
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
The three-dimensional (3D) structure of RNA usually plays an important role in the recognition with RNA-binding protein. Along with the discovering of RNAs, several RNA databases are developed to study the functions of RNA based on sequence, secondary structure, local 3D structural motif and global structure. Based on RNA function and structure, different RNAs are classified and stored in SCOR and DARTS, respectively. The classification of RNA structures is useful in RNA structure prediction and function annotation. However, the SCOR and DARTS are not updated any more. In this study, we present an RNA classification database RR3DD based on RNA fold with the global 3D structural similarity. The RR3DD includes 13,601 RNA chains from PDB and mmCIF format structures which are classified into 780 RNA folds. The RNA chains from PDB and mmCIF format structures are aligned and clustered into 675 and 220 RNA folds, respectively. By analysing the RNA structure in RR3DD, we find that there are 11 clusters with more than 50 members. These clusters include rRNAs, riboswitches, tRNAs and so on. By mapping RR3DD into Rfam, we found that some RNAs without annotation by Rfam can be annotated through structural alignment. For example, we analysed tRNAs and found that tRNA were successfully grouped in RR3DD for which Rfam did not classify them into one family. Finally, we provide a web interface of RR3DD offering functions of browsing RR3DD, annotating RNA 3D structure and finding templates for RNA homology modelling.
Introduction
RNA is an essential molecule in vivo, and it can perform its function by binding protein. To investigate the relationship between RNA sequence, structure and function, many function-based and structure-based RNA databases have been developed. Rfam is a structural database for collecting and classifying non-coding RNAs [Citation1]. In Rfam, RNAs are clustered into a family through the seed alignment generated by experts with RNA secondary structures. The secondary structures of RNAs for classification in Rfam are derived from the computational prediction and experimental data. Based on Rfam, Boccaletto et al. proposed RNArchitecture, a four-level classification system, focusing on RNA secondary structure by defining the RNA fold similar to protein fold in SCOP or CATH [Citation2]. In RNArchitecture, the central level of classification is Family, which is based on RNA secondary structure similarity. The second level is Superfamily consisting of Families with similar structures and functions (evolutionarily related). The third level is Architecture grouped by the Superfamilies sharing a similar core structure. The highest level is Class which only consisted of two members. In DARTS [Citation3], Abraham et al. classified 1333 RNA structures into 94 clusters. Because they found dissimilar RNA tertiary structures with similar 2D structures, which can be classified into the same family by Rfam or RNArchitecture. Besides Rfam and RNArchitecture, several databases focus on 3D structural motif in RNA structure. SCOR is a global structural database which is based on function. It also classified 3D motifs based on local structure [Citation4]. In SCOR, Motifs are extracted from RNA 3D structures and then classified with Directed Acyclic Graph (2.0 later) or hierarchical clustering (1.2 before). In SCOR2.0, it contains 511 RNA structures downloaded from PDB. And total 5880 secondary structural motifs from these RNA structures are grouped into 2104 hairpin loops and 3776 internal loops. These loops are also annotated as special 2D structural motifs. However, DARTS and SCOR are no longer maintained. Since most of the RNA functional sites are resided in the RNA loop region, there are three databases that based on the loop. RNA 3D Motif Atlas database includes 3D motifs extracted from RNA 3D structure in PDB with FR3D [Citation5,Citation6]. The motifs are classified based on the geometric similarity and structural annotations. Like the classification of motifs in RNA 3D Motif Atlas, RNAMCS takes the effect of base pairing on structural similarity into account [Citation7]. In RNAMCS, motifs are extracted from the RNA tertiary structure and grouped into 191 clusters. Besides, in DART, motifs are classified with the RNA 3D structural alignment approach ARTS, which takes the sequence and secondary structure into account at the same time [Citation3,Citation8]. The classification of these three databases [Citation3,Citation5,Citation7] depends on the motifs discovered by the motif searching algorithms. RNA Tetraloop database only includes the motif consisting of 4-nt nucleotide [Citation9]. Other motifs, such as G-quadruplex, are missing if the database only includes local structure. Besides RNA Tetraloop, InterRNA collected and classified the RNA motifs including quadruples, quintuples and sextuples [Citation10]. These databases mentioned above don’t take the molecules that interact with RNA motif into account. RNA Bricks presents a molecular environment in which RNA, protein, metal ions, water molecules and ligands are included [Citation11]. In order to avoid bias about the parameters obtained from statistically, Leonti et. al produced nonredundant 3D RNA structure datasets [Citation12]. All these databases provide a wealth of data to study the relationship between RNA function and structure. But so far, there is no classification database based on global similarity of RNA 3D structure, which RNAs perform their biological function by folding into a special 3D structure [Citation13].
In this manuscript, we present RR3DD, an RNA global structure-based RNA 3D classification Database. The RNAs are clustered with the similar matrix by average linkage hierarchical clustering algorithm. The similar matrix is generated by all-to-all alignment of RNA with RMalign, an RNA 3D structural alignment method based on RMscore [Citation14], which has been applied in protein-RNA complex structure modelling [Citation15]. By analysing RR3DD, we study the relationship between sequence, structure and function of all RNAs in PDB. By comparing with Rfam, tRNAs that have not been annotated by Rfam are classified into one family in RR3DD. Therefore, the classification in RR3DD may afford a way to annotate RNA in PDB. With the cluster in RR3DD mapped to the family in Rfam, RR3DD can provide templates for annotating a new RNA 3D structure. Finally, we provide a web interface that is convenient for users to browse RR3DD, annotate RNA folds and search templates for RNA 3D modelling.
Materials and methods
RNA 3D structure preparation
RNA chains are extracted from RNA-containing PDB format files which were downloaded from PDB (29 July 2021 released) [Citation16]. The RNAs with length shorter than 5-nts are filtered out for that RMalign can’t compare these RNAs with others. Finally, 7466 RNA chains are kept for classification. In addition to the structures with PDB format, it includes 950 downloadable RNA-containing structures with mmCIF format in PDB. In order to use RMalign to align these structures, we downloaded these structures with PDB format-like files from PDB website. Finally, we get 6135 RNA chains from these structures. We classified these RNA chains by aligning them with the centre structures obtained from the classification of 7466 RNA chains with PDB format. The monomers which have no base-pairing are ignored to study the relationship between RNA sequence and structure [Citation3].
Rfam is a non-coding RNA classification database and have been mapped to PDB database. In Rfam 14.6, there are 14,412 items and 7079 items that have been mapped to PDB in pdb_full_region.txt.gz (http://ftp.ebi.ac.uk/pub/databases/Rfam/CURRENT/database_files/pdb_full_region.txt.gz) and Rfam.pdb.gz (http://ftp.ebi.ac.uk/pub/databases/Rfam/CURRENT/Rfam.pdb.gz), respectively. To investigate the relationship between RNA structure and function, we compared RR3DD with Rfam on these structures (the details of the comparison are listed in Tables S1 and S2).
RMalign
In 2019, our group revealed the liner relationship between the logarithmic length of RNA and the logarithmic radius of gyration (Rg) of RNA, and described a complex function relation between the aligned correlation coefficient (ACC) and RMSD. Based on these, we have developed a novel RNA 3D structural alignment approach RMalign with a size independent scoring function RMscore [Citation14]. The RMscore returned by RMalign is used to measure the structural similarity of two RNAs. The value of RMscore ranges from 0 to 1, and 1 represent that two structures are total identical and vice versa.
RNA global structure clustering
To cluster all RNAs, we use RMalign to align RNA global structures and construct a similarity matrix based on RMscore, which contained results of all-to-all alignments for 7466 RNA chains. The average linkage hierarchical clustering algorithm is used to cluster all RNA structures, which minimizes the average distance between all members of paired clusters. The average distance between two clusters is calculated by formula (1).
The and
represent two clusters.
and
represent the number of cluster
and cluster
.
represent the distance between two elements of two clusters.
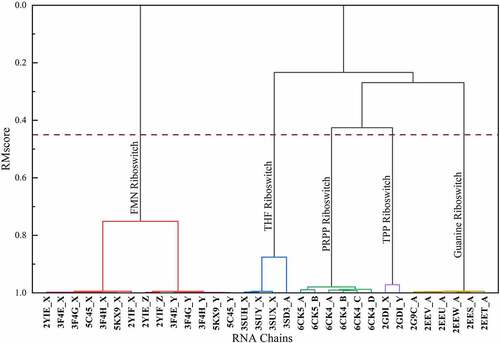
The hierarchical clustering results of 32 riboswitch structures are shown in (). The RMscore cut-off is set as 0.45 to cluster the RNA structures. It is because that in our previous study, RMalign achieved the highest MCC (MCC = 0.73) while the RMscore cut-off is set as 0.45 in RNA functional classification [Citation14].
Figure 1. An example of hierarchical clustering results of riboswitches. We calculated the similarity matrix of 32 riboswitch structures from 10 riboswitch-containing structures and grouped them by hierarchical clustering. The hierarchical clusters are reported as a tree (or dendrogram). The x axis represents RNA structure ID. The y axis is the distance between two clusters. The dashed line represents the cut-off of clustering
Results
Clustering analysis
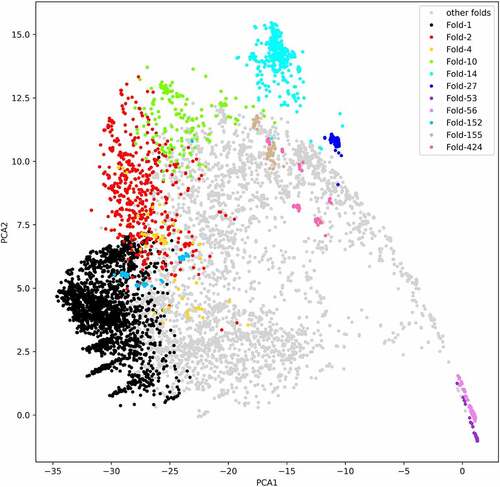
These 7466 RNA chains were grouped into 675 clusters by clustering. After clustering, all the RNA chains are clustered into 675 folds based on the global 3D structural similarity. The centre RNA has the minimum average distance (1.0 – RMscore) between itself and the other RNAs in the same cluster. For each cluster, the centre RNA structure is chosen as the representative structure and defined as RNA fold. These clusters are named as fold-x (x ranges from 1 to 675). In (), it shows the number of members in fold-x. And all the clusters of RNAs are listed in (Table S1). The number of RNA chains in most folds (69.0%) are less than 5. Clusters containing only one RNA chain account for 30.1%. There are 11 clusters that include more than 50 members. The largest cluster named fold-1 includes 2099 RNA chains. To evaluate whether these RNA chains are classified into the same fold is reasonable or not, similar to previous study [Citation9], we utilize PCA (principal component analysis) method to analyse the structural similarity matrix. The structural similarity matrix is built based on all-to-all RNA structural alignments. As is shown in (), the RNAs with similar structure are classified into the same fold. Since tRNAs have special structure, they were clustered into Fold-14 shown in (). The PCA analysis reveals that RNAs in the same fold are close to each other. The tRNAs are not annotated by Rfam, but they are clustered into fold-14. The other clusters including more than 50 RNA chains are Fold-1, Fold-2, Fold-4, Fold-10, Fold-14, Fold-27, Fold-53, Fold-56, Fold-152, Fold-155, Fold-424, respectively. The centroid structure of Fold-27, Fold-53, Fold-56 and Fold-155 are 5s rRNA, 23s rRNA, 16s rRNA and guanine riboswitch, respectively.
Figure 2. The number of the members in folds. The folds with the number of members less than 30 are not shown
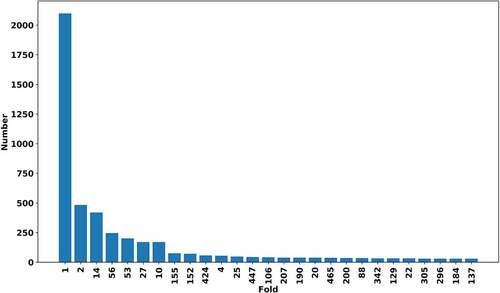
Figure 3. The principal components analysis of similarity matrix from all-to-all RNA structure alignments. The different colour except lightgrey represents different clusters in RR3DD. We coloured the clusters with the number of members more than 50. The fold-14 includes 419 tRNAs. The fold-53 includes 201 23s ribosomal RNAs. The fold-27 includes 170 5s ribosomal RNAs. The fold-56 includes 246 16s ribosomal RNAs. The fold-155 includes 75 guanine riboswitches. RNA fold is determined by a hierarchical clustering algorithm and a similar matrix based on RMscore, which is generated by all-to-all (7466x7466) alignments of RNA with RMalign. The PCA is only used to analysis the distribution of different RNA in PCA space. The PCA analysis reveals that RNAs in the same fold are close to each other in PCA space
Why fold-1 containing the largest number of members?
The largest cluster in RR3DD is fold-1 including 2099 RNA chains with an average length of 15-nt. The most members of this cluster form complex with RNA or DNA, and these complexes are annotated as ‘SYNTHETIC CONSTRUCT’ (such as 5us2:B) [Citation17] or RNA fragment. The centroid structure (PDB ID:4k4s, chain ID: G) of this cluster is annotated as ‘SYNTHETIC’ [Citation18]. Most members in this cluster are synthetized to analysis the structure and function. Such as PDB ID: 8drh, chain ID: B, Bachelin et al. studied this structure for it adapts A-form while interacting with phosphorothioate modified DNA. This double helix DNA/RNA hybrid structure can be recognized and digested by RNase H similar to other unmodified DNA/RNA hybrid for they have similar global structure [Citation19]. Besides, Fedoroff et al. presented a DNA/RNA complex structure (PDB ID:124d) [Citation20]. By analysing this structure, they found that the protein and the substrate have close surface complementary and explained the mechanism for RNase H recognize DNA/RNA duplex. Since these short oligoribonucleotides form duplex with other molecules, they have the similar 3D structure.
Why is Fold-1 close to Fold-152 in space?
We find that Fold-1 and Fold-152 relatively close in space shown in (), but these two folds are not classified into one cluster. It’s because that the average length of members is different. Fold-152 includes 71 RNA chains with average length 21-nt. The core structure (PDB ID: 2bcz, chain ID: D) of Fold-152 interacted with other single-strand RNAs function as all-RNA hairpin ribozyme [Citation21]. This fold also includes other single-strand RNAs that form hairpin ribozyme, such as PDB ID:2p7d, chain ID: C [Citation22]. As shown in (), Fold-1 closes to Fold-152 because both their members form single-strand chain with nearly number of nucleotides.
Classification of G-quadruplex-forming RNA
G-rich sequences can adapt a special helical fold known as G-quadruplex under appropriate conditions [Citation23]. G-quadruplexes have diverse forms, including propeller loop, lateral loop and diagonal loop [Citation24]. In RNA world, the G-quadruplex structure plays an import role in mRNA translation, such as translation inhibition [Citation25] or cap-independent translation [Citation26]. Since the G-quadruplex is complicated structure, it can be formed by one, two or four RNA chains. For example, a G-quadruplex-containing RNA aptamer (PDB ID: 4kzd, chain ID: R) that can activate green fluorescence is different with other G-quadruplex RNA because it only contains two layers of G-tetrads [Citation23]. We analysed the classification of G-quadruplex-formed RNA chains. There are 42 RNA complex structures with G-quadruplexes in PDB. After splitting these complexes into monomers, there are 86 RNA chains. As shown in (), 86 RNA chains are classified into 16 clusters in RR3DD. Among them, the G-quadruplex formed with one RNA chain belongs to the Fold-660. Fold-411 consists of four RNA chains (PDB ID: 4kzd, 4kze, 4q9q, 4q9r) with similar 3D structure. Fold-208, Fold-321, Fold-227, Fold-439 Fold-220, Fold-520, Fold-429 and Fold-514 consists of 2, 3, 4, 4, 7, 8, 15 and 15 G-quadruplex monomers, respectively. The other G-quadruplex monomers are classified into other folds and shared a fold with the other non-G-quadruplex RNAs. The classifications of G-quadruplex RNAs indicate that G-quadruplex can be formed by dissimilar monomers. Therefore, these G-quadruplex-forming RNA chains are classified into different fold.
Table 1. Classification of G-quadruplex (43 G-quadruplex containing RNA structures)
The relationship between sequence and structure of RNA
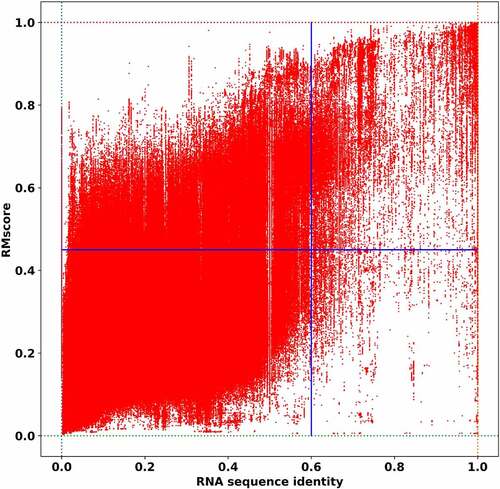
In the previous section, we analyse the clustering results in RR3DD briefly. The clusters in RR3DD shows the relationship between structure and function are not a simple one-to-one. To uncover the relationship between sequence and structure in RNA, we plot the structural similarity vs sequence identity of the RNA and the result is shown in (), of which structural similarity is calculated by RMalign and sequence identity is calculated by needle [Citation27]. The figure is divided into four areas by the line x = 0.6 (the relationship between sequence and structure conservation will be weakened if the sequence identity below this cut-off, ref [Citation28].) and the line y = 0.45 (this cut-off is set as a transition point between similar and dissimilar RNA 3D structure). The sequence and structure are both dissimilar in the lower left in (), which contains 93.42% of all alignments. These points indicate that these two RNAs have dissimilar structure and dissimilar sequence. The pairwise alignments between them are random, which indicates they are not homologs. The points in upper left represent the alignments with similar structure and dissimilar sequence, which contains 5.44% of all alignments. It also indicates that these highly conserved RNA structure can only be found by structural alignment. These points in the upper right represent the alignments that both RNA sequence and structure are similar, which contains 1.02% of all alignments (these structures with sequence identity and RMscore are listed in Table S2). Since the number of structure-based alignments (with RMScore >0.45) are 5 times more than that of sequence-based alignments, it indicated that structure-based classification method can find more homology structure than sequence-based classification method. The almost empty area, which contains 0.12% of all alignments, located in lower right in () suggest that two RNAs tend to have similar structure when they have similar sequence. These results indicate that there is a transition point (sequence identity = 0.6) between similar and dissimilar in RNA, which is consistent with the previous researches in sequence and structure conservation (sequence identity cut-off = 0.6) of RNA [Citation28], binding modes (TMm = 0.4) of protein–protein complex [Citation29], binding modes (complex structure score = 0.45) of protein-RNA complex [Citation30] and protein [Citation31]. Over all, the percentage of RNA with dissimilar sequence and dissimilar structure, with dissimilar sequence and similar structure, and with similar sequence and similar structure are 93.42%, 5.44% and 1.02%, respectively. Structure-based alignment approach can find more remote homology structure templates than sequence-based approach, which benefits for template-based RNA, RNA-RNA complex and RNA-protein complex structure prediction. These observations agree with what we have found in our template-based protein-RNA modelling method PRIME [Citation30].
Figure 4. The relationship between RNA structure similarity and sequence identity. The sequence identity of the RNA is plotted against the structural similarity in all-to-all pairwise comparison of RNA structures in RR3DD. The RNAs without base-pairing are ignored. The sequence identity is calculated by needle. The RNA structure similarity is measured by RMscore. The lines separate quadrants below and above a sequence (identity = 0.6) and a structure-based threshold (RMscore = 0.45). It indicates that 5.44% RNAs have similar structure with low sequence identity in the second quadrant. 1.02% RNAs are homologs with similar structure and similar sequence. The number of structure-based alignments (with RMScore > 0.45) are 5 (=5.44%/1.02%) times more than that of sequence-based alignments
Comparing RR3DD with Rfam
Based on the mapping Rfam to PDB, we compare RR3DD with Rfam. To illustrate the effect of sequence, secondary and 3D structure on RNA classification, the relationship between RR3DD and Rfam are analysed. Since the classification of Rfam for RNA in PDB is based on the results of Infernal, one RNA may belong to more than one family (such as 1c2w:B belongs to RF02543, RF02541 and RF02540) and some RNAs do not belong to any family (such as 2zni:D). Finally, a total of 1861 RNA chains in Rfam are mapped to RR3DD (Table S1), which include 170 clusters in RR3DD and 79 families in Rfam. The mapping results are listed in (Table S1) and the results of top 10 folds more than 30 members are shown in (), respectively. In (), it shows that each RNA fold is mapped into several families in Rfam. Here, a case about tRNA is analysed in detail to illustrate the difference between RR3DD and Rfam. 419 RNAs with similar 3D structures are assigned to Fold-14 in RR3DD. And these tRNAs are assigned to RF00005 (annotated as tRNA), RF01852 (annotated as tRNA-sec), RF00390 (annotated UPSK RNA), RF00233 (annotated tRNA-like elements) and RF02340 (annotated Dengue virus SLA) in Rfam. RNAs without annotation in Rfam share the similar with fold-14 in RR3DD (Figure S1). For example, an RNA chain annotated with bacterial tRNA (PDB ID: 2zni chain ID: D) is not assigned to any family in Rfam. And RR3DD can find the similar structure with different annotation in the same fold. For example, 6mj0 is a tRNA-like structure that includes tRNA-like structure (TLS) and upstream pseudoknot domain (UPD), which has similar functions to tRNA [Citation32]. Therefore, the difference between RR3DD and Rfam is that RR3DD can annotate RNA function by structural alignment. Since RNAs with similar secondary structure may have dissimilar 3D structures (Figure S2) and RNAs with similar structure may have different functions (Figure S3), a structure-based classification database, RR3DD, can be used to study the relationship between RNA structure and function.
Figure 5. Mapping RNA fold database RR3DD into Rfam. The RNA folds of RR3DD are mapped to Rfam. Top 9 folds with more than 30 members are shown. The x-axis represents the RNA folds. The y-axis represents that the number of the structures in each RNA fold. The same colour belongs to the same families in Rfam. It can be seen that each RNA fold is mapped into several families in Rfam. For example, the Fold-27, Fold-156, Fold-188 and Fold-356 are mapped into one family in Rfam and the Fold-59 is mapped into four families in Rfam. The different fold may perform the same function. For example, both Fold-26 and Fold-55 are mapped into RF02540 (wheat), RF02541 (mintcream) and RF02543 (deeppink) in Rfam. These data come from pdb_full_region.txt in Rfam
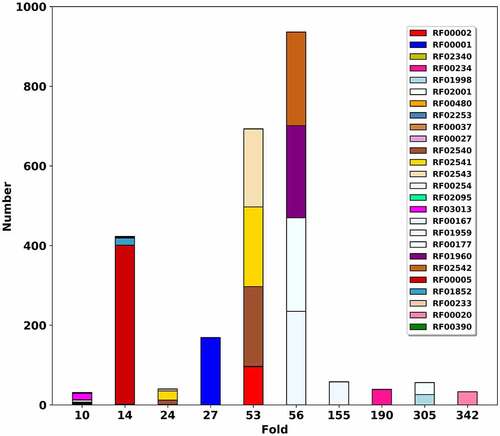
Classification of tRNA
tRNA adopts cloverleaf structure transports amino acid to ribosome [Citation33], and its length varied from 76 to 90-nt [Citation34]. Since these RNAs have the same functions, most tRNA have been classified into one family (RF00005) in Rfam. According to the mapping from Rfam to PDB, there are 410 RNA chains annotated with tRNA or tRNA-sec. In RR3DD, these tRNA chains belong to 6 clusters. Among them, 419 tRNA chains are classified in Fold-14 while the other 22 chains belong to other five different clusters. The PDB ID of centre structure of Fold-14 is 3wfs (chain ID:B) annotated as tRNA in complex with enzyme [Citation35]. Since these RNAs annotated with tRNA have not been classified into one cluster in RR3DD, we want to figure out the reason. By aligning these structures to the members of Fold-14, we found that the average distance between these tRNAs and members of Fold-14 are lower than 0.45. These tRNA chains have a lower similarity to the fold of Fold-14 because these tRNAs miss nucleotides or undergo a big conformational change in forming complexes [Citation36,Citation37] [e.g. 5u4j:x, 4zdp:E]. Although tRNAs perform the same function, they may have dissimilar 3D structures as expected (Figure S4). Overall, 419 tRNAs are classified into one cluster in RR3DD, which may afford a way to annotate RNA functions.
Classification of riboswitch
Riboswitches play vital role in the mechanism of bacteria and archaea [Citation38,Citation39]. Its length ranges from 50 to 250-nt [Citation40]. The typical structure includes two function domains: aptamer and expression platform. Riboswitch can directly interact with ligands without the involvement of protein factors to regulate gene expression [Citation38]. At present, riboswitches have been classified into nearly 40 groups based on their functions [Citation41]. Here, we analysed the classification of riboswitch RNA in RR3DD. These riboswitches have been classified into 31 clusters. Since these riboswitches can bind different ligands, these riboswitches have different structure in general. Fold-155 have the most riboswitches. In Fold-155, the centroid structure is 6uc8 (chain ID: B), which is annotated as guanine riboswitch and bind to 8-aminoguanine [Citation42]. The fold of riboswitches and the functions of centroid structure are listed in ().
Table 2. The classification of riboswitches
Classification of RNA chains in mmCIF files
After filtering out the structure that RMalign can’t align, there are 6135 RNA chains in mmCIF format structures finally. In PDB website, it also gives us a link to get PDB-like format for these mmCIF format structures. To check whether these structures in PDB-like format are similar to the folds obtained from the RNA chains in PDB-format or not, we aligned these structures with the centre structures defined before. If the structure has the most similar centre structure and the RMscore between them larger than 0.45, we classified the structure into the fold that the centre structure belongs to. After that, we found that 96.12% (5897/6135) of these structures can be classified into 115 folds defined before. And then, we classified the left 238 RNA chains into 105 folds as we do before and named these folds with from Fold-676 to Fold-780. About 54% of these RNAs are annotated as mRNA. Many mRNAs are linear structures for they have been unwound into single strand chains, which can be translated by ribosome [Citation43][Citation44] . Therefore, they are grouped into different folds due to conformational difference. The classification of RNA chains from mmCIF format structures and the comparison to Rfam 14.6 are listed in (Table S3).
Web interface of RR3DD
We implement a web interface that is convenient for users to browse RR3DD, annotate RNA fold and search templates for RNA 3D modelling. By mapping the RR3DD cluster to Rfam family, web interface of RR3DD can provide template structure for RNA. In web interface of RR3DD, the user can submit the Rfam ID or 3D structure of an RNA. If the user submits the family ID of Rfam, the webserver will query RR3DD and return the results corresponding to the family ID, and then provide the centroid structure of the cluster as the fold. If the user submits the 3D structure of an RNA, the webserver will employ RMalign to search and output the most similar RNA 3D structure in RR3DD.
Discussion and conclusion
In this manuscript, we present RR3DD, which is currently the only database that classifies RNA structures based on the similarity of RNA global 3D structures. RMalign was employed to generate a similarity matrix for all RNA chains in the PDB. The RNAs are then clustered with the average linkage hierarchical clustering algorithm. A total of 13,601 RNA chains are clustered into 780 folds. In RR3DD, by analysing the relationship between RNA structure and sequence, we find that the RNA structure is more conserved than sequence. The classification of tRNA and G-quadruplex in RR3DD illustrates that the relationship between RNA structure and RNA function is not simple one-to-one. By comparing RR3DD with Rfam, we found that each RNA fold is mapped into several families in Rfam. Therefore, RR3DD can provide templates for RNA 3D structure modelling. Finally, we provide the web interface of RR3DD, which provides the services of browsing database, annotating RNA fold and providing 3D templates for RNA 3D homology modelling.
Accession codes
The web interface can be accessed at http://rnabinding.com/RR3DD.
Author contributions
Conceptualization, XH, JFZ and SYL; Investigation JX, JFZ, XXT, XH and SYL; RR3DD software development, JFZ, XH and SYL; Data Curation, XH and JFZ; Writing - Original Draft, XH; Writing - Review & Editing, JX, JFZ, XH, XXT, XDL, QS, SL and SYL; Funding Acquisition, SYL; Supervision, SYL.
Supplemental Material
Download Zip (3.5 MB)Disclosure statement
The authors declare no competing interests.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Griffiths-Jones S, Bateman A, Marshall M, et al. Rfam: an RNA family database. Nucleic Acids Res. 2003;31(1):439–441.
- Boccaletto P, Magnus M, Almeida C, et al. RNArchitecture: a database and a classification system of RNA families, with a focus on structural information. Nucleic Acids Res. 2018;46:D202–D5.
- Abraham M, Dror O, Nussinov R, et al. Analysis and classification of RNA tertiary structures. RNA. 2008;14(11):2274–2289.
- Tamura M, Hendrix DK, Klosterman PS, et al. SCOR: structural Classification of RNA, version 2.0. Nucleic Acids Res. 2004;32(90001):D182–4.
- Petrov AI, Zirbel CL, Leontis NB. Automated classification of RNA 3D motifs and the RNA 3D Motif Atlas. RNA. 2013;19(10):1327–1340.
- Sarver M, Zirbel CL, Stombaugh J, et al. FR3D: finding local and composite recurrent structural motifs in RNA 3D structures. J Math Biol. 2008;56(1–2):215–252.
- Ge P, Islam S, Zhong C, et al. De novo discovery of structural motifs in RNA 3D structures through clustering. Nucleic Acids Res. 2018;46(9):4783–4793.
- Dror O, Nussinov R, Wolfson H. ARTS: alignment of RNA tertiary structures. Bioinformatics. 2005;21(Suppl 2):ii47–53.
- Bottaro S, Lindorff-Larsen K. Mapping the universe of RNA tetraloop folds. Biophys J. 2017;113(2):257–267.
- Appasamy SD, Hamdani HY, Ramlan EI, et al. InterRNA: a database of base interactions in RNA structures. Nucleic Acids Res. 2016;44(D1):D266–71.
- Chojnowski G, Walen T, Bujnicki JM, et al. database of RNA 3D motifs and their interactions. Nucleic Acids Res. 2014;42(D1):D123–31.
- Leontis NB, Zirbel CL. Nonredundant 3D structure datasets for RNA knowledge extraction and benchmarking. Nucleic Acids and Mol Bio. 2012;27:281–298.
- Phan AT, Kuryavyi V, Darnell JC, et al. Structure-function studies of FMRP RGG peptide recognition of an RNA duplex-quadruplex junction. Nat Struct Mol Biol. 2011;18(7):796–804.
- Zheng J, Xie J, Hong X, et al. RMalign: an RNA structural alignment tool based on a novel scoring function RMscore. BMC Genomics. 2019;20(1):276.
- Zheng J, Hong X, Xie J, et al. P3DOCK: a protein-RNA docking webserver based on template-based and template-free docking. Bioinformatics. 2020;36(1):96–103.
- Berman HM, Westbrook J, Feng Z, et al. The protein data bank. Nucleic Acids Res. 2000;28(1):235–242.
- Dias R, Kolaczkowski B. Improving the accuracy of high-throughput protein-protein affinity prediction may require better training data. BMC Bioinformatics. 2017;18(S5):102.
- Gong P, Kortus MG, Nix JC, et al. Structures of coxsackievirus, rhinovirus, and poliovirus polymerase elongation complexes solved by engineering RNA mediated crystal contacts. PLoS One. 2013;8(5):e60272.
- Bachelin M, Hessler G, Kurz G, et al. Structure of a stereoregular phosphorothioate DNA/RNA duplex. Nat Struct Biol. 1998;5(4):271–276.
- Fedoroff O, Salazar M, Reid BR. Structure of a DNA:RNA hybrid duplex. Why RNase H does not cleave pure RNA. J Mol Biol. 1993;233(3):509–523.
- Salter J, Krucinska J, Alam S, et al. Water in the active site of an All-RNA hairpin ribozyme and effects of Gua8 base variants on the geometry of phosphoryl transfer. Biochemistry. 2006;45(3):686–700.
- Torelli AT, Krucinska J, Wedekind JE. A comparison of vanadate to a 2ʹ-5ʹ linkage at the active site of a small ribozyme suggests a role for water in transition-state stabilization. RNA. 2007;13(7):1052–1070.
- Huang H, Suslov NB, Li NS, et al. A G-quadruplex-containing RNA activates fluorescence in a GFP-like fluorophore. Nat Chem Biol. 2014;10(8):686–691.
- Yang D. G-Quadruplex DNA and RNA. Methods Mol Biol. 2019;2035:1–24.
- Arora A, Dutkiewicz M, Scaria V, et al. Inhibition of translation in living eukaryotic cells by an RNA G-quadruplex motif. RNA. 2008;14(7):1290–1296.
- Bonnal S, Schaeffer C, Creancier L, et al. A single internal ribosome entry site containing a G quartet RNA structure drives fibroblast growth factor 2 gene expression at four alternative translation initiation codons. J Biol Chem. 2003;278(41):39330–39336.
- Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48(3):443–453.
- Capriotti E, Marti-Renom MA. Quantifying the relationship between sequence and three-dimensional structure conservation in RNA. BMC Bioinformatics. 2010;11(1):322.
- Kundrotas PJ, Zhu Z, Janin J, et al. Templates are available to model nearly all complexes of structurally characterized proteins. Proc Natl Acad Sci U S A. 2012;109(24):9438–9441.
- Zheng J, Kundrotas PJ, Vakser IA, et al. Template-based modeling of Protein-RNA interactions. PLoS Comput Biol. 2016;12(9):e1005120.
- Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5(4):823–826.
- Hartwick EW, Costantino DA, MacFadden A, et al. Ribosome-induced RNA conformational changes in a viral 3ʹ-UTR sense and regulate translation levels. Nat Commun. 2018;9(1):5074.
- Kim SH, Suddath FL, Quigley GJ, et al. Three-dimensional tertiary structure of yeast phenylalanine transfer RNA. Science. 1974;185(4149):435–440.
- Sharp SJ, Schaack J, Cooley L, et al. Structure and transcription of eukaryotic tRNA genes. CRC Crit Rev Biochem. 1985;19(2):107–144.
- Yamashita S, Takeshita D, Tomita K. Translocation and rotation of tRNA during template-independent RNA polymerization by tRNA nucleotidyltransferase. Structure. 2014;22(2):315–325.
- Barraud P, Schmitt E, Mechulam Y, et al. A unique conformation of the anticodon stem-loop is associated with the capacity of tRNAfMet to initiate protein synthesis. Nucleic Acids Res. 2008;36(15):4894–4901.
- Zeng F, Chen Y, Remis J, et al. Structural basis of co-translational quality control by ArfA and RF2 bound to ribosome. Nature. 2017;541(7638):554–557.
- Pavlova N, Kaloudas D, Penchovsky R. Riboswitch distribution, structure, and function in bacteria. Gene. 2019;708:38–48.
- Gupta A, Swati D. Riboswitches in Archaea. Comb Chem High Throughput Screen. 2019;22(2):135–149.
- Beyene SS, Ling T, Ristevski B, et al. A novel riboswitch classification based on imbalanced sequences achieved by machine learning. PLoS Comput Biol. 2020;16(7):e1007760.
- McCown PJ, Corbino KA, Stav S, et al. Riboswitch diversity and distribution. RNA. 2017;23(7):995–1011.
- Matyjasik MM, Hall SD, Batey RT. High affinity binding of N2-Modified guanine derivatives significantly disrupts the ligand binding pocket of the guanine riboswitch. Molecules. 2020;25(10):2295.
- Xie P, Chen H. Mechanism of ribosome translation through mRNA secondary structures. Int J Biol Sci. 2017;13(6):712–722.
- Sehnal D, Bittrich S, Deshpande M, et al. Mol* viewer: modern web app for 3D visualization and analysis of large biomolecular structures. Nucleic Acids Res. 2021;49(W1):W431–W7.