
Abstract
Mitochondria serve as membrane sources and signaling platforms for regulating autophagy. Accumulating evidence has also shown that damaged mitochondria are removed through both selective mitophagy and general autophagy in response to mitochondrial and oxidative stresses. Protein ubiquitination through mitochondrial E3 ligases plays an integrative role in mitochondrial outer membrane protein degradation, mitochondrial dynamics, and mitophagy. Here we showed that MUL1, a mitochondria-localized E3 ligase, regulates selenite-induced mitophagy in an ATG5 and ULK1-dependent manner. ULK1 partially translocated to mitochondria after selenite treatment and interacted with MUL1. We also demonstrated that ULK1 is a novel substrate of MUL1. These results suggest the association of mitochondria with autophagy regulation and provide a new mechanism for the beneficial effects of selenium as a chemopreventive agent.
Abbreviations
| ACTB | = | actin, β |
| ATG5 | = | autophagy-related 5 |
| BECN1 | = | Beclin 1, autophagy-related |
| BAF | = | bafilomycin A1 |
| CHX | = | cycloheximide |
| FCCP | = | carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone GST, glutathione S-transferase |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 β |
| MFN1/2 | = | mitofusin 1/2 |
| MG132 | = | carbobenzoxy-L-leucyl-L-leucyl-L-leucinal |
| MUL1 | = | mitochondrial E3 ubiquitin protein ligase 1 |
| MUL1ΔR | = | mitochondrial E3 ubiquitin protein ligase 1 lacking the RING finger domain |
| PARK2 | = | parkin RBR E3 ubiquitin protein ligase |
| PINK1 | = | PTEN-induced putative kinase 1 |
| ROS | = | reactive oxygen species |
| SQSTM1/p62 | = | sequestosome 1 |
| TIMM23 | = | translocase of inner mitochondrial membrane 23 homolog (yeast) |
| TOMM20 | = | translocase of outer mitochondrial membrane 20 homolog (yeast) |
| Ub | = | ubiquitin |
| ULK1 | = | unc-51 like autophagy activating kinase 1. |
Mitochondria utilize oxygen and nutritional sources to produce ATP.Citation1 These organelles also produce ample reactive oxygen species (ROS) as an inevitable byproduct of oxidative phosphorylation.Citation2 Mitochondria are highly sensitive to the changes of free radical status and intracellular bioenergetics, and function as sentinel to these changes to regulate distinct signaling pathways such as HIF1α and AMPK, which are key regulators for autophagy.Citation3,4 AMPK, a conserved cellular and mitochondrial energy sensor, can phosphorylate ULK1 which functions as a master regulator for the initiation of both general autophagy and also mitophagy.Citation5 The genetic ablation of ULK1 in mice reveals an unremarkable phenotype, except for a defect in the clearance of mitochondria and ribosomes during red blood cell maturation.Citation6 Mitochondria-derived ROS increase autophagy.Citation7,8 For example, ROS activate autophagy through the oxidation of ATG4, leading to the inactivation of this protein, thereby facilitating LC3 conjugation to autophagosomes.Citation9 In addition, mitochondria provide a membrane source for autophagosomes through mitochondria-associated membranes (MAM).Citation10,11
On the other hand, mitochondria are also subjected to destruction through autophagy in response to bioenergetics crises and oxidative stresses.Citation12,13 Eukaryotes have evolved elaborate mechanisms to selectively remove damaged and unwanted mitochondria through mitochondrial autophagy or mitophagy, in which double-membrane vesicles, called autophagosomes, enclose damaged or unwanted mitochondria and are subsequently transported to lysosomes for degradation.Citation14 How these cellular homeostatic events are integrated in response to cellular or environmental cues remains unknown. Mitochondrial outer membrane protein ubiquitination likely plays an integrative role in stress responses.Citation15,16 It is well documented that mitochondrial outer membrane protein ubiquitination determines proteasomal degradations under mild oxidative stress, and the selective mitophagy upon chronic and severe stresses that cause the loss of mitochondrial membrane potential.Citation17,18 Mitochondria-localized E3 ligases and deubiquitinases have been implicated in mitochondrial dynamics and mitochondrial autophagic activities.Citation19,20 In an effort to understand the mechanism of selenite-induced mitophagy, we have demonstrated that ULK1 is a novel substrate of MUL1. These data suggest a new link between mitochondria and the regulation of autophagy.
Results
MUL1 is involved in selenite-induced mitophagy
Mitophagy is activated through mitochondrial stresses, such as oxidative stress and the loss of membrane potential. Previous studies have shown that sodium selenite induces mitochondrial oxidative stress and the loss of membrane potential.Citation21,22 At low doses, selenite effectively induces GFP-LC3 aggregation and the mitochondrial protein degradation (Fig. S1A–C), hallmarks for mitophagy as we previously reported,Citation23 but does not induce apoptosis (Fig. S2). We reasoned that mitochondria-localized E3 ligases might play a role in selenite-induced mitophagy. Several E3 ligases, such as RNF185, AMFR, MARCH5, SMURF1, PARK2, and MUL1, mediate mitochondrial outer membrane protein ubiquitination, mitochondrial dynamics, and/or mitophagy.Citation19,20,24,25 To identify roles for these E3 ligases in selenite-induced mitophagy, we synthesized 2 siRNAs for each of these proteins and examined the effects of genetic silencing on selenite-induced mitophagy based on mitochondrial marker protein degradation. The results showed that siRNAs targeted to MUL1 in HeLa cells, but not to other mitochondrial E3 ligases, inhibited the degradation of the mitochondrial marker proteins, TIMM23 and TOMM20 ( Fig. S3A, B). However, MUL1 siRNA minimally affected mitochondrial protein degradation induced through FCCP, amino acid starvation or hypoxia, well-known inducers of autophagy or mitophagy (). Further experiments of immunofluorescence microscopy demonstrating GFP-LC3 puncta formation colocalizing with mitochondria, confirmed the function of MUL1 in selenite-induced mitophagy (). Although the degradation of mitochondrial marker proteins was detected after selenite treatment, MUL1, a mitochondria outer-membrane protein, remained relatively stable (), reflecting the transcriptional upregulation of MUL1 through selenite (Fig. S1D). MUL1 expression also enhanced mitochondrial protein degradation, which could be inhibited with bafilomycin A1 (BAF), an autophagy flux blocker,Citation26 and pepstatin, a protease inhibitor, but not with the proteasome inhibitor MG132 (). As shown in and Figure S1E, an accumulation of the sequestered mitochondria could be clearly observed in double-membrane autophagic vesicles in the cells transfected with plasmids expressing MUL1-MYC under the electron microscope. To demonstrate that MUL1 is a specific regulator of selenite-induced mitophagy, we performed rescue experiments using MUL1 knockdown cells. As expected, wild-type MUL1 restored the reduction of TIMM23 and TOMM20, and SQSTM1, a marker for general autophagy (). Taken together, these data indicate that MUL1 is involved in selenite-induced mitophagy.
Figure 1 (See previous page). Identification of MUL1 in selenite-induced mitophagy. (A) HeLa cells transfected with siRNAs specifically targeting RNF185, AMFR, MARCH5 or MUL1 were treated with 5 μM of selenite for 12 h, followed by western blot analysis of the indicated proteins. (B) Quantitative analysis of the TOMM20 protein level described in (A). TOMM20 protein level was determined by dividing the intensity of TOMM20 with the intensity of tubulin on the blot. (The intensity of bands was measured with ImageJ software; mean ±SEM, from 3 independent experiments, 2-way ANOVA, ***P < 0.001, N.S., not significant.) (C) HeLa cells transfected with siMUL1 or scrambled RNA (Scr) were treated with the indicated stresses (5 μM FCCP, 6 h; EBSS, 6 h; hypoxia with 1% O2, 12 h), followed by the analysis of the indicated protein levels. (D) Quantitative analysis of the TOMM20 protein level described in (C). TOMM20 protein level was determined by dividing the intensity of TOMM20 with the intensity of tubulin on the blot (the intensity of bands was measured with ImageJ software; mean ±SEM, from 3 independent experiments, 2-way ANOVA, ***P < 0.001). (E) HeLa cells expressing GFP-LC3 were transfected with scrambled RNA or siMUL1-CY5, followed by treatment with 5 μM of selenite for 12 h and immunofluorescence microscopy to detect GFP-LC3 puncta. (F) Quantification of GFP-LC3 punctate structures associated with the mitochondria (TOMM20) described in (E) (mean ±SEM; n = 50 cells from 3 independent experiments, one-way ANOVA, **P < 0.01, ***P < 0.001). (G) HeLa cells transfected with MUL1-MYC were treated with 5 μM of selenite for 12 h with or without 10 nM BAF, 5 μM MG132 or 10 mg/mL pepstatin for 6 h and subjected to western blotting analysis of the indicated protein levels. (H) Quantitative analysis of the TOMM20 protein level described in (G). TOMM20 protein level was determined by dividing the intensity of TOMM20 with the intensity of tubulin on the blot (The intensity of bands was measured with Image J software. mean ±SEM, from 3 independent experiments, 2-way ANOVA, *P < 0.05, N.S., not significant). (I) HeLa cells were overexpressed with MUL1-MYC for 24 h, and then the samples were analyzed by electron microscopy. Arrows indicate that mitochondria are enclosed within autophagosomes.
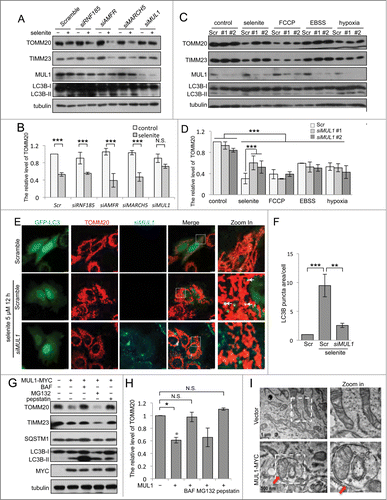
Figure 2 (See previous page). Both ULK1 and ATG5 are required for selenite-induced mitophagy. (A) HeLa cells with MUL1 was knocked down were rescued by exogenous wild-type MYC-tagged MUL1, followed by treatment with selenite for 12 h before western blotting analysis of the indicated proteins. (B) Wild-type MEFs, Atg5−/−, Ulk1−/− and park2−/− MEFs transfected with GFP-LC3 were treated with or without 5 μM of selenite for 12 h, followed by staining with MitoTracker Red for mitochondria and immunofluorescence microscopy to visualize GFP-LC3 puncta. (C) Quantification of GFP-LC3 punctate structures associated with mitochondria (TOMM20) described in (B) (mean ±SEM; n = 50 cells from 3 independent experiments, one-way ANOVA, ***P < 0.001, N.S., not significant). (D) The MEFs cells described in (B) were treated with or without selenite for 12 h, followed by subjected to western blotting analysis of the indicated proteins.
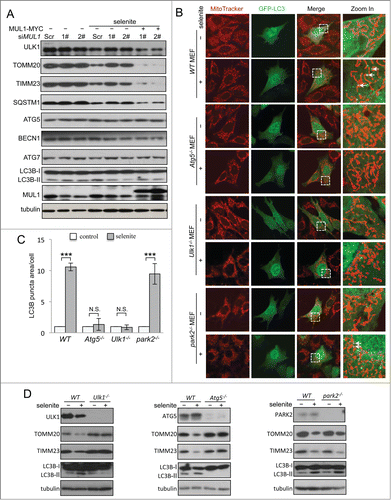
Both ULK1 and ATG5 are required for selenite-induced mitophagy
Next we determined whether MUL1 regulates selenite-induced mitophagy in a ULK1- or ATG5-dependent manner. ULK1 is the most upstream ATG protein for autophagy initiation in response to stress signals, and the ATG12–ATG5 system mediates ubiquitin-like conjugation for autophagosomal membrane expansion.Citation27-29 The results obtained from both immunofluorescence and western blotting analysis showed that selenite-induced mitophagy was abrogated in Ulk1- or Atg5-deficient MEFs (). PARK2, an E3 ubiquitin ligase associated with Parkinson disease, is required for mitophagy after mitochondrial damage.Citation17,30 However, in the absence of PARK2, selenite induced the appearance of LC3B-II and the reduction of mitochondrial marker proteins (, right panel). These results suggest that selenite induced mitophagy through ULK1 and ATG5, but not PARK2. In addition, the ectopic expression of MUL1 induced mitophagy in a similar fashion (Fig. S4).
ULK1 partially translocates onto mitochondria and interacts with MUL1
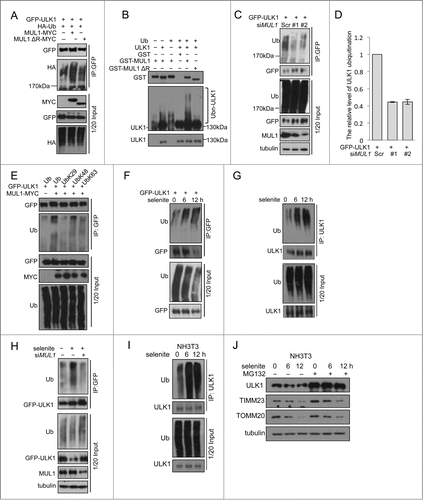
The results of the above rescue experiments revealed ULK1 degradation following selenite treatment, which was blocked when MUL1 was knocked down using siRNA. However, this effect was regained when MUL1 was reintroduced into the knockdown cells. Similar results were not observed for other ATG proteins, such as ATG5, ATG7, and BECN1 (). As MUL1 is an E3 ligase that mediates protein degradation,Citation20 we wanted to determine whether MUL1 interacts with ULK1 to regulate ULK1 protein stability. Immunofluorescence microscopy analysis showed that ULK1 was partially recruited to mitochondria in response to selenite treatment (). Subcellular fractionation analysis further confirmed ULK1 translocation to mitochondria (). The translocation of ULK1 to mitochondria was dynamic, as the levels of mitochondrial and cytosolic ULK1 were reduced at later time points, likely reflecting protein degradation. Coimmunoprecipitation revealed that MUL1 interacts with ULK1 (; Fig. S5A, B), which is enhanced by treatment of selenite (). Purified GST-MUL1 protein pulled down endogenous ULK1 from HeLa cell lysates, suggesting that the ULK1 complex physically interacted with MUL1 (). Since ULK1 forms a stable functional complex with ATG13 in mammalian cells,Citation31,32 we further examined whether MUL1 interacted with the ULK1-ATG13 complex. The result showed that both ULK1-MYC and GFP-MUL1 were coimmunoprecipitated with Flag-ATG13 (). Truncations of ULK1 were constructed according to its functional domains and further immunoprecipitation analyses revealed that this interaction with MUL1 is mediated through the C-terminal domain of ULK1 ().
Figure 3 (See previous page). MUL1 interacts with the ULK1 kinase in mammalian cells. (A) HeLa cells transfected with GFP-ULK1 (Green) were treated with 5 μM of selenite for the indicated time followed by staining with MitoTracker Red for mitochondria and DAPI (blue) for the nucleus. (B) HeLa cells were treated with 5 μM of selenite for the indicated times. Mitochondria were isolated and subsequently subjected to western blotting analysis of the indicated proteins. (C) Immunoprecipitation was performed with an ULK1 antibody. Coimmunoprecipitated endogenous MUL1 was detected through western blotting with an anti-MUL1 antibody. (D) ULK1-MYC was transfected into HEK 293T cells, and immunoprecipitation was performed with an anti-MYC antibody. Coimmunoprecipitated endogenous MUL1 was detected through western blotting with an anti-ULK1 antibody. (E) MUL1-MYC was transfected into HEK 293T cells, and immunoprecipitation was performed with an anti-MYC antibody. Coimmunoprecipitated endogenous ULK1 was detected through western blotting with an anti-ULK1 antibody. (F) HeLa cells transfected with MYC-vector or MUL1-MYC were treated with selenite for the indicated time and immunoprecipitation was performed with an anti-MYC antibody. Coimmunoprecipitated endogenous ULK1 level was detected through western blotting with an anti-ULK1 antibody. (G) Purified GST and GST-tagged MUL1 protein were used for the GST affinity isolation of endogenous ULK1, and blotted with an anti-ULK1 antibody. (H) GFP-MUL1 and ULK1-MYC were cotransfected with Flag-ATG13 or Flag vector, and immunoprecipitation was performed with an anti-Flag antibody. Coimmunoprecipitated ULK1 and MUL1 were detected through western blotting with anti-GFP and anti-MYC antibodies respectively. (I) Truncated forms of ULK1-MYC were constructed based on its functional domains. GFP-MUL1 was cotransfected with full-length or truncated forms of ULK1-MYC, and immunoprecipitation was performed with an anti-MYC antibody. Coimmunoprecipitated MUL1 was detected using an anti-GFP antibody. (J) Ulk1−/− MEFs were transfected with exogenous wild-type ULK1 or truncation ULK1Δ CTD, followed by treatment with selenite for 12 h before western blotting analysis of the indicated proteins.
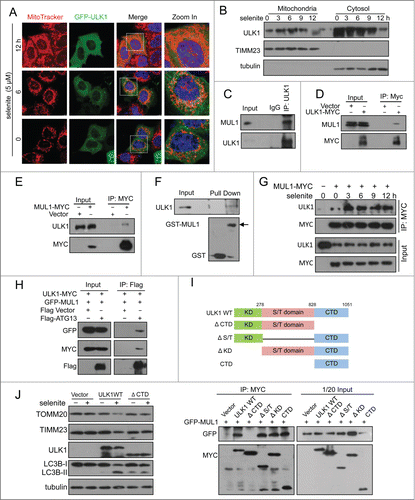
To clarify whether the interaction between MUL1 and ULK1 was crucial in selenite-induced mitophagy, rescue experiments were carried out in Ulk1−/− MEFs using wild-type ULK1 (ULK1WT) and truncated ULK1 with the absence of C-terminal domain (ULK1Δ CTD). Western blotting results revealed that ULK1WT, but not ULK1Δ CTD, restored mitophagy and LC3B-II transformation in Ulk1−/− MEFs, suggested that the interaction between MUL1 and ULK1 was important in mitophagy ().
ULK1 is a novel substrate of MUL1 in selenite-induced mitophagy
Cycloheximide (CHX) chase experiments showed that selenite-induced ULK1 degradation was inhibited by MG132 (). This effect was not detected when cells were treated with FCCP, hypoxia, or EBSS (; Fig. S5C). As MUL1 is a mitochondria-localized E3 ligase, we further tested if MUL1 promoted the degradation of ULK1 in a proteasome-dependent manner. To this end, we examined the ULK1 protein level when MUL1 is overexpressed. As shown in the expression of wild-type MUL1 significantly reduced exogenous and endogenous ULK1, and these effects could be partially abrogated with both BAF and MG132, suggesting that both proteasomes and autolysosomes may be involved in MUL1-mediated ULK1 degradation and mitophagy,Citation33,34 similar to other mitophagy paradigms (i.e., PARK2- and CCCP-induced mitophagy).Citation35,36 Conversely, the deletion of the RING-domain of the MUL1 E3 ligase showed no effect (), suggesting that ULK1 degradation is dependent on the E3 ligase activity of MUL1. Next, we examined the effect of MUL1 on ULK1 stability using a CHX chase assay. MUL1 overexpression significantly facilitated ULK1 degradation, which was blocked by MG132 (). These results suggested that MUL1 mediates ULK1 degradation through a proteasome-dependent pathway. The results of a subsequent ubiquitination assay showed that MUL1 enhanced the ubiquitination of ULK1 depending on its E3 ligase activity (). In vitro ubiquitination assays further substantiated this finding (). Importantly, knockdown of MUL1 expression reduced the ubiquitination and degradation of ULK1 (). Lys48-linked polyubiquitination is a canonical targeting signal for proteasome degradation.Citation37 Indeed, MUL1 induced the polyubiquitination of ULK1 in the presence of wild-type or Lys48 ubiquitin (). We also showed that selenite effectively promoted the ubiquitination of ULK1, and this effect was attenuated through siMUL1 (). Similar experiments were performed using normal NIH-3T3 cells; the results showed that selenite effectively induced ULK1 ubiquitination and subsequent degradation through proteasome pathway as well as mitophagy just as it did in HeLa cells (). Taken together, these results demonstrated that ULK1 is a candidate substrate for MUL1, which regulates selenite-induced mitophagy.
Figure 4. Overexpression of MUL1 and treatment with selenite promotes ULK1 degradation through the proteasome pathway. (A) HeLa cells were treated with CHX (10 μM, 12 h) and selenite (5 μM) for the indicated time, with or without MG132, and subjected to western blotting analysis of the ULK1. (B) Quantification of ULK1 protein levels in (A) (mean ±SEM, from 3 independent experiments). (C) HeLa cells were treated with the indicated agents (FCCP 5 μM; hypoxia with 1% O2; selenite 5 μM), and then subjected to western blotting analysis of ULK1 (The intensity of indicated bands was measured with ImageJ software). (D) After transfection with plasmids as indicated, HeLa cells were treated with BAF (10 nM, 6 h) or MG132 (5 μM, 6 h) prior to harvesting, followed by western blotting analysis of the GFP-ULK1 level. (E) After transfection with MUL1-MYC as indicated, HeLa cells were treated with BAF (10 nM, 6 h) or MG132 (5 μM, 6 h), followed by western blotting analysis of the ULK1 protein level. (F) HeLa cells were transfected with MUL1-MYC or MUL1ΔR-MYC (MUL1 with absence of the RING finger domain) for 24 h, and subjected to western blotting analysis of the indicated protein levels (The intensity of indicated bands was measured with ImageJ software). (G) HeLa cells transfected with MUL1-MYC for 24 h, followed by treatment with CHX (10 μM) for the indicated time, with or without MG132, and subjected to western blotting analysis of the ULK1 protein level. (H) Quantification of ULK1 protein levels in (G). Mean ±SEM, from 3 independent experiments.
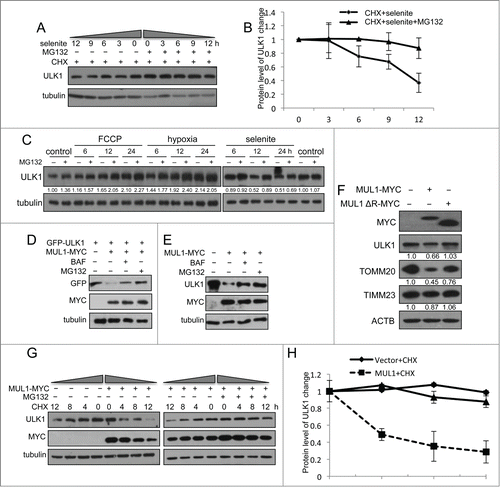
Figure 5 (See previous page). MUL1 promotes ubiquitination of ULK1. (A) HEK 293T cells were transfected with MUL1-MYC, MUL1ΔR-MYC, or the empty MYC-vector together with GFP-ULK1 and HA-Ub. Ubiquitination assays were performed as described in Materials and Methods. The ubiquitination level of GFP-ULK1 was detected using an anti-HA antibody. (B) In vitro ubiquitination assays were performed as described in Materials and Methods. The ubiquitinated form of ULK1 was immunoblotted using an anti-ULK1 antibody. (C) MUL1 knockdown cells transfected with GFP-ULK1 were subjected to ubiquitination assays for analysis with an anti-Ub antibody. (D) Quantitative analysis of ubiquitinated GFP-ULK1 level as described in (C) (The intensity of bands was measured with Image J software. mean±SEM; from 3 independent experiments). (E) HEK 293T cells were transfected with the indicated plasmids for 24 h, and subsequently a ubiquitination assay was performed for analysis with an anti-Ub antibody. (F) HeLa cells transfected with GFP-ULK1 were treated with 5 μM of selenite for the indicated time, followed by ubiquitination assays for analysis with an anti-Ub antibody. (G) HeLa cells were treated with 5 μM of selenite for the indicated time, followed by ubiquitination assays for analysis with an anti-Ub antibody. (H) Scrambled RNA- or siMUL1-transfected cells were treated with or without selenite for 12 h, followed by ubiquitination assays for analysis with an anti-Ub antibody. (I) NIH-3T3 cells were treated with 5 μM of selenite for the indicated time, followed by ubiquitination assays for analysis with an anti-Ub antibody. (J) NIH-3T3 cells were treated with selenite for the indicated time, with or without MG132, and subjected to western blotting analysis of the ULK1 protein level.
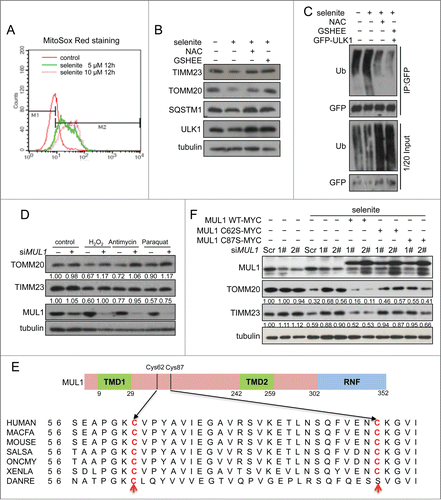
Selenite-induced mitophagy was inhibited by NAC and GSH-EE
Previous reports including our own studies have suggested that the perturbation of the redox system and subsequent ROS generation are causally linked to the effects induced by selenite. Also, ROS were able to trigger both general autophagy and mitophagy. We thus were prompted to test if ROS are important for selenite-induced MUL1-ULK1-dependent mitophagy. We first detected selenite-induced ROS production by MitoSox staining and found that selenite induced ROS production in a dose-dependent manner (). Moreover, selenite-induced mitochondrial protein degradation and ULK1 ubiquitination were significantly attenuated by the ROS scavenging agent NAC (N-Acetyl-L-cysteine) and GSH-EE (glutathione reduced ethylester), respectively (). Knockdown of MUL1 by siMUL1 prevented mitochondrial protein degradation by selenite and other known ROS inducers (Fig. 6D). These results suggested that MUL1 was able to sense the mitochondrial ROS signal. The alignment among species from human to zebrafish show 2 conserved cysteines at Cys62 and Cys87 on the intermembrane loop of MUL1 (). MYC-tagged mutants C62S and C87S were then constructed and transfected into HeLa cells, and both mutants located well on mitochondria as wild-type MUL1-MYC did (Fig. S8). It is interesting to find that these 2 mutants failed to restore selenite-induced mitophagy compared with that of wild-type MUL1 in MUL1 knockdown cells (). Collectively, our data suggest that mitochondrial ROS are involved in selenite-induced MUL1-ULK1-dependent mitophagy, which was probably facilitated by the conserved cysteine on MUL1.
Figure 6. MUL1 response to selenite-induced ROS stress depending on conserved cysteines 62 and 87. (A) HeLa cells treated with selenite as indicated were stained with MitoSox Red and subjected to flow cytometry. (B) HeLa cells were pretreated with NAC (10 mM) or GSH-EE (10 mM) for 30 min, followed by treatment with 5 μM of selenite for 12 h. Cell lysates were subjected to western blotting analysis of the indicated proteins. (C) HeLa cells transfected with GFP-ULK1 were pretreated with NAC (10 mM) or GSH-EE (10 mM) for 30 mins, followed by treatment of selenite (12 h). The ubiquitination assays were performed as described in Materials and Methods. (D) Alignment of the MUL1 amino acids in different species. (E) HeLa cells with MUL1 knockdown were transfected with exogenous MYC-tagged wild-type MUL1 or MUL1 with cysteine mutations (C62S, C87S), followed by treatment with selenite (5 μM, 12 h) before western blotting analysis of the indicated proteins (The intensity of the indicated bands was measured with ImageJ software.).
Discussion
The results of the present study demonstrated that ULK1 is a novel substrate of the mitochondria-localized E3 ligase MUL1. Here, we demonstrated that selenite-induced mitophagy is dependent on ULK1 and ATG5, but not PARK2 (). We showed that MUL1 mediates the ubiquitination of ULK1, for the subsequent degradation of this protein. Notably, the knockdown of MUL1 or expression of the RING mutant, lacking E3 ligase activity, did not show these effects. As ULK1 is a key regulator for intracellular energy sensing and the initiation of both autophagy and mitophagy, these results suggest a novel association between mitochondria and autophagy regulation.
It is rather paradoxical that MUL1 enhances mitophagy and promotes ULK1 degradation. Indeed, we have observed that mitophagy is inhibited in Ulk1-deficient MEF cells after selenite treatment or MUL1 overexpression. In addition, ULK1 partially translocated to mitochondria. It is likely that under normal conditions, MUL1, as an E3 ligase, monitors the mitochondrial outer membrane qualityCitation20,38,39 and prevents ULK1 from initiating mitophagy. However, under stress conditions, higher amounts of ROS within the intermembrane space promote ULK1 translocation to mitochondria to initiate mitophagy. In addition, we showed that MUL1-mediated mitophagy is highly sensitive to mitochondrial ROS, and both NAC and GSH-EE diminish intracellular ROS and prevent selenite-induced mitophagy (). We also identified 2 highly conserved cysteine residues in MUL1, and the mutation of these 2 cysteine rendered cells insensitive to ROS (). MUL1 is also induced through ROS, likely mediated the redox-sensitive FOXO1 transcription factor.Citation20 In mammalian cells, BNIP3L/NIX,Citation40 FUNDC1Citation23 and PINK1-PARK2 pathway participate in selective mitophagy. Our results suggested that MUL1-ULK1 mediated mitophagy pathway was independent with PARK2 and FUNDC1 pathway (; Fig. S7).
Our results showed that ULK1 is a novel substrate of MUL1, and the ULK1 protein level is regulated by MUL1 E3 ligase-dependent proteasomal pathway (). We have shown that ULK1 is translocated onto mitochondria where it interacts with MUL1 upon selenite treatment. Similarly, it was reported that both FCCP and hypoxia induce ULK1 transloaction and mitophagy.Citation41 As a master regulator of autophagy and mitophagy, ULK1 activity is tightly regulated by AMPK and MTOR, which are the key energy sensing kinases. AMPK is able to directly activate ULK1 through phosphorylation of Ser317 and Ser777 while MTOR prevents ULK1 activation by phosphorylating ULK1 at Ser757.Citation42 Activated ULK1 (a mammalian ortholog of yeast Atg1) can also phosphorylate ATG9Citation43 and BECN1 for the initiation of autophagy.Citation28 Previous studies have also shown that ULK1 stability is regulated through its interaction with AMBRA1, which recruits the E3-ligase TRAF6 and promotes ubiquitination of Lysine-63-linked chains.Citation44 To our knowledge, MUL1 is the first mitochondrial E3 ligase for regulating the stability of ULK1, which play a critical role for selenite-induced mitophagy. Our work further supports mitochondria as a platform for both general autophagy and mitophagy through regulating ULK1 stability.
MUL1 has been widely implicated in regulating mitochondrial dynamics.Citation20,45 The E3-active, C-terminal RING finger of MUL1 faces the cytosol, where this domain has access to the components of the Ub system and other cytosolic substrates.Citation39 MUL1 promotes the sumoylation of DNM1L/Drp1 to promote mitochondrial fission,Citation38,46 and retards mitochondrial fusion through the ubiquitination and degradation of MFN2, involved in mitophagy during muscle wasting.Citation20 Our result showing that MUL1-induced mitochondrial fragmentation was not restrained by merely expressing MFNs also supported the duel function of MUL1 in both MFN and Drp1-dependent mitochondrial morphology control (Fig. S6). It also remains to be examined whether MUL1 can sumoylate ULK1 that may protect it from degradation and this may also account for the paradoxical role of MUL1 in ULK1-mediated autophagy initiation and termination. Additionally, the ubiquitination sites of ULK1 need to address and will help us to understand the roles of ULK1 in both mitophagy and autophagy. Mitochondria undergo constant fission and fusion, providing a sorting mechanism for dysfunctional mitochondria to undergo mitophagy.Citation47 These results suggest that MUL1 plays integrative roles in both mitochondrial segregation and subsequent mitophagy. The precise molecular details of these roles warrant further investigation.
Supranutritional levels of selenium have benefits in preventing several types of cancer, including lung, colorectal, and prostate cancers. Previous studies have shown that selenite induces mitochondrial superoxide flash,Citation48 autophagyCitation49,50 and apoptosisCitation21 in response to graded oxidative stress. MUL1-mediated protein ubiquitination likely plays an integrative role in graded stress responses, including proteasomal protein degradation, mitochondrial segregation, autophagy, and apoptosis.Citation20,39 The results obtained in the present study suggested potential roles for MUL1 and ULK1 in selenite-induced mitophagy, representing novel aspects of the beneficial effects of selenium for chemoprevention.
Materials and Methods
Reagents and antibodies
The following antibodies were purchased from commercial manufacturers as indicated. BD Biosciences: TIMM23 (611222), TOMM20 (612278), SQSTM1 (610832), BECN1 (612113). Santa Cruz Biotechnology: MYC (SC-40), GFP (SC-9996), HA (SC-7392), ULK1 (SC-33182). Sigma: polyclonal anti-MYC (C3956), Flag (F7425), GFP (G1544), ATG5 (A0731), ATG7 (A2856). MBL: LC3B (PM036). Sungene: tubulin (KM9003). Biomol: monoclonal anti-Ub (PW8805). Polyclonal MUL1 antiserum was generated after immunizing rabbits with affinity-purified peptides MUL1.
The following fluorescent secondary antibodies were purchased from Invitrogen: goat anti-mouse IgG-FITC (1:200; 626312); goat anti-mouse-Cy3 (1:200; M30010); and goat anti-mouse-Cy5 (1:500; M35011). BAF was purchased from Sangon Biotech (BF1116), and MG132 was purchased from Selleck Chem (S2619).
Cell culture and plasmid transfection
The Ulk1−/− MEFs used in this study was kindly provided by Prof. Li Yu (Tsinghua University, Beijing, China). Ulk1−/− MEFs, Atg5−/− MEFs, park2−/− MEFs, HeLa, and HEK 293T cells were grown in DMEM (GIBCO, 11965-118) supplemented with 10% fetal bovine serum (HyClone, SV30087.02) and 1% penicillin/streptomycin at 37°C under 5% CO2. Full-length MUL1 and ULK1 cDNA were cloned into the pcDNA4/TO/myc-His B (Invitrogen, V103020) and pEGFP-C1(BD Biosciences Clontech, 6084-1) vectors, respectively. Truncations and mutations of MUL1 were generated through PCR using different primers and the plasmids pcDNA4TO- MUL1 and pEGFP-C1-MUL1 as templates. Truncations of ULK1 were generated through PCR using different primers and the plasmids pcDNA4TO-ULK1 as templates. DNA transfections were performed using PEI (Polysciences, 23966) according to the manufacturer's instructions.
RNA interference
The siRNA directed against MUL1 and the scrambled, nontargeting control RNA, was obtained from Guangzhou RiboBio Co., Ltd. For the siRNA-mediated knockdown of MUL1 expression in HeLa cells, 1.3 × 10Citation5 cells/mL were transfected with siRNA against human MUL1 at a final concentration of 50 nM using Lipofectamine RNAiMAX (Invitrogen, 13778075), according to the manufacturer's instructions, in serum-free medium and changed into DMEM containing 10% fetal bovine serum at 4 h after transfection. Subsequently, the cells were incubated at 37°C under 5% CO2 and harvested at the indicated times.
Western blotting, immunoprecipitation, and GST affinity isolation
Following treatment, the cells were lysed in lysis buffer (20 mM Tris, pH 7.4, 137 mM NaCl, 2 mM EDTA, 10% glycerol, 1% NP-40 (NEW industry, 728601), and protease inhibitor cocktail (Roche, 04693132001) for 40 min on ice as previously described.Citation51 The gray scale values of the bands were measured using ImageJ software. For immunoprecipitation, the cells were lysed for 40 min on ice in lysis buffer containing protease inhibitors (Roche Applied Science, 04693132001). The cell lysates were incubated with the indicated antibodies, followed by incubation with protein A/G-agarose beads (Abmart, A10001) overnight at 4°C. The beads were extensively washed with lysis buffer. The immune complexes were dissolved in sample buffer containing 1% SDS for 5 min at 95°C analyzed through SDS-PAGE and western blotting. For GST affinity isolation using HeLa cell lysates, 4 μg of GST-MUL1 protein was incubated with glutathione Sepharose 4 fast flow beads (Amersham Biosciences, 17-5132-01) for 2 h at 4°C, followed by washing 5 times with 1 mL lysis buffer. The beads were incubated with 500 μL (∼1,000 μg) of whole HeLa cell lysates at 4°C overnight, followed by washing 5 times with 1 mL lysis buffer. The precipitate complex was eluted in sample buffer, followed by boiling for 5 min at 95°C and analysis through SDS-PAGE and western blotting.
Recombinant protein purification and protein expression in vitro
Bacterial expression constructs (pGEX-4T-1; Amersham Pharmacia biotech, 27-4580-01) containing the indicated genes were transformed into Trans BL21 (DE3) (Transgen Biotech, CD601-01). Protein expression was induced with 0.05 mM IPTG (isopropyl β-d-1-thiogalactopyranoside; Sangon Biotech, SD1007) at 18°C for 12 h. The cells were resuspended in phosphate buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4) containing 1% FOS-CHOLINE-12 ANAGRADE (Affymetrix, F308), followed by ultrasonication. The recombinant proteins were isolated and eluted using Glutathione-Sepharose 4 Fast Flow beads according to the manufacturer's instructions. The purified proteins were dialyzed against 20 mM Tris, pH 8.0. ULK1 was translated in vitro using the TNT® SP6 High-Yield Wheat Germ Protein Expression System (Promega, L3261) according to the manufacturer's instructions.
Real-time PCR analysis
RNA was prepared using Trizol reagent (Invitrogen, H10522). cDNA was synthesized using the SuperScript VILOTM cDNA synthesis kit (Invitrogen, 11754-050). Real-time PCR was performed using a FastStart Universal SYBR® Green Master (Rox) (Roche Applied Science, 13800300) probe and the thermal cycler (Mastercycler; Eppendorf, 22331 Hamburg, Germany). Result analysis was performed using software RealPlex Service Real-time PCR System (Eppendorf).
Immunofluorescence microscopy
The cells were grown to 60% confluence on coverslips. After treatment, the cells were fixed with freshly prepared 3.7% formaldehyde at 37°C for 15 min, followed by washing 3 times with PBS. Antigen accessibility was increased after treatment with 0.2% Triton X-100 (ROTH, 3010244) on ice for 10 min. The cells were subsequently incubated with primary antibodies at room temperature for 1 h or 4°C overnight, followed by washing 4 or 5 times with 0.05% Triton X-100 and staining with the secondary antibody for 1 h at room temperature. The cell images were captured with TCS SP5 (Leica Microsystems Inc., Buffalo Grove, IL) confocal and Axio Imager Z1 (ZEISS, Regionalbüro, Hamburg,) microscopes. Colocalization analysis was performed as previously describedCitation41 using software ImageJ.
Subcellular fractionation
The cells were collected and resuspended in hypotonic buffer. After gentle homogenization using a Dounce homogenizer, the cell lysates were subjected to differential and gradient centrifugation. The resulting membrane fractions were lysed and analyzed through western blotting.
Ubiquitination assays
HEK 293T cells were transfected with the indicated tagged constructs in each experiment using PEI. The cells were treated with 5 μM MG132 for 6 h, followed by harvesting. The cells were subsequently lysed in denaturing buffer containing 50 mM Tris (pH 7.4), 70 mM β-mercaptoethanol and 0.1% sodium dodecyl sulfate (SDS; Sangon Biotech, SS0228), with protease inhibitors. The cell lysate was incubated with anti-GFP antibody, followed by immunoprecipitation with protein A/G-agarose beads. The precipitates were subjected to western blotting with an antibody against HA or Ub. An in vitro ubiquitination assay was performed as previously described.Citation52 Briefly, 2 μg of GST, GST-MUL1, or GST-MUL1ΔR (MUL1 lacking the RING finger domain), purified using an E. coli expression system, was incubated with in vitro translated ULK1 (2 μg) in 50 μL of ubiquitination reaction buffer, containing 50 mM Tris-HCl (pH 7.5), 5 mM MgCl2, 2 mM DTT, 2 mM ATP (Promega, P113B), 10 μg ubiquitin (Millipore, R0704D003B), 100 ng E1 (Upstate, 0611046111) and 200 ng E2 (UbcH5; Upstate, 0611046121). The reaction was incubated for 2 h at 25°C and terminated through the addition of SDS loading buffer. The reaction products were subsequently analyzed through western blotting with an anti-ULK1 antibody.
Electron microscopy analysis
For electron microscopy, HeLa cells transfected with MUL1 for 24 h were fixed with 2.5% glutaraldehyde for 2 h, followed by postfixation in 2% osmium tetroxide. Subsequently, the cells were dehydrated with sequential washes in 50, 70, 90, 95, and 100% ethanol. The ultrathin sections were collected on copper grids (smdj-0000; KYKY Technology) and counterstained using uranyl acetate and lead citrate. The images were captured using a transmission electron microscope (JEM-2100; JEOL, 3-1-2 Musashino, Akishimashi, Tokyo, Japan).
Statistical analysis
For quantitative analyses of cultured cells represented as histograms, values were obtained from 3 independent experiments and expressed as the mean±SEM. Statistical analyses were performed using one- and 2-way ANOVAS with Tukey's HSD as post-hoc test or the Student t test. Significance levels of * P < 0.05,**P < 0.01,***P < 0.001 vs. the corresponding controls are indicated.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1017180_Supplemental_Material.zip
Download Zip (2.7 MB)Acknowledgments
We are grateful to Professor Li Yu from Tsinghua University for generously providing Ulk1−/− MEFs.
Funding
This research was supported by the 973 program project (2011CB910903 to Q.C and No 2010CB91220 to Y.Z) from MOST and the Natural Science Foundation of China (31471300, 81130045, 31271529).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Wallace DC, Brown MD, Melov S, Graham B, Lott M. Mitochondrial biology, degenerative diseases and aging. BioFactors 1998; 7:187-90; PMID:9568243; http://dx.doi.org/10.1002/biof.5520070303
- Green DR, Reed JC. Mitochondria and apoptosis. Science 1998; 281:1309-12; PMID:9721092; http://dx.doi.org/10.1126/science.281.5381.1309
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283:10892-903; PMID:18281291; http://dx.doi.org/10.1074/jbc.M800102200
- Russell RC, Yuan HX, Guan KL. Autophagy regulation by nutrient signaling. Cell Res 2014; 24:42-57; PMID:24343578; http://dx.doi.org/10.1038/cr.2013.166
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 2008; 112:1493-502; PMID:18539900; http://dx.doi.org/10.1182/blood-2008-02-137398
- Petrosillo G, Ruggiero FM, Di Venosa N, Paradies G. Decreased complex III activity in mitochondria isolated from rat heart subjected to ischemia and reperfusion: role of reactive oxygen species and cardiolipin. FASEB J 2003; 17:714-6; PMID:12586737; http://dx.doi.org/10.1096/fj.03-0012com
- Djavaheri-Mergny M, Amelotti M, Mathieu J, Besancon F, Bauvy C, Codogno P. Regulation of autophagy by NFkappaB transcription factor and reactives oxygen species. Autophagy 2007; 3:390-2; PMID:17471012; http://dx.doi.org/10.4161/auto.4248
- Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J 2007; 26:1749-60; PMID:17347651; http://dx.doi.org/10.1038/sj.emboj.7601623
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010; 141:656-67; PMID:20478256; http://dx.doi.org/10.1016/j.cell.2010.04.009
- Sugiura A, Nagashima S, Tokuyama T, Amo T, Matsuki Y, Ishido S, Kudo Y, McBride HM, Fukuda T, Matsushita N, et al. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol Cell 2013; 51:20-34; PMID:23727017; http://dx.doi.org/10.1016/j.molcel.2013.04.023
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9-14; PMID:21179058; http://dx.doi.org/10.1038/nrm3028
- Zhu J, Wang KZ, Chu CT. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 2013; 9:1663-76; PMID:23787782; http://dx.doi.org/10.4161/auto.24135
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005; 8:3-5; PMID:15798367; http://dx.doi.org/10.1089/rej.2005.8.3
- Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 2011; 286:19630-40; PMID:21454557; http://dx.doi.org/10.1074/jbc.M110.209338
- Neutzner A, Youle RJ, Karbowski M. Outer mitochondrial membrane protein degradation by the proteasome. Novartis Found Symp 2007; 287:4-14; discussion 14-20; PMID:18074628; http://dx.doi.org/10.1002/9780470725207.ch2
- Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 2008; 183:795-803; PMID:19029340; http://dx.doi.org/10.1083/jcb.200809125
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119-31; PMID:20098416; http://dx.doi.org/10.1038/ncb2012
- Nakamura N, Kimura Y, Tokuda M, Honda S, Hirose S. MARCH-V is a novel mitofusin 2- and Drp1-binding protein able to change mitochondrial morphology. EMBO Rep 2006; 7:1019-22; PMID:16936636; http://dx.doi.org/10.1038/sj.embor.7400790
- Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, Sharma M, Kambadur R. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab 2012; 16:613-24; PMID:23140641; http://dx.doi.org/10.1016/j.cmet.2012.10.005
- Huang F, Nie C, Yang Y, Yue W, Ren Y, Shang Y, Wang X, Jin H, Xu C, Chen Q. Selenite induces redox-dependent Bax activation and apoptosis in colorectal cancer cells. Free Radic Biol Med 2009; 46:1186-96; PMID:19439215; http://dx.doi.org/10.1016/j.freeradbiomed.2009.01.026
- Kralova V, Benesova S, Cervinka M, Rudolf E. Selenite-induced apoptosis and autophagy in colon cancer cells. Toxicol Vitro 2012; 26:258-68; PMID:22200533; http://dx.doi.org/10.1016/j.tiv.2011.12.010
- Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 2012; 14:177-85; PMID:22267086; http://dx.doi.org/10.1038/ncb2422
- Tang F, Wang B, Li N, Wu Y, Jia J, Suo T, Chen Q, Liu YJ, Tang J. RNF185, a novel mitochondrial ubiquitin E3 ligase, regulates autophagy through interaction with BNIP1. PloS One 2011; 6:e24367; PMID:21931693; http://dx.doi.org/10.1371/journal.pone.0024367
- Fu M, St-Pierre P, Shankar J, Wang PT, Joshi B, Nabi IR. Regulation of mitophagy by the Gp78 E3 ubiquitin ligase. Mol Biol Cell 2013; 24:1153-62; PMID:23427266; http://dx.doi.org/10.1091/mbc.E12-08-0607
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23:33-42; PMID:9639028; http://dx.doi.org/10.1247/csf.23.33
- Karanasios E, Stapleton E, Manifava M, Kaizuka T, Mizushima N, Walker SA, Ktistakis NT. Dynamic association of the ULK1 complex with omegasomes during autophagy induction. J Cell Sci 2013; 126(Pt 22):5224-38; PMID:24013547
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741-50; PMID:23685627; http://dx.doi.org/10.1038/ncb2757
- Mizushima N, Sugita H, Yoshimori T, Ohsumi Y. A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J Biological Chem 1998; 273:33889-92; PMID:9852036; http://dx.doi.org/10.1074/jbc.273.51.33889
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392:605-8; PMID:9560156; http://dx.doi.org/10.1038/33416
- Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell 2009; 20:1981-91; PMID:19211835; http://dx.doi.org/10.1091/mbc.E08-12-1248
- Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem 2009; 284:12297-305; PMID:19258318; http://dx.doi.org/10.1074/jbc.M900573200
- Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Øvervatn A, Kirkin V, Johansen T. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem 2012; 287:39275-90; PMID:23043107; http://dx.doi.org/10.1074/jbc.M112.378109
- Joo JH, Dorsey FC, Joshi A, Hennessy-Walters KM, Rose KL, McCastlain K, Zhang J, Iyengar R, Jung CH, Suen DF, et al. Hsp90-Cdc37 chaperone complex regulates Ulk1- and Atg13-mediated mitophagy. Mol Cell 2011; 43:572-85; PMID:21855797; http://dx.doi.org/10.1016/j.molcel.2011.06.018
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 2011; 20:1726-37; PMID:21296869; http://dx.doi.org/10.1093/hmg/ddr048
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 2010; 191:1367-80; PMID:21173115; http://dx.doi.org/10.1083/jcb.201007013
- Tanaka K, Suzuki T, Hattori N, Mizuno Y. Ubiquitin, proteasome and parkin. Biochim Biophys Acta 2004; 1695:235-47; PMID:15571819; http://dx.doi.org/10.1016/j.bbamcr.2004.09.026
- Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep 2009; 10:748-54; PMID:19407830; http://dx.doi.org/10.1038/embor.2009.86
- Bae S, Kim SY, Jung JH, Yoon Y, Cha HJ, Lee H, Kim K, Kim J, An IS, Kim J, et al. Akt is negatively regulated by the MULAN E3 ligase. Cell Res 2012; 22:873-85; PMID:22410793; http://dx.doi.org/10.1038/cr.2012.38
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454:232-5; PMID:18454133; http://dx.doi.org/10.1038/nature07006
- Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 2012; 125:1488-99; PMID:22275429; http://dx.doi.org/10.1242/jcs.094110
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, et al. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell 2014; 53:471-83; PMID:24440502; http://dx.doi.org/10.1016/j.molcel.2013.12.011
- Nazio F, Strappazzon F, Antonioli M, Bielli P, Cianfanelli V, Bordi M, Gretzmeier C, Dengjel J, Piacentini M, Fimia GM, et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat Cell Biol 2013; 15:406-16; PMID:23524951; http://dx.doi.org/10.1038/ncb2708
- Li W, Bengtson MH, Ulbrich A, Matsuda A, Reddy VA, Orth A, Chanda SK, Batalov S, Joazeiro CA. Genome-wide and functional annotation of human E3 ubiquitin ligases identifies MULAN, a mitochondrial E3 that regulates the organelle's dynamics and signaling. PloS One 2008; 3:e1487; PMID:18213395; http://dx.doi.org/10.1371/journal.pone.0001487
- Scorrano L, Liu D. The SUMO arena goes mitochondrial with MAPL. EMBO Rep 2009; 10:694-6; PMID:19525920; http://dx.doi.org/10.1038/embor.2009.141
- Feng D, Liu L, Zhu Y, Chen Q. Molecular signaling toward mitophagy and its physiological significance. Exp Cell Res 2013; 319:1697-705; PMID:23603281; http://dx.doi.org/10.1016/j.yexcr.2013.03.034
- Ma Q, Fang H, Shang W, Liu L, Xu Z, Ye T, Wang X, Zheng M, Chen Q, Cheng H, et al. Superoxide flashes: early mitochondrial signals for oxidative stress-induced apoptosis. J Biol Chem 2011; 286:27573-81; PMID:21659534; http://dx.doi.org/10.1074/jbc.M111.241794
- Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, Choi KS. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res 2007; 67:6314-24; PMID:17616690; http://dx.doi.org/10.1158/0008-5472.CAN-06-4217
- Mehta SL, Kumari S, Mendelev N, Li PA. Selenium preserves mitochondrial function, stimulates mitochondrial biogenesis, and reduces infarct volume after focal cerebral ischemia. BMC Neurosci 2012; 13:79; PMID:22776356; http://dx.doi.org/10.1186/1471-2202-13-79
- Zhu Y, Li M, Wang X, Jin H, Liu S, Xu J, Chen Q. Caspase cleavage of cytochrome c1 disrupts mitochondrial function and enhances cytochrome c release. Cell Res 2012; 22:127-41; PMID:21577235; http://dx.doi.org/10.1038/cr.2011.82
- Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 2011; 286:11649-58; PMID:21292769; http://dx.doi.org/10.1074/jbc.M110.144238