
ABSTRACT
Circular RNA is progressively reported to occur in various species including mammals where it is thought to be involved in the post-transcriptional regulation of gene expression, partly via interactions with microRNA. Here, we asked whether the circular topology causes functional differences to linear forms when interacting with short RNA strands in vitro and in human cells. Kinetic studies with human bladder cancer-derived synthetic circular RNA versus linear transcripts, respectively, with short oligoribonucleotides showed similar association rates for both topologies. Conversely, a substantial topology-related difference was measured for the activation entropy and the activation enthalpy of RNA–RNA annealing. This finding strongly indicates a significant difference of the mechanism of RNA–RNA interactions. To investigate whether these characteristics of circular RNA are biologically meaningful we performed transient transfection experiments with a microRNA-regulated expression system for luciferase in bladder cancer-derived cells. We co-transfected linear or circular RNA containing one microRNA binding site for the target-suppressing microRNA mlet7a. Here, the circular isoform showed a strongly increased competition with microRNA function versus linear versions. In summary, this study suggests novel topology-related characteristics of RNA–RNA interactions involving circRNA in vitro and in living cells.
Introduction
Since circular RNAs (circRNA) moved into the focus of biomedical research, most of the increasing numbers of reports describe their (bio-)synthesis, their origin, and their presumed fundamental biological role [Citation1–5]. The majority of these studies contain in silico analyses derived from transcriptome profiling data [Citation6–10]. Many of the published data on circRNA suggest their central biological role in the regulation of cellular gene expression and metabolism, and also their potential use as specific disease biomarkers [Citation11]. Conversely, biochemical characteristics of circRNA and mechanisms underlying its interactions with complementary RNA are almost unknown. As experimental mechanistic insights are missing, there remains a lack of models explaining crucial biological roles hypothesized for circRNAs including binding of cellular RNAs, the regulation of gene expression via microRNA (miRNA), a storage function, and a role in the localization of RNA or RNA-binding proteins [Citation12,Citation13]. In summary, circRNAs are thought to be involved in a highly complex structural and functional intracellular RNA network which consists of mRNAs, circRNAs, regulatory RNAs, and interacting molecules [Citation14]. The concerted action of all kinds of RNAs involved seems to provide a fine-tuned, temporal, spatial, and developmental stage-specific mode of regulated gene expression. The ´sponge model´ assumes interactions between redundant binding sites of circRNA for cognate miRNA species thereby serving as a reservoir for microRNA, termed ´sponge´, by which the intracellular level and activity of miRNAs can be regulated [Citation12,Citation13].
To avoid artificial test systems and to establish a biologically relevant model system we chose sequences of circRNA that occurs in human bladder cancer cells (BCa). Further, we favoured a strand that is short enough for efficient in vitro transcription followed by efficient enzymatic circularization. From a more biomedical viewpoint, we focused on a potential circular RNA that has one binding site for a miRNA which was reported by at least two independent references [Citation12,Citation14–16]. These considerations were met by circRNA ‘hsa_circ_0001406’ according to (www.circbase.org/alias hsa_circ_000391 [according to http://gyanxet-beta.com/circdb/], length 176 nt, NM_006345, SLC30A9, binds miR-101-3p). In order to make use of T7 RNA polymerase, we added the six nucleotides GGGAGA at the 5´terminus, to guarantee high in vitro transcription yields. The addition led to the 182 nt long BCa associated start molecule for our model system.
Here we describe experimental studies that indicate major influences of RNA topology on RNA–RNA interactions. We established an experimental platform for the biochemical understanding of interactions of circRNAs with complementary RNA including antisense RNA or partially complementary RNA such as microRNA. The presumed network of dynamic intracellular RNA–RNA interactions involves circRNAs and endogenous RNA. With regard to the tight relationship between kinetics of RNA–RNA annealing in vitro and biological phenotypes in vivo [Citation17,Citation18], it seemed to be reasonable to start with kinetic studies in vitro at physiological conditions. Thus, we elucidated the kinetics of interactions between in vitro transcripts representing the naturally occurring bladder carcinoma (BCa)-associated human circRNA hsa_circ_000391 with matching single-stranded oligomeric RNA. This includes a 92mer truncated version and three longer homologs.
Regarding the RNA species used in vitro for biochemical studies and for cell culture experiments, respectively, please note the following nomenclature. In vitro, we study 92mers, termed lin-92 and circ-92, respectively, and the complementary oligonucleotide as101 which has no binding motif for mlet7a. When checking whether in vitro observations are biologically meaningful, we used a mlet7a-regulated indicator system. Thus, we had to modify lin-92 and circ-92 such that the resulting derivatives contain a binding motif for mlet7a. The resulting constructs were termed lin-mlet7a-92 and circ-mlet7a-92, respectively.
Results and discussion
To shed light on the role of the topology of circular RNA, we studied the natural hsa_circ_1406 system for which potential binding of microRNAs was analysed by RNA hybrid 2.2[Citation19]. and by the data base ´circ2traits´[Citation20]. The results indicate that this circular RNA can interact with miR-101-3p. For the following studies we increased the sequence complementarity with circRNA which resulted in as101, a complementary 21mer based on miR-101-3p. Further, we attempted to exclude phenomena which could be specific for a given individual circRNA species, e.g. length dependency. Thus, we investigated a set of four related homologous circRNAs of 92, 122, 152, and 182 nts (Table S1 and Table S2). All of these linear and circular RNAs were synthesized using protocols as described recently [Citation21]. Briefly, two 3´-overlapping oligodeoxyribonucleotides (sequences in Table S3) were filled in by Klenow fragment. The resulting duplex contained a T7 promoter element at one terminus by which transcripts were produced in vitro. Transcripts served as linear target RNA or were ligated intra-molecularly, and used as circular target RNA.
We characterized linear and circular RNA species by using three different methodologies (shown for lin-92 and circ.92). (i) 1D- PAGE (7 M urea, circular RNA; ), (ii) 2D-PAGE (no urea; & E), and (iii) cleavage by RNase H which linearizes circ-92 and produces two fragments of lin-92 ().
Figure 1. Secondary structure models of the linear and circular isoforms of the 92mer, lin-92 (A) and circ-92 (B), respectively. The segments that are complementary to the microRNA model as101 are indicated by yellow colour. (C) Polyacrylamide gel showing the linear and circular isoforms of the 92mer, termed circ-92 and lin-92, respectively. The lane on the left shows size markers. (D) Two-dimensional polyacrylamide gel on which linear RNA species are separated. 1st dimension, denaturing conditions (7 M urea) 15% polyacrylamide; 2nd dimension: 17.5% denaturing polyacrylamide or 15% native polyacrylamide., xxxxx; 2nd dimension, xxxxxx. (E) Two-dimensional polyacrylamide gel on which linear RNA species together with circ-92 are separated. 1st dimension, xxxxx; 2nd dimension, xxxxxx. The linear RNA species form quasi lines of signals while circ-92 migrates clearly outside this line. The position of circ-92 on the lower left of the gel is indicated by a white circle. (F) Separation of RNase H cleavage products of lin-92 and circ-92. Left lane, size markers; middle left lane, oligodeoxyribonucleotide (ODN) forming a substrate for RNase H together with both 92mers; middle right lane, mixture of circ-92, lin-92; right panel, cleavage products
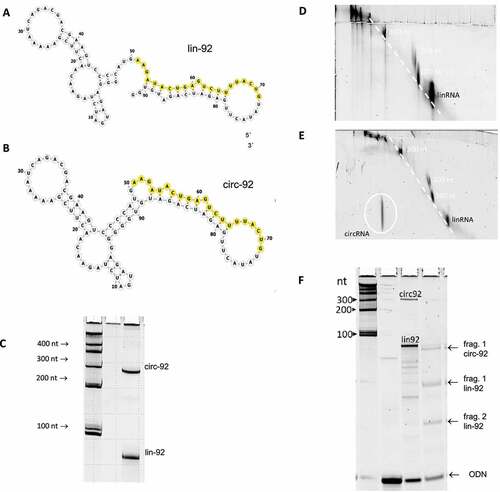
It is important to note that intermolecular RNA–RNA interactions are sensitive to local RNA structures and to accessibility for RNA–RNA interactions [Citation22]. Thus, we first modelled secondary structures of the RNA molecules used in this study by extensive in silico analyses in the use of the programms RNAfold and forna that are accessible via the ViennaRNA package 2.4.1 [Citation23,Citation24]. Analyses included the temperature-dependency of the linear and circular RNAs of the series of four homologs 92mer, 122mer, 152mer, and 182mer (Supplementary Fig. 1, Supplementary ). The 2D models of the linear version of the 92mer (lin-92), respectively, indicate a similar structural context of the binding sites for as101 of both topological isoforms at 37°C (). Thus, we assume that the as101 binding sites and their structural context have no major influence on biochemical studies of the interactions between the 92mers and as101.
Table 1. Relationship between RNA topology and activation energies of the association of 92mer with as101 at 37°C. For detailed experimental data see suppl. table 5
First, we studied the annealing kinetics of linear or circular 92mers with as101. The RNA–RNA association kinetics were measured at pseudo-first-order conditions at 37°C, physiological ionic strength, and physiological pH value, essentially as described [Citation25]. The association rate constants ranged between 1.1 × 105 (M−1 s−1) and 2.1 × 105 (M−1 s−1) for all RNAs regardless of their topology and size (Figure S2, Table S4). It is noteworthy, that a slight length-dependent increase of association rates by a factor of approximately 1.6 and 1.8, respectively, occurs in the length range between 92 nts and 182 nts for the linear and for the circular versions (Table S4). This observation is consistent with earlier systematic studies on the length-dependent increase of RNA–RNA annealing kinetics [Citation26].
To investigate in more depth the role of the covalently closed circular topology of circRNA for RNA–RNA interactions, we studied two major characteristics. Firstly, we examined the activation energies of the association of as101 with linear or circular RNA (lin-92 or circ-92; , ).
Figure 2. Arrhenius-plots of the association of as101 with lin-92 or circ-92, respectively (A). Dissociation studies in vitro. (B) Schematic depiction of strand displacement of the labelled as101 strand (as101*) within the complex with lin-92 (Iin-92•as101*) by an excess of unlabelled as101. (C) Gel analysis of the time-dependent dissociation kinetics. The complex Iin-92•as101* is composed of lin-92mer and the fluorescently labelled as101* (label indicated by*). Dissociation is driven by an excess of unlabelled as101 resulting in decreasing amounts of the visible complex Iin-92•as101*and increasing signals for as101* with increasing time. Lane pc, positive control (pre-formed complex after 90 min incubation; at 37°C). Lane nc, negative control (as101*) (D) Observed half-life of complexes containing lin-92 or circ-92 as a function of the molar excess of unlabelled as101. Dissociation of as101* from the linear complex is fast (short half-life) and increases at increasing concentrations of the ´displacer strand´ as101 (open circles). Conversely, release of as101* from a complex with circular RNA is slow and more reduced at increasing concentration of as101 (filled circles)
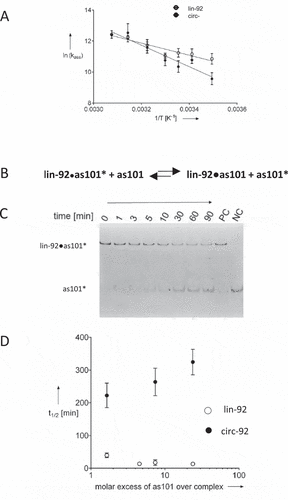
The temperature dependency of the annealing between as101 and the linear or the circular version of the 92mer, respectively, gave rise to Arrhenius plots (), and the activation energies of this reaction (). Since the annealing rate constants are similar between the as101 and the two topologies of the 92mer, the values for the Gibbs energy of activation (ΔG‡) are also similar (). Conversely, the Arrhenius activation energy differs substantially which is reflected by the considerable difference of the enthalpy of activation (ΔH‡) and the entropy of activation (ΔS‡) which constitute ΔG‡. This suggests major mechanistic differences of the RNA–RNA annealing mechanisms as a function of the topology of the long-chain RNA strand (). While Ea/ΔH‡ is increased for circRNA this thermodynamic disadvantage is compensated by the activation entropy.
With regard to the hypothesis that circRNAs act like ‘sponges’ for miRNAs in which also the controlled release of miRNA occurs, we further focused on RNA–RNA dissociation. Hence, dissociation kinetics of a partially double-stranded complex between as101 and the 92mer was studied at physiological conditions (compare methods section). The experimental design is schematically depicted in .
A pre-formed complex containing the 92mer and a fluorescently labelled ORN (as101*) was mixed with unlabelled as101 at varying excesses. The time-dependent release of the labelled strand was monitored by withdrawing samples, quenching the reaction, and analysing complex formation by polyacrylamide gel electrophoresis (). Note that RNA-RNA dissociation may occur via two different pathways, an associative or a dissociative pathway [Citation27] (Figure S3). Since no information related to these pathways is available, we measured the release of as101* from pre-formed complexes and provide the half-life of the apparent dissociation reactions in . Overall the release of as101* is slower in case of circRNA-containing complexes compared to linear RNA. Further, the data in show that the concentration dependency of the dissociation kinetics from the unlabelled ‘displacer strand’ is fundamentally different between the two complexes differing in the topology of the long-chain RNA strand. The release of the short RNA strand from the linear complex seems to be enhanced at increasing concentrations of the ´displacer strand´ which is consistent with an associative mechanism that has been observed for a number of RNA–RNA complexes containing linear RNA [Citation27]. Conversely, in case of a complex formed of circular RNA and an oligoribonucleties, the dissociation kinetics decrease at increasing concentrations of ´displacer strand´ which rather favours a non-associative mechanism of RNA–RNA dissociation ().
The kinetic data described here show crucial differences of RNA–RNA recognition of linear and circular RNA. These findings provide a second independent line of evidence for a fundamental role of RNA topology for RNA–RNA interactions in vitro at physiological conditions. At a first glance it is surprising that for circRNA, dissociation becomes slower at increasing concentration of as101, which is not compatible with the dissociative nor the associative mechanism of RNA–RNA dissociation. However, considering similar association rate constants this observation is not contradicting the sponge hypothesis as at increasing as101 concentrations (modelling microRNA), the equilibrium shifts towards the complex, i.e. towards ´sponge characteristics´. The underlying mechanism needs to be resolved which, however, is beyond the scope of this initial investigation.
With regard to interactions of RNA with microRNA we like to draw the attention to the complexity of test systems in vitro and in in living cells. A large number of bioinformatics studies, genetic studies, and phenotypic functional studies indicate complexes between microRNA binding sites and microRNAs with partial sequence complementarity. Conversely, in studies in vitro at physiological conditions, RNA–RNA complexes between RNA strands and only partly complementary miRNA are extremely difficult to detect. Thus, studying those interactions in vitro seems to be quite a challenge and we had to use model systems with complete sequence complementarity.
To investigate whether the biochemical findings described above are biologically relevant for the action of circRNA in living cells, we established a cell culture model that is based on human bladder carcinoma (ECV-304) cells. This system makes use of a mlet7a-regulated recombinant luciferase-coding target mRNA harbouring mlet7a binding sites within its 3´-terminal region [Citation28]. Thus, we adapted lin-92 and circ-92 to mlet7a. The resulting RNAs are also 92nts in length and RNA structure predictions according to the in vitro system with as101 resulted in RNA models which show binding sites and a structural context that is similar for both topologies (). Although the RNA sequences of this cell culture system are not identical to the in vitro studies described above, the overall structure seems to be enough closely related to allow to test whether the biology of a competitor RNA for mlet7a is dependent on its linear or circular topology.
Figure 3. Evidence for competing effects of lin-mlet7a-92 and circ-mlet7a-92 in human cells. (A) Secondary structure models of the linear and circular isoforms of the RNA mlet7a-92, respectively. This 92mer RNA contains a binding site for the microRNA mlet7a (indicated by red colour). (B) Schematic depiction of components of co-transfection experiments with mlet7a, the target mRNA encoding Renilla luciferase, and lin-mlet7a-92 or circ-mlet7a-92, respectively. (C) The RNA mlet7a-92 reduces mlet7a-mediated suppression of Renilla luciferase expression in ECV-304 cells. The co-transfected ´competitor´ RNAs are, lane 1, circ-mlet7a-92; lane 2, circ-92; lane 3, lin-mlet7a-92; lane 4, lin-92. The control RNAs (circ-92 and lin-92) have identical lengths with mlet7a-92 and similar sequences but lack intact mlet7a binding sites. In all transfection experiments the same amount of nucleic acids was used (see, Materials & Methods). (D) Concentration dependency of competitive effects of linear and circular forms of mlet7a-92 RNA, respectively. ECV-304 cells were transiently transfected with mixtures containing pGL3 (internal standard, expressing Firefly luciferase), pHmga2m7 (expressing Renilla luciferase controlled by mlet7a), and the RNAs indicated on the lower left panel at the concentrations indicated in the lower panel. The symbols ‘+’ and “-“ indicate co-transfection of mlet7a, control RNA (si-scr), lin-mlet7a-92, or lin-mlet7a-92, respectively. The numbers on top of the bars indicate mean values and the standard deviation of four measurements. All values of the activity of Renilla luciferase (Y-axis) are standardized to the levels of Firefly luciferase activity in the dual luciferase assay. Expression of Renilla luciferase in the presence of control RNA (3rd bar from left) is set 100%
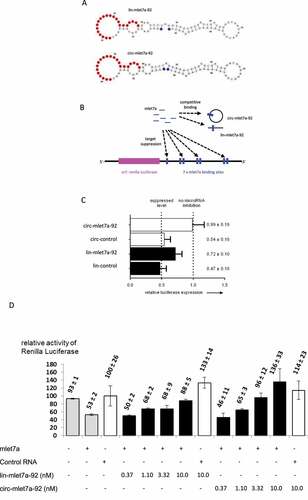
In this cell culture model, microRNA, ´competitor RNA´ (lin-mlet7a-92 and circ-mlet7a-92) and two recombinant plasmids are co-transfected (). One plasmid encodes the target transcript containing seven functional binding sites for mlet7a and the second plasmid contains an indicator gene serving as control for transfection efficiency. While one can define amounts of all nucleic acids that are added to cells, their intracellular fate is complex. The transfected RNAs are thought to be internalized by an endocytotic pathway followed by intracellular release from endosomes and by intracellular trafficking. This makes it very difficult to derive ´real´ endogenous concentrations at the site of action. Plasmid-encoded RNA is transcribed after nuclear uptake of plasmids. It is reasonable to assume that their transcripts have to be exported to the cytoplasm in order to encounter microRNAs. Further, there are other relevant unknown critical parameters like, for example, metabolic stability of all RNAs and their intracellular localization. For this reason, we study in cell culture a side-by-side comparison of the biological functions of linear versus circular RNA species.
After transient transfection of the plasmid-coded recombinant luciferase gene and mlet7a, luciferase activity is suppressed which is thought to occur via RNA–RNA interactions between mlet7a and its 3´-UTR-located binding sites [Citation28]. To investigate the effects of co-transfected potentially ´competing RNA´, also termed ´sponge RNA´ by some laboratories [Citation12,Citation13,Citation29–32] we added linear or circular versions of a 92mer containing a mlet7a binding site termed mlet7a-92. Based on the overall structure we designed a derivative of the 92mer () that contains one defined binding site for mlet7a, assuming this can act as a decoy for mlet7a in human cells, thereby decreasing mlet7a-mediated down-regulation of luciferase activity. If the topology of this RNA would influence its interactions with mlet7a, then different strengths of effects should result in the use of the linear versus the circular form of this RNA.
This transient cell culture experiment () shows a counteracted down-regulation of luciferase expression by mlet7a by both co-transfected versions of mlet7a-92 transcripts. The circular version of the mlet7a-binding RNA (circ-mlet7a-92) is a significantly more potent competitor for mlet7a function than the linear isoform (lin-mlet7a-92, ). This indicates that, in functional terms, circ-mlet7a-92 binds to mlet7a more efficiently than the linear version. In order to describe this behaviour more quantitatively, we titrated the functional competition studies with circular or linear RNA, respectively, in the cell culture set-up schematically depicted in Supplementary Figure S5. Here, the binding of mlet7a to Hmga2m7 transcripts in transfected cells is competed with co-transfected in vitro synthesized linear or circular 92mers, respectively, both of which contain one mlet7a binding site (). Reproductions of this experiment showed a robust concentration-dependent increase of luciferase activity in the presence of mlet7a-92mers. This effect was reproducibly stronger in the use of the circular version circ-mlet7a-92 over the linear homolog lin-mlet7a-92. It is noteworthy that luciferase activity is increased a little at varying amplitude over the control when only lin-mlet7a-92 or circ-mlet7a-92, respectively, are co-transfected with the expression plasmid for Hmga2m7 in the absence of mlet7a (, bars 8 & 13).
A crucial issue of this study is the conceivable possibility of interactions between mlet7a and linear or circular 92mers, respectively, prior to their intracellular localization, e.g. during the preparation of transfection mixtures. Thus, we asked whether interactions between mlet7a and linear or circular 92mer, respectively, could occur in the course of transient transfection. To study these possibilities, we performed transfection experiments in which the components (i) lipofectamine, (ii) mlet7a, (iii) lin-92 or circ-92, respectively, and (iv) plasmids were mixed with different compositions and orders of transfection (Supplementary Figure S5). This control experiment suggests that critical RNA–RNA interactions do not occur before all interacting RNA molecules and endogenous plasmid-encoded transcripts are present inside cells (Supplementary Figure S5).
In general, a higher nuclease resistance and, hence, higher intracellular stability is postulated for circular RNAs. Thus, we investigated the amounts of transfected linear and circular RNAs in this transient transfection assay by Northern analyses and by liquid hybridization (Supplementary Figure S4). Both detection methods provide rough quantitative data. Both methods were applied to measure lin-mlet7a-92 and circ-mlet7a-92 in experiments that show the functional phenotypes shown in &D. The probe used in either assay was equally sensitive for lin-mlet7a-92 and circ-mlet7a-92 (Supplementary Figure S4A). Either RNA detection method indicates comparable amounts of both isoforms of the 92mers. Thus, potentially different intracellular stability of both isoforms does not seem to explain the differential competition properties of lin-mlet7a-92 versus circ-mlet7a-92.
Conversely, a complex network of RNA–RNA interactions involving two microRNAs, one long ncRNA, and circular RNA is noteworthy. Genetic studies in mice suggest that the circular RNA ´Cdr1as´ which contains large numbers of binding sites for miR-7 does not act as a typical microRNA sponge for miR-7. Although functional interactions between miR-7 and Cdr1as seem stop be very likely, regulatory pathways might involve spatial or temporal properties [Citation14]. While these findings by Kleaveland et al. [Citation14] nicely show the highly complex regulatory intracellular network of functional RNAs involving circRNA and microRNAs, the system is different from the in vitro and cell culture studies described here.
In summary, this study strongly suggests a significant role of circular RNA topology for RNA–RNA interactions, i.e. the biology of microRNA in mammalian cells. Secondly, the findings described here are consistent with a hypothetical kind of `RNA sponge´ or `miRNA reservoir ´ suggested and discussed by others [Citation12,Citation13,Citation33]. However, more direct evidence for the existence of circular RNA sponge characteristics is beyond this study. Finally, we like to note that transient transfection studies do not necessarily monitor cellular metabolism and regulation at steady state conditions.
Considering the differences of thermodynamic characteristics of interactions between linear versus circular RNA and complementary short RNAs (see , Figure S1, , and ), we hypothesize that the regulation of RNA–RNA interactions is different in mechanistic terms that might also be true in living cells. Thus, we expect that further detailed mechanistic studies reveal new characteristics of circRNA and, as a consequence, more yet unknown functions of circRNA. To gain progressively more knowledge is of major importance as the number of newly identified circRNAs is rapidly expanding and evidences for circRNA involvement in human diseases are frequently found [Citation34–36] .
In conclusion, kinetics and thermodynamics of interactions between linear or circular RNA and complementary short-chain RNA suggest a topology-related fundamental mechanistic difference of RNA–RNA interactions at physiological conditions in vitro which is reflected by the different behaviour of circular versus linear RNA in human cells.
Methods
Chemicals and enzymes
Unlabelled dNTPs, DNase I, T7 RNA polymerase, Klenow fragment exo-, Klenow reaction buffer, RiboLock™, RNA size standards, and T4 polynucleotide kinase were purchased from ThermoFisher Scientific (Schwerte, Germany). Radioactively labelled ATP ([γ-[Citation32]P]ATP) was obtained from PerkinElmer® (Groningen, Netherlands), T4 RNA Ligase I (T4 RnL) and the appropriate buffer from New England Biolabs (Frankfurt am Main, Germany). DNA primers were purchased from eurofins Genomics (Ebersberg, Germany), RNA oligonucleotides (21mers) were provided by IBA (Goettingen, Germany). RNase R including the required buffer was obtained from epicentre (Oldendorf, Germany). All chemicals and reagents were of analytical grade and filtered through a 0.2 µm polyvinyl difluoride membrane before use. Upon electrophoresis, polyacrylamide (PAA) gels were either stained for 10 min with SYBR®Gold (1:50.000 in Tris/borate/EDTA (TBE) buffer (pH 8.0 at 37°C)). Alternatively, a 3ʹ-Carboxyfluorescein-(6-FAM-) residue or a [Citation32]P-labelled 5´-terminus of the short-chain oligoribonucleotide was used to detect and to quantify educts and products of annealing reactions. For quantification, a Typhoon FLA 9500 (GE Healthcare Life Sciences, Little Chalfont, Britain) was used. Agarose gels were stained with ethidium bromide (EtBr) at a final concentration of 0.5 µg/ml in TBE and visualized using the Variocam gel documentation system. All UV spectra were recorded on a NanoDrop ND 1000 spectrophotometer.
Synthesis of RNA
Klenow primers (sequences listed in Table S3, Supporting Information) were used in Klenow reactions in a total volume of 500 µl according to the manufacturer’s protocol. After precipitation of double-stranded DNAs using ice-cold ethanol at room temperature, product formation was controlled using native agarose gels (1.5%, EtBr stained). Since only one product was detectable after Klenow reaction, 1/10 volume of the resolved DNA pellet was used in GMP-primed in vitro transcription without further purification.
In vitro synthesis of RNA was performed essentially as follows. RNAs were formed by GMP-primed in vitro transcription of Klenow DNA templates with T7 RNA polymerase in the presence of ATP, UTP and CTP (2 mM each), GTP (1.25 mM), GMP (6 mM) and 1 U/µl RiboLock™ in 1x HEPES buffer (50 mM Na-HEPES, 12 mM MgCl2 hexahydrate, 2 mM spermidine pH 7.5 at 37°C) in a total reaction volume of 50 µl at 37°C for 3 h. DNA template was hydrolysed at 37°C for 30 min by DNase I (2 U) directly added to the in vitro transcription reaction mixture. The reaction was stopped via precipitation using ethanol at room temperature. Subsequent electrophoresis on 12% denaturing polyacrylamide gels (for composition see subchapter PAGE analysis below) was followed by elution of the product-containing bands with sodium acetate buffer (0.3 M, pH 7.0, shaking at 500 rpm, at 10°C). The procedure was done twice for at least 2 hours and overnight for the final elution step. Combined product-containing solutions were filtrated to remove PAA fibres and RNA was desalted by precipitation using 250 vol.-% ethanol at −20°C overnight.
Polyacrylamide gelelectrophoresis (PAGE)
For single RNA species analysis or purification, denaturing (7 M or 8 M urea) polyacrylamide gel electrophoresis (acrylamide: bisacrylamide 19:1, ammonium persulphate 0.1% w/v, N,N,N′,N′-Tetramethylethane-1,2-diamine 0.1% v/v) was applied, using 1x TBE buffer as running buffer and stop mix (formamide, bromophenol blue and xylene cyanol each 5 vol.-%) for sample loading. RNAs were denatured at 90°C for 2 min and directly loaded onto the gel.
Analysis of RNA–RNA complex formations required semi-denaturing (4 M urea) or non-denaturing conditions for 10% v/v PAA gels. Loading buffer of semi-denaturing gels was composed of formamide, 40 mM EDTA, 0.05% SDS and 7 M urea, loading buffer for non-denaturing gels consisted of 20 mM Tris-HCl (pH 8), 40 mM EDTA, 0.05% w/v SDS in 20% v/v glycerol.
Ligation of RNA
In vitro transcription mixture was used for end-joining by T4 RNA Ligase I in a total reaction volume of 18 µl. Either 1/10 volume of the resolved RNA pellet or 10 pmol purified transcript was used. Ligation was performed in 50 mM Tris-HCl (pH 7.5 at 25°C), 10 mM MgCl2, 1 mM DTT, 10 mM ATP, and PEG4000 (10% vol/vol) after RNA was denatured and re-folded at 90°C, 50°C, and 37°C at a duration per step of at least 5 min. Subsequently, 1 U or 4 U of T4 RNA Ligase I was added and ligation was performed at 37°C for 1–5 hours.
Two-dimensional gel electrophoresis
All ligated constructs were analysed using two-dimensional PAGE (for polyacrylamide gel composition, buffers and staining see above PAGE analysis).
- first dimension: denaturing conditions (7 M urea) 15% polyacrylamide;
- second dimension: 15% native polyacrylamide.
Ligation reaction mixtures including linear RNAs of several sizes due to former uncompleted in vitro transcription were used for unambiguously identification of circular species. Mixtures were separated in the first dimension by gel electrophoresis. Subsequently, the gel lane was excised and used for electrophoresis in the second dimension.Here, linear RNAs compose a diagonal line. Covalently closed cyclic RNAs possess reduced degrees of freedom, displaying a non-linear dependency of migration manner and occur outside the diagonal [Citation21,Citation37,Citation38].
Exoribonuclease treatment of RNA
Hydrolysis of linear RNAs within a mixture of linear and circular RNAs was performed using 5 pmol RNA. The reaction buffer was 0.2 M Tris-HCl (pH 8.0), 1 M KCl, and 1 mM MgCl2 and MgCl2 concentration was increased to 5 mM. The reaction mixture was heated at 90°C for 5 min cooled to 50°C before addition of 0.1 U of RNase R. Hydrolysis occurred at 50°C for 60 min. The reaction was stopped using an equal volume of 7 M urea and 50 mM EDTA, which is also used as loading buffer for electrophoresis, and the mixture was immediately frozen in liquid nitrogen. Products were analysed using PAGE in 15% denaturing PAA gels.
Citation32P-labelling of RNA.
Radioactive labelling of as101 was performed using [γ-[Citation32]ATP] (30 µCi), 10 pmol of RNA, reaction buffer (50 mM Tris-HCl (pH 7.6 at 25°C), 10 mM MgCl2, 5 mM DTT, 0.1 mM spermidine) and 10 U of T4 polynucleotide kinase. Reactions took place for 1 hour at 37°C and were stopped by heat inactivation at 75°C for 10 min, followed by product isolation using G-50 Sephadex column and ethanol precipitation for 1 hour at −20°C. Phosphorylation efficiency was checked by liquid scintillation measurement and denaturing PAGE (15%, 7 M urea).
Determination of association rates
RNA–RNA association was measured at a 5 fold excess of linear or circular RNAs over the 21mers in 20 mM Tris-HCl (pH 7.4), 100 mM NaCl, and 10 mM MgCl2 at 37°C and stopped at time points 0, 1, 2, 4, 8, or 16 min. To stop the reaction, aliquots were mixed with loading buffer and analysed via 10% native PAGE (see subchapter PAGE). Band intensities were quantified by using ImageJ and rate constants were calculated by linear regression analysis.
Dissociation studies
Prior to analysis of dissociation, a complex consisting of linear or circular RNAs with an appr. 5.6 fold excess over the labelled 21mers had to be generated for at least 2 hours at 37°C in hybridization buffer (see determination of association rates). Subsequently, different concentrations (37 nM, 110 nM, 370 nM, 1110 nM) of unlabelled 21mers were added. At indicated time points (compare ) reaction aliquots were withdrawn and immediately mixed with loading buffer. Further analysis was performed with 15% native PAGE (see subchapter PAGE). ImageJ was used for the determination of band intensities. Rate constants and half-life were calculated using linear regression analysis.
Liquid hybridization
Hybridization of total RNA (50% of RNA isolated from one well of 96 well plates) with the probe 5ʹ-FAM-as101 (0.25 μM) was performed in a total volume of 20 μL in 20 mM Tris/HCl, pH 7.4, and 0.1 M NaCl at a temperature of 37°C. First, RNAs were denatured at 90°C for 5 min followed by a 30 min incubation. Samples were kept at on ice prior to gel electrophoresis.
RNAse H treatment
Reactions were performed in a total volume of 20 μL containing 15 mM Na-HEPES (pH 7.4) 50 mM KOAc, 1 mM Mg(OAc)2, total RNA (0,04 μM), and oligonucleotide probes (1,2 μM) at 37°C for 30 min. 2 U of RNase H were added per reaction. The probe sequences were: as26, 5‘-GGACTTCGTCGTCTGATTTT-3‘ and as46, 5‘-AAGACTCAGTATCTTCATGG–3‘.
Transfection
ECV-304 cells were transfected using 5 µg/ml Lipofectamine 2000 (Invitrogen, Karlsbad, California) in 96-well plates (2.0 x 105 cells/well) with 10 ng pGL3 control (Promega, Fitchburg, USA), 100 ng Hmga2m7, 0.5 nM ds mlet7a, or 0.5 nM ds scramble and 2.5 nM l/c-92 or LRa/CRa-mlet7a following the manufacturer’s instructions (50 µl final volume of the transfection solution). Hmga2 3ʹUTR m7 luciferase (Luc-m7) was a gift from D. Bartel (Addgene plasmid # 14,788) [Citation28]. Additionally, all cells were transfected with stocking DNA (comp. Figure S2) to ensure equal amounts of nucleic acids in each transfection mix. After 16–40 hours cells were washed with PBS and lysed with passive lysis buffer (Promega).
Luciferase assay
A non-commercial dual-luciferase enzyme assay described by Dyer et al., 2000 was used to determine the luciferase activities [Citation39–42] . Light emission was measured directly in a 96-well micro-titre plate in a FLUOstar Omega BM6 Labtech. The ratio of the Renilla and the Firefly activities serves as a control for transfection efficiency. In order to determine the effect of LRa-mlet7a or CRa-mlet7a on the miRNA-regulating system the obtained values were normalized to those of the values from cells, which were transfected with lin-92 or circ-92.
Supplemental Material
Download PDF (2.8 MB)Acknowledgments
We cordially thank Rosel Kretschmer-Kazemi Far for helpful discussions and for carefully reading this manuscript, Patrick Lamm for initial support, Evelyn Knappe, and Fenja Fahrig for excellent assistance with the synthesis of circular RNA and for initial studies on RNA–RNA complex formation. We are also grateful to Jana Kochmann and Moritz Lübcke for support with RNA detection studies.
Disclosure statement
No conflict-of-interest declared.
Supplementary material
Supplemental data for this article can be accessed here
References
- Rong D, Sun H, Li Z, et al. An emerging function of circRNA-miRNAs-mRNA axis in human diseases. Oncotarget. 2017;8(42):73271–73281.
- Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: identification, biogenesis and function. Biochim Biophys Acta. 2016;1859(1):163–168.
- Petkovic S, Muller S. RNA circularization strategies in vivo and in vitro. Nucleic Acids Res. 2015;43(4):2454–2465.
- Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. RNA. 2013;19(2):141–157.
- Welden JR, Pawluchin A, Van Doorn J, et al. 2020. Use of alu element containing minigenes to analyze circular RNAs. J Vis Exp. 157. 10.3791/59760.
- Metge F, Czaja-Hasse LF, Reinhardt R, et al. FUCHS-towards full circular RNA characterization using RNAseq. PeerJ. 2017;5:e2934.
- Ivanov A, Memczak S, Wyler E, et al. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Rep. 2015;10(2):170–177.
- Chuang TJ, Wu CS, Chen CY, et al. NCLscan: accurate identification of non-co-linear transcripts (fusion,trans-splicing and circular RNA) with a good balance between sensitivity and precision. Nucleic Acids Res. 2016;44(3):e29.
- Luo Y, Liu S, Yao K. Transcriptome-wide Investigation of mRNA/circRNA in miR-184 and Its r.57c > u mutant type treatment of human lens epithelial cells. Mol Ther Nucleic Acids. 2017;7:71–80.
- Raz O, Granot G, Pasmanik-Chor M, et al. Profiling and bioinformatics analyses reveal chronic lymphocytic leukemia cells share a unique circular RNA expression pattern. Exp Hematol. 2020;85:8–12.
- Haque S, Ames RM, Moore K, et al. Islet-expressed circular RNAs are associated with type 2 diabetes status in human primary islets and in peripheral blood. BMC Med Genomics. 2020;13(1):64.
- Barrett SP, Salzman J. Circular RNAs: analysis, expression and potential functions. Development. 2016;143(11):1838–1847.
- Kristensen LS, Andersen MS, Stagsted LVW, et al. The biogenesis, biology and characterization of circular RNAs, Nature reviews. Genetics. 2019;20:675–691.
- Kleaveland B, Shi CY, Stefano J, et al. A network of noncoding regulatory rnas acts in the mammalian brain. Cell. 2018;174(2):350–362.e317.
- Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338.
- Salzman J, Chen RE, Olsen MN, et al. Cell-type specific features of circular RNA expression. PLoS Genet. 2013;9(9):e1003777.
- Rybak-Wolf A, Stottmeister C, Glažar P, et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870–885.
- Maass PG, Glažar P, Memczak S, et al. A map of human circular RNAs in clinically relevant tissues. J Mol Med (Berl). 2017;95(11):1179–1189.
- Rittner K, Burmester C, Sczakiel G. In vitro selection of fast-hybridizing and effective antisense RNAs directed against the human immunodeficiency virus type 1. Nucleic Acids Res. 1993;21(6):1381–1387.
- Persson C, Wagner EG, Nordstrom K. Control of replication of plasmid R1: formation of an initial transient complex is rate-limiting for antisense RNA–target RNA pairing. EMBO J. 1990;9(11):3777–3785.
- Rehmsmeier M, Steffen P, Hochsmann M, et al. Fast and effective prediction of microRNA/target duplexes. RNA. 2004;10(10):1507–1517.
- Ghosal S, Das S, Sen R, et al. Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Front Genet. 2013;4:283.
- Petkovic S, Badelt S, Block S, et al. Sequence-controlled RNA self-processing: computational design, biochemical analysis, and visualization by AFM. RNA. 2015;21(7):1249–1260.
- Patzel V, Sczakiel G. Theoretical design of antisense RNA structures substantially improves annealing kinetics and efficacy in human cells. Nat Biotechnol. 1998;16(1):64–68.
- Lorenz R, Bernhart SH, Siederdissen HZ, et al. ViennaRNA Package 2.0. Algorithms for molecular biology: AMB. 2011;6(1):26
- Kerpedjiev P, Hammer S, Hofacker IL. Forna (force-directed RNA): simple and effective online RNA secondary structure diagrams. Bioinformatics. 2015;31(20):3377–3379.
- Wunsche W, Sczakiel G. The activity of siRNA in mammalian cells is related to the kinetics of siRNA-target recognition in vitro: mechanistic implications. J Mol Biol. 2005;345(2):203–209.
- Patzel V, Sczakiel G. Length dependence of RNA-RNA annealing. J Mol Biol. 1999;294(5):1127–1134.
- Homann M, Nedbal W, Sczakiel G. Dissociation of long-chain duplex RNA can occur via strand displacement in vitro: biological implications. Nucleic Acids Res. 1996;24(22):4395–4400.
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science. 2007;315(5818):1576–1579.
- Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388.
- Fang X, Bai Y, Zhang L, et al. Silencing circSLAMF6 represses cell glycolysis, migration, and invasion by regulating the miR-204-5p/MYH9 axis in gastric cancer under hypoxia. Biosci Rep. 2020;40(6). 10.1042/BSR20201275
- Ye M, Hou H, Shen M, et al., Circular RNA circFOXM1 Plays a Role in Papillary Thyroid Carcinoma by Sponging miR-1179 and Regulating HMGB1 Expression, Molecular therapy. Nucleic acids. 2020; 19:741–750.
- Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4(9):721–726.
- Franco-Zorrilla JM, Valli A, Todesco M, et al. Target mimicry provides a new mechanism for regulation of microRNA activity. Nat Genet. 2007;39(8):1033–1037.
- Hansen TB, Kjems J, Damgaard CK. Circular RNA and miR-7 in cancer. Cancer Res. 2013;73(18):5609–5612.
- Chen S, Li T, Zhao Q, et al. Using circular RNA hsa_circ_0000190 as a new biomarker in the diagnosis of gastric cancer. Clin Chim Acta. 2017;466:167–171.
- Nair AA, Niu N, Tang X, et al. Circular RNAs and their associations with breast cancer subtypes. Oncotarget. 2016;7:80967–80979.
- Lavenniah A, Luu TDA, Li YP, et al. Engineered circular RNA sponges act as mirna inhibitors to attenuate pressure overload-induced cardiac hypertrophy. Mol Ther. 2020;28(6):1506–1517.
- Pasman Z, Been MD, Garcia-Blanco MA. Exon circularization in mammalian nuclear extracts. RNA. 1996;2:603–610.
- Petkovic S, Muller S. RNA self-processing: formation of cyclic species and concatemers from a small engineered RNA. FEBS Lett. 2013;587(15):2435–2440.
- Dyer BW, Ferrer FA, Klinedinst DK, et al. A noncommercial dual luciferase enzyme assay system for reporter gene analysis. Anal Biochem. 2000;282(1):158–161.