
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.ABSTRACT
In most bacteria, the three ribosomal RNAs (rRNAs) are encoded together in each of several near-identical operons. As soon as the nascent precursor rRNA emerges from RNA polymerase, ribosome assembly begins. This process entails ribosomal protein binding, rRNA folding, rRNA modification, and rRNA processing. In the model organisms Escherichia coli and Bacillus subtilis, rRNA processing results in similar mature rRNAs, despite substantial differences in the cohort of RNAses involved. A recent study of Flavobacterium johnsoniae, a member of the phylum Bacteroidota (formerly Bacteroidetes), revealed that helix H1 of 23S rRNA is absent from ribosomes, apparently a consequence of rRNA maturation. In this work, we mined RNA-seq data from 19 individual organisms and ocean metatranscriptomic samples to compare rRNA processing across diverse bacterial lineages. We found that mature ribosomes from multiple clades lack H1, and typically these ribosomes also lack an encoded H98. For all groups analysed, H1 is predicted to form in precursor rRNA as part of a longer leader-trailer helix. Hence, we infer that evolutionary loss of H98 sets the stage for H1 removal during 50S subunit maturation.
Introduction
Biogenesis of the ribosome entails folding of ribosomal RNA (rRNA), binding of ribosomal (r) proteins, modification of rRNA, and processing of rRNA (reviewed in [Citation1]). In Escherichia coli, all three rRNA molecules (16S, 23S, 5S) are encoded (along with one or two tRNAs) in each of seven operons. Subunit assembly begins co-transcriptionally, as primary r proteins interact with the nascent pre-rRNA. This facilitates rRNA folding and subsequent r protein-binding events, allowing ribonucleoparticles of increasing size and complexity to form, until complete subunits are ultimately made. In the context of subunit formation, the long pre-rRNA transcript, which includes structures important for assembly [Citation2–4], is processed by various RNases [Citation5]. Sequences flanking the 16S rRNA form a > 40 basepair (bp) leader-trailer (LT) helix [Citation6], while sequences flanking the 23S rRNA form a ~ 30 bp LT helix [Citation3]. RNase III, a double-stranded endonuclease, recognizes each LT helix and cleaves both strands, generating the pre-16S (17S) and pre-23S molecules. For 30S subunit maturation, subsequent cleavage events by RNases E and G remove the remaining leader strand of the 17S precursor, while YbeY and/or 3ʹ-to-5ʹ exonucleases remove the remaining trailer strand [Citation5]. For 50S subunit maturation, RNase T and an unknown nuclease further trim the 5ʹ and 3ʹ ends of pre-23 rRNA but leave the 8 bp helix H1 (formed by nucleotides 1–8 and 2895–2902) intact. RNase E excises pre-5S rRNA from the primary transcript, and RNase T and an unknown nuclease further trim the precursor molecule to generate the mature 3ʹ and 5ʹ ends, respectively [Citation5].
In most other bacteria, rRNA genes are similarly arranged in operons. Sequences flanking the 16S and 23S regions typically show complementarity [Citation7], consistent with LT helices akin to those of E. coli, and RNase III is present in nearly all bacteria [Citation8]. While these observations suggest common aspects of rRNA processing across bacteria, there are also clear indications of variation. For example, intergenic regions of rRNA operons exhibit a high degree of sequence diversity [Citation9–11]. Moreover, studies of Bacillus subtilis have revealed a unique cast of RNases involved in rRNA maturation [Citation5]. Following initial cleavage of pre-rRNA by RNase III, the double-stranded endonucleases Mini-III and RNase M5 generate the mature ends of 23S and 5S rRNAs, respectively [Citation12,Citation13]. For 30S subunit maturation, the 5ʹ-to-3ʹ exonuclease RNase J1 removes the 5ʹ leader strand, while YqfG (homologous to E. coli YbeY) cuts off the remaining 3ʹ trailer. Notably, B. subtilis completely lacks RNase E, G, and T, underscoring mechanistic differences in rRNA processing between E. coli and B. subtilis [Citation5].
Recently, Jha et al. mapped the 5ʹ and 3ʹ ends of the rRNAs in mature ribosomes of Flavobacterium johnsoniae, a representative of the Bacteroidota (formerly Bacteroidetes) [Citation14]. They found that, in 5 of 6 cases, the actual termini differ from those predicted in the annotated genome. Most notably, nucleotides 1–8 and 2891–2902 of 23S rRNA, which include the two strands of helix H1, are missing in the mature ribosome. Covariation analysis indicates that H1 is a conserved feature of bacterial 23S rRNA [Citation15], and complementary strands corresponding to H1 are encoded in the F. johnsoniae genome. Hence, it was inferred that, in F. johnsoniae, H1 forms in the precursor rRNA and is later removed during subunit maturation [Citation14].
In this work, we mined RNA-seq data from 19 individual bacteria and SMART-seq data from the ocean microbiome to compare rRNA processing in diverse lineages. We found that helix H1 is absent from mature 50S subunits in more than half of the bacteria analysed, and loss of H1 generally coincides with the absence of helix H98 from 23S rRNA.
Results
High-throughput sequencing methods have been developed to globally identify the 5ʹ ends (dRNA-seq/TSS-seq), 3ʹ ends (Term-seq), or both ends (Rend-seq) of RNA molecules. Ribosomal RNA is highly abundant in growing bacteria, and the lion’s share of rRNA in the cell is fully processed, contained in ribosomes. To identify the ends of mature 23S, 16S, and 5S rRNA in various lineages, we analysed Rend-seq data from four organisms, dRNA-seq/TSS-seq data (-TEX/-RppH) from 16 organisms, and Term-seq data from 2 organisms (). For each organism, 5ʹ read end coverage was plotted with respect to the annotated 5ʹ ends of the rRNA genes, and 3ʹ read end coverage (when obtained) was plotted with respect to the annotated 3ʹ ends of the rRNA genes. In most plots, a single large peak was observed, corresponding to the mature end of the rRNA (, Figures S1-S23). Assignments of the mapped ends, determined here and reported previously [Citation14], are shown in . Throughout this report, we use the universal numbering of rRNA nucleotides, based on structural conservation, with E. coli as the reference model [Citation15].
Table 1. Bacteria analysed in this study
Figure 1. Use of Rend-seq data to map the mature ends of rRNA in E. coli. Coverage of 5ʹ read ends (A, C, E) or 3ʹ read ends (B, D, F) is plotted with respect to genome NC_000913.3 position. Position zero (dashed red line) marks the genome-annotated terminus of 23S (a-b), 16S (c-d), and 5S (e-f) rRNA
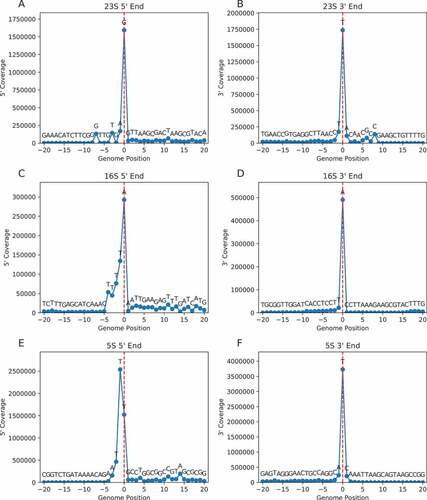
Figure 2. Mapping the ends of 23S rRNA in various bacteria. An alignment of RNA sequences near the ends of 23S rRNA is shown, comparing mapped 5ʹ and 3ʹ nucleotides (blue font) to genome-annotated predictions (red font). Cases of congruence between experimental data and genome annotation are indicated with purple font. Nucleotide numbers are shown above, regions of complementarity are indicated with underscores, and nucleotides forming helix H1 in the mature ribosome based on solved structures are highlighted in yellow. The organisms analysed represent GTDB classes Gammaproteobacteria (Gamma), Alphaproteobacteria (Alpha), Campylobacteria (Campyl), Chlorobia (Chloro), Bacteroidia (Bacter), Cyanobacteriia (Cyano), Actinomycetia (Actino), Bacilli, and Clostridia (Clostr). Species names are given in

Figure 3. Mapping the ends of 16S rRNA in various bacteria. An alignment of RNA sequences near the ends of 16S rRNA is shown, comparing mapped 5ʹ and 3ʹ nucleotides (blue font) to genome-annotated predictions (red font). Cases of congruence between experimental data and genome annotation are indicated with purple font. Nucleotides of the anti-Shine-Dalgarno (ASD) sequence are shaded in grey. See legend for a description of other annotations

Figure 4. Mapping the ends of 5S rRNA in various bacteria. An alignment of RNA sequences near the ends of 5S rRNA is shown, comparing mapped 5ʹ and 3ʹ nucleotides (blue font) to genome-annotated predictions (red font). Cases of congruence between experimental data and genome annotation are indicated with purple font. See legend for a complete description of the annotations
One known caveat to Rend-seq is that 5ʹ end mapping cannot always achieve nucleotide resolution [Citation16]. This stems from the ability of reverse transcriptase to incorporate an additional adenosine after reaching the end of the RNA template. If the resulting cDNA happens to match the genome sequence, such reads will be included and can shift the coverage peak by one nucleotide. To help mitigate this, we also analysed independent dRNA-seq datasets, when possible.
Mapping rRNA ends in individual organisms
23S rRNA
The experimentally deduced ends of 23S rRNA matched those predicted from the genome in surprisingly few cases (3 of 20 for 5ʹ end; 1 of 7 for 3ʹ end) (). Only for E. coli did the mapping data agree with the genome annotation for both ends. Genomes CP009977.1 of Vibrio natriegens and CP000487.1 of Campylobacter foetus are wildly misannotated, based on comparisons with other strains of the same species, which helps explain the large discrepancies seen in these cases. Notably, the mapping data indicate that helix H1 of 23S rRNA is missing from mature ribosomes of several lineages. A9 represents the 5ʹ nucleotide of Bacteroides thetaiotamicron 23S rRNA, just as observed in F. johnsoniae. Nucleotides 7 and 2890 represent the 5ʹ and 3ʹ ends of 23S rRNA in Zymomonas mobilis and Caulobacter cresentus, indicating equivalent removal of both strands of H1. A6 represents the 5ʹ end of 23S rRNA in both Campylobacter foetus and Helicobacter pylori, consistent with severe truncation (if not removal) of H1. In all organisms analysed, complementary strands corresponding to H1 are encoded in the genome (, underscored regions). Yet, ribosomes of Alphaproteobacteria (Z. mobilis, C. cresentus) and Bacteroidia (B. thetaiotamicron, F. johnsoniae) lack H1, while ribosomes of Campylobacteria (C. foetus, H. pylori) lack or mostly lack H1. We infer that, in these organisms, H1 forms during 50S biogenesis and is subsequently removed or processed.
16S rRNA
The large number of discrepancies between the mapped and annotated ends of 23S rRNA prompted us to examine the other rRNAs as well. The mapped 5ʹ end of 16S rRNA matched the genome annotation in 6 of 19 cases, while the mapped 3ʹ end matched the genome annotation in 2 of 7 cases (). The 5ʹ termini of 16S rRNA from C. fritschii and F. thermalis could not be mapped, because the corresponding genomes (obtained via shotgun sequencing) are incomplete and lack data just upstream of the 16S genes. For most bacteria, one to six additional 5ʹ nucleotides are present, as compared to the reference E. coli molecule. On the 3ʹ end of 16S rRNA, 2–4 nucleotides beyond 1542 are often seen, with the terminal sequence UUUCU (nt 1540–1544) being most common. These additional 3ʹ nucleotides (1543–1544) can potentially pair with mRNA as part of an extended SD-ASD helix [Citation14]; hence, even small differences in 3ʹ tail length may affect ribosome function.
5S rRNA
When the 5ʹ read ends of the E. coli Rend-seq data were mapped back to the genome, the peak of coverage was seen at position −1 ()). Fairly high coverage was also seen at position 0, corresponding to U1 of 5S rRNA. These data might be explained by some natural length heterogeneity of this molecule. However, analogous data from a related organism, V. natriegens, showed a sharp peak corresponding to U1 (Figure S2, ), and independent dRNA-seq data from E. coli mapped U1 as the 5ʹ nucleotide (Figure S1). Hence, we suspect that the Rend-seq peak at −1 is most likely due to the RT-dependent artefact discussed above. The 3ʹ end of E. coli 5S rRNA, as determined by Rend-seq, precisely matched the genome annotation, as expected ()).
For other bacteria analysed, the mapped 5ʹ end of 5S rRNA matched the genome annotation in 5 of 19 cases, while the mapped 3ʹ end matched the genome annotation in 2 of 6 cases (). In the case of Bordetella pertussis, there were two peaks of read coverage – one corresponding to C1 and another larger peak corresponding to A10 (Figure S3). This raises the possibility that A10, which lies near a helical junction in 5S rRNA, is vulnerable to RNase cleavage in this organism.
Loss of H1 of 23S rRNA is largely concentrated in specific groups of the taxonomic tree
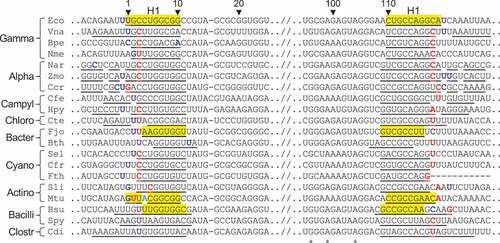
The apparent removal of H1 from ribosomes of several lineages prompted us to investigate the presence and absence of H1 in 23S rRNA more broadly across Bacteria. To this end, we exploited the fact that RNA sequenced using the SMART-seq approach is captured all the way to the 5ʹ end. In addition, the strand transfer of the polymerase at the 5ʹ end upon recognition of the Template Switching Oligo (TSO) during library preparation results in the true 5ʹ ends of RNAs being marked in such SMART-seq data by a non-encoded GGG that is complementary to the 5ʹ-most locked cytosine bases of the TSO. We thus identified all reads from the ribosomal section of a massive ocean metatranscriptomics data set starting with a GGG, and aligned them against a database of 2940 representative bacterial 23S rRNA sequences. The position of each alignment provided information on the location of the 5ʹ end of the RNA while the full alignment identified the species of origin among our reference 23S rRNA sequences. Stringent quality filtering resulted in 498 species with assigned 5ʹ ends. A histogram of the locations of these 5ʹ ends relative to the true 5ʹ end of E. coli shows a clearly bimodal distribution ()), one peak of which (around zero offset relative to E. coli) corresponds to species in which H1 is present, and the other peak of which (around an offset of 6 relative to E. coli) corresponds to species in which H1 is absent, with only 36 species among the 498 featuring offsets that are difficult to interpret as either a proper H1 or a proper 23S rRNA without H1 and thus classified as outliers. We conclude that even though this automatic approach of assigning 5ʹ ends to 23S rRNA en masse from the byproducts of a metatranscriptomics study is likely more noisy than manual inspection of individual data sets specifically constructed to reveal 5ʹ ends, it is able to provide reliable information on the presence and absence of H1 for hundreds of bacterial species.
Figure 5. Bacterial ribosomes contain or lack 23S rRNA helices H1 and H98. (a) Shown is a histogram of the offsets of 23S rRNA 5ʹ ends relative to the E. coli 23S rRNA 5ʹ end. The bimodal nature of the histogram allows classification of species into those containing H1 (offset between – 4 and 4, brown range), those lacking H1 (offset between 5 and 12, grey range), and a small number, for which H1 status remains unknown. (b) Shown is a histogram of the distances between two conserved sequence motifs surrounding H98 in 23S rRNA. The bimodal nature of the histogram allows classification of species into those containing H98 (distance between 45 and 55, brown range), those lacking H98 (distance between 30 and 40, grey range), and a small number, for which H98 status remains unknown
When projecting the presence and absence of H1 onto the GTDB taxonomic tree (Figure S24), we find that the vast majority (278 out of 284) of all species within Gammaproteobacteria, for which we can make a determination about H1, contain H1. Interestingly, the Gammaproteobacteria include two species, Actinobacillus equuli and Actinobacillus suis, within the same genus (red frame in Figure S24) with differing behaviour concerning H1 (each of which is strongly supported by the alignment data, see Figure S25). On the other hand, none of the 39 species within Bacteroidia or 109 species within Alphaproteobacteria contain H1. Members of Saccharimonadia and Fusobacteriia also lack H1, whereas the one member of Clostroidia (C. difficile) contains H1. Because these latter groups contain few representatives, little can be inferred about the consistency of H1 retention in these clades. Only within the Actinomycetia is there a lot of variability, with 11 out of 18 species that do not contain H1 and 7 that do. Notably, five organisms analysed by the automatic approach were also analysed manually (B. pertussis, E. coli, F. johnsoniae, N. meningitidis, and Z. mobilis), and H1 calls were consistent in all cases. We conclude that presence and absence of H1 largely follows the groups of the bacterial tree but that there are a few interesting outliers deserving future investigation. We also note that the pattern of presence and absence of H1 indicates that removal of H1 may have independently arisen multiple times in evolution.
Loss of H1 of 23S rRNA correlates with the absence of H98
One difference between the E. coli and F. johnsoniae 23S rRNA is that the latter naturally lacks H98, a poorly conserved helix (). In the E. coli ribosome, H98 lies across nucleotides 10–12 and 2889–2894, and A9 and G2890 correspond to the terminal nucleotides of 23S rRNA in the F. johnsoniae ribosome. This raises the possibility that H98 might prevent H1 removal by protecting these rRNA regions from RNase cleavage. To investigate this idea, we scored for the presence of H98 in all the organisms analysed. For the manually investigated organisms, we found that, with few exceptions, loss of H1 occurs in those organisms which also lack H98 (). The Bacteroidota lack H98, and ribosomes of F. johnsoniae and B. thetaiotamicron lack H1. The Campylobacterota lack H98, and ribosomes of H. pylori and C. foetus lack (or exhibit a severely truncated) H1. The Alphaproteobacteria lack H98, and ribosomes of Z. mobilis and C. cresentus lack H1, as do ribosomes from R. palustris [Citation17]. One apparent exception to this trend entails N. aromaticivorans, another Alphaproteobacteria, whose ribosomes lack H98 and contain H1. Ribosomes of C. tepidum, a member of the Chlorobia, similarly retain H1 despite the absence of H98. For other groups of bacteria, including the Firmicutes, Actinobacteriota, Cyanobacteria, and Gammaproteobacteria, 23S rRNA contains H98. In 12 of 12 cases analysed, ribosomes of these organisms retain H1. Overall, the co-occurrence of these elements, deemed significant by Fisher’s exact test (p = 0.0003), suggests that retention of H1 in the mature ribosome generally depends on the presence of H98. This pattern remains true for the automatically annotated organisms, with 442/457 containing both or lacking both helices (Figure S24). In 11 species, H98 is present without H1 (all in Actinomycetia or Gammaproteobacteria); and in 4 species, H1 is present without H98 (all in Gammaproteobacteria). Formally, this corresponds to a Fisher’s exact p-value of 1.3 · 10−103 although this exaggerates the significance given that many species are closely related and thus cannot be treated as independent. Nevertheless, it is clear that the removal of H1 and the absence of H98 go hand in hand.
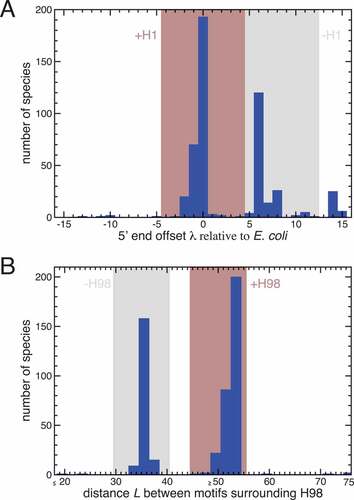
Figure 6. Comparison of 23S rRNA structure in ribosomes containing or lacking H1. (a) Shown is a superimposition of E. coli (cyan) and F. johnsoniae (tan) 23S rRNA in the vicinity of the 5ʹ and 3ʹ termini (red nucleotides, as indicated). Helices H2, H25, H94, and H99 (as indicated) are common features, whereas H1 and H98 are missing in the F. johnsoniae ribosome. Ribosomal proteins have been computationally omitted for clarity. This image was generated in PyMOL using PDB files 2QAM and 7JIL. (b) Comparison of secondary structure elements (E. coli, cyan, left; F. johnsoniae, tan, right) in the relevant portion of domain VI of 23S rRNA, with helices and terminal nucleotides indicated
Discussion
In this study, we mined RNA-seq datasets to identify the ends of rRNA in many bacteria. We found that helix H1 of 23S rRNA is absent from mature ribosomes in about half the organisms analysed. Removal of H1 correlates strongly with the absence of H98, suggesting that retention of H1 generally depends on the presence of H98. Helix H98 is positioned across nucleotides 10–12 and 2890–2896 and hence may protect these strands from cellular nucleases, enabling retention of H1. Loss of H98 by chromosomal deletion may have little consequence on ribosome function [Citation18,Citation19] but allow for H1 excision during subunit maturation. This could explain the multiple independent losses of H98 during evolution and the parallel removals of H1.
Structures of ribosomes from various bacteria, including representatives of Firmicutes (B. subtilis, S. aureus, E. faecalis), Deinococcota (T. thermophilus, D. radiodurans), Actinobacteriota (M. smegmantis), and Gammaproteobacteria (E. coli, P. aeroginosa, A. baumannii), have now been solved [Citation20–28]. All of these ribosomes contain H1 and H98, which adopt similar conformations regardless of the source organism. To our knowledge, the only solved structure of a bacterial ribosome that lacks H1 and H98 is that of F. johnsoniae [Citation14], a representative of Bacteroidota. In this structure, the 3ʹ terminal nucleotide (G2890) is tucked behind nucleotides 2790–2791, components of a small loop that replaces H98. As for the 5ʹ end of 23S rRNA, the path of nucleotides 9–12 diverges (by nearly 180°) from that seen in H98/H1-containing ribosomes. This enables contacts between C9 (the 5ʹ terminal nucleotide) and uL22, an interaction which may protect the rRNA from exonucleases. Whether other ribosomes that lack H1/H98 exhibit similar or analogous interactions remains to be determined.
The absence of H1 and H98 from mature ribosomes of numerous bacteria indicates that these elements are unnecessary for ribosome function. Previous studies have shown that base substitutions in either strand of H1 results in severely diminished levels of active 50S subunits in the cell [Citation3]. Compensatory mutations in the opposite strand restore active subunits to normal levels, implying the importance of H1 secondary structure in 50S biogenesis and/or stability. Given that the sequences flanking 23S rRNA exhibit complementarity in diverse bacteria [Citation7] (), we infer that H1 contributes to 50S biogenesis in the context of the larger LT structure. In most lineages that contain H98, processing of the LT structure leaves a remnant, H1, behind. On the other hand, in most lineages that lack H98, the entire LT structure, including H1, is removed.
Finally, our work exemplifies precise mapping of RNA 5ʹ ends from metatranscriptomic datasets. By taking advantage of the GGG tag generated in SMART-seq library construction, we are able to determine the true 5ʹ ends of the 23S rRNA for hundreds of species in parallel. While the ability of the strand-switching polymerase in the SMART-seq protocol to identify 5ʹ ends is well recognized in general [Citation29,Citation30], the approach taken here is to our knowledge unique in that it does not involve a specialized library preparation protocol but can be applied to standard SMART-seq data. Its main caveats are that, since 5ʹ ends are not strongly enriched, it requires a high abundance RNA (which 23S clearly is naturally) and an approximate knowledge of the correct location of the 5ʹ end (in our case provided by the highly conserved 24–60 nt region of 23S rRNA). These conditions are not unique to 23S rRNA but hold for other ribosomal and high abundance messenger and noncoding RNAs as well, so the method should be applicable more broadly. Another caveat that we observe is that the method sometimes detects spurious 5ʹ ends associated with genomically encoded runs of Gs, presumably due to internal priming by the template switching oligo. For example, a run of three Gs close to the true 5ʹ end of the cyanobacterial 23S rRNA causes all Cyanobacteria to fail our quality control criteria, explaining their absence from our results in spite of their abundance in ocean communities. A detailed exploration of the capabilities of the method will be left for a future publication.
MATERIALS and METHODS
Mapping of rRNA ends
The mature ends of each rRNA molecule were identified from publicly available data. Because processed 5ʹ ends were sought, control dRNA-seq/TSS-seq datasets (e.g. minus terminator 5ʹ-phosphate-dependent exonuclease, -TEX; or minus RNA 5ʹ pyrophosphohydrolase, -RppH) were analysed. For dRNA-seq, TSS-seq, and Term-seq data, raw reads were downloaded from SRA and converted to fastq using the SRA toolkit [Citation31]. 5ʹ adapters were identified by minion [Citation32] and trimmed using skewer [Citation33] where necessary. Reads were aligned using Bowtie2 [Citation34], converted to bam format using samtools [Citation35], and read coverage was obtained using the bedtools genomecov command [Citation36]. For rend-seq data, WIG format coverage files were used when available, and raw data analysed as above otherwise.
5ʹ and/or 3ʹ read coverage were each plotted with respect to the corresponding genome-annotated gene ends using in-house python scripts. For each 5′ and/or 3′ coverage plot, we observed a large peak corresponding to the mature 5′ and/or 3′ ends, due to the end enrichment protocols used.
Sequence alignments and helix calling
were generated from multiple alignments of the entire RNA made in clustalw2 [Citation37], with gaps removed manually. Helix H1 was called as missing if three or fewer predicted base pairs remained, and H98 was determined to be missing by inspection of the multiple alignment. The genomes of Vibrio natriegens (CP009977.1) and Campylobacter foetus (CP000487.1) were compared to strains of the same species (NZ_CP016351.1 and CP059443.1 respectively), which confirmed misannotations of rRNA in the former cases.
Computational approach to bulk mapping of 23S 5ʹ ends
Query sequence acquisition
Metatranscriptomic reads were obtained for 551 samples of a study published by the TARA Ocean’s consortium [Citation38]. These libraries had been prepared with the SMARTer Stranded RNA-Seq Kit (Clontech) following Ribo-Zero depletion and only the reads designated by SortMeRNA as ribosomal in origin were provided by the authors of the TARA Ocean’s study.
Query sequence selection
The forward ends of the paired end samples were selected. Since the last 3 bps of the Template Switch Oligo, GGG, were present at the beginning of reads starting at the 5ʹ end of rRNAs, reads were down selected to only those beginning with GGG and the GGG was stripped from these reads with CutAdapt v3.4 (-g ^GGG -discard-untrimmed) [Citation39].
An alignment of the 23S rRNA sequences from the species of was used to define a conserved motif (GNGGATGCCTTGGCNNNNNNAGNCGANGAAGGACGTG) at positions 24–60. This motif was used to only retain reads containing this motif (thus predicted to originate from the vicinity of the 5ʹ end of 23S rRNA) using CutAdapt v3.4 (-a GNGGATGCCTTGGCNNNNNNAGNCGANGAAGGACGTG -e 0.25 -action=none -discard-untrimmed -O 37). Following both CutAdapt steps, 103,968,502 reads remained from all samples.
23S rRNA sequence acquisition
A list of all organisms with NCBI type data and designated as species representative was obtained from the Genome Taxonomy Database (GTDB) [Citation40]. Their annotated genomes were downloaded and the 23S rRNA sequence was extracted based on the annotation. Finally, 23S rRNA sequences were added for the five species (S. pyogenes, C. tepidum, C. difficile, B. thetaiotaomicron, and S. lividans) of , which were not already included. 23S rRNA subject sequences, with their accompanying NCBI accession and genome positions, were collected and are available in Table S1.
Normalization of 23S rRNA annotations
The annotated 5ʹ end of 23S rRNA sequences varied significantly between different NCBI assembled genomes, and even genomes sharing the same species presented significantly different (and often errant) annotated 23S 5ʹ ends. Thus, a 5ʹ end normalization offset was generated for each collected 23S rRNA subject sequence by comparing the number of nucleotides upstream of the above-mentioned conserved motif (GNGGATGCCTTGGCNNNNNNAGNCGANGAAGGACGTG). E. coli was used as a template to normalize ∆ for all 23S rRNA subject sequences, since the location of H1 on E. coli is well known. For each collected 23S rRNA subject sequence, i, the offset ∆i was calculated as:
Query mapping and normalized 23S 5ʹ end shift
Metatranscriptomic reads were aligned to the 23S rRNA subject sequences using LAMBDA2 [Citation41]. Alignments were considered valid if their percent identity was above 95%, their query alignment start and/or subject alignment start was 1 (i.e. the alignment occurs at the beginning of the query and/or subject sequence), and the query alignment end equalled the query sequence length (i.e. the 3ʹ end of the query is completely aligned to the subject). The predicted normalized 5ʹ end shift for the read, , was calculated as
. 1,398,885 reads had at least one valid alignment.
Species normalized 5ʹ shift prediction
All values were grouped based on the species of the subject sequence. For each unique species, a histogram of the distribution of
values was generated.
values outside the range from −15 to 15 were discarded as implausible indicators of true 5ʹ ends. To determine if (i) there was enough data, and (ii) the data had high agreement, two quality control criteria were imposed: (1) at least 20 of the remaining reads must map to the species, and (2) one 3 nucleotide sliding window from x-axis values −15 to 15 must contain at least 70% of the reads mapping to the species. Histograms passing both screening protocols were subsequently evaluated for their mode
value, which became the predicted normalized 5ʹ end shift value for the species, which is defined as
.
Identification of helix H1
The predicted values from all species passing both screening protocols were collected. Based on a histogram of these values ()) species whose value was between −4 and 4 were deemed to contain H1, species whose
value was between 5 and 12 were deemed to lack H1. No call was made for species with
values outside the −4 to 12 range. Values and H1 statuses of all species passing the quality control criteria are given in Table S1.
Identification of helix H98
H98 is present near the 3ʹ end of 23S rRNA. From the multiple sequence alignment of 23S rRNA of the 20 species of , two conserved motifs around the H98 position, one upstream (AGANNANNNNNTTGATAGGNNNNNNNTG), and one downstream(GATAANNGCTGAAAGCATCTAAGNNNGAANC) were identified. CutAdapt v3.4 (-a ‘AGANNANNNNNTTGATAGGNNNNNNNTG;min_overlap=28ʹ -g ‘GATAANNGCTGAAAGCATCTAAGNNNGAANC;min_overlap=31ʹ -discard-untrimmed -n 2 -e 0.25) was used to extract the portion of the subject sequence between both motifs, the length of which was defined as .
A histogram of all values revealed two groups of species: those containing H98 (the right group, i.e. those sequences with more nucleotides between the motifs, implying the presence of H98) and those without H98 (the left group) ()). Based on this histogram, species whose
value was between 45 and 55 were deemed to contain H98, species whose
value was between 30 and 40 were deemed to lack H98, and the H98 status of all other species remained unknown. The number of nucleotides between the motifs and the H98 status of all taxonomies passing the quality control criteria are given in Table S1.
Visualization of taxonomic tree
The GTDB taxonomic classification for each species passing the quality control criteria and having known H1 and H98 status was retrieved and converted to Newick format. The H1 and H98 status information for each of these taxonomies was converted into an iTOL annotation file using a custom script and the tree and its annotation visualized in iTOL [Citation42].
Supplemental Material
Download Zip (4.6 MB)Acknowledgments
We thank M. Sullivan, C. Da Silva, and TARA Oceans for providing us metatranscriptomic data; P. Yan and L. Walker for discussions about using the TSO as a marker of 5ʹ ends; and B. Warner for comments on the manuscript
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed here
Additional information
Funding
Related Research Data
References
- Shajani Z, Sykes MT, Williamson JR. Assembly of bacterial ribosomes. Annu Rev Biochem. 2011;80(1):501–526.
- Theissen G, Thelen L, Wagner R. Some base substitutions in the leader of an Escherichia coli ribosomal RNA operon affect the structure and function of ribosomes. Evidence for a transient scaffold function of the rRNA leader. J Mol Biol. 1993;233(2):203–218.
- Liiv A, Remme J. Base-pairing of 23 S rRNA ends is essential for ribosomal large subunit assembly. J Mol Biol. 1998;276(3):537–545.
- Mangiarotti G, Turco E, Perlo C, et al. Role of precursor 16S RNA in assembly of E. coli 30S ribosomes. Nature. 1975;253:569–571.
- Bechhofer DH, Deutscher MP. Bacterial ribonucleases and their roles in RNA metabolism. Crit Rev Biochem Mol Biol. 2019;54(3):242–300.
- Young RA, Steitz JA. Complementary sequences 1700 nucleotides apart form a ribonuclease III cleavage site in Escherichia coli ribosomal precursor RNA. Proc Natl Acad Sci U S A. 1978;75(8):3593–3597.
- Saito R, Ozawa Y, Kuzuno N, et al. Computer analysis of potential stem structures of rRNA operons in various procaryote genomes. Gene. 2000;259(1–2):217–222.
- Condon C. The phylogenetic distribution of bacterial ribonucleases. Nucleic Acids Res. 2002;30(24):5339–5346.
- Perez Luz S, Rodriguez-Valera F, Lan R, et al. Variation of the ribosomal operon 16S-23S gene spacer region in representatives of salmonella enterica subspecies. J Bacteriol. 1998;180(8):2144–2151.
- Gurtler V, Stanisich VA. New approaches to typing and identification of bacteria using the 16S-23S rDNA spacer region. Microbiology (Reading). 1996;142(Pt 1):3–16.
- Anton AI, Martinez-Murcia AJ, Rodriguez-Valera F. Sequence diversity in the 16S-23S intergenic spacer region (ISR) of the rRNA operons in representatives of the Escherichia coli ECOR collection. J Mol Evol. 1998;47(1):62–72.
- Stahl DA, Pace B, Marsh T, et al. The ribonucleoprotein substrate for a ribosomal RNA-processing nuclease. J Biol Chem. 1984;259(18):11448–11453.
- Redko Y, Bechhofer DH, Condon C. Mini-III, an unusual member of the RNase III family of enzymes, catalyses 23S ribosomal RNA maturation in B. subtilis. Mol Microbiol. 2008;68(5):1096–1106.
- Jha V, Roy B, Jahagirdar D, et al. Structural basis of sequestration of the anti-shine-dalgarno sequence in the bacteroidetes ribosome. Nucleic Acids Res. 2021;49(1):547–567.
- Cannone JJ, Subramanian S, Schnare MN, et al. The comparative RNA web (CRW) site: an online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinf. 2002;3(1):2.
- Lalanne JB, Taggart JC, and Guo MS, et al. Evolutionary convergence of pathway-specific enzyme expression stoichiometry. Cell. 2018;173:749–761 e38.
- Zahn K, Inui M, Yukawa H. Characterization of a separate small domain derived from the 5ʹ end of 23S rRNA of an alpha-proteobacterium. Nucleic Acids Res. 1999;27(21):4241–4250.
- Spahn CM, Grassucci RA, Penczek P, et al. Direct three-dimensional localization and positive identification of RNA helices within the ribosome by means of genetic tagging and cryo-electron microscopy. Structure. 1999;7(12):1567–1573.
- Youngman EM, Green R. Affinity purification of in vivo-assembled ribosomes for in vitro biochemical analysis. Methods. 2005;36(3):305–312.
- Flygaard RK, Boegholm N, Yusupov M, et al. Cryo-EM structure of the hibernating Thermus thermophilus 100S ribosome reveals a protein-mediated dimerization mechanism. Nat Commun. 2018;9(1):4179.
- Halfon Y, Jimenez-Fernandez A, La Rosa R, et al. Structure of Pseudomonas aeruginosa ribosomes from an aminoglycoside-resistant clinical isolate. Proc Natl Acad Sci U S A. 2019;116(44):22275–22281.
- Hentschel J, Burnside C, Mignot I, et al. The complete structure of the mycobacterium smegmatis 70S ribosome. Cell Rep. 2017;20(1):149–160.
- Kaminishi T, Schedlbauer A, Fabbretti A, et al. Crystallographic characterization of the ribosomal binding site and molecular mechanism of action of Hygromycin A. Nucleic Acids Res. 2015;43(20):10015–10025.
- Khusainov I, Vicens Q, Bochler A, et al. Structure of the 70S ribosome from human pathogen Staphylococcus aureus. Nucleic Acids Res. 2016;44(21):10491–10504.
- Morgan CE, Huang W, and Rudin SD, et al. Cryo-electron microscopy structure of the acinetobacter baumannii 70s ribosome and implications for new antibiotic development. mBio. 2020 11(1) ;e03117–19.
- Murphy EL, Singh KV, Avila B, et al. Cryo-electron microscopy structure of the 70S ribosome from Enterococcus faecalis. Sci Rep. 2020;10(1):16301.
- Noeske J, Wasserman MR, Terry DS, et al. High-resolution structure of the Escherichia coli ribosome. Nat Struct Mol Biol. 2015;22(4):336–341.
- Sohmen D, Chiba S, Shimokawa-Chiba N, et al. Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat Commun. 2015;6(1):6941.
- Cole C, Byrne A, Beaudin AE, et al. Tn5Prime, a Tn5 based 5′ capture method for single cell RNA-seq. Nucleic Acids Res. 2018;46(10):e62.
- Machida RJ, Lin YY, Oudejans C. Four methods of preparing mRNA 5ʹ end libraries using the Illumina sequencing platform. PLoS One. 2014;9:e101812.
- Leinonen R, Sugawara H, Shumway M. International nucleotide sequence database C. The sequence read archive. Nucleic Acids Res. 2011;39:D19–21.
- Davis MP, van Dongen S, Abreu-Goodger C, et al. Kraken: a set of tools for quality control and analysis of high-throughput sequence data. Methods. 2013;63(1):41–49.
- Jiang H, Lei R, Ding SW, et al. Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinformatics. 2014;15(1):182.
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9(4):357–359.
- Li H, Handsaker B, Wysoker A, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079.
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26(6):841–842.
- Larkin MA, Blackshields G, Brown NP, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948.
- Salazar G, Paoli L, Alberti A, et al. Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell. 2019;179(5):1068–83 e21.
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 2011; 17(1): 10–12 .
- Parks DH, Chuvochina M, Chaumeil PA, et al. A complete domain-to-species taxonomy for bacteria and archaea. Nat Biotechnol. 2020;38(9):1079–1086.
- Hauswedell H, Singer J, Reinert K. Lambda: the local aligner for massive biological data. Bioinformatics. 2014;30(17):i349–55.
- Letunic I, Bork P. Interactive Tree of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021;49:W293–W6.
- Thomason MK, Bischler T, Eisenbart SK, et al. Global transcriptional start site mapping using differential RNA sequencing reveals novel antisense RNAs in Escherichia coli. J Bacteriol. 2015;197(1):18–28.
- Amman F, D’Halluin A, Antoine R, et al. Primary transcriptome analysis reveals importance of IS elements for the shaping of the transcriptional landscape of Bordetella pertussis. RNA Biol. 2018;15:967–975.
- Heidrich N, Bauriedl S, Barquist L, et al. The primary transcriptome of Neisseria meningitidis and its interaction with the RNA chaperone Hfq. Nucleic Acids Res. 2017;45:6147–6167.
- Myers KS, Vera JM, and Lemmer KC, et al. Genome-wide identification of transcription start sites in two alphaproteobacteria, rhodobacter sphaeroides 2.4.1 and novosphingobium aromaticivorans DSM 12444. Microbiol Resour Announc. 2020;9(36):e00880–20.
- Vera JM, Ghosh IN, and Zhang Y, et al. Genome-scale transcription-translation mapping reveals features of zymomonas mobilis transcription units and promoters. mSystems. 2020;5(4) ;e00250–20.
- Frohlich KS, Forstner KU, Gitai Z. Post-transcriptional gene regulation by an Hfq-independent small RNA in Caulobacter crescentus. Nucleic Acids Res. 2018;46:10969–10982.
- Kienesberger S, Sprenger H, Wolfgruber S, et al. Comparative genome analysis of Campylobacter fetus subspecies revealed horizontally acquired genetic elements important for virulence and niche specificity. PLoS One. 2014;9(1):e85491.
- Bischler T, Tan HS, Nieselt K, et al. Differential RNA-seq (dRNA-seq) for annotation of transcriptional start sites and small RNAs in Helicobacter pylori. Methods. 2015;86:89–101.
- Hilzinger JM, Raman V, and Shuman KE, et al. Differential RNA sequencing implicates sulfide as the master regulator of S(0) metabolism in chlorobaculum tepidum and other green sulfur bacteria. Appl Environ Microbiol. 2018;84(3) ;e01966–17.
- Ryan D, Jenniches L, Reichardt S, et al. A high-resolution transcriptome map identifies small RNA regulation of metabolism in the gut microbe bacteroides thetaiotaomicron. Nat Commun. 2020;11(1):3557.
- Tan X, Hou S, Song K, et al. The primary transcriptome of the fast-growing cyanobacterium synechococcus elongatus UTEX 2973. Biotechnol Biofuels. 2018;11(1):218.
- Koch R, Kupczok A, Stucken K, et al. Plasticity first: molecular signatures of a complex morphological trait in filamentous cyanobacteria. BMC Evol Biol. 2017;17(1):209.
- Lee Y, Lee N, Jeong Y, et al. The transcription unit architecture of streptomyces lividans TK24. Front Microbiol. 2019;10:2074.
- Shell SS, Wang J, Lapierre P, et al. Leaderless transcripts and small proteins are common features of the mycobacterial translational landscape. PLoS Genet. 2015;11(11):e1005641.
- Lecrivain AL, Le Rhun A, Renault TT, et al. In vivo 3′-to-5′ exoribonuclease targetomes of Streptococcus pyogenes. Proc Natl Acad Sci U S A. 2018;115(46):11814–11819.
- Soutourina O, Dubois T, Monot M, et al. Genome-wide transcription start site mapping and promoter assignments to a sigma factor in the human enteropathogen clostridioides difficile. Front Microbiol. 2020;11:1939.