
Abstract
The Toll-like receptor (TLR) adaptor protein MyD88 is integral to airway inflammatory response to microbial-enriched organic dust extract (ODE) exposures. ODE-induced airway neutrophil influx and release of pro-inflammatory cytokines was essentially abrogated in global MyD88-deficient mice, yet these mice demonstrate an increase in airway epithelial cell mucin expression. To further elucidate the role of MyD88-dependent responses specific to lung airway epithelial cells in response to ODE in vivo, the surfactant protein C protein (SPC) Cre+ embryologic expressing airway epithelial cells floxed for MyD88 to disrupt MyD88 signaling were utilized. The inducible club cell secretory protein (CCSP) Cre+, MyD88 floxed, were also developed. Using an established protocol, mice were intranasally instilled with ODE or saline once or daily up to 3 weeks. Mice with MyD88-deficient SPC+ lung epithelial cells exhibited decreased neutrophil influx following ODE exposure once and repetitively for 1 week without modulation of classic pro-inflammatory mediators including tumor necrosis factor (TNF)-α, interleukin (IL)-6, and neutrophil chemoattractants. This protective response was lost after 3 weeks of repetitive exposure. ODE-induced Muc5ac mucin expression at 1 week was also reduced in MyD88-deficient SPC+ cells. Acute ODE-induced IL-33 was reduced in MyD88-deficient SPC+ cells whereas serum IgE levels were increased at one week. In contrast, mice with inducible MyD88-deficient CCSP+ airway epithelial cells demonstrated no significant difference in experimental indices following ODE exposure. Collectively, these findings suggest that MyD88-dependent signaling targeted to all airway epithelial cells plays an important role in mediating neutrophil influx and mucin production in response to acute organic dust exposures.
Introduction
Environmental agriculture organic dust exposures are associated with workplace asthma and chronic bronchitis (Kelly and Poole Citation2019). The Toll-like receptor (TLR)/interleukin-1 receptor (TIR) adaptor protein MyD88-dependent signaling pathway is fundamental in the airway inflammatory response to complex agriculture-related organic dust extract (ODE) exposures (Bauer et al. Citation2013; Dickinson et al. Citation2019; Johnson et al. Citation2020). Global Myd88 knockout mice demonstrate a profound reduction in neutrophil influx and release of pro-inflammatory cytokines/chemokines following acute and repetitive ODE exposure as compared to wild-type mice (Bauer et al. Citation2013; Johnson et al. Citation2020). In contrast, Muc5ac expression following ODE stimulation was found to be increased in mice globally deficient in MyD88 (Dickinson et al. Citation2019). Further work broadly investigating the role of hematopoietic vs. lung structural/resident cells using MyD88 bone marrow chimera strategies highlighted the importance of lung resident cells in mediating ODE-induced airway inflammatory responses, neutrophil influx, and to some degree cytokine responsiveness (Poole et al. Citation2015b). The impact, however, of specifically targeting MyD88 signaling in lung airway epithelial cells in vivo in response to ODE exposure has yet to be elucidated. Such information could inform future therapeutic approaches aimed at reducing epithelial damage and lung remodeling to reduce organic dust and/or environmental toxin-induced lung disease.
Targeted deletion of alleles can be achieved using Cre/Lox technology, wherein LoxP sites are inserted flanking an exon of interest and Cre recombinase is expressed under the control of cell-specific promoter (Wang Citation2009). This approach circumvents potential confounding issues of conventional gene-targeting approaches, including the unknown impact that global gene deletion may have on numerous cell and tissue types outside the area of study, and the possibility of overexpression in global transgenic approaches. The major lung epithelial cell types include ciliated cells, secretory cells, serous cells, and basal cells in the airways, and Type I and Type II pneumocytes in the alveoli (Singh and Katyal Citation2000). Although expression of surfactant protein C protein (SPC) is a marker Type II pneumocytes in postnatal mice, when under control of the Sftpc promoter, Cre recombinase is expressed in all these cell types during embryogenesis, allowing deletion of alleles (i.e. alleles that are floxed) throughout the lung epithelium. In comparison, the club cell-specific protein (CCSP) Cre mouse targets secretory epithelial cells diffusely expressed throughout the airways (Rawlins et al. Citation2009). Thus, using genetically engineered mouse lines, MyD88 deficiency can be localized to airway epithelial cells such that studies could then be undertaken to inform the specific roles of airway epithelial cells throughout the lung compartments.
In the study presented here it was hypothesized that ODE-mediated airway inflammatory consequences were mediated by MyD88-dependent signaling in lung epithelial cells. To test this hypothesis, constitutive deletion of MyD88 in SPC expressing cells using Myd88fl/fl; SPCCre/+ mice was first utilized. These mice were challenged with ODE exposure once and/or daily for a period of up to 3 weeks. The findings generated highlighted how airway epithelial cell MyD88 was indispensable for acute ODE-mediated neutrophil recruitment, but not required for classical pro-inflammatory mediator release and that inflammatory mucin (Muc5ac) production was regulated by MyD88. Interestingly, these findings could not be ascribed to mid-airway club cells.
Materials and methods
Animals
To investigate the effect of the airway epithelial cell MyD88 signaling pathway, two airway epithelial cell-specific murine strains employing Cre-floxed (fl) strategies were obtained. Specifically, SPCCre± vs. SPCCre−/−, Myd88fl/fl mice were generated as previously described (Cleaver et al. Citation2014). Tamoxifen-inducible CCSPCre± vs. CCSPCre−/− mice were purchased from The Jackson Laboratory (Bar Harbor, ME), and then bred with the SPCCre± MyD88fl/fl mice to create CCSPCre± Myd88fl/fl mice. Genotyping for SPC and CCSP Cre− vs. Cre+ was performed to confirm genetic strains with details in the Supplementary Methods.
To induce CCSP-specific deletion of MyD88 in adult mice, CCSPCre± Myd88fl/fl mice were administered 200 μl tamoxifen in corn oil (at 20 mg/ml concentration; 4 mg/injection) intraperitoneally (IP) daily for 5 days (Mon–Fri). Control CCSPCre−/− Myd88fl/fl littermates were also administered tamoxifen daily for 5 days. All mice were then rested 4 days prior to intra-nasal treatments with ODE.
All mice were housed in pathogen-free facilities maintained at 20–24 °C with a 40% relative humidity and a 12-h light:dark cycle. All mice had ad libitum access to alfalfa-free chow (Harlan Laboratories, Indianapolis, Indiana) and filtered tap water, as recommended by The Jackson Laboratory. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Nebraska Medical Center and conducted according to the U.S. National Institutes of Health Guidelines for the Use of Rodents. For all these studies, both male and female mice (6-12-wk-of-age) were utilized in the experimental procedures.
Organic dust extract
Aqueous organic dust extract (ODE) was prepared from settled dust collected from horizontal surfaces 3 ft above the floor of moderately-sized swine confinement feeding operations (≈500–700 animals) located in Colfax County, NE (population density approximates 25 people/square mile). Extracts were batch-prepared and filter (0.22 μm)-sterilized, as previously described (Poole et al. Citation2009, Citation2017). Stock ODE was diluted to a concentration of 12.5% (v/v) in sterile phosphate-buffered saline (PBS, pH 7.4); this represents a concentration previously shown (Poole et al. Citation2009, Citation2017) to elicit optimal lung inflammation in mice. Pilot studies showed the 12.5% ODE contained ≈3–4 mg/ml total protein (measured by spectrophotometry; NanoDrop Technologies, Wilmington, DE) and endotoxin at levels ranging from 22.1 to 91.1 EU/ml (measured by limulus amebocyte lysate assay; Sigma, St. Louis, MO).
Murine ODE airway inflammatory exposure model
Under light sedation with isoflurane inhalation, mice were administered 50 μl of the 12.5% ODE or sterile PBS by intranasal inhalation as previously described (Poole et al. Citation2009, Citation2017). Exposures were either just once (acute exposure modeling) or daily for 1 and 3 weeks (repetitive exposure modeling). All mice were euthanized (anesthetizing with isoflurane followed by cervical dislocation) 5 h after the single or final exposure for use in determining the experimental endpoints outlined below.
Bronchoalveolar lavage fluid studies
Bronchoalveolar lavage fluid (BALF) was collected by instilling 1 ml PBS into the airways of each host three times using previously reported techniques (Poole et al. Citation2009); the first lavage was reserved for cytokine measures before being pooled for total lung cell collections. Cytokines implicated in agriculture respiratory disease, including interleukin (IL)-6, neutrophil chemoattractants CXCL1 and CXCL2, and tumor necrosis factor (TNF)-α (Kelly and Poole Citation2019) were measured in the cell-free supernatant of the first lavage by ELISA (R&D Systems, Minneapolis, MN). The kits had a minimal detectable dose (MDD) of 2.0 pg IL-6/ml, 1.5 pg CXCL1/ml, 7.2 pg CXCL2/ml, and 1.88 pg TNFα/ml. Total number of cells recovered from the three pooled lavages were enumerated using a TC20 automated cell counter (BioRad, Hercules, CA, USA) and differential cell counts performed on cytospin-prepared slides (Cytopro Cytocentrifuge; Wescor, Logan, UT, USA) that were stained with DiffQuick (Siemens, Neward, DE, USA).
Lung cell staining and flow cytometry
Following BALF removal, the chest cavity was opened, and the right ventricle was infused with 10 ml of sterile PBS with heparin to remove blood from the pulmonary vasculature. The harvested lungs (right lungs) were subjected to an automated dissociation procedure using a gentleMACS Dissociator instrument (Miltenyi Biotech, Auburn, CA, USA) (Poole et al. Citation2017) and total lung cells present were enumerated using a TC20 automated cell counter (BioRad). The liberated lung cells were then processed for multiparametric flow cytometry using previously reported protocols (Ghosn et al. Citation2008; Poole et al. Citation2017; Mikuls et al. Citation2021). Lung cells from each animal were incubated with a LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen, Carlsbad, CA, USA) and CD16/32 (Fc Block, Biolegend, San Diego, CA, USA) to minimize non-specific anti-body staining. Cells were stained with monoclonal antibody (mAb) against rat anti-mouse CD45 BV605 (clone: 30-F11; BD Biosciences), CD11b peridinin–chlorophyll–protein (PerCP) Cy5.5 (clone: M1/70; BD Biosciences), Ly6G AF700 (clone 1A8; BD Biosciences, Franklin Lakes, NJ, USA), CD11c phycoerythrin (PE) eFluor 610 (clone: N418; Invitrogen), CD4 fluorescein-5-isothiocyanate (FITC) (clone RM4-5, BD Biosciences), CD8a PE (clone 53-6.7, BD Biosciences), CD19 allophycocyanin (APC) eFluor 780 (clone 1D3, Invitrogen), and hamster anti-mouse CD3e PE-Cy7 (clone 145-2C11, BD Biosciences). Compensation was performed with antibody capture beads (BD Biosciences) stained separately with individual mAb used in the test samples. The cells were examined in a LSR II cytometer system with FACSDiva 8.01 software (both BD Biosciences, Franklin Lakes, NJ, USA) using gating strategies for Ly6G+ neutrophils, CD3+CD4+ T-cells, CD3+CD8+ T-cells, CD19+ B-cells, CD11c+CD11blo alveolar macrophages, CD11c+CD11bhi activated macrophages, CD11cintCD11bhi transitioning monocytes-macrophages, and CD11c−CD11bhi monocytes. The percentages of all respective cell populations were determined from live CD45+ lung leukocytes after excluding debris and doublets. A minimum of 50 000 events/sample was acquired each time.
Immunostaining and microscopy of mouse lungs
After BALF collection, the remaining left lung of each mouse was harvested and inflated with 1 ml 10% formalin under a pressure of 20 cm H2O for 24 h for optimal preservation of lung parenchymal architecture. Thereafter, the formalin-fixed lung tissue was embedded in paraffin and cut to 5 μm sections. Tissue sections were microscopically reviewed and semi-quantitatively scored for severity of peri-bronchiolar/peri-vascular and alveolar inflammation by experimental pathologist (TAW) blinded to treatment conditions, as previously described (Bates et al. Citation2018). This scoring system evaluates the spectrum of inflammatory changes and independently assigns a value from 0 to 5 with a higher score indicating greater inflammatory changes in the lung using the following criteria (% of total pulmonary tissue examined): (0) no changes compared to control mice; (1) minimal (<10%); (2) slight (10–25%); (3) moderate (26–50%); (4) severe (51–75%); or (5) very severe (>75%) of total area affected.
Mucin immunostaining
Mucin immunostaining within random sections of each left lung was performed as in Dickinson et al. (Citation2018). In brief, de-paraffinization of random slides was performed with three sequential 5-min rinses with xylene and isopropyl alcohol. Antigen retrieval was then performed by boiling the slides in Trilogy solution (Cell Marque [920P-09], Rocklin, CA, USA) for 20 min. The slides were then coated with 5% donkey serum (Sigma) in 0.1% Triton/PBS solution (blocking buffer) and incubated at room temperature for 60 min to block non-specific binding events. For the actual staining, anti-Lectin-UEA-1 (#RL 1062, Vector Labs, Burlingame, CA, USA) conjugated with a fluorophore that reacts at 555 nm (1:500 dilution in blocking buffer) was utilized for staining Muc5ac. A mouse anti-Muc5b (3E1, courtesy of Dr. Burton Dickey) antibody (1:500 dilution in blocking buffer) was used to stain for Muc5b. Slides were coated with primary antibodies and then incubated overnight at 4 °C. The slides were then gently rinsed with PBS before a solution of blocking buffer containing fluorophore-labeled species-specific donkey secondary antibody (at 2 mg/ml in blocking buffer; 1:1000 dilution) (#A-31570, #A21202, #21206; Vector Labs) was applied. The slides were then incubated for 1 h at room temperature before being gently washed with PBS. To counterstain nuclei and quantify number of cells per airway, DNA staining with 4′,6-diamidino-2-phenylindole (DAPI) was included in Vectashield mounting medium (H-1200; Vector Labs). A minimum of 10 fields/lungs - with a minimum of 5 mice/group - was generated in each study.
Immunofluorescent images were then captured using an Axio Observer Z.1 microscope and analyzed with Zen software (Zeiss, San Diego, CA, USA). Intracellular mucin from mouse airways were quantified with a common signal threshold with multiple fields at 20X magnification, using ImageJ (v2.0) software (NIH, Bethesda, MD, USA) as previously described (Dickinson et al. Citation2016, Citation2018).
Muc5ac immunoblot
In separate studies from those described above, excised whole mouse lungs (non-lavaged, but perfused) were recovered from the mice, snap frozen in liquid nitrogen, and later homogenized in PBS with protease inhibitors (Roche, Maneheim, Germany) using the gentleMACS Dissociator as described Sweeter et al. (Citation2021). Muc5ac assessed using UEA-1 detection as detailed in the Supplementary Methods. Blots were scanned using an Odyssey DLx Scanner (LiCor, Lincoln, NE, USA). Mucin levels in each blot were normalized for quantification using total protein levels as previously described (Dickinson et al. Citation2019).
Statistics
All data are presented as means ± SEM. A sample-size calculation was estimated from a previous single ODE exposure in WT and MyD88 KO murine study (Bauer et al. Citation2013), whereby it was calculated a sample size of N = 3 in each group would allow one to achieve 80% power at the 0.05 level of significance in order to detect a difference in airway neutrophil influx, assuming a mean (SD) of 11.15 × 105 (4.5 × 105) for WT + ODE and a mean of 0.02 × 105 (0.0065 × 105) for KO + ODE. The experimental groups consisted of sex- and age-matched litter mates as available. Sample sizes for each outcome listed in the figure legends reflect sample quantity and quality available from the pooled studies. To detect significant differences among three or more groups, a one-way analysis of variance (ANOVA) was performed; in the event a significance across groups of p < 0.05 was evident with the ANOVA, a non-parametric Mann–Whitney test was used to determine statistical difference between two groups. In all cases, Prism software (v.7.0c; GraphPad, La Jolla, CA, USA) was used. Significance was accepted at p < 0.05.
Results
Mice deficient in lung epithelial MyD88 demonstrate decreased influx of total cells and neutrophils after a single ODE exposure, without attenuation in pro-inflammatory cytokines/chemokines
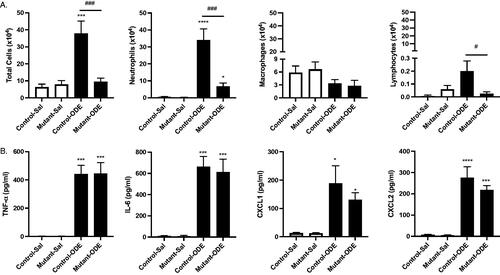
A one-time treatment with ODE induced BALF influx of total cells, neutrophils, and lymphocytes in ‘control’ (SPCCre−/− MyD88fl/fl) mice as compared to saline treatment (). This acute-ODE induced influx of total cells and neutrophils was profoundly decreased in the lung epithelial MyD88-deficient (SPCCre± MyD88fl/fl) mice, termed ‘mutant’ mice. There was also no significant increase in lymphocytes in the mutant mice following the one-time ODE exposure as compared to saline. Eosinophils were rarely detected and levels did not differ among the groups (data not shown). The one-time ODE exposure also induced release of pro-inflammatory TNFα and IL-6 and the neutrophil chemoattractants CXCL1 and CXCL2 in the control mice, and these responses were not reduced in the mutant mice ( 1(B)).
Figure 1. Mice deficient in lung epithelial MyD88 exhibit decreased influx of total cells and neutrophils following a single ODE exposure, without modulation in pro-inflammatory cytokines/chemokines. Control (SPCCre−/− MyD88fl/fl) and mutant (SPCCre± MyD88fl/fl) mice were treated once with ODE or saline; bronchoalveolar lavage fluid (BALF) was collected 5 h following the exposure. Bars depict mean ± SEM of (A) total cells, neutrophils, macrophages, and lymphocytes enumerated and (B) levels of TNFα, IL-6, and neutrophil chemoattractants CXCL1 and CXCL2. There are no differences in cytokines/chemokines between ODE-treated Cre+ vs. Cre− mice. N = 5 mice/group (saline), N = 7 (control-ODE), N = 8 (Mutant-ODE) from at least two independent experimental runs. Statistical differences vs. saline-treated mice are depicted as *p < 0.05; ***p < 0.001 and between groups as #p < 0.05; ###p < 0.001 by ANOVA.
Mice deficient in lung epithelial MyD88 demonstrated decrease influx of total cells, neutrophils, macrophages, and TNFα release following repetitive ODE for 1 week, but not for 3 weeks
Mice were treated daily with ODE for 8 days (1 week) or daily (weekends excluded) for 3 weeks whereupon BALF was collected to determine cellular influx and cytokine/chemokine release patterns between the control (SPCCre−/− MyD88fl/fl) and mutant (SPCCre± MyD88fl/fl) mice. After 1 week of repetitive ODE exposure, there were significantly decreased numbers of total cells, neutrophils, and macrophages (but not lymphocytes) in the lungs of the mutant mice as compared to in those of the control animals (). In these same 1-week exposed mice, ODE-induced TNFα was present at significantly decreased levels in the mutant mice as compared to in the control animals, but there were no differences in IL-6, CXCL1, nor CXCL2 levels between groups.
Figure 2. Mice deficient in lung epithelial MyD88 exhibit decreased influx of total cells, neutrophils, macrophages, and TNFα release after repetitive 1-week but not 3-week exposures. Control (SPCCre−/− MyD88fl/fl) and mutant (SPCCre± MyD88fl/fl) mice were treated daily with ODE for 1 or 3 weeks, whereupon bronchoalveolar lavage fluid (BALF) was collected 5 h after the final exposure. Saline control is pooled samples of control- and mutant-saline mice for reference. Bar graphs depict mean ± SEM of total cells, neutrophils, macrophages, and lymphocytes and levels of TNFα, IL-6, and neutrophil chemoattractants CXCL1 and CXCL2 in the lungs of (A) 1-week and (B) 3 weeks treated mice. N = 10 mice/group (saline), N = 7 (control and mutant, 1 week), N = 7 (control, 3 weeks), and N = 8 (mutant, 3 weeks) from at least two independent experimental runs. Statistical differences vs. saline treated mice are depicted as *p < 0.05, **p < 0.01, ***p < 0.001, and between groups as ##p < 0.01 by (A) ANOVA and (B) Mann–Whitney.
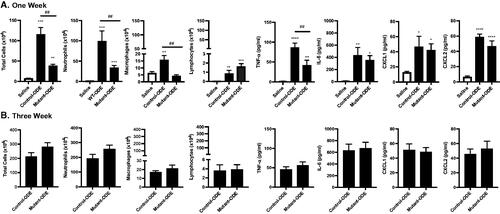
By extending ODE exposure to 3 weeks, ODE-induced cellular differences dependent on MyD88 signaling in lung epithelial cells were lost. Specifically, there were no differences in total cells, neutrophils, macrophages, or lymphocytes found in the BALF isolated from control and mutant mice (). Moreover, there were no differences between the mutant and control mice with regard to ODE-induced levels of TNFα, IL-6, CXCL1, or CXCL2 ().
Mice deficient in lung epithelial MyD88 exhibit decreased lung neutrophil infiltrates and Muc5ac mucin levels following 1 week of repetitive ODE exposure
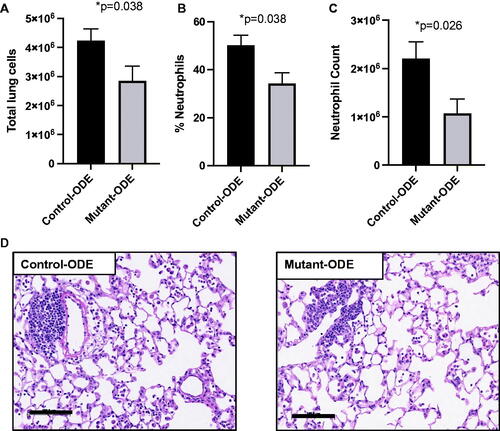
Based on the strong reduction in ODE-induced neutrophil influx at 1, but not 3, weeks, the following studies were focused at 1-week (8 days) of repetitive ODE exposure in the control (SPCCre−/− MyD88fl/fl) and mutant (SPCCre± MyD88fl/fl) mice. It was seen that there was a reduction in ODE-induced total lung cell infiltrates enumerated in lungs of the mutant mice as compared to in the lungs of the controls (). This response was seemingly driven by reduction in lung neutrophils, as determined by flow cytometry. The only cell population that was found to be significantly different between the groups was Ly6G+ neutrophils, as evident by a decrease in both the percentage (% of live CD45+ cells) and number of neutrophils in lungs of the mutant mice as compared to in the lungs of the control animals treated with ODE (, and data not shown). Lung sections from the treatment groups were also H&E-stained; there was no difference in semi-quantitative lung inflammatory scores (range 0–4, low to high) between the ODE-treated control and mutant mice (control 2.7 [±0.2] and mutant 2.5 [±0.4], p = 0.75) ().
Figure 3. Mice deficient in lung epithelial MyD88 exhibit decreased lung neutrophil infiltrates following 1 week of repetitive ODE exposure. Control (SPCCre−/− MyD88fl/fl) and mutant (SPCCre± MyD88fl/fl) mice were treated daily with ODE for 1 week before lung tissue was processed by flow cytometry and histopathology. Bar graphs depict mean ± SEM of (A) total lung cells enumerated by hematocytometer; (B) Ly6G+ neutrophils determined as percentage of CD45+ lung cells by flow cytometry analysis, and (C) this percentage multiplied by total cells to determine total lung neutrophils count. A representative H&E-stained lung section from each group is shown. N = 7 (control and mutant, 1 week) from two independent experimental runs. Scale = 100 μm. Statistical differences determined by Mann–Whitney.
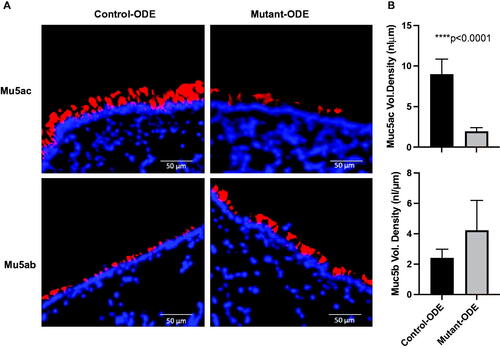
Lung tissue sections were also stained with lectin UEA-1 to detect Muc5ac or with anti-Muc5b to help evaluate total mucin levels () and airway-associated intracellular mucin. The data indicate that after 1 week of ODE exposure, mutant mice with lung epithelial MyD88 deficiency (SPCCre−/+ MyD88fl/fl) demonstrated significantly less Muc5ac staining compared to control mice (SPCCre−/− MyD88fl/fl), but no significant difference in Muc5b staining ().
Figure 4. ODE-induced inflammatory mucin levels of Muc5ac were significantly reduced in mice deficient in lung epithelial MyD88. (A) Lung sections from repetitive (1 week) ODE treated control (SPCCre−/− MyD88fl/fl) and mutant (SPCCre± MyD88fl/fl) mice were stained for mucins (red) with lectin UEA-1 for Muc5ac and mouse anti-Muc5b with nuclei staining (DAPI, blue). (B) Airway-associated intracellular mucin was quantified (ImageJ); bars mean ± SEM of volume densities between treatment groups (N = 3 control, N = 5 mutant). Statistical differences determined by Mann–Whitney.
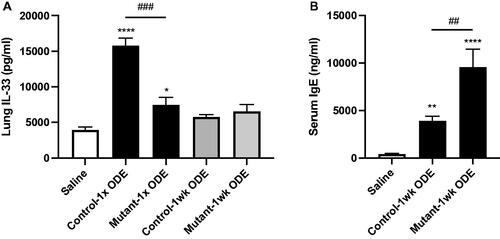
Single ODE exposure-induced IL-33 release was reduced in mice deficient in lung epithelial MyD88 but serum IgE levels were increased
Although classic allergic responses (e.g. increases in presence of eosinophils, T-helper cell type 2 [TH2] cytokines) have not been associated with ODE-induced airway inflammatory responses in past studies, increased lung expression of alarmin IL-33 and increased serum IgE have been demonstrated with ODE exposures (Johnson et al. Citation2020). The results here indicated that lung homogenate levels of IL-33 were significantly reduced in mutant (SPCCre± MyD88fl/fl) mice compared to in control mice (SPCCre−/− MyD88fl/fl) following a one-time ODE exposure, but not with 1 week of repetitive ODE exposures (). Interestingly, as compared to in control animals, serum IgE levels were increased in the mutant mice following 1 week of ODE treatment (), similar to findings with prior global MyD88 KO and ODE studies (Johnson et al. Citation2020).
Figure 5. Single (1×) ODE induced-IL-33 release was reduced in mice deficient in lung epithelial MyD88, but serum IgE levels were increased with a 1-week repetitive exposure. (A) Bar graphs depict mean ± SEM of IL-33 levels in lung homogenates from pooled saline animals, control (SPCCre−/− MyD88fl/fl) and mutant (SPCCre± MyD88fl/fl) mice treated once or daily with ODE for 1 week. (B) Serum IgE levels from 1-week treated animals. N = 10 mice/group (saline), N = 7 (control and mutant, 1 week) from at least two independent experimental runs. Statistical differences vs. saline-treated mice are depicted by *p < 0.05, **p < 0.01, ***p < 0.001, and between groups by ##p < 0.01, ###p < 0.001 by ANOVA.
MyD88 deficiency in CCSP-expressing cells does not alter responses to ODE
In another set of studies, the role of MyD88 signaling specific to CCSP+ secretory cells in the conducting airways of adult mice—in an inducible manner—was investigated. Utilizing the tamoxifen-treated, promoter-inducible control (CCSPCre−/− MyD88fl/fl) and mutant (CCSPCre± MyD88fl/fl) mice to target club cells, animals were treated once or daily for 1 week with ODE whereupon inflammatory cellular influx and cytokine/chemokine release was investigated. The results revealed there were no significant differences in influx of total cells, neutrophils, macro-phages, or lymphocytes, nor in release of TNFα, IL-6, and CXCL1, nor in Muc5ac expression between the groups (Supplementary Table 1).
Discussion
Organic dusts from agricultural environments contain a wide variety and abundance of bacterial motifs recognized by several Toll-like receptors (TLR), inducing inflammatory host responses via the shared adaptor protein MyD88 (Poole et al. Citation2011; Bauer et al. Citation2013). In the study here, it was shown that deletion of MyD88 in lung epithelial cells robustly reduced acute ODE-mediated neutrophil influx and reduced Muc5ac expression without substantial impact on classic ODE-associated pro-inflammatory cytokines/chemokines. Collectively, it appeared that lung epithelial cell MyD88-dependent signaling pathway played a selective role in mediating neutrophil influx and inflammatory mucin production in response to acute organic dust exposures.
Genetic modeling to target ‘all’ airway epithelial cells led to the development of the surfactant protein C (SPC)-Cre mouse model because of SPC expression during embryogenesis on all epithelial cells types (Okubo et al. Citation2005; Cleaver et al. Citation2014). In mature animals, SPC itself is synthesized exclusively in alveolar Type II cells and absence/mutation in SPC has been associated with inflammatory lung diseases, particularly interstitial lung diseases and pulmonary fibrosis (Sitaraman et al. Citation2021). In this current work, the SPC-Cre murine strain was utilized as a tool to specifically understand the role of MyD88-dependent airway epithelial cell responses in vivo to ODE exposure. The data obtained herein strongly support a critical role for airway epithelial cells in regulating airway neutrophil influx to ODE exposures because neutrophil influx following acute ODE exposures (single and 1-week repetitive) was profoundly reduced in these mutant mice (SPCCre± MyD88fl/fl). As this protection was lost with extended (i.e. 3-week repetitive) exposures, this suggests an involvement of compensatory signaling pathways and/or engagement of other resident or recruited lung cell populations.
Further, based on the data here, it seemed that the classic pro-inflammatory mediators including TNFα, IL-6, and neutrophil chemoattractants associated with agriculture exposure-induced lung disease were not dependent on airway epithelial cell MyD88-dependent signaling pathways. It was anticipated these mediators would have been reduced as prior bone marrow chimera studies demonstrate that MyD88 signaling in lung structural/resident cells are partially responsible for ODE-induced IL-6 and neutrophil chemoattractant release (Poole, Wyatt, et al. 2015).
Although epithelial cells comprise a large compartment of lung resident cells, fibroblasts, myofibroblasts, and epithelial-mesenchymal transitional cells are also essential lung resident cells with capacity to initiate pro-inflammatory responses to various organic dust exposures (Nordgren et al. Citation2014). It is also recognized that TLR/MyD88-independent airway epithelial cell responses to organic dust exposures are important, including protease activated receptor (PAR1 and PAR2) signaling (Romberger et al. Citation2015), protein kinase C activation (Poole, Romberger, et al. 2012; Wyatt et al. Citation2014, Citation2020), scavenger receptor pathways (Poole, Anderson, et al. 2015; Wyatt et al. Citation2020), STAT-3 activation (Natarajan et al. Citation2019), amphiregulin signaling (Poole et al. 2020), omega-3-fatty acid derivative pathways (Nordgren et al. Citation2015; Ulu et al. Citation2021), and high mobility group box-1 activation (Bhat et al. Citation2019). Additionally, innate immune cells, including alveolar macrophages, are also important mediators of pro-inflammatory mediator release as well as neutrophil influx following organic dust exposures (Poole, Gleason, et al. 2012).
Paralleling the reduction of neutrophil influx in response to acute ODE, Muc5ac levels were found to be decreased in the lungs of the lung epithelial MyD88-deficient mice treated with ODE. This was consistent with results of other studies demonstrating attenuation of MUC5AC gene expression through NF-κB inhibition, downstream of MyD88 signaling (Fujisawa et al. Citation2009). However, the current findings contrast with results seen in global MyD88 KO mice whereby Muc5ac epithelial cell expression was strikingly increased, not decreased, following in vivo and in vitro ODE exposures (Dickinson et al. Citation2019). One potential explanation for this is that signaling crosstalk occurred, but exploratory studies into potential crosstalk signals, including SA-pointed domain-containing Ets-like factor (SPDEF), have been unrevealing (Dickinson et al. Citation2019). Janssen et al. (Citation2016) have proposed interaction links between mucin and alveolar macrophage defensive functions in regulating homeostatic, inflammatory, and resolving immune functions. In the model in the present study, the intact macrophage MyD88 signaling pathway, and thus the defensive function of macrophages, might have allowed for compensatory clearance of secreted airway mucous. Thus, a physical clearance mechanism may reduce a positive feedback loop of inspissated mucous secretions in blocked airways causing further increase in mucin production. Future studies of the impact of a macrophage-specific MyD88 deletion are thus warranted.
The alarmin IL-33 affects mucus cell biology; here, there was decreased ODE-induced IL-33 release rapidly following a one-time ODE exposure in the lungs of lung epithelial MyD88-deficient mice. Whereas this finding could be contributing to the decrease in mucus expression after 1 week of repetitive exposures, the ODE-induced IL-33 attenuated response was lost after 1 week. ODE exposure does not induce allergic or TH2 cytokines (i.e. IL-4, IL-3) or eosinophil influx, but is associated with increased serum IgE levels; such a response is augmented in global MyD88 KO animals (Johnson et al. Citation2020). Serum IgE levels were also increased here in the lung epithelial MyD88-deficient mice. Spatio-temporally secreted factors from airway epithelial cells are known to be necessary for regulating IgE production (Moon et al. Citation2018); further studies may be warranted to clarify what role such secreted factors might have in the mechanisms underlying the outcomes seen in the present study.
Lastly, in the current study, the findings of an importance of the MyD88-dependent signaling pathway was attributed to all airway epithelial cells, but could not be further ascribed to club (CCSP+) cells (non-ciliated, non-mucoid cells) of the respiratory bronchioles (Singh and Katyal 200 l; Rawlins et al. Citation2009). Such outcomes would serve to highlight the roles of the central and upper airways, or even alveolar epithelial cells, in ODE-induced responses in vivo. Alternatively, it is also recognized that hematopoietic cell MyD88-dependent signaling is also important as prior bone marrow chimera studies demonstrated that MyD88 action in hematopoietic cells is mainly responsible for acute ODE-induced TNF-⍺ release and partially responsible for IL-6 and neutrophil chemoattractant release and neutrophil influx (Poole, Wyatt, et al. 2015). However, the hematopoietic cell-specific (e.g. monocytes, macrophages, lymphocytes, dendritic cells) MyD88-signaling dependence in regulating the lung response to acute and/or repetitive ODE exposures remains unknown and future studies including use of cell-specific Cre/Lox strategies could be investigated.
Conclusions
In summary, based on the data here, one may conclude that MyD88-dependent signaling of lung epithelial cells governs several organic dust-induced airway inflammatory consequences, including neutrophil recruitment and mucin production. By delineating lung epithelial cell-specific MyD88-dependent responses, these studies also uncovered potential links in mucin production/clearance potentially involving macrophages. The upper airway responses appear to be driving responses as there was a loss in MyD88-depenent protection against inflammation in mid-airway epithelial cell club cells, which may have implications in development of lung disease. These findings might have implication in the design of future of cell-targeted therapeutic approaches for agriculture workers.
Author contributions
Conceptualization: ANJ, JD, JAP. Methodology: ANJ, JD, AN, RG, KK, SE, KJ, TAW, JAP. Investigation: ANJ, JD, AN, RG, KK, KJ, TAW, JAP. Visualization: ANG, JD, AN, RG, KK, SE, KJ, TAW, JAP. Funding acquisition: JAP, TAW. Project administration: ANJ, AN, JAP. Supervision: JAP, AN.
Supplemental Material
Download MS Word (77 KB)Acknowledgments
The Authors acknowledge members of the Tissue Sciences Facility at the Department of Pathology and Microbiology (University of Nebraska Medical Center [UNMC] Omaha, NE, USA) for assistance with tissue processing and staining. The Authors thank Victoria B. Smith and Craig Semerad in the Flow Cytometry Research Core Facility at UNMC for aiding with flow cytometry studies. The Authors also thank Lisa Chudomelka for MS submission assistance.
Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest. JAP has received research products from AstraZeneca and is a site investigator for human clinical research studies from GlaxoSmithKline, Takeda, and AstraZeneca, all unrelated to this project. No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Bates M, Akbari P, Gilley K, Wagner J, Li N, Kopec A, Wierenga K, Jackson-Humbles D, Brandenberger C, Holian A, et al. 2018. Dietary docosahexaenoic acid prevents silica-induced development of pulmonary ectopic germinal centers and glomerulonephritis in the lupus-prone NZBWF1 mouse. Front Immunol. 9:2002.
- Bauer C, Kielian T, Wyatt T, Romberger D, West W, Gleason A, Poole J. 2013. Myeloid differentiation factor 88-dependent signaling is critical for acute organic dust-induced airway inflammation in mice. Am J Respir Cell Mol Biol. 48(6):781–789.
- Bhat S, Massey N, Karriker L, Singh B, Charavaryamath C. 2019. Ethyl pyruvate reduces organic dust-induced airway inflammation by targeting HMGB1-RAGE signaling. Respir Res. 20(1):27.
- Cleaver J, You D, Michaud D, Pruneda F, Juarez M, Zhang J, Weill P, Adachi R, Gong L, Moghaddam S, et al. 2014. Lung epithelial cells are essential effectors of inducible resistance to pneumonia. Mucosal Immunol. 7(1):78–88.
- Dickinson J, Alevy Y, Malvin N, Patel K, Gunsten S, Holtzman M, Stappenbeck T, Brody S. 2016. IL-13 activates autophagy to regulate secretion in airway epithelial cells. Autophagy. 12(2):397–409.
- Dickinson J, Sweeter J, Staab E, Nelson A, Bailey K, Warren K, Jaramillo A, Dickey B, Poole J. 2019. MyD88 controls airway epithelial Muc5ac expression during TLR activation conditions from agricultural organic dust exposure. Am J Physiol Lung Cell Mol Physiol. 316(2):L334–L347.
- Dickinson J, Sweeter J, Warren K, Ahmad I, de Deken X, Zimmerman M, Brody S. 2018. Autophagy regulates DUOX1 localization and superoxide production in airway epithelial cells during chronic IL-13 stimulation. Redox Biol. 14:272–284.
- Fujisawa T, Velichko S, Thai P, Hung L, Huang F, Wu R. 2009. Regulation of airway MUC5AC expression by IL-1β and IL-17A; The NF-κB paradigm. J Immunol. 183(10):6236–6243.
- Ghosn E, Yang Y, Tung J, Herzenberg L, Herzenberg L. 2008. CD11b expression distinguishes sequential stages of peritoneal B-1 development. Proc Natl Acad Sci USA. 105(13):5195–5200.
- Janssen W, Stefanski A, Bochner B, Evans C. 2016. Control of lung defense by mucins and macrophages: Ancient defense mechanisms with modern functions. Eur Respir J. 48(4):1201–1214.
- Johnson A, Harkema J, Nelson A, Dickinson J, Kalil J, Duryee M, Thiele G, Kumar B, Singh A, Gaurav R, et al. 2020. MyD88 regulates a prolonged adaptation response to environmental dust exposure-induced lung disease. Respir Res. 21(1):97.
- Kelly K, Poole J. 2019. Pollutants in the workplace: Effect on occupational asthma. J Allergy Clin Immunol. 143(6):2014–2015.
- Mikuls T, Gaurav R, Thiele G, England B, Wolfe M, Shaw B, Bailey K, Wyatt T, Nelson A, Duryee M, et al. 2021. The impact of airborne endotoxin exposure on rheumatoid arthritis-related joint damage, autoantigen expression, autoimmunity, and lung disease. Int Immunopharmacol. 100:108069.
- Moon H, Kim S, Jeong J, Han S, Jarjour N, Lee H, Abboud-Werner S, Chung S, Choi H, Natarajan V, et al. 2018. Airway epithelial cell-derived colony stimulating factor-1 promotes allergen sensitization. Immunity. 49(2):275–287 e5.
- Natarajan K, Meganathan V, Mitchell C, Boggaram V. 2019. Organic dust induces inflammatory gene expression in lung epithelial cells via ROS-dependent STAT-3 activation. Am J Physiol Lung Cell Mol Physiol. 317(1):L127–L140.
- Nordgren T, Bauer C, Heires A, Poole J, Wyatt T, West W, Romberger D. 2015. Maresin-1 reduces airway inflammation associated with acute and repetitive exposures to organic dust. Transl Res. 166(1):57–69.
- Nordgren T, Friemel T, Heires A, Poole J, Wyatt T, Romberger D. 2014. The omega-3 fatty acid docosahexaenoic acid attenuates organic dust-induced airway inflammation. Nutrients. 6(12):5434–5452.
- Okubo T, Knoepfler P, Eisenman R, Hogan B. 2005. Nmyc plays an essential role during lung development as a dosage-sensitive regulator of progenitor cell proliferation and differentiation. Development. 132(6):1363–1374.
- Poole J, Wyatt T, Oldenburg P, Elliott M, West W, Sisson J, von Essen S, Romberger D. 2009. Intranasal organic dust exposure-induced airway adaptation response marked by persistent lung inflammation and pathology in mice. Am J Physiol Lung Cell Mol Physiol. 296(6):L1085–1095.
- Poole J, Mikuls T, Duryee M, Warren K, Wyatt T, Nelson A, Romberger D, West W, Thiele G. 2017. A role for B-cells in organic dust induced lung inflammation. Respir Res. 18(1):214.
- Poole J, Wyatt T, Kielian T, Oldenburg P, Gleason A, Bauer A, Golden G, West W, Sisson J, Romberger D. 2011. Toll-like receptor 2 regulates organic dust-induced airway inflammation. Am J Respir Cell Mol Biol. 45(4):711–719.
- Poole J, Gleason A, Bauer C, West W, Alexis N, van Rooijen N, Reynolds S, Romberger D, Kielian T. 2012a. CD11c+/CD11b+ cells are critical for organic dust-elicited murine lung inflammation. Am J Respir Cell Mol Biol. 47(5):652–659.
- Poole J, Romberger D, Bauer C, Gleason A, Sisson J, Oldenburg P, West W, Wyatt T. 2012b. Protein kinase C epsilon is important in modulating organic-dust-induced airway inflammation. Exp Lung Res. 38(8):383–395.
- Poole J, Anderson L, Gleason A, West W, Romberger D, Wyatt T. 2015a. Pattern recognition scavenger receptor A/CD204 regulates airway inflammatory homeostasis following organic dust extract exposures. J Immunotoxicol. 12(1):64–73.
- Poole J, Wyatt T, Romberger D, Staab E, Simet S, Reynolds S, Sisson J, Kielian T. 2015b. MyD88 in lung resident cells governs airway inflammatory and pulmonary function responses to organic dust treatment. Respir Res. 16:111.
- Rawlins E, Okubo T, Xue Y, Brass D, Auten R, Hasegawa H, Wang F, Hogan B. 2009. The role of Scgb1a1+ Clara cells in the long-term maintenance and repair of lung airway, but not alveolar, epithelium. Cell Stem Cell. 4(6):525–534.
- Romberger D, Heires A, Nordgren T, Souder C, West W, Liu X, Poole J, Toews M, Wyatt T. 2015. Proteases in agricultural dust induce lung inflammation through PAR-1 and PAR-2 activation. Am J Physiol Lung Cell Mol Physiol. 309(4):L388–399.
- Singh G, Katyal S. 2000. Clara cell proteins. Ann N Y Acad Sci. 923:43–58.
- Sitaraman S, Martin E, Na CL, Zhao S, Green J, Deshmukh H, Perl A, Bridges J, Xu Y, Weaver T. 2021. Surfactant protein C mutation links post-natal type 2 cell dysfunction to adult disease. JCI Insight. 6(14):e142501.
- Sweeter J, Kudrna K, Hunt K, Thomes P, Dickey B, Brody S, Dickinson J. 2021. Autophagy of mucin granules contributes to resolution of airway mucous metaplasia. Sci Rep. 11(1):13037.
- Ulu A, Burr A, Heires AJ, Pavlik J, Larsen T, Perez P, Bravo C, DiPatrizio N, Baack M, Romberger D, et al. 2021. A high docosahexaenoic acid diet alters lung inflammation and recovery following repetitive exposure to aqueous organic dust extracts. J Nutr Biochem. 97:108797.
- Wang X. 2009. Cre transgenic mouse lines. Meth Mol Biol. 561:265–273.
- Wyatt T, Nemecek M, Chandra D, DeVasure JM, Nelson A, Romberger D, Poole J. 2020. Organic dust-induced lung injury and repair: bi-directional regulation by TNFα and IL-10. J Immunotoxicol. 17(1):153–162.
- Wyatt T, Poole J, Nordgren T, DeVasure J, Heires A, Bailey K, Romberger D. 2014. cAMP-dependent protein kinase activation decreases cytokine release in bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 307(8):L643–651.