleavage site of the pre-5S substrate. Even though the structure was obtained at 200x excess of M5 to ribosome, no density for M5 was observed at the 5ʹ-strand, indicating that the 3ʹ-extension is removed prior to the 5ʹ-extension. Cleavage of the 5ʹ-strand after removal of the 3ʹ-strand cannot occur via simple re-localization of the NTD to the 5ʹ-strand, but instead takes place via an induced-fit mechanism involving disruption of NTD interaction with CTD α7, likely accompanied by rearrangements of the RNA substrate towards the NTD. A scheme outlining the mechanistic route taken by M5 for pre-5S rRNA processing, and the role of the r-protein, uL18, in forming the RNP particle substrate, is outlined in .
Figure 6. Mechanistic route for pre-5S rRNA maturation by M5. The pre-5S rRNA (orange/yellow) is bound to uL18 (purple) forming the RNP substrate of M5 within the 50S ribosomal subunit. M5 (green/red) recognizes the RNP particle and removes, initially, the 3ʹ-extension. Subsequent induced-fit type rearrangements in both M5 and pre-5S rRNA allows removal of the 5ʹ-extension, to obtain a fully mature 5S rRNA within the 50S ribosomal subunit
The crystal data for the M5 NTD reveals a long loop with two small helical segments (residues 82–85, 95–97) that protrudes out from the structure between strand β4 and helix α4. In other Toprim proteins such as human topoisomerase IA (PDB: 5ZEN), S. aureus gyrase (PDB: 6FQM), bacteriophage T7 primase (PDB: 1NUI), and E. coli DnaG primase (PDB: 1DD9), this loop is involved in binding of substrate DNA or in protein:protein interactions with other complex domains. In the cryoEM structure of M5 bound to the 3ʹ-strand (PDB 6TPQ), this region presented with poor density suggesting flexibility and thus a lack of stabilizing interactions with the 3ʹ-strand. The structure of the NTD solved here crystallized in an alternative space group to that of the previously published NTD structure (PDB: 6TG6) in which the protruding loop adapts an entirely new, extended conformation. When superimposing this new extended conformation onto the NTD of the cryoEM structure, the loop in the new conformation locates close to the substrate helix and could bind the 5ʹ-strand when this rearranges after 3ʹ-strand removal. In support of this, the loop contains several basic residues (R82, K86, R88, K91), one of which (K91) can form hydrogen bonds to nt G-1 and C-2 on the 3ʹ-strand in the superimposition. Interestingly, previous studies showed that the nature of the nt immediately 5ʹ of the 5‘-strand cleavage site (G-1) is important for activity [Citation29]. This base is G-C base-paired in the intact pre-5S rRNA, but initial removal of the 3ʹ-strand would open the 5ʹ-strand for base-recognition of G-1 by the NTD loop. Thus, rearrangements of the rRNA substrate subsequent to 3ʹ-strand cleavage, to accommodate the 5ʹ-strand in the active site, could well explain this base dependency.
Methods/materials
Cloning, expression and purification of RNase M5 and uL18
DNA fragments encoding Geobacillus stearothermophilus full-length (FL) M5 (residues 1–187), constructs for the NTD (residues 1–80, 1–110, or 1–113) or the CTD (residues 105–187), were subcloned into the pET28a vector modified to incorporate an N-terminal His6-tag followed by a Tobacco Etch Virus protease (TEV) cleavage site. Mutations were generated using the QuikChange Site-Directed Mutagenesis Kit (Agilent), and verified by sequencing. Plasmids were transformed into E. coli BL21(DE3) CodonPlus RIL competent cells, cultured in Lysogenic Broth at 37°C, and protein expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 3 h growth at 37°C. A cell pellet from 2 L culture was lysed by sonication in a buffer containing 20 mM Tris pH 8.0, 1 M NaCl, 5% (v/v) glycerol, 1 mM DTT, 20 mM imidazole, and clarified by centrifugation. The clarified lysate was applied to a 5 mL HisTrap HP column (GE Healthcare) and eluted with a gradient of 20–500 mM imidazole. FL M5 protein was buffer exchanged into 50 mM HEPES pH 7.5, 200 mM NaCl, 5% (v/v) glycerol, 1 mM DTT and applied to a 5 mL HiTrap Heparin HP column (GE Healthcare). The protein was eluted with a gradient of 200–1000 mM NaCl. All proteins were treated with TEV for 12 h at room temperature and applied to a 5 mL HisTrap HP column (GE Healthcare) to remove the His6-tag. Purified proteins were buffer exchanged into 20 mM Tris pH 7.4, 200 mM NaCl, 2 mM MgCl2, 1 mM β-mercaptoethanol before storage.
Native mass spectrometry (native MS)
Prior to native mass spectrometry analyses, all samples were first buffer exchanged against 200 mM ammonium acetate pH 8.0, using 2 minute cycles of centrifugation at 1500 G with 7 K MWCO 0.5 mL ZebaTM Spin desalting columns (Thermo Fisher Scientific, Waltham, MA USA). All samples were diluted in 200 mM AcONH4 to a final concentration of 5 µM and infused with an automated chip-based nano-electrospray device (Triversa Nanomate, Advion Bioscience, Ithaca, USA) operating in positive ion mode coupled to a quadrupole-time-of-flight Synapt G2 HDMS mass spectrometer (Waters, Manchester, UK). Prior to non-denaturing MS analysis of the samples, the mass spectrometer was calibrated using singly charged ions produced by a 2 g/L solution of caesium iodide (Acros organics, Thermo Fisher Scientific, Waltham, MA USA) in 2-propanol/water (50/50 v/v). Instrumental parameters were optimized by raising the backing pressure to 6 mbar and the cone voltage to 150 V. Then, data treatment was performed on MassLynx 4.1 software (Waters, Manchester, UK).
NMR spectroscopy
NMR spectra were measured at 25°C on a Bruker AVIII-HD 700 MHz spectrometer equipped with a TCI 5-mm cryoprobe with 3-mm tubes. 1D 1H NMR spectra in the NMR buffer (20 mM Tris pH 7.4, 300 mM NaCl, 10 mM MgCl2, 1 mM beta-mercaptoethanol, D2O 5% (v/v)) were recorded at 25°C using the 3-9-19 WATERGATE sequence for the elimination of water [Citation30] with a spectral width of 15 ppm. Standard 2D (1H,15N)-HSQC experiments were measured at 25°C in the NMR buffer for M5 and M5 CTD concentrated to 0.8 and 0.3 mM, respectively.
Differential scanning fluorimetry (DSF)
DSF was performed in a 96-well plate using a CFX96Touch real-time PCR detection system (Bio-Rad) with excitation and emission filters of 450–490 and 515–530 nm, respectively. The precision on temperature is ±0.2°C on this apparatus. Each well consisted of 2 µL protein in a buffer of 20 mM HEPES pH 7.5, 150 mM NaCl to a final concentration of 5 µM, 2 µL of SYPRO ORANGE diluted 5000-fold in buffer from the manufacturer’s stock (Invitrogen), and (if applicable) 2 µL MgCl2 to 1 mM. Fluorescence intensities were measured from 25°C to 95°C with a ramp rate of 1°C/min. Tm-values were determined by curve-fitting using GraphPad Prism v.5.01 software [Citation31] from three technical replicates.
Crystallization, data collection and structure determination
Crystals of G. stearothermophilus M5 NTD bound to Mg2+ were grown by vapour diffusion at 20°C from sitting drops composed of 50 nL of protein (24.4 mg/ml) supplemented with 25 mM MgCl2 and 100 nL of reservoir solution containing 0.2 M ammonium acetate, 0.1 M sodium acetate trihydrate pH 4.6, 30% (w/v) polyethylene glycol 4000. Crystals were cryo-protected with reservoir solution supplemented with 20% (v/v) glycerol, and flash-cooled in liquid nitrogen. Diffraction data were collected to 2.14 Å. Phases were determined by molecular replacement using a previous model for G. stearothermophilus M5 NTD1-113 (PDB 6TG6). Further modelling and refinement was carried out using Refmac5 [Citation32] and COOT [Citation33]. The asymmetric unit (ASU) contained four protein molecules for which residues were assigned as follows: K2 to E83 and G94 to E113 for chain A, S-1 (from the vector) to E113 for chain B, M1 to P81 and I93 to E113 for chain C, and M1 to R82 and G94 to E113 for chain D. The coordination of Mg2+ ions modelled into chain B was checked using the CheckMyMetal server [Citation34]. Final statistics are reported in .
B. subtilis strain constructions and isolation of 50S ribosomal subunits
The construction of the B. subtilis strain CCB038 (∆rnmV) has been described previously [Citation5]. Pre-50S subunits were isolated from 200 mL cultures of CCB038 and CCB1262 grown in 2xYT medium. Cells were pelleted, washed in buffer A (20 mM Tris-HCl pH 7.5, 200 mM NH4Cl, 6 mM β-mercaptoethanol) supplemented with 10 mM MgCl2. Pellets were resuspended in 1.7 mL buffer A containing 10 mM MgCl2 and lysed by French Press (Glen Mills; 20,000 Psi). The lysate was clarified by centrifugation and layered on top of a 38 mL 10–30% sucrose gradient in buffer A containing 3 mM MgCl2 (4°C), prepared in ultracentrifuge tubes (Seton), with a Biocomp Gradient Master 108 (Serlabo) gradient maker. Gradients were centrifuged 16 h at 22,100 rpm in a Beckmann SW28 rotor at 4°C. Fractions were collected, and those containing pre-50S subunits were pooled, concentrated, and resuspended in 20–40 μL of a buffer containing 20 mM Tris-HCl pH 7.4, 100 mM NaCl, 100 mM KCl, 6 mM β-mercaptoethanol and 10 mM MgCl2.
RNase activity assays
Assays were performed in 10 µL reactions containing 0.8 µg (0.7 pmol) pre-50S subunits prepared from CCB038 cells and 25 ng (1.2 pmol) M5 protein in buffer A containing 10 mM MgCl2 at 37°C. M5 samples were phenol extracted, precipitated with 0.1 volumes of 20 mg/mL glycogen, 0.1 volumes 10 mM LiCl, and 3 volumes of 95% EtOH, resuspended in 10 µL H2O, and added 10 µL running dye. Samples were denatured at 95°C for 5 mins before migration on 5% polyacrylamide/7 M urea gels, and electro-transferred (300 mA) to hybond-N (GE Healthcare) membranes. Membranes were UV-cross-linked and probed with oligo HP246 (5ʹ-ATC GGC GCT GAA GAG CTT AAC TTC C-3ʹ) complementary to the mature 5S rRNA sequence as described previously [Citation5].
Size exclusion chromatography-coupled small-angle X-ray scattering (SEC-SAXS)
Proteins were subjected to SEC-SAXS on a Bio SEC-3 300 column (Agilent) at 1 mM in 20 mM Tris-HCl pH 7.4, 150 mM KCl, 150 mM NaCl, 5 mM MgCl2, 1 mM β-mercaptoethanol, by flowing sample through an in-vacuum quartz capillary of 1.6 mm diameter at 0.16 mL/min. Data were collected using a Pilatus2M detector (Dectris, CH) with a sample-detector distance of 3914 mm and a wavelength of λ = 1 Å. The range of momentum transfer of 0.0041 < s < 0.41 Å −1 was covered (s = 4πsinθ/λ, where θ is the scattering angle). The data covering the peak were radially averaged and the scattering of buffer was subtracted using Scatter (v.3.0) (Robert Rambo (www.bioisis.net)). The radius of gyration, Rg, and Kratky plot were obtained using Scatter (v. 3.0).
Modelling of M5 solution structure
A complete model of M5 including the linker was generated under SAXS constraints using DADIMODO [Citation35], a genetic algorithm-based refinement program. The cryoEM model of M5 was used as input, and the NTD (residues 4–109) and CTD (residues 123–186) were kept as rigid bodies. The minimum and maximum scattering angle (q) were set to 0.006 and 0.54, respectively, and the SAXS data were set to put 100% weight on the goodness of fit (χ2). A comparison of the predicted scattering profile from a solution model with the SAXS scattering data was created using the FoXs server [Citation36].
In vitro transcription of precursor 5S rRNA
Single-stranded DNA templates for G. stearothermophilus pre-5S-rRNA lacking the 3ʹ-end extension (5ʹ- GmCmC TAG CAG CGA CCT ACT CAC ATC GGC GCT GGA GGG CTT AAC TTC CGT GTT CGG GAT GGG AAC GGG TGT TTC CCC TCC GCT ATC ACC ACT AGG CAA TCC C TA TAG TGA GTC GTA TTA – 3ʹ) (Eurogentec) was used for in vitro transcription with a T7 RNA polymerase primer (5ʹ TAA TAC GAC TCA CTA TAG 3ʹ). RNA was transcribed in a 5 mL reaction with 0.2 μM DNA template in a buffer of 20 mM Tris-HCl pH 8.0, 0.5 mM spermidine, 2.5 mM DTT, 0.005% Triton X-100, supplemented with 5 mM of each rNTPs, 34.3 mM MgCl2, 5.6 mM GMP, and T7 RNA polymerase. The reaction was incubated for 4 h at 37°C and stopped with 100 mM EDTA. The reaction mixture was purified on an 8 mL MonoQ 10/100 GL anion exchange column (GE Healthcare) in a buffer of 25 mM sodium phosphate pH 6.5, 50 mM NaCl, and eluted in a similar buffer with a 50 mM to 1 M NaCl gradient. The RNA was buffer exchanged into 1.7 M (NH4)2SO4 and applied to an 8 mL Phenyl superose HR 10/10 column (GE Healthcare) and eluted in H2O with a 1.7 M to 0 M (NH4)2SO4 gradient.
Hydrogen-deuterium exchange coupled to mass spectrometry (HDX-MS)
Proteins and RNA samples were prepared with a 20 mM Tris, 200 mM NaCl, 2 mM MgCl2, 1 mM β-mercaptoethanol, 5% glycerol pH 7.4 buffer. For M5D56A experiments, the sample was diluted to 16 µM while M5:5S rRNA:uL18 experiment involved a dilution to 8 µM for M5D56A and to 16 µM for 5S rRNA and uL18 protein (molar ratio of 1:2:2). Preparation and injection of the samples were automatically conducted using a LEAP HDX Automation Manager (Waters, Manchester, UK), and chromatography was carried out on Acquity UPLC system with HDX technology (Waters, Manchester, UK): samples were incubated at 20°C for 5 deuteration time-points (0.5, 2, 10, 30 and 60 minutes) in 20 mM Tris, 200 mM NaCl, 2 mM MgCl2, 1 mM β-mercaptoethanol, pD 7.4 deuterated buffer. Exchange reactions were then stopped by adding 1:1 (v/v) of quench buffer (2 M guanidine-HCl, 100 mM glycine pH 2.35) in a 1°C refrigerated drawer for 0.5 minute. Quenched samples were then were digested through a pepsin-immobilized cartridge for 3 min in 0.1% aqueous formic acid solution at a flow rate of 200 µL/min. Digested peptides were first trapped and desalted on a UPLC pre-column (ACQUITY UPLC BEH C18 VanGuard pre-column, 2.1 mm I.D. x 5 mm, 1.7 µM particle diameter, Waters) and then separated on a UPLC reversed-phase column (ACQUITY UPLC BEH C18, 1.0 mm I.D. x 100 mm, 1.7 µM particle diameter, Waters) at 0.1°C with a gradient elution of solvent A (0.1% aqueous formic acid) and solvent B (acetonitrile with 0.1% formic acid) [2–40% B (7 min), 40–85% B (0.5 min), and 85% B (1 min)] at a flow rate of 40 µL/min. MS/MS analyses were acquired on a Synapt G2SI HDMS (Waters, Manchester, UK) in positive polarity and resolution mode initially calibrated and using a lock-mass correction with glufibrinogen peptide. Data were acquired in MSE acquisition mode with the following parameters: capillary voltage, 3 kV; sampling cone voltage, 40 V; source temperature, 80°C; desolvation gas, 150°C and 600 L/h; acquisition range, acquisition range, 50 − 2000 m/z; scan time, 0.3 s; trap collision energy, 15 to 40 eV. MSE data were processed using Waters ProteinLynx Global Server 2.5.3 (Waters) with a home-made protein sequence library containing M5 and pepsin sequences, where peptide and fragment tolerances were automatically adjusted by PLGS, and oxidized methionine was set as variable modification. Data were then filtered with DynamX 3.0 (Waters) as follows: each experiment was carried out in triplicate and only peptides identified in all replicates were kept with a minimum fragment of 0.2 per amino acid, a minimum intensity at 103, a length between 5 and 30 residues and a file threshold of 3. Deuterium uptakes for all identified peptides were manually checked and validated. Deuterium uptakes were not corrected for back-exchange, represented as relative. HDX-MS results were statistically validated using Deuteros software [Citation37] with a confidence interval of 99%. The mass spectrometry proteomics data (including HDX uptake plots) have been deposited to the ProteomeXchange Consortium via the PRIDE [Citation38] partner repository with the dataset identifier PXD019202.
Supplemental Material
Download MS Word (13 MB)Acknowledgments
We thank the staff at the Synchrotron Soleil beamline Proxima-2 (PX2) and SWING for support during data collection. In particular, we wish to thank Javier Perez for valuable discussions on the DADIMODO software. We also acknowledge access to the biomolecular NMR platform of the IBPC that is supported by the CNRS, the Labex DYNAMO (ANR-11-LABX-0011), the Equipex CACSICE (ANR-11-EQPX-0008) and the Conseil Régional d’Île-de-France (SESAME grant). We thank Clément Dégut for initial studies of the M5 using small-angle X-ray scattering.
Disclosure statement
The authors report no conflict of interest.
Data Availability
Atomic coordinates and structure factors for the reported crystal structure of M5 NTD:Mg2+ have been deposited in the Protein Data Bank (PDB) under accession number 6Z2B. HDX-MS data have been deposited in the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD019202.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Nikolaev N, Silengo L, Schlessinger D. Synthesis of a large precursor to ribosomal RNA in a mutant of escherichia coli. Proc Natl Acad Sci. 1973;70:3361–3365.
- Zingales B, Colli W. Ribosomal RNA genes in Bacillus subtilis Evidence for a cotranscription mechanism. BBA Sect Nucleic Acids Protein Synth. 1977;474:562–577.
- Schlessinger D. Mechanism and regulation of bacterial ribosomal RNA processing. Annu Rev Microbiol. 1990;44:105–129.
- Baumgardt K, Gilet L, Figaro S, et al. The essential nature of YqfG, a YbeY homologue required for 3ʹ maturation of Bacillus subtilis 16S ribosomal RNA is suppressed by deletion of RNase R. Nucleic Acids Res. 2018;46:8605–8615.
- Condon C, Brechemier-Baey D, Beltchev B, et al. Identification of the gene encoding the 5S ribosomal RNA maturase in bacillus subtilis: mature 5S rRNA is dispensable for ribosome function. RNA. 2001;7:242–253.
- Sogin ML, Pace NR. In vitro maturation of precursors of 5S ribosomal RNA from bacillus subtilis. Nature. 1974;252:598–600.
- Sogin ML, Pace NR. Nucleotide sequence of 5 S ribosomal RNA precursor from bacillus subtilis. J Biol Chem. 1976;251:3480–3488.
- Meyhack B, Pace B, Pace NR. Involvement of precursor-specific segments in the in vitro maturation of bacillus subtilis precursor 5S ribosomal RNA. Biochemistry. 1977;16:5009–5015.
- Redko Y, Bechhofer DH, Condon C. Mini-III, an unusual member of the RNase III family of enzymes, catalyses 23S ribosomal RNA maturation in B. subtilis. Mol Microbiol. 2008;68:1096–1106.
- Oerum S, Dendooven T, Catala M, et al. Structures of B. subtilis maturation RNases captured on 50S ribosome with Pre-rRNAs. Mol Cell. 2020;80:1–10.
- Stahl DA, Pace B, Marsh T, et al. The ribonucleoprotein substrate for a ribosomal RNA-processing nuclease. J Biol Chem. 1984;259:11448–11453.
- Pace B, Stahl DA, Pace NR. The catalytic element of a ribosomal RNA-processing complex*. J Biol Chem. 1984;259:11454–11458.
- Redko Y, Condon C. Ribosomal protein L3 bound to 23S precursor rRNA stimulates its maturation by Mini-III ribonuclease. Mol Microbiol. 2009;71:1145–1154.
- Aravind L, Leipe DD, Koonin EV. Toprim - A conserved catalytic domain in type IA and II topoisomerases, DnaG-type primases, OLD family nucleases and RecR proteins. Nucleic Acids Res. 1998;26:4205–4213.
- Dong KC, Berger JM. Structural basis for gate-DNA recognition and bending by type IIA topoisomerases. Nature. 2007;450:1201–1205.
- Zhang H, Barcelo JM, Lee B, et al. Human mitochondrial topoisomerase I. Proc Natl Acad Sci. 2002;98:10608–10613.
- Cao N, Tan K, Annamalai T, et al. Investigating mycobacterial topoisomerase I mechanism from the analysis of metal and DNA substrate interactions at the active site. Nucleic Acids Res. 2018;46:7296–7308.
- Schiltz CJ, Lee A, Partlow EA, et al. Structural characterization of class 2 OLD family nucleases supports a two-metal catalysis mechanism for cleavage. Nucleic Acids Res. 2019;47:9448–9463.
- Harding MM. Geometry of metal-ligand interactions in proteins. Acta Crystallogr Sect D Biol Crystallogr. 2001;57:401–411.
- Nowotny M, Gaidamakov SA, Crouch RJ, et al. Crystal structures of RNase H bound to an RNA/DNA hybrid: substrate specificity and metal-dependent catalysis. Cell. 2005;121:1005–1016.
- Nowotny M, Yang W. Stepwise analyses of metal ions in RNase H catalysis from substrate destabilization to product release. Embo J. 2006;25:1924–1933.
- Allemand F, Mathy N, Brechemier-Baey D, et al. The 5S rRNA maturase, ribonuclease M5, is a Toprim domain family member. Nucleic Acids Res. 2005;33:4368–4376.
- Holm L, Laakso LM. Dali server update. Nucleic Acids Res. 2016;44:W351–W355.
- Negi SS, Schein CH, Oezguen N, et al. InterProSurf: a web server for predicting interacting sites on protein surfaces. Bioinformatics. 2007;23:3397–3399.
- Wang H, Di Gate RJ, Seeman NC. An RNA topoisomerase. Proc Natl Acad Sci. 2002;93:9477–9482.
- Ahmad M, Xue Y, Lee SK, et al. RNA topoisomerase is prevalent in all domains of life and associates with polyribosomes in animals. Nucleic Acids Res. 2016;44:6335–6349.
- Xu D, Shen W, Guo R, et al. Top3β is an RNA topoisomerase that works with Fragile X syndrome protein to promote synapse formation. Nat Neurosci. 2013;16:1238–1247.
- Corbett KD, Berger JM. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu Rev Biophys Biomol Struct. 2004;33:95–118.
- Stahl DA, Meyhack B, Pace NR. Recognition of local nucleotide conformation in contrast to sequence by a rRNA processing endonuclease. Proc Natl Acad Sci U S A. 1980;77:5644–5648.
- Piotto M, Saudek V, Sklenar V. Gradient-tailored excitation for single-quantum NMR spectroscopy of aqueous solutions. J Biomol NMR. 1992;2:661–665.
- Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat Protoc. 2007;2:2212–2221.
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr Sect D Biol Crystallogr. 1997;53:240–255.
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr Sect D Biol Crystallogr. 2004;60:2126–2132.
- Heping Zheng WM. Validating metal binding sites in macromolecule structures using the CheckMyMetal web server. Nat Protoc. 2014;9:156–170.
- Evrard G, Mareuil F, Bontems F, et al. DADIMODO: a program for refining the structure of multidomain proteins and complexes against small-angle scattering data and NMR-derived restraints. J Appl Crystallogr. 2011;44:1264–1271.
- Schneidman-Duhovny D, Hammel M, Tainer JA, et al. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys J. 2013;105:962–974.
- Lau AMC, Ahdash Z, Martens C, et al. Deuteros: software for rapid analysis and visualization of data from differential hydrogen deuterium exchange-mass spectrometry. Bioinformatics. 2019;35:3171–3173.
- Perez-Riverol Y, Csordas A, Bai J, et al. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47:D442–D450.