
Abstract
Autophagy inhibition has been widely accepted as a promising therapeutic strategy in cancer, while the lack of effective and specific autophagy inhibitors hinders its application. Here we found that liensinine, a major isoquinoline alkaloid, inhibits late-stage autophagy/mitophagy through blocking autophagosome-lysosome fusion. This effect is likely achieved via inhibiting the recruitment of RAB7A to lysosomes but not to autophagosomes. We further investigated the effects of autophagy inhibition by liensinine on the therapeutic efficacy of chemotherapeutic drugs and found that cotreatment of liensinine markedly decreased the viability and increased apoptosis in breast cancer cells treated with various chemotherapeutic agents. Mechanistically, we found that inhibition of autophagy/mitophagy by liensinine enhanced doxorubicin-mediated apoptosis by triggering mitochondrial fission, which resulted from dephosphorylation and mitochondrial translocation of DNM1L. However, blocking autophagosome/mitophagosome formation by pharmacological or genetic approaches markedly attenuated mitochondrial fission and apoptosis in cells with combinatatorial treatment. Moreover, liensinine was synergized with doxorubicin to inhibit tumor growth in MDA-MB-231 xenograft in vivo. Our findings suggest that liensinine could potentially be further developed as a novel autophagy/mitophagy inhibitor, and a combination of liensinine with classical chemotherapeutic drugs could represent a novel therapeutic strategy for treatment of breast cancer.
Abbreviations
| 3-MA | = | 3-methyladenine |
| ATG | = | autophagy-related |
| Baf | = | bafilomycin A1 |
| CDDP | = | cisplatin |
| COX4I1 | = | cytochrome c oxidase subunit IV isoform 1 |
| CQ | = | chloroquine |
| DNM1L | = | dynamin 1-like |
| Dox | = | doxorubicin |
| EGFP | = | enhanced green fluorescent protein |
| HCQ | = | hydroxychloroquine |
| H&E | = | hemotoxylin and eosin |
| HSPD1/HSP60 | = | heat shock 60kDa protein 1 (chaperonin) |
| LAMP | = | lysosomal-associated membrane protein |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| Lien | = | liensinine |
| mRFP | = | modified red fluorescent protein |
| Rapa | = | rapamycin |
| RFP | = | red fluorescent protein |
| siRNA | = | small interfering RNA |
| SQSTM1 | = | sequestosome 1 |
| TEM | = | transmission electron microscopy |
| tfLC3 | = | tandem fluorescent LC3 |
| TUNEL | = | TdT-mediated dUTP nick-end labeling. |
Introduction
Autophagy is a homeostatic cellular degradation system that is responsible for degrading damaged or unnecessary cellular organelles such us the endoplasmic reticulum, mitochondria, and Golgi apparatus.Citation1 Mitophagy, selective mitochondrial degradation through autophagy, is an evolutionarily conserved cellular process used for the elimination of excessive and damaged mitochondria in eukaryotes.Citation2 During autophagy, parts of the cytoplasm and cellular organelles are engulfed within a double-membraned vesicle called the autophagosome. The autophagosome fuses with a lysosome to form an autolysosome, which results in the degradation of the sequestered materials by various lysosomal hydrolytic enzymes.Citation1 Autophagy plays an important role in various human diseases, including cancer.Citation3,4 It is becoming increasingly clear that altered autophagy activity is associated with tumor formation and progression as well as with altered response to cancer therapy.Citation5,6 Since autophagy is often a prosurvival response to chemotherapeutic drugs in cancer cells, suppression of autophagy during chemotherapy has been proposed as a novel therapeutic strategy.Citation7,8 At present, only chloroquine (CQ) and its derivative hydroxychloroquine (HCQ) are currently used in patients as inhibitors of autophagy and recent reports demonstrated the feasibility and beneficial effects of using CQ or HCQ, either alone or in combination in cancer therapy.Citation9-11 Therefore, development of a new type of autophagy inhibitor for the treatment of cancer has important clinical significance.
Liensinine, a major isoquinoline alkaloid (), extracted from the seed embryo of Nelumbo nucifera Gaertn, has a wide range of biological activities, including anti-arrhythmias, anti-hypertension, anti-pulmonary fibrosis, relaxation on vascular smooth muscle, etc.Citation12,13 As liensinine and neferine share a similar pharmacophore, they exhibit anticancer activity. For instance, neferine has been reported to inhibit the proliferation in acute leukemic cells, and increase the sensitivity of imatinib (STI 571) and doxorubicin to K562 cells.Citation14 More recently, natural compounds from alkaloids including liensinine exhibit anticancer effects through the modulation of MTOR-dependent autophagy.Citation15 However, the exact mechanism by which liensinine regulates autophagy in human breast cancer cells remains unclear.
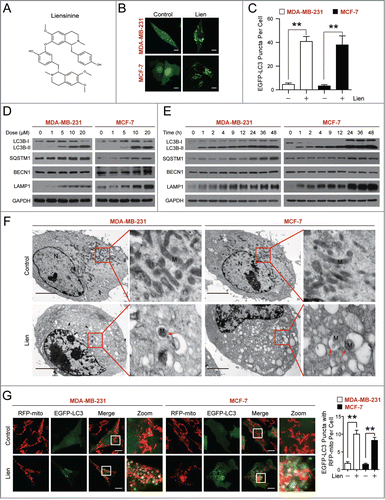
Figure 1 (See previous page). Liensinine induces autophagic/mitophagic alterations in MDA-MB-231 and MCF-7 cells. (A) The chemical structure of liensinine. (B) EGFP-LC3 expressing MDA-MB-231 and MCF-7 cells were treated without or with liensinine (Lien, 20 μM) for 24 h, the EGFP-LC3 puncta were observed under confocal microscopy; scale bars: 10 μm. (C) Quantification of average EGFP puncta per cell in (B) from 3 independent experiments. Data was presented as mean ± SD (**P < 0.01); 50 cells were analyzed per treatment condition. (D and E) Cells were exposed to various concentrations of Lien for 24 h, or treated with 20 μM Lien for different time intervals as indicated. The expression of autophagy-related proteins (LC3B-I/LC3B-II, SQSTM1, BECN1 and LAMP1) was detected by western blot analysis. GAPDH was used as a loading control. (F) Representative TEM images depicting ultrastructure of MDA-MB-231 and MCF-7 cells treated without or with Lien (20 μM) for 24 h. N, nucleus; M, mitochondria; red arrows indicates autophagic vacuoles. Scale bars: 2 μm. (G) Confocal microscopy images of MDA-MB-231 and MCF-7 cells treated without or with Lien (20 μM) for 24 h after co-expressing RFP-mito and EGFP-LC3; scale bars: 10 μm. Quantitation of EGFP puncta with RFP-mito per cell. Data was presented as mean ± SD (**P < 0.01); 50 cells were analyzed per treatment condition.
In the present study, we investigated the effect of liensinine on modulation of autophagy in human breast cancer cells. We found for the first time that liensinine potently inhibited autophagosome-lysosome fusion, leading to the accumulation of autophagosomes/mitophagosomes. This effect is likely due to inhibiting the recruitment of RAB7A to lysosomes but not to autophagosomes. We also investigated the effects of autophagy inhibition by liensinine on anticancer potency of a number of chemotherapeutic drugs. Cotreatment of liensinine markedly decreased the viability and increased apoptosis in cells treated with chemotherapy. Importantly, liensinine is more potent to reduce cell viability in combination with doxorubicin as compared to chloroquine or bafilomycin A1. Mechanistically, we found that inhibition of autophagy/mitophagy by liensinine enhanced doxorubicin-mediated apoptosis by excessive accumulation of autophagosomes/mitophagosomes (“autophagic stress”)Citation16-18 and triggering mitochondrial fission, which resulted from dephosphorylation and mitochondrial translocation of DNM1L. In addition, inhibition of autophagosome/mitophagosome formation at an early stage by pharmacological or genetic approaches significantly attenuated mitochondrial fission and apoptosis in cells treated with the combinatorial therapy. These findings suggest that the accumulation of autophagosomes/mitophagsomes is implicated in the combination-treatment mediated DNM1L dephosphorylation and mitochondrial translocation, resulting in mitochondrial fission and apoptosis. The synergistic effect of liensinine and doxorubicin was further confirmed in vivo using a mouse xenograft model. Our findings thus demonstrate that inhibition of autophagy/mitophagy with liensinine potently enhances the efficacy of chemotherapy, and that such a combination may represent a novel therapeutic strategy for treatment of breast cancer.
Results
Liensinine enhances LC3B-II stability and puncta formation in multiple cancer cells
To determine whether liensinine affects autophagy in human breast cancer cells, we first used MDA-MB-231 and MCF-7 cells transiently expressing EGFP-LC3. Autophagosome accumulation can be detected with a confocal laser-scanning microscope. Treating cells with liensinine resulted in a marked increase in EGFP-LC3 puncta formation in MDA-MB-231 and MCF-7 cells (). During autophagy, LC3B is cleaved by ATG4 to generate the cytoplasmic form LC3B-I (18 kDa), which can be further modified and converted to the phagophore-associated LC3B-II form (16 kDa) through conjugating with the lipid phosphatidylethanolamine.Citation19 The conversion of LC3B-I to LC3B-II is commonly used to evaluate autophagy activity. Next, we examined the effect of liensinine on LC3B conversion in both MDA-MB-231 and MCF-7 cells. Western blot analysis showed that liensinine resulted in dose- and time-dependent accumulation of LC3B-II in both cell lines (). Besides LC3B, levels of other autophagy-related (ATG) proteins such as LAMP1 (lysosomal-associated membrane protein 1), SQSTM1 (a ubiquitin-binding receptor protein), and BECN1 (a critical autophagy-regulating protein) were determined using western blot analysis. As shown in , liensinine treatment caused the pronounced accumulation of LAMP1 and SQSTM1 in dose- and time-dependent manners. In contrast, the protein level of BECN1 remained unchanged in response to liensinine. Since SQSTM1 serves as a link between LC3 and ubiquitinated substrates, inhibition of autophagy correlates with increased levels of SQSTM1 in mammals.Citation20 Thus, these observations indicate that liensinine is probably a potent autophagic flux inhibitor. To investigate whether the effect of liensinine in LC3 and SQSTM1 were breast-cancer specific, we also tested the protein level of LC3 and SQSTM1 in U937, LN229, and A549 cells with liensinine treatment. As with MCF-7 cells, liensinine treatment caused the marked accumulation of LC3B-II and SQSTM1 in these cell lines (Fig. S1).
To further determine the characteristics of autophagy in MDA-MB-231 and MCF-7 cells treated with liensinine, transmission electron microscopy (TEM) was used to observe the ultrastructural changes in these cells. The TEM studies revealed an increased number of autophagic vacuoles in the liensinine-treated cells compared with control cells (). Interestingly, we clearly observed some partially divided mitochondria () and some mitophagy structures suggesting that abnormal mitochondria were fused with autophagic vesicles or surrounded by double membranes of typical autophagosomes. To further confirm whether liensinine affects mitophagy, we used fiuorescent-labeled markers for autophagosomes (EGFP-LC3) and mitochondria (RFP-mito) in MDA-MB-231 and MCF-7 cells to assess colocalization of autophagosomes with mitochondria. We found the increased colocalization of EGFP-LC3 and RFP-mito in liensinine-treated cells compared to that in control cells (), indicating that liensinine could influence the process of mitophagy in MDA-MB-231 and MCF-7 cells.
Liensinine suppresses autophagic degradation
Our earlier observations showed that treatment with liensinine increased the levels of SQSTM1 in multiple cancer cells, suggesting that liensinine may act as a potent autophagic flux inhibitor and inhibit autophagic degradation. To further confirm these observations, we examined the effects of liensinine on the EGFP fluorescence intensity in MDA-MB-231 cells with stable expression of EGFP-LC3 in the presence or absence of bafilomycin A1, a vacuolar-type ATPase inhibitor that blocks autophagic degradation.Citation21 Flow cytometry analysis revealed that treatment with either liensinine or bafilomycin A1 alone significantly increased the total EGFP intensity, and combined treatment with liensinine and bafilomycin A1 did not show any further significant changes in EGFP intensity compared with bafilomycin A1 treatment alone (). We also examined the effects of liensinine on accumulation of LC3B-II and SQSTM1 in the presence or absence of bafilomycin A1 using western blot analysis. As shown in , liensinine treatment resulted in accumulation of LC3B-II and SQSTM1, which was similar to the result caused by bafilomycin A1. However, compared with liensinine or bafilomycin A1 treatment alone, combined treatment with liensinine and bafilomycin A1 did not show any significant changes in accumulation of LC3B-II and SQSTM1, suggesting that liensinine-mediated accumulation of LC3B-II through an autophagic-lysosomal blockade of LC3B-II degradation. In contrast, treatment with rapamycin (Rapa), which is known to induce autophagy, resulted in a slight increase in levels of LC3B-II that were further enhanced by bafilomycin A1 (). Furthermore, treatment with rapamycin led to decreased SQSTM1 levels that were markedly reversed by bafilomycin A1 (). These results indicate that liensinine acts similar to bafilomycin A1 by blocking autophagic degradation and is different from rapamycin, which promotes autophagic degradation.
Figure 2. Liensinine inhibits autophagic degradation in MDA-MB-231 cells. (A) MDA-MB-231 cells stably expressing EGFP-LC3 were treated without or with Lien (20 μM) in the presence or absence of 25 nM Baf for 24 h, and total EGFP intensity was measured by flow cytometry. Data are presented as mean ± SD from 3 independent experiments (n.s, not significant, **P < 0.01). (B) Cells were treated without or with Lien (20 μM), or Rapa (0.25 μM) in the presence or absence of 25 nM Baf for 4 h or 24 h; the expression of SQSTM1 and LC3B-II was analyzed by western blot. (C) Cells were transfected with a tandem reporter construct (tfLC3), and were exposed to Lien (20 µM), Baf (20 nM) and Rapa (0.25 μM) as indicated. The colocalization of EGFP and mRFP-LC3 puncta was examined by confocal microscopy. Scale bars: 10 μm. (D) Cells were treated without or with Rapa (0.25 μM) in the presence or absence of 20 μM Lien for 24 h, the expression of SQSTM1 and LC3B-II was analyzed by western blot. Comparisons of the intensities were statistically estimated and represented as mean ± SD for 3 independent experiments (n.s, not significant; **P < 0.01).
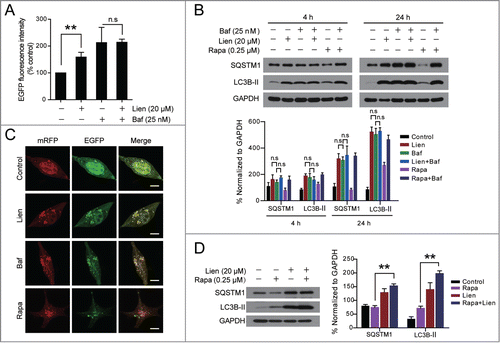
To further examine whether the effects of liensinine are due to suppressed autophagic flux, cells transfected with a tandem reporter construct (tfLC3) were treated with liensinine (20 µM, 24 h) followed by assessment of EGFP-LC3 and mRFP-LC3 puncta colocalization. EGFP-fluorescence is quenched in acidic environments (such as that encountered in the autolysosome), whereas mRFP is more stable under acidic conditions.Citation20 Indeed, exposure to liensinine caused pronounced formation of LC3 puncta that displayed both green and red fluorescence intensity producing a yellow overlay (). Furthermore, cells exposed to bafilomycin A1 displayed an LC3-fluorescence pattern (puncta formation with yellow overlay) that was mimicked by liensinine treatment (). In contrast, cells exposed to rapamycin led to the production of large amounts of red-only puncta (). Thus, these data clearly demonstrate that liensinine suppresses autophagy at a late stage. We examined the effects of liensinine on LC3B-II degradation induced by rapamycin. Exposing cells to rapamycin resulted in a slight increase in levels of LC3B-II that were further enhanced by liensinine (). In addition, treatment with rapamycin led to decreased SQSTM1 levels that were markedly reversed by liensinine (). Together, these finding suggest that liensinine inhibits a late stage of autophagy, thereby resulting in a marked accumulation of autophagosomes.
Liensinine blocks autophagosome-lysosome fusion
In the late stage of autophagy, fusion of autophagosomes with lysosomes leads to the formation of autolysosomes. Inhibition of this process impairs autophagic degradation.Citation6 To address whether liensinine affects autophagosome-lysosome fusion, we examined the colocalization of EGFP-LC3 and LysoTracker Red, a specific dye for live cell lysosome labeling. Notably, EGFP-LC3 puncta were greatly increased in liensinine-treated cells and did not colocalize with LysoTracker Red, which was similar to the cells treated with bafilomycin A1, which blocks the fusion of autophagosomes with lysosomes (). In contrast, rapamycin treatment induced a remarkable increase of EGFP-LC3 puncta, which were well colocalized with LysoTracker Red (). The intensity of LysoTracker Red dye can be changed by pH alteration, and therefore we examined the colocalization of mRFP-LC3 and LAMP1-mGFP, a marker for endosomal and lysosomal membranes. As shown in , liensinine-treated cells exhibited separation of mRFP-LC3 and LAMP1-mGFP, which was similar to that in cells treated with bafilomycin A1. In contrast, mRFP-LC3 was well colocalized with LAMP1-mGFP during rapamycin treatment. Examination of the endogenous colocalization of LC3 and LAMP1 by immunofluorescence confirmed our discoveries (Fig. S2). These findings indicate that liensinine inhibits autophagy through blockade of autophagosome-lysosome fusion.
Figure 3. Liensinine blocks autophagosome-lysosome fusion by preventing RAB7A recruitment to lysosomes. (A) MDA-MB-231 cells were transiently transfected with EGFP-LC3 and treated with vehicle, Lien (20 µM), Baf (20 nM) or Rapa (0.25 μM) for 24 h. The fluorescent signals were detected by confocal microscopy after staining with LysoTracker Red. Scale bars: 10 µm. (B) Cells cotransfected with LAMP1-mGFP and mRFP-LC3, were treated and detected for fluorescent signals as in (A). Scale bars: 10 µm. (C) Cells were treated without or with 20 μM Lien for different time intervals as indicated; the expression of LAMP2 and RAB7A in whole cell lysates were determined by western blot. The intensities of band normalization to GAPDH is represented as mean ± SD for 3 independent experiments. (D and E) Cells were treated without or with Lien (20 µM) or Rapa (0.25 µM) for 24 h, whole cell lysate was prepared and subjected to immunoprecipitation using anti-LAMP1 (left panel) or anti-RAB7A (right panel), and the associated LC3B-I/LC3B-II, RAB7A, and LAMP1 were determined using immunoblotting. (F and G) The representative images of MDA-MB-231 cells stained for RAB7A (green), LAMP1 (red) or transfected with mRFP-LC3 after treating with Lien (20 µM) or Rapa (0.25 µM) for 24 h. The Pearson's correlation coefficient (Rr) of RAB7A and LAMP1 or mRFP-LC3 colocalization were represented as mean ± SD (n.s, not significant; *P < 0.05; **P < 0.01), 50 cells. Scale bars: 10 μm.
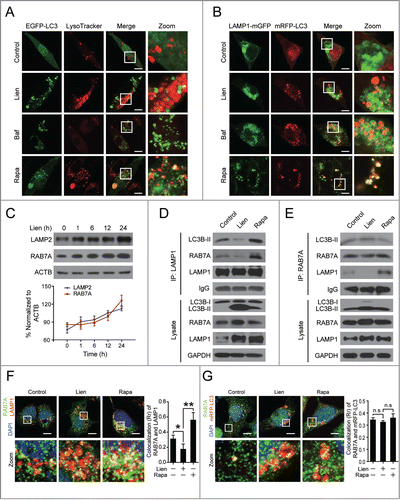
Since RAB7A, LAMP1, and LAMP2 are critical for autophagosome-lysosome fusion,Citation7 we examined the effects of liensinine on the expression of RAB7A, LAMP1, and LAMP2 using western blot analysis. Treating cells with liensinine resulted in increases in LAMP1 protein levels in a time-dependent manner (). Liensinine treatment also caused an increase of RAB7A protein and LAMP2 protein (), suggesting that the mechanism by which liensinine blocked autophagosome-lysosome fusion was not due to reduced expression of RAB7A, LAMP1, and LAMP2. Thus, we further assessed the recruitment of RAB7A to lysosomes or autophagosomes. Immunoprecipitation analysis confirmed that the interaction of LAMP1 with LC3B-II or RAB7A was decreased by liensinine treatment, whereas these interactions were increased by rapamycin treatment (). Intriguingly, no changes in the interaction of RAB7A and LC3B-II were observed in liensinine-treated cells (). Similarly, immunofluorescence analysis revealed that the colocalization of LAMP1 and RAB7A was decreased by liensinine treatment, but increased by rapamycin treatment, while the colocalization of EGFP-LC3 and RAB7A was not changed (). Our findings thus suggest that the liensinine-mediated blockade of autophagosome-lysosome fusion was due to impaired recruitment of RAB7A to lysosomes but not autophagosomes.
Liensinine influences the endocytic pathway and lysosomal cathepsin maturation but not lysosomal pH
The recruitment of RAB7A to lysosomes is clearly required for the process of multiple membrane fusion events including lysosome fusion with late endosomes and autophagosomes.Citation22,23 To determine whether liensinine blocks late endosome-lysosome fusion as well, we examined the effect of liensinine on the expression of CTSB, CTSD and CTSL, which are largely dependent on the endocytic pathway for transport into the lysosome for maturation.Citation24-26 As shown in , treating cells with liensinine resulted in increases in the levels of the precursor/pro-forms of CTSB, CTSD, and CTSL, but marked decreases in the levels of the mature forms. In contrast, treating with rapamycin resulted in marked increases in the levels of mature CTSB, CTSD, and CTSL, but decreased the expression of the pro-cathepsins ().
Figure 4. Liensinine blocks cathepsin maturation without alteration of lysosomal pH. (A and B) MDA-MB-231 cells were treated without or with various concentrations of Lien or Rapa (0.25 µM) for 24 h, total cellular extracts were prepared and subjected to western blot using antibodies against CTSB, CTSD and CTSL. Quantification represents the relative protein levels of the cells normalized to GAPDH (mean ± SD for 3 independent experiments). (C) MDA-MB-231 cells were treated without or with Lien (20 µM) or Baf (25 nM) for 24 h, after which cells were stained with acridine orange and analyzed by flow cytometry. The quantification shows the relative fluorescence intensity ratio normalized to control (mean ± SD for 3 independent experiments; **P < 0.01). (D) MDA-MB-231 cells were treated without or with Lien (20 µM) or Baf (25 nM) for 24 h, and stained with LysoSensor Yellow/Blue DND-160 (1 µM) for 5 min; the fluorescence of live cells was detected by confocal microscopy exciting at 365 nm, and the overlays of the blue (450 nm), and green (510 nm) fluorescence and bright field (DIC) of each treatment are shown. Scale bars: 10 μm. (E) MDA-MB-231 cells were treated without or with Lien (20 µM) or Baf (25 nM) for 24 h; the pH in lysosome was determined by using LysoSensor Yellow/Blue DND-160 staining and a microreader. (F) Confocal microscopy images of MDA-MB-231 cells immunostained for CTSD (green) and LAMP1 (red) after treatment with Lien (20 μM) or Rapa (0.25 µM). The average Pearson's correlation coefficient of CTSD and LAMP1 colocalization are marked. Scale bars: 10 μm.
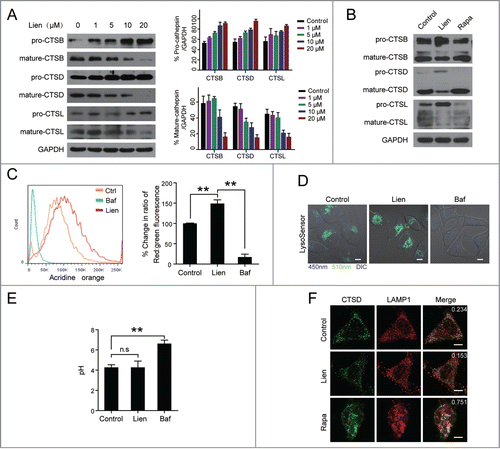
Since acidic pH in the lysosomal lumen is required for proteolytic activation of lysosomal proteases including cathepsins, we next assessed the effect of liensinine on the lysosomal pH by the acridine orange method (AO), which was used to detect acidic vesicles. As shown in , treating cells with liensinine resulted in a significant increase in acidic vesicles which display red fluorescence compared to the control. In contrast, treating with bafilomycin A1 resulted in a considerable decrease in acidic vesicles. To further monitor alterations in the lysosomal pH caused by liensinine, LysoSensor Yellow/Blue DND-160, a ratiometric fluorescence probe, was used to selectively label the acidic organelles of living cells. This dye preferentially partitions into acidic compartments (excluding the nucleus) and appears blue when the pH is neutral or alkaline and green/yellow when the pH is acidic. Similar to that in the control, liensinine-treated cells display punctate green/yellow staining of lysosomes throughout the cytoplasm. In contrast, a shift in fiuorescence was observed from green/yellow to blue in cells-treated with bafilomycin A1, indicating an elevated pH (). Using a fluorescence reader to quantitatively measure the pH of lysosomes stained with LysoSensor Yellow/Blue DND-160, there was no significant difference between control and liensinine treatment (pH ∼4.5, ). Thus, our data provide clear evidence that liensinine treatment does not affect the lysosomal pH.
To confirm whether liensinine affects the lysosomal pro-CTSD, we examined the colocalization of CTSD with lysosomes using immunofluorescent staining in MDA-MB-231 cells treated without or with liensinine. As shown in , the colocalization of CTSD and LAMP1 was observed in the control cells, whereas the colocalization patterns were nearly abolished in the liensinine-treated cells. Collectively, these data demonstrate that the blockage of the endocytic pathway-dependent cathepsin transport to lysosomes by liensinine is mostly due to the reduced recruitment of RAB7A to lysosomes and subsequently impaired lysosome-endosome fusion.
Autophagy inhibition in combination with chemotherapy is more effective than monotherapy in reducing viability and inducing apoptosis in breast cancer cells via the mitochondrial pathway
Increasing evidence indicates that inhibiting autophagy enhances the efficacy of chemotherapy by abolishing chemoresistance and increasing cancer cell death.Citation27,28 We therefore determined whether autophagy inhibition by liensinine would sensitize breast cancer cells to chemotherapy, including several clinically available anticancer drugs (i.e., doxorubicin, paclitaxel, vincristine, and cisplatin) and the mitochondrial damage agents (e.g., staurosporine). Cotreatment of MDA-MB-231 and MCF-7 cells with a nontoxic concentration of liensinine (20 μM) and minimal toxic concentrations of doxorubicin, paclitaxel, vincristine, cisplatin, and staurosporine resulted in decreases in cell viability compared to monotherapy alone ().
Figure 5. Liensinine sensitizes to a variety of anticancer drug-induced cell death in MDA-MB-231 and MCF-7 cells. (A) MDA-MB-231 and MCF-7 cells were treated with various concentrations of doxorubicin (Dox), paclitaxel, vincristine, cisplatin (CDDP), and staurosporine (STS) in the presence or absence of 20 μM Lien for 48 h, and MTT assays were performed to assess cell proliferation. (B) Cells were treated with increasing doses of Dox in the presence or absence of chloroquine (CQ, 20 μM), Baf (25 nM) or Lien (20 μM) for 48 h, and cell proliferation were measured by MTT assay (mean ± SD for 3 independent experiments; **P < 0.01, n.s, not significant). (C) Cells were cotreated with 20 μM Lien and Dox (0.2 μM for MDA-MB-231 and 0.4 μM for MCF-7 cells) for 48 h, and apoptosis was determined by ANXA5-FITC/PI staining and flow cytometry (mean ± SD for 3 independent experiments; **P < 0.01 compared with control or Lien and Dox treatment alone). (D) Cells were treated as in (C), total cellular extract and cytosol fractions were prepared and subjected to western blot using antibodies against PARP1, cleaved-CASP9 (C-CASP9), cleaved-CASP3 (C-CASP3), and CYCS/cytochrome c (cytosol fraction).
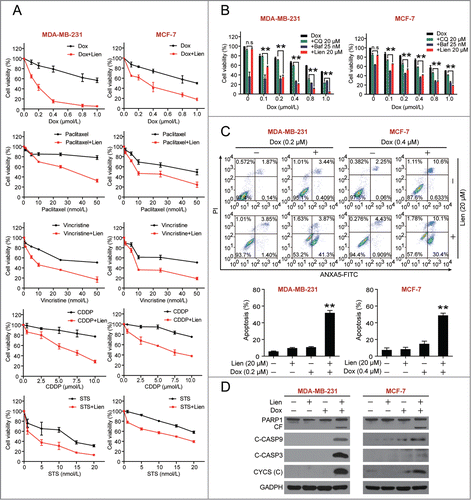
CQ is a potent autophagy inhibitor that is FDA-approved and available for clinical trial,Citation29 and bafilomycin A1 is a specific V-ATPase inhibitor that blocks autophagic degradation. Similar to liensinine, both CQ and bafilomycin A1 have synergistic effects with doxorubicin (). Moreover, liensinine exhibited more intense accumulation of LC3B-II compared with CQ at the same concentration (Fig. S3). The synergistic effect of liensinine and doxorubicin was more potent than that of CQ and doxorubicin at the same concentrations (Fig. S4).
Since liensinine exhibited the potent combination effect with doxorubicin, we focused on this combination for the consequent studies to identify the underlying molecular mechanisms of this synergism. We assessed the synergistic effect between liensinine and doxorubicin in breast cancer cells on apoptosis by using the ANXA5 (annexin V)-FITC propidium iodide (PI) staining assay. Treating cells with liensinine or doxorubicin alone minimally increased apoptosis (∼5% to ∼10%), whereas the combined treatment markedly increased apoptosis (∼50%) (). Consistent with these findings, combined treatment with liensinine and doxorubicin resulted in increases in cleavage/activation of CASP9 and CASP3, as well as PARP1 degradation (). Similar results were also found with combined treatment of cells with doxorubicin and CQ or bafilomycin A1 (Fig. S5).
We further assessed the effects of the combination of liensinine and doxorubicin on mitochondrial integrity. Whereas exposure of MDA-MB-231 cells to liensinine or doxorubicin alone for 48 h had no evident effects, a combination of liensinine and doxorubicin resulted in a pronounced increase in release of CYCS/cytochrome c from the mitochondria to the cytosol, a hallmark of apoptosis induction via the mitochondrial pathway ().
Liensinine sensitizes doxorubicin-induced cell death via excessive accumulation of autophagosomes/mitophagosomes
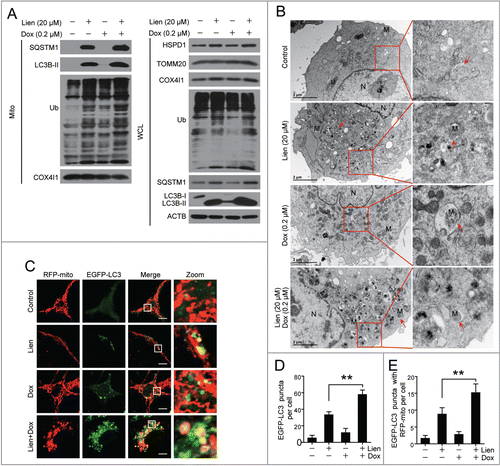
Our earlier data indicate that autophagy/mitophagy serves as a prosurvival function in doxorubicin-induced cell death, whereas liensinine suppresses autophagic/mitophagic flux. We therefore tested whether the synergistic effect of the combination treatment with liensinine and doxorubicin was due to the suppression of autophagic/mitophagic flux by liensinine. Liensinine treatment alone increased accumulation of LC3B-II, SQSTM1 and ubiquitin in the whole cellular extract and the mitochondrial fraction, and a combination of liensinine and doxorubicin further enhanced LC3B-II, SQSTM1 and ubiquitin accumulation (, left panel). Furthermore, treating with liensinine alone resulted in increased levels of HSPD1 and COX4I1, the mitochondrial markers, which were enhanced by the combination of liensinine and doxorubicin (, right panel). Furthermore, the combination of liensinine and doxorubicin increased the amounts of autophagic/mitophagic vacuoles, and damaged swollen mitochondria with altered morphology and aberrant inner-membrane cristae (). Similarly, immunofluorescence microscopy analysis showed that the combination of liensinine and doxorubicin increased the autophagosome/mitophagosome quantity, evidenced by an increase in EGFP-LC3 puncta and colocalization of EGFP-LC3 puncta with RFP-mito compared to liensinine treatment alone (). This colocalization was also observed between EGFP-LC3 puncta and MitoTracker Red CMXRos (Fig. S6). These findings indicate that liensinine is able to sensitize breast cancer cells to doxorubicin-induced cell death probably through the suppression of autophagy/mitophagy and the excessive accumulation of autophagosomes/mitophagosomes.
Figure 6. Excessive accumulation of autophagosomes/mitophagosomes mediated by combination of liensinine and doxorubicin in MDA-MB-231 cells. (A) Cells were treated with vehicle, Lien (20 μM), Dox (0.2 μM) alone or combined treated with Lien and Dox for 48 h; whole cell lysates (WCL) and mitochondrial fractions (Mito) were prepared and subjected to western blot using antibodies against LC3B-II, SQSTM1, ubiquitin (Ub), HSPD1, TOMM20, COX4I1, and ACTB. (B) Representative TEM images of cells treated as in (A), N, nucleus; M, mitochondria; red arrows indicates autophagic vacuoles. Scale bars: 2 μm. (C) Cells were transiently cotransfected with RFP-mito and EGFP-LC3 then treated indicated as in (A), and fluorescence images were determined by confocal microscopy. Scale bars: 10 μm. (D and E) Quantification of EGFP-LC3 puncta, RFP-mito and EGFP-LC3 colocalization puncta in 50 cells of 3 independent experiments. (mean ± SD **P < 0.01 compared with Lien treatment alone).
Liensinine combined with doxorubicin induces mitochondrial fission via dephosphorylation and mitochondrial translocation of DNM1L
A number of studies have demonstrated that mitochondrial fission participates in Bax-mediated permeabilization of the outer mitochondrial membrane and CYCS release.Citation30,31 Our data shown above indicate that liensinine markedly enhances doxorubicin-induced apoptosis through the mitochondrial pathway. We therefore examined the effects of the combination of liensinine and doxorubicin on mitochondrial dynamics using MitoTracker Red CMXRos. As shown in , control cells had filamentous mitochondria. Liensinine or doxorubicin treatment alone caused a slight increase in the proportion of cells with fragmented mitochondria. Notably, the combination of liensinine and doxorubicin resulted in a significant increase in the proportion of cells with fragmented mitochondria (). Similar results were also found in cells treated with doxorubicin in combination with chloroquine or bafilomycin A1 ().
Figure 7. Autophagic inhibition combined with doxorubicin induces mitochondrial fission via dephosphorylation and mitochondrial translocation of DNM1L. (A and D) MDA-MB-231 cells were treated with vehicle, Lien (20 μM), Dox (0.2 μM) alone, or combined treated with Lien plus Dox, CQ plus Dox, and Baf plus Dox for 48 h; mitochondrial morphology was observed by MitoTracker Red CMXRos staining and confocal microscopy. Scale bars: 10 μm in (A) and 20 μm in (D). (B and C) Mitochondrial length and percentage of cells with mitochondrial fragmentation were measured with ImageJ software. 50 cells of 3 independent experiments (mean ± SD *P < 0.05, **P < 0.01 compared with vehicle treatment alone). (E) After treatment in (A), whole cell lysates (WCL), mitochondrial (Mito) and cytosolic (Cyto) fractions were prepared and subjected to western blot using antibodies against DNM1L, and p-DNM1L (Ser616 or Ser637). (F) Cells were exposed to Lien, CQ, and Baf in the presence or absence of Dox for 48 h; WCL, Mito, and Cyto fractions were prepared and subjected to western blot using antibodies as indicated. (G) After treatment in (A), cells were double stained with MitoTracker Red and DNM1L (Alexa Fluor 647, green). Fluorescence images were collected using confocal microscopy. The average Pearson's correlation coefficient of MitoTracker Red and DNM1L colocalization are marked. Scale bars: 10 μm.
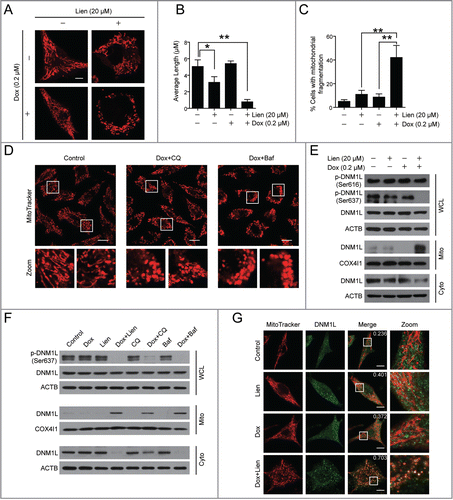
Increasing evidence has revealed that DNM1L plays a critical role in the regulation of mitochondrial fission through its translocation from the cytosol to the mitochondria.Citation31 We then asked whether mitochondrial translocation of DNM1L is involved in mitochondrial fission found in cells treated with a combination of liensinine and doxorubicin. Western blot analysis showed that the combination of liensinine and doxorubicin increased the levels of DNM1L in mitochondria and decreased that in the cytosol, whereas liensinine or doxorubicin treatment alone had little or no effect (). Similarly, a combination of doxorubicin with chloroquine or bafilomycin A1 also increased the levels of DNM1L in the mitochondria and decreased the levels of DNM1L in the cytosol, whereas chloroquine or bafilomycin A1 treatment alone had little effect (). Consistent results were found from the immunofluorescence analysis showing DNM1L signal at the mitochondria in cells treated with a combination of liensinine and doxorubicin ().
It has been reported that dephosphorylated DNM1L (Ser637) or phosphorylated DNM1L (Ser616) promotes DNM1L translocation from the cytosol to mitochondria and increases mitochondrial fission.Citation32 We then examined the effect of a combination of liensinine and doxorubicin on phosphorylation of DNM1L at Ser637 and Ser616. Combination of liensinine and doxorubicin resulted in a decrease in the levels of phospho-DNM1L at Ser637, but had no effect on phosphorylation of DNM1L at Ser616 (). Similarly, combined treatment of doxorubicin with chloroquine or bafilomycin A1 also markedly decreased the levels of phospho-DNM1L at Ser637 (). Taken together, these findings suggest that dephosphorylation and mitochondrial translocation of DNM1L are involved in mitochondrial fission induced by liensinine in combination with doxorubicin.
Pharmacological or genetic DNM1L inhibition blocks mitochondrial fission and apoptosis induced by liensinine in combination with doxorubicin
To further address the causative role of DNM1L in mitochondrial fission observed in cells treated with a combination of liensinine and doxorubicin, Mdivi-1, a selective inhibitor of DNM1L,Citation33 was employed. Pretreatment with Mdivi-1 markedly reduced DNM1L translocation from the cytosol to the mitochondria (). Interestingly, pretreatment with Mdivi-1 also reduced accumulation of LC3B-II and SQSTM1 in the mitochondrial fraction (). Consistently, immunofluorescence microscopy revealed that pretreatment with Mdivi-1 markedly blocked the mitochondrial localization of DNM1L in cells treated with a combination of liensinine and doxorubicin (). Subsequently, pretreatment with Mdivi-1 blocked mitochondrial fission ().
Figure 8 (See previous page). Pharmacological or genetic inhibition of DNM1L blocks mitochondrial fission and apoptosis induced by liensinine in combination with doxorubicin. (A) MDA-MB-231 cells were co-exposed to Lien (20 μM) and Dox (0.2 μM) in the presence or absence of Mdivi-1 (10 μM) for 48 h; whole cell lysates (WCL), cytosol fractions (Cyto) and mitochondrial fractions (Mito) were prepared and subjected to western blot analysis. (B) Cell were treated as indicated in (A), and the intracellular localization of TOMM20 (red), DNM1L (green), EGFP-LC3 and RFP-mito were evaluated by immunofluorescence confocal microscopy. Scale bars: 10 μm. (C) Quantification of colocalization of puncta of EGFP-LC3 with RFP-mito and average mitochondrial length. 50 cells were counted in 3 independent experiments (mean ± SD **P < 0.01). (D) Cells were treated as indicated in (A), apoptosis were evaluated by flow cytometry (mean ± SD **P < 0.01), and the expression of C-CASP3 and CYCS (cytosol fraction) were determined by western blot. (E) MDA-MB-231 cells were transfected with control siRNA (Sc-siRNA) and DNM1L siRNA; the expression of DNM1L was determined by western blot. (F) After combined treatment with Lien (20 μM) and Dox (0.2 μM), WCL and Mito fractions were prepared and subjected to western blot using antibodies against LC3B-I/LC3B-II and SQSTM1. (G) The localization of TOMM20 (red) and DNM1L (green), or RFP-mito and EGFP-LC3 in cells treated indicated as in (F). Scale bars: 10 μm. (H) Quantification of average mitochondrial length and the colocalization of puncta corresponding to EGFP-LC3 and RFP-mito. 50 cells were counted in 3 independent experiments (mean ± SD **P < 0.01). (I) Treatment indicated as in (F); apoptosis was determined by flow cytometry (mean ± SD **P < 0.01), and the expression of C-CASP3 and CYCS (cytosol fraction) were determined by western blot. (J) MDA-MB-231 cells transfected with DNM1LWT-mCherry, DNM1LS637A-mCherry and DNM1LS637D-mCherry were treated without or with doxorubicin (0.2 μM) in the presence or absence of liensinine (20 μM), western blot was used to detect DNM1L, p-DNM1L (Ser 637), C-CASP3 in whole cell lysate and CYCS in cytosol fractions. Apoptosis was determined by ANXA5-FITC/PI staining and flow cytometry (mean ± SD for 3 independent experiments; *P < 0.05, **P < 0.01).
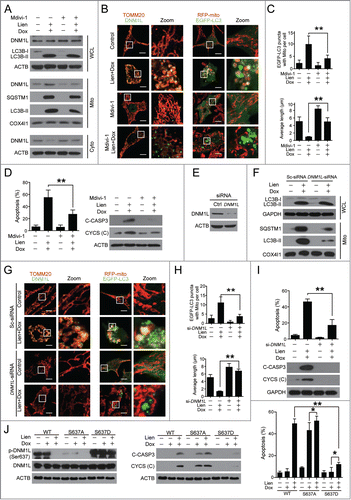
Since mitochondrial fission participates in mitophagy,Citation34 we therefore asked whether inhibition of mitochondrial fission by Mdivi-1 blocks the autophagosome/mitophagosome formation mediated by the combination of liensinine and doxorubicin. Immunofluorescence microscopy analysis showed that the combination of liensinine and doxorubicin increased the colocalization of EGFP-LC3 puncta and RFP-mito, which was blocked by pretreatment with Mdivi-1 ().
Last, we determined whether inhibition of mitochondrial fission by Mdivi-1 blocked apoptosis induced by the combination of liensinine and doxorubicin. Pretreatment with Mdivi-1 significantly reduced combination-induced apoptosis (). Consistent with these results, pretreatment with Mdivi-1 also blocked combination-mediated CASP3 cleavage and CYCS release ().
To exclude nonspecific effects of Mdivi-1, we next tested whether specific suppression of DNM1L with small interfering RNA (siRNA) affects combination-induced mitochondrial fission and apoptosis. Transfecting cells with DNM1L siRNA reduced DNM1L expression by more than 50% (). Knockdown of DNM1L reduced the combination-mediated accumulation of LC3B-II in mitochondria (). Immunofluorescence microscopy indicated that knockdown of DNM1L markedly blocked the combination-mediated localization of DNM1L at the mitochondria and mitochondrial fission (). In addition, knockdown of DNM1L markedly reduced the colocalization of EGFP-LC3 puncta and RFP-mito (). Lastly, knockdown of DNM1L significantly reduced combination-induced apoptosis, CASP3 cleavage, as well as CYCS release ().
To further investigate the role of dephosphorylation of DNM1L at Ser637 in apoptosis mediated by the combination of doxorubicin and liensinine, we generated a mutant of DNM1LS637D-mCherry to mimic the phosphorylated form or a mutant of DNM1LS637A-mCherry to mimic the dephosphorylated form (). Overexpression of DNM1LS637D significantly decreased apoptosis and caspase activation induced by doxorubicin at higher concentration (5 μM) (Fig. S7). We further investigated the synergistic effect of liensinine and doxorubicin on apoptosis in a DNM1LS637A mutant or a DNM1LS637D mutant, and found that cotreatment with liensinine slightly enhanced doxorubicin-mediated apoptosis, CASP3 activation, and CYCS release in the DNM1LS637A mutant. In addition, overexpression of the DNM1LS637D mutant obviously attenuated apoptosis, activated/cleaved CASP3 (C-CASP3), and CYCS release mediated by the combination of doxorubicin and liensinine (). We also found that DNM1LS637D mutant-mediated suppression of apoptosis, which was induced by doxorubicin, could be slightly overcome by liensinine (). Similar results were obtained by autophagy inhibition with CQ (Fig. S8). Taken together, these findings indicate that dephosphorylation and mitochondrial translocation of DNM1L play a critical role in mitochondrial fission and cell death induced by combination treatment with liensinine and doxorubicin.
Excessive accumulation of autophagosomes/mitophagsomes is implicated in the combination-mediated mitochondrial fission and apoptosis
As indicated above, doxorubicin induces the formation of autophagosomes, whereas liensinine suppresses autophagic flux by blocking autophagosome/mitophagosome-lysosome fusion. The combination of liensinine and doxorubicin resulted in excessive accumulation of autophagosomes/mitophagosomes. We therefore aimed to further test the involvement of excessive accumulation of autophagosomes/mitophagosomes in mitochondrial fission and apoptosis in cells treated with a combination of liensinine and doxorubicin. We treated cells with 3-methyladenine (3-MA, a widely used autophagy inhibitor) to inhibit formation of autophagosomes, then assessed the cell sensitivity to a combination of doxorubicin and liensinine. 3-MA pretreatment reduced combination-mediated LC3B-II accumulation () and autophagosome/mitophagosome formation (). Furthermore, pretreatment with 3-MA markedly inhibited combination-mediated dephosphorylation (Ser637) () and mitochondrial translocation of DNM1L (). Inhibition of autophagosome formation by 3-MA also prevented combination-induced mitochondrial fission (), apoptosis, CYCS release, and CASP3 activation ().
Figure 9 (See previous page). Autophagosome/mitophagosome accumulation contributed to the combination effect of liensinine and doxorubicin. MDA-MB-231 cells were co-exposed to Lien (20 μM) and Dox (0.2 μM) in the presence or absence of 3-MA (5 mM) for 48 h. (A) Whole cell lysate (WCL), cytosolic fractions (Cyto) and mitochondrial fractions (Mito) were prepared and subjected to western blot analysis. (B) The intracellular localization of EGFP-LC3 and RFP-mito were evaluated by confocal microscopy. Scale bars: 10 μm. The EGFP-LC3 puncta and EGFP-LC3 puncta with mitochondria were counted and presented as mean ± SD **P < 0.01. (C) The colocalization and Pearson's correlation coefficient (Rr) of TOMM20 (red) and DNM1L (green) treated as in (A). Scale bars: 10 μm. (D) Quantification of average mitochondrial length; 50 cells were counted in 3 independent experiments (mean ± SD **P < 0.01). (E) Apoptosis was determined by flow cytometry (mean ± SD **P < 0.01), and the expression of C-CASP 3 and CYCS (cytosol fraction) were detected by western blot. (F) MDA-MB-231 cells stably expressing Scramble-siRNA (si-Sc), ATG5-siRNA (si-ATG5) and ATG7-siRNA (si-ATG7) were cotreated without or with Lien (20 μM) and Dox (0.2 μM), WCL, Mito and Cyto fractions were prepared and subjected to western blot. (G) The localization of RFP-mito and EGFP-LC3 were evaluated by confocal microscopy. Scale bars: 10 μm. The EGFP-LC3 puncta and colocalization of puncta were counted and presented as mean ± SD **P < 0.01. (H) The colocalization and Rr of TOMM20 (red) and DNM1L (green) treated as in (F). Scale bars: 10 μm. (I) Quantification of average mitochondrial length. 50 cells were counted in 3 independent experiments (mean ± SD **P < 0.01). (J) Apoptosis was determined by flow cytometry (mean ± SD **P < 0.01), and the expression of C-CASP 3 and CYCS (cytosol fraction) were determined by western blot.
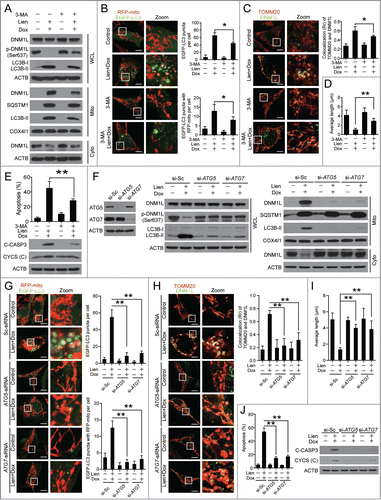
To exclude the possibility of nonspecific effects of 3-MA, a siRNA approach was used to stably knock down ATG5 or ATG7 expression. Knockdown of ATG5 or ATG7 reduced combination-mediated LC3B-II accumulation () and autophagosome/mitophagosome formation (). Knockdown of ATG5 or ATG7 also markedly reduced combination-mediated dephosphorylation (Ser637) () and mitochondrial translocation of DNM1L (). Furthermore, knockdown of ATG5 or ATG7 significantly reduced combination-mediated mitochondrial fission (), apoptosis, CASP3 activation, and CYCS release (). To avoid a misleading conclusion based solely on alterations of cell death kinetics,Citation35 clonogenic growth assays were performed to demonstrate long-term protection from cell death by genetic inhibition of ATG5 or ATG7. Knockdown of ATG5 or ATG7 exhibited long-term protection against inhibition of cell growth by a combination of liensinine and doxorubicin (Fig. S9). Taken together, these findings indicated that the excessive accumulation of autophagosomes/mitophagosomes is implicated in mitochondrial fission and apoptosis mediated by combination drug treatment in breast cancer cells.
Liensinine enhances the therapeutic efficacy of doxorubicin in a mouse xenograft model in vivo
To determine whether our in vitro findings that suppression of autophagy by liensinine can sensitize to doxorubicin-induced cell death could be replicated in vivo, we next examined the effect of liensinine on the therapeutic efficacy of doxorubicin in vivo using a mouse xenograft model. Nude mice were inoculated subcutaneously with MDA-MB-231 cells, followed by receiving liensinine (60 mg/kg, intraperitoneal) or doxorubicin (2 mg/kg, intraperitoneal) treatment alone, and a combination of liensinine with doxorubicin for 30 d starting one wk after tumor inoculation. As shown in , liensinine or doxorubicin treatment alone had minimal effect on the growth of tumors, whereas a combination of liensinine with doxorubicin led to a significant reduction in tumor growth compared to untreated controls (P < 0.01 for combination treatment vs. control, liensinine, or doxorubicin alone). However, no significant changes in body weight were observed among vehicle control, liensinine or doxorubicin treatment alone, and combination of liensinine with doxorubicin ().
Figure 10. The combination of liensinine and doxorubicin inhibits tumor growth in a MDA-MB-231 mouse xenograft model. (A) Average tumor volume in vehicle control mice, mice treated with Lien (60 mg/kg) or Dox (2 mg/kg) or a combination of Lien and Dox. Error bars represent means ± SD. **P < 0.01 compared with control, Lien or Dox alone. (B) Body weight changes of mice during 30 d of exposure. Statistical analysis of body weight changes showed no significant differences between combination of Lien with Dox and other groups including control, Lien or Dox treatment alone. (C) Tumor tissues were sectioned and subjected to H&E staining, TUNEL assay, and immunohistochemistry for evaluating histological morphology, apoptosis and expression of C-CASP3. Scale bars: 50 μm. (D) and (E) Tumor tissues were sectioned and subjected to immunofluorescence staining for determination of the colocalization of LC3 and HSPD1, or DNM1L and TOMM20 by confocal microscopy. The average Pearson's correlation coefficient of LC3 and HSPD1, or DNM1L and TOMM20 colocalization are marked. Scale bars: 50 μm.
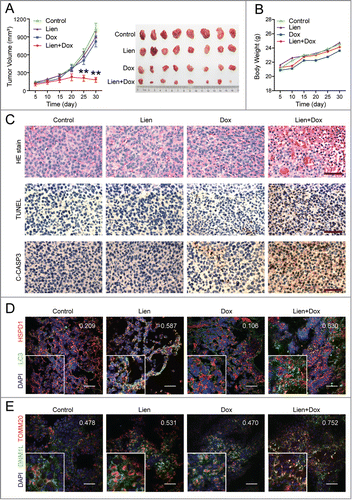
To evaluate whether the combination of liensinine with doxorubicin results in a morphological change and induction of apoptosis in MDA-MB-231 cells in vivo, excised tumor samples were sectioned and stained with hemotoxylin and eosin (H&E), TdT-mediated dUTP nick-end labeling (TUNEL), and analyzed by immunohistochemistry. H&E staining of sections of the tumors from mice treated with the liensinine-doxorubicin combination exhibited significant changes in morphology, with signs of necrosis with infiltration of inflammatory cells and fibrosis (, top panels). Either liensinine or doxorubicin treatment alone caused a minimal increase in the number of TUNEL-positive cells (brown color) compared with control. However, the liensinine and doxorubicin combination dramatically increased the number of TUNEL-positive cells compared with either control or treatment alone (, middle panels). In addition, the combination of liensinine with doxorubicin caused a marked increase in immunoreactivity for cleaved CASP3, indicative of apoptosis (, bottom panels).
To confirm whether liensinine sensitizes doxorubicin-induced cell death via suppression of autophagy/mitophagy in vivo, immunofluorescence microscopy analysis was employed. The combination of liensinine with doxorubicin increased the autophagosome/mitophagosome number measured by LC3 puncta and colocalization of LC3 puncta with HSPD1 compared to liensinine or doxorubicin treatment alone (). Our earlier findings have implied that mitochondrial translocation of DNM1L plays an important role in combination-induced mitochondrial fission and apoptosis in vitro. To test this possibility, immunofluorescence microscopy was performed to evaluate the subcellular localization of DNM1L before and after combination of liensinine with doxorubicin. As shown in , combination of liensinine with doxorubicin greatly increased the colocalization of DNM1L and TOMM20. Taken together, these findings suggest that liensinine potently enhances the therapeutic activity of doxorubicin in vivo by promoting DNM1L-dependent mitochondrial fission and cell death.
Discussion
In this report, we provide clear evidence, for the first time, that liensinine induced the accumulation of autophagosomes/mitophagosomes by inhibiting autophagosome-lysosome fusion. Autophagy is the basic catabolic metabolism that involves cell degradation of unnecessary or dysfunctional cellular components through the actions of lysosomes.Citation36 In eukaryotic cells, the autophagic pathway is initiated when parts of the cytoplasm and cellular organelles are engulfed within a double-membraned vesicle called the autophagosome. The autophagosome fuses with a lysosome to form an autolysosome, leading to sequestration and degradation of intracellular material. A great number of studies have demonstrated that autophagy can be inhibited pharmacologically through targeting the different stage of the autophagic process. The inhibitors that act at the early stage of autophagy like 3-MA and LY294002 inhibit the class III phosphatidylinositol 3-kinase (PIK3C3) and block the formation of autophagosomes.Citation37 The inhibitors that target a late stage of autophagy like chloroquine, bafilomycin A1, leupeptin, etc. either inhibit the fusion of autophagosomes with lysosomes or the degradation capacity of autolysosomes.Citation37 Similar to chloroquine, hydroxychloroquine, and bafilomycin A1, our studies demonstrated that liensinine induced the accumulation of autophagosomes/mitophagosomes by inhibiting autophagosome-lysosome fusion based on the following findings: (i) liensinine suppresses autophagic degradation, leading to accumulation of autophagosomes; (ii) liensinine blocks autophagosome-lysosome fusion; (iii) liensinine notably accumulates the substrates of autophagy; (iv) liensinine markedly attenuates the enzymatic maturation of several important lysosomal hydrolases. Although the effect of liensinine was similar to chloroquine, hydroxychloroquine, and bafilomycin A1, the mechanism of liensinine-mediated blockade of autophagosome-lysosome fusion was different from that of chloroquine, hydroxychloroquine, and bafilomycin A1. The latter agents alkalinized lysosomal pH to suppress the fusion between autophagosomes with lysosomes and interfere with the action of lysosomal hydrolases. However, the lysosomal pH was not changed in response to liensinine treatment, suggesting that the lysosomal pH is not necessary for inhibition of autophagosome-lysosome fusion and cathepsin maturation mediated by liensinine. The small GTP binding protein RAB7A has a role in the late endocytic pathway, lysosome biogenesis, and the final maturation of late autophagic vacuoles.Citation22 Based on our observation, we proposed that liensinine reduced recruitment of RAB7A to lysosomes thus inhibiting the endocytic pathway-dependent cathepsins transport to lysosomes, and blocking autophagosome-lysosome fusion.
In this study, we identified liensinine as a novel inhibitor of autophagy/mitophagy, which inhibits the fusion of autophagosomes with lysosomes. These data are in conflict with another report that indicated that the alkaloid compounds liensinine, isoliensinine, dauricine, and cepharanthine are able to induce autophagic-related cell death in cancer cells by inducing autophagy.Citation15 Unexpectedly, this report showed a controversial result indicating that the alkaloid compounds induced the accumulation of SQSTM1, which is commonly used as a marker of the inhibition of autophagy.Citation38 These findings are consistent with our results, which indicated that treating cells with liensinine caused the pronounced accumulation of SQSTM1, which could not be enhanced by bafilomycin A1 pretreatment. By using transmission electron microscopy, we did not observe the single-membrane enclosed structures containing electron-dense osmiophilic inclusions, which is a standard marker of microscopy-based examination of autolysosome structures,Citation20 after liensinine treatment. Furthermore, using immunofluorescence, we did not observe the fusion events between autophagosomes and lysosomes after liensinine treatment. Therefore, our findings suggest that liensinine functions as an inhibitor of autophagy/mitophagy by blocking autophagosome-lysosome fusion.
Therapeutic targeting of the autophagy pathway as a novel anticancer strategy has been under extensive investigation. Numerous studies suggest that the inhibition of autophagy enhances the efficacy of chemotherapy and targets therapy against cancer.Citation7 Several phase I/II clinical studies involving autophagy inhibition using FDA-approved chloroquine or hydroxychloroquine in combination with chemotherapy for the treatment of different cancers, including breast cancer, are currently under way.Citation9-11 Therefore, development of a new type of autophagy inhibitor for the treatment of cancer has important clinical significance. Inhibiting autophagy enhances the efficacy of chemotherapy, which is associated with increased tumor cell death. In the present study, we provide evidence that liensinine exhibits an inhibitory effect on autophagy/mitophagy that markedly enhances chemotherapeutic drugs-induced cell death. To the best of our knowledge, this is the first report to demonstrate the effectiveness of autophagy/mitophagy inhibition by liensinine in combination with doxorubicin in breast cancer cells in vitro and in vivo.
Notably, we demonstrated that inhibition of autophagy/mitophagy by liensinine sensitized breast cancer cells to doxorubicin-induced cell death through DNM1L-dependent mitochondrial fission. It is well documented that DNM1L translocates from the cytosol to the mitochondria to execute the mitochondrial fission process.Citation31 Mitochondrial fission leads to CYCS release and activation of caspases, which can ultimately lead to cell death. Increasing evidence reveals that mitochondrial translocation of DNM1L is regulated by various post-translational modifications.Citation32 Importantly, dephosphorylation of DNM1L at Ser637 regulates its translocation to mitochondria and subsequent mitochondrial fission, which leads to an increased response to apoptotic stimuli.Citation39 In this study, we demonstrate that dephosphorylation and mitochondrial translocation of DNM1L is necessary for the combination of liensinine and doxorubicin to induce mitochondrial fission and subsequent apoptosis. This conclusion is supported by several important observations: (i) Combination treatment of liensinine and doxorubicin induced dephosphorylation of DNM1L at Ser637; (ii) the combination of liensinine and doxorubicin caused pronounced translocation of DNM1L from the cytosol to the mitochondria; (iii) pretreatment with Mdivi-1, a selective inhibitor of DNM1L, markedly reduced combination-mediated DNM1L translocation from the cytosol to the mitochondria and blocked combination-induced mitochondrial fission and apoptosis; (iv) knockdown of DNM1L with siRNA potently reduced combination-mediated mitochondrial translocation of DNM1L, mitochondrial fission, and apoptosis; (v) overexpression of the DNM1LS637A mutant sensitized cells to doxorubicin-induced apoptosis, which was slightly enhanced by liensinine or CQ, and constant expression of the DNM1LS637D mutant attenuated doxorubicin-mediated apoptosis, which was slightly overcome by liensinine or CQ.
Currently the role of autophagy/mitophagy in cell survival and cell death remains controversial.Citation40 Several studies suggested that doxorubicin induced LC3 puncta formation, indicating that doxorubicin is capable of inducing autophagy in cancer cells.Citation41,42 We found that liensinine potently inhibited fusion of autophagosomes with lysosomes, leading to the accumulation of autophagosomes/mitophagosomes. We also found that inhibition of autophagy/mitophagy by liensinine potently enhanced doxorubicin-induced mitochondrial fission and apoptosis in breast cancer cells in vitro and in a mouse xenograft model in vivo. However, it is not yet clear how inhibition of autophagy/mitophagy by liensinine sensitizes tumor cells to doxorubicin-induced apoptosis. It is likely that the excessive autophagosome/mitophagosome accumulation, which may be called “autophagic stress,” could be involved in these sensitization effects based on the following observations: First, partially blocking autophagosome/mitophagosome formation with 3-MA or siRNA against ATG5 or ATG7 markedly inhibited combination-mediated dephosphorylation and mitochondrial translocation of DNM1L and mitochondrial fission. Second, partially blocking autophagosome/mitophagosome formation with 3-MA or siRNA against ATG5 or ATG7 markedly attenuated combination-mediated apoptosis, CASP3 activation, and CYCS release. These findings suggest that excessive accumulation of autophagosomes/mitophagosomes is responsible for mitochondrial fission and apoptosis mediated by combination treatment in breast cancer cells. However, the exact mechanism of “autophagic stress”-mediated dephosphorylation and mitochondrial translocation of DNM1L needs further details to explain.
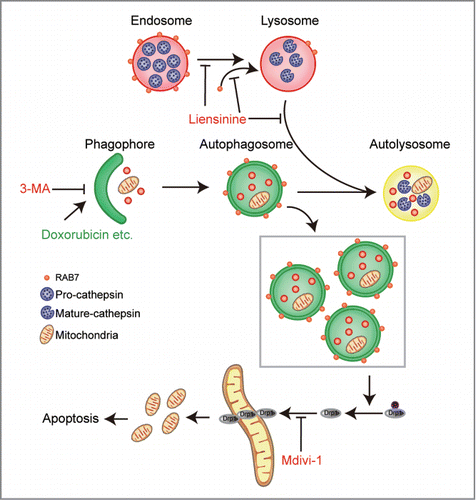
In summary (), the present findings demonstrate for the first time that liensinine potently inhibited autophagosomal-lysosomal fusion, leading to the accumulation of autophagosomes/mitophagosomes. This effect is likely due to inhibiting the recruitment of RAB7A to lysosomes but not to autophagosomes. We also found that inhibition of autophagy/mitophagy by liensinine enhanced doxorubicin-mediated apoptosis by triggering mitochondrial fission, which resulted from dephosphorylation and mitochondrial translocation of DNM1L. Our findings suggest that liensinine could potentially be further developed as a novel autophagy/mitophagy inhibitor, and a combination of liensinine with classical chemotherapeutic drugs could represent a novel therapeutic strategy for treatment of breast cancer.
Figure 11. The proposed mechanisms of liensinine-inhibited autophagy and the synergistic effects of liensinine and chemotherapeutic agents.
Materials and Methods
Cell lines and reagents
MDA-MB-231, MCF-7 and A549 cells were obtained from the American Type Culture Collection (ATCC, HTB-26, HTB-22, CCL-185) and cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS; Gibco, 10100–147) in 5% CO2 at 37°C, U937 cells (ATCC, CRL-1593.2) were cultured in RMPI 1640 (Gibco, 22400) with 10% FBS, LN229 cells (ATCC, CRL-2611) were cultured in DMEM/F-12 (Gibco, 10565) with 10% FBS. 293FT cells (Invitrogen, R700–07) were cultured in DMEM containing 10% FBS, 0.5 mg/ml G418 (Sigma-Aldrich, A1720), 4 mM L-glutamine, 0.1 mM MEM nonessential amino acids (Gibco, 11140), 1 mM sodium pyruvate (Gibco, 11360). MDA-MB-231 cells stably expressing EGFP-LC3 were maintained in DMEM with 10% FBS and 200 μg/ml G418. MDA-MB-231 cells with a stable knockdown of ATG5 or ATG7 were maintained in DMEM with 10% FBS and 15 μg/ml puromycin (Sigma-Aldrich, P9620). Liensinine (2586–96–1) was purchased from MUST bio-technology, 3-methyladenine (M9281), rapamycin (R117), acridine orange (158550), chloroquine diphosphate salt (C6628), Thiazolyl Blue Tetrazolium Blue (MTT, M2128), paclitaxel (T7191), vincristine (V8879) and cisplatin (P4394) were purchased from Sigma-Aldrich, bafilomycin A1 (11038) was purchased from Cayman Chemical, doxorubicin (sc-200923) and staurosporine (sc-360258) were purchased from Santa Cruz Biotechnology, and Mdivi-1 (s7162) was from Selleck Chemicals.
Cell viability (MTT) assay
Cells were seeded in 96-well plates at 2,000 to 5,000 and were treated as indicated for 48 to 72 h dependent on experimental conditions. Twenty μl MTT (5 mg/ml) was added per well and incubated for 4 h. Each well was supplemented with 150 μl DMSO to dissolve the formazan before being measured by microplate reader at 490 nm. The cell viabilities were normalized to the control group.
Transmission electronic microscopy
Cells were treated as indicated and were fixed in 2.5% glutaraldehyde at 4°C overnight and postfixed with 2% osmium tetroxide for 1.5 h at room temperature. After fixation, cells were embedded and stained with uranyl acetate/lead citrate. The sections were examined under a transmission electron microscope (JEM-1400PLUS, Japan) at 60 kV.
Transfections and RNA interference, site mutant
Transfection was achieved using Lipofectamine 3000 Transfection Reagent (Invitrogen, L3000) according to the manufacturer's protocol. Cells were transfected with plasmids encoding mRFP-LC3 (21075), tfLC3 (21074) and LAMP1-mGFP (34831) from Addgene. EGFP-LC3, RFP-mito, ATG5-siRNA and ATG7-siRNA plasmids were constructed by GeneChem Co. Ltd (Shanghai, China). After 24 h to 48 h incubation, the transfection mixture was removed and replaced with fresh complete medium. For RNA interference, MDA-MB-231 cells were transfected with Lipofectamine 3000 Transfection Reagent using DNM1L siRNA (Santa Cruz Biotechnology, sc-43732) as described by the manufacturer. For RNA interference by lentiviral vectors, 293FT cells were transfected with pLP1, pLP2, pLP/VSVG (Invitrogen, K4975) and ATG5-siRNA or ATG7-siRNA plasmid.
Site mutant plasmids were prepared from WT DNM1L-mCherry (Addgene, 49152) by site directed point mutagenesis.Citation43 To generate DNM1L-mCherry mutants site directed mutagenesis was performed using the following primer sequences: S637A (FW: 5′-CCA GTT CCT GTT GCA CGA AAA CTA GCT GCT CGG GAA CAG CGA GAT TGT GAG-3′; RV: 5′-CTC ACA ATC TCG CTG TTC CCG AGC AGC TAG TTT TCG TGC AAC AGG AAC TGG-3′); S637D (FW: 5′-CCA GTT CCT GTT GCA CGA AAA CTA GAT GCT CGG GAA CAG CGA GAT TGT GAG-3′; RV: 5′-CTC ACA ATC TCG CTG TTC CCG AGC ATC TAG TTT TCG TGC AAC AGG AAC TGG-3′). Constructs were verified by sequencing.
Flow cytometry
The extent of apoptosis was determined by flow cytometry using an ANXA5-FITC/PI staining kit (BD Biosciences, 559763) according to the manufacturer's instructions. Acidic vesicles were detected by staining cells with acridine orange at a final concentration of 1.5 µg/ml for 15 min at 37°C. Cells were harvested, washed and analyzed by flow cytometry using the ratio of red (660 nm) to green (530 nm) fluorescence. MDA-MB-231 cells stably expressing EGFP-LC3 were established as described above; cells were washed and resuspended in phosphate-buffered saline (PBS; Santa Cruz Biotechnology, sc-362183), and analyzed by flow cytometry immediately.
Mitochondrial and cytosolic fractionation
Mitochondrial and cytosolic fractions were obtained as previously described.Citation44 MDA-MB-231 and MCF-7 cells were treated and genetically modified as indicated, then were harvested and washed twice with cold PBS, resuspended in 5 × buffer A (20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4, 2 mM leupeptin (Santa Cruz Biotechnology, sc-215242), 1 mM PMSF (Santa Cruz Biotechnology, sc-482875), 1 mM DTT, 2 mM pepstatin A (Sigma-Aldrich, P5318), and 250 mM sucrose (Santa Cruz Biotechnology, sc-204311). Cells were homogenized by passing through a 22-gauge needle 30 times. The homogenate was centrifuged at 3,500 g, at 4°C, and the pellet fraction was considered the “mitochondrial” fraction; the supernatant fraction was centrifuged at 120,000 g at 4°C, and the supernatant fraction was considered the “cytosolic” fraction.
Western blots and immunoprecipitation
For western blot, cells were treated and lysed in 1 × NuPAGE LDS sample buffer (Invitrogen, NP0007) to obtain total protein lysates. Mitochondrial and cytosolic fractions were obtained as described and the concentration measured using an Enhanced BCA protein assay kit (Beyotime Biotechnology, P0011), then cell lysates were loaded onto SDS-PAGE gels and transferred to PVDF membranes, after blocking with 5% fat-free dry milk in Tris-buffered saline (TBS; 10 mM Tris-Base, 150 mM NaCl, pH 7.6), containing 0.1% Tween-20 (Santa Cruz Biotechnology, sc-29113), and membranes were incubated with primary antibodies as follows: LC3B (L7543) and BECN1 (B6186) were from Sigma. SQSTM1 (5114), CTSD (2284), cleaved-CASP9 (9505), cleaved-CASP3 (9661), phospho-DNM1L Ser616 (3455), phospho-DNM1L Ser637 (4867), COX4I1 (4850), HSPD1 (12165) and ubiquitin (3933) were from Cell Signaling Technology. LAMP1 (20011), LAMP2 (18822), RAB7A (10767), CTSB (13985), CTSL (32801), CYCS/cytochrome c (13156), DNM1L (271583) and TOMM20 (11415) were from Santa Cruz Biotechnology. PARP1 (1078) was purchased from Epitomics. Horseradish peroxidase-conjugated species-specific secondary antibodies (Kirkegaard and Perry Laboratories, 4741506, 4741806) were used to detect primary antibodies. Bands were visualized with Clarity Western ECL Substrate (Bio-Rad, 1705063). For Immunoprecipitation, total protein lysates was obtained as described, and equal quantities of proteins were incubated with primary antibodies at 4°C on a rocking platform. Immune complexes were collected with protein A/G agarose beads (Pierce, 88802) followed by 5 washes in PBS. Samples were then subjected to SDS-PAGE and western blot. Quantification relative to ACTB or GAPDH by densitometric analysis used Quality One software (Bio-Rad).
Immunofluorescence
Cells or histological sections were plated on coverslips, for different experimental conditions, and mitochondria and lysosomes were stained with MitoTracker Red CMXRos (Molecular Probes, M7512), LysoTracker Red DND 99 (Molecular Probes, L7528) or LysoSensor Yellow/Blue DND-160 (Molecular Probes, L7545) respectively, according to the manufacturer's instructions. Cells and tissue sections were fixed in 4% paraformaldehyde for 30 min, and permeabilized with 0.1% Triton X-100 (Amresco, 0694) in PBS for 5–10 min. After blocking in PBS containing 10% donkey serum (Jackson ImmunoResearch Europe, 017000121), cells were incubated overnight with antibodies, followed by the appropriate secondary antibodies. Cells were viewed using a laser-scanning confocal microscope (Zeiss, Germany). All images were analyzed by ImageJ software (MD, USA)
Lysosomal pH measurements
Lysosomal pH was measured by LysoSensor Yellow/Blue DND-160 (Molecular Probes) staining as reported previously.Citation45 Briefly, MDA-MB-231 cells were labeled with 5 µM LysoSensor Yellow/Blue DND-160 for 5 min at 37°C and washed twice using PBS. The labeled cells were treated for 2 min with 10 µM monensin (Santa Cruz Biotechnology, sc200109) and 10 µM nigericin (Invitrogen, N1495) in ice-cold 25 mM 2-(N-morpholino) ethanesulfonic acid (MES; Amresco, X218) calibration buffer containing 5 mM NaCl, 115 mM KCl and 1.2 mM MgSO4, pH 3.5–6.0. Light emitted at 535 nm in response to excitation at 340 nm and 380 nm was measured. The pH calibration curve was generated by the ratio of light emitted with 340 nm and 380 nm excitation against the pH values. The pH of lysosomes in MDA-MB-231 cells was evaluated with a pH calibration curve.
Clonogenic growth assays
MDA-MB-231 cells stably expressing ATG5-siRNA, ATG7-siRNA or Scramble-siRNA were co-treated without or with liensinine and doxorubicin for 48 h, then 5,000 cells were reseeded in 6-well plates, cultured for 1 to 7 d, and cells were counted with a Z2 cell counter (Beckman).
Xenograft assay
Female nude mice (5–7 wk of age) were purchased from Vital Rrver Laboratories (VRL, Beijing, China), and fed a standard animal diet and water. Animal studies were approved by the university institutional animal care and use committee. MDA-MB-231 cells were suspended in a 1:1 ratio in serum-free DMEM medium with a Matrigel basement membrane matrix (Sigma, E1270). Cells (1 × 107) were inoculated in the right legs of mice. After tumor inoculation, the mice were randomly assigned into 4 treatment groups (8 mice per group), liensinine (60 mg/kg) was administered daily by intraperitoneal injection in tumor-bearing mice, and doxorubicin (2 mg/kg) was injected at intervals of 4 d. The tumor diameters were measured and the tumor volume (mm3) was calculated as follows: volume= (shortest diameter)2 × (longest diameter)/2. The animals were sacrificed 30 d after medication. Mice were given 200 μl of pentobarbital sodium (1%) by intraperitoneal injection 30 min before sacrifice, organs were collected and tumors were excised, and either formalin-fixed or flash-frozen at −20°C until further use. TUNEL, histological and immunohistochemical assay were performed as previously described.Citation46
Statistical analysis
Statistical analyses were performed with SAS. Some data were normalized to vehicle or scrambled control and analyzed with one-sample t-tests. n.s., not significant; 0.01 ≤ *P <0 .05; **P < 0.01.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1056970_supplemental_files.doc
Download MS Word (1 MB)Acknowledgments
The authors would like to thank Professor Tamotsu Yoshimori (Department of Biochemistry, Graduate School of Medicine, Osaka University), Professor Esteban Dell'Angelica (Department of Human Genetics, David Geffen School of Medicine, University of California) for providing mRFP-LC3, tfLC3, LAMP1-mEGFP plasmids, and Professor Gia Voeltz (University of Colorado Boulder) for providing DNM1L-mCherry plasmids.
Funding
This work was funded by Chongqing Natural Science Foundation (CSTC2013jjB10007), National Natural Science Foundation of China (81402202; 81173141; 81402970; 81402013), and Chongqing Postdoctoral Science Foundation (Xm2014126).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res 2013; 73:3-7; PMID:23288916; http://dx.doi.org/10.1158/0008-5472.CAN-12-2464
- Macintosh RL, Ryan KM. Autophagy in tumour cell death. Semin Cancer Biol 2013; 23:344-51; PMID:23774296; http://dx.doi.org/10.1016/j.semcancer.2013.05.006
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov 2007; 6:304-12; PMID:17396135; http://dx.doi.org/10.1038/nrd2272
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Koren I, Kimchi A. Cell biology. Promoting tumorigenesis by suppressing autophagy. Science 2012; 338:889-90; PMID:23161981; http://dx.doi.org/10.1126/science.1230577
- Rosenfeldt MT, Ryan KM. The role of autophagy in tumour development and cancer therapy. Expert Rev Mol Med 2009; 11:e36; PMID:19951459; http://dx.doi.org/10.1017/S1462399409001306
- Janku F, McConkey DJ, Hong DS, Kurzrock R. Autophagy as a target for anticancer therapy. Nat Rev Clin Oncol 2011; 8:528-39; PMID:21587219; http://dx.doi.org/10.1038/nrclinonc.2011.71
- Rubinsztein DC, Codogno P, Levine B. Autophagy modulation as a potential therapeutic target for diverse diseases. Nat Rev Drug Discov 2012; 11:709-30; PMID:22935804; http://dx.doi.org/10.1038/nrd3802
- Mahalingam D, Mita M, Sarantopoulos J, Wood L, Amaravadi RK, Davis LE, Mita AC, Curiel TJ, Espitia CM, Nawrocki ST, et al. Combined autophagy and HDAC inhibition: a phase I safety, tolerability, pharmacokinetic, and pharmacodynamic analysis of hydroxychloroquine in combination with the HDAC inhibitor vorinostat in patients with advanced solid tumors. Autophagy 2014; 10:1403-14; PMID:24991835; http://dx.doi.org/10.4161/auto.29231
- Rangwala R, Chang YC, Hu J, Algazy KM, Evans TL, Fecher LA, Schuchter LM, Torigian DA, Panosian JT, Troxel AB, et al. Combined MTOR and autophagy inhibition: phase I trial of hydroxychloroquine and temsirolimus in patients with advanced solid tumors and melanoma. Autophagy 2014; 10:1391-402; PMID:24991838; http://dx.doi.org/10.4161/auto.29119
- Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, et al. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy 2014; 10:1380-90; PMID:24991834; http://dx.doi.org/10.4161/auto.29264
- Kashiwada Y, Aoshima A, Ikeshiro Y, Chen YP, Furukawa H, Itoigawa M, Fujioka T, Mihashi K, Cosentino LM, Morris-Natschke SL, et al. Anti-HIV benzylisoquinoline alkaloids and flavonoids from the leaves of Nelumbo nucifera, and structure-activity correlations with related alkaloids. Bioorg Med Chem 2005; 13:443-8; PMID:15598565; http://dx.doi.org/10.1016/j.bmc.2004.10.020
- Dong ZX, Zhao X, Gu DF, Shi YQ, Zhang J, Hu XX, Hu MQ, Yang BF, Li BX. Comparative effects of liensinine and neferine on the human ether-a-go-go-related gene potassium channel and pharmacological activity analysis. Cell Physiol Biochem 2012; 29:431-42; PMID:22508050; http://dx.doi.org/10.1159/000338497
- Qin Q, Chen XP, Yang ZS, Xiao YH, Min H, Li YJ. Neferine increases STI571 chemosensitivity via inhibition of P-gp expression in STI571-resistant K562 cells. Leuk Lymphoma 2011; 52:694-700; PMID:21261505; http://dx.doi.org/10.3109/10428194.2010.550075
- Law BY, Chan WK, Xu SW, Wang JR, Bai LP, Liu L, Wong VK. Natural small-molecule enhancers of autophagy induce autophagic cell death in apoptosis-defective cells. Sci Rep 2014; 4:5510; PMID:24981420; http://dx.doi.org/10.1038/srep05510
- Yang YP, Hu LF, Zheng HF, Mao CJ, Hu WD, Xiong KP, Wang F, Liu CF. Application and interpretation of current autophagy inhibitors and activators. Acta Pharmacol Sin 2013; 34:625-35; PMID:23524572; http://dx.doi.org/10.1038/aps.2013.5
- Chu CT. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol 2006; 65:423-32; PMID:16772866; http://dx.doi.org/10.1097/01.jnen.0000229233.75253.be
- Marino G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol 2014; 15:81-94; PMID:24401948; http://dx.doi.org/10.1038/nrm3735
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014; 157:65-75; PMID:24679527; http://dx.doi.org/10.1016/j.cell.2014.02.049
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC. Does bafilomycin A(1) block the fusion of autophagosomes with lysosomes? Autophagy 2008; 4:849-50; PMID:18758232; http://dx.doi.org/10.4161/auto.6845
- Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci 2004; 117:4837-48; PMID:15340014; http://dx.doi.org/10.1242/jcs.01370
- Hyttinen JM, Niittykoski M, Salminen A, Kaarniranta K. Maturation of autophagosomes and endosomes: a key role for Rab7. Biochim Biophys Acta 2013; 1833:503-10; PMID:23220125; http://dx.doi.org/10.1016/j.bbamcr.2012.11.018
- Press B, Feng Y, Hoflack B, Wandinger-Ness A. Mutant Rab7 causes the accumulation of cathepsin D and cation-independent mannose 6-phosphate receptor in an early endocytic compartment. J Cell Biol 1998; 140:1075-89; PMID:9490721; http://dx.doi.org/10.1083/jcb.140.5.1075
- Coutinho MF, Prata MJ, Alves S. Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol Genet Metab 2012; 105:542-50; PMID:22266136; http://dx.doi.org/10.1016/j.ymgme.2011.12.012
- Appelqvist H, Waster P, Kagedal K, Ollinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol 2013; 5:214-26; PMID:23918283; http://dx.doi.org/10.1093/jmcb/mjt022
- Townsend KN, Hughson LR, Schlie K, Poon VI, Westerback A, Lum JJ. Autophagy inhibition in cancer therapy: metabolic considerations for antitumor immunity. Immunol Rev 2012; 249:176-94; PMID:22889222; http://dx.doi.org/10.1111/j.1600-065X.2012.01141.x
- Chittaranjan S, Bortnik S, Dragowska WH, Xu J, Abeysundara N, Leung A, Go NE, DeVorkin L, Weppler SA, Gelmon K, et al. Autophagy inhibition augments the anticancer effects of epirubicin treatment in anthracycline-sensitive and -resistant triple-negative breast cancer. Clin Cancer Res 2014; 20:3159-73; PMID:24721646; http://dx.doi.org/10.1158/1078-0432.CCR-13-2060
- Maes H, Rubio N, Garg AD, Agostinis P. Autophagy: shaping the tumor microenvironment and therapeutic response. Trends Mol Med 2013; 19:428-46; PMID:23714574; http://dx.doi.org/10.1016/j.molmed.2013.04.005
- Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 2012; 337:1062-5; PMID:22936770; http://dx.doi.org/10.1126/science.1219855
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 2012; 46:265-87; PMID:22934639; http://dx.doi.org/10.1146/annurev-genet-110410-132529
- Otera H, Mihara K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J Biochem 2011; 149:241-51; PMID:21233142; http://dx.doi.org/10.1093/jb/mvr002
- Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell 2008; 14:193-204; PMID:18267088; http://dx.doi.org/10.1016/j.devcel.2007.11.019
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 2008; 27:433-46; PMID:18200046; http://dx.doi.org/10.1038/sj.emboj.7601963
- Liu Y, Levine B. Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 2014; 22(3):367-76; PMID:25257169
- Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013; 14:283-96; PMID:23609508; http://dx.doi.org/10.1038/nrm3565
- Kreuzaler P, Watson CJ. Killing a cancer: what are the alternatives? Nat Rev Cancer 2012; 12:411-24; PMID:22576162; http://dx.doi.org/10.1038/nrc3264
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Labbe K, Murley A, Nunnari J. Determinants and functions of mitochondrial behavior. Annu Rev Cell Dev Biol 2014; 30:357-91; PMID:25288115; http://dx.doi.org/10.1146/annurev-cellbio-101011-155756
- Liu EY, Ryan KM. Autophagy and cancer–issues we need to digest. J Cell Sci 2012; 125:2349-58; PMID:22641689; http://dx.doi.org/10.1242/jcs.093708
- Kim DG, Jung KH, Lee DG, Yoon JH, Choi KS, Kwon SW, Shen HM, Morgan MJ, Hong SS, Kim YS. 20(S)-Ginsenoside Rg3 is a novel inhibitor of autophagy and sensitizes hepatocellular carcinoma to doxorubicin. Oncotarget 2014; 5:4438-51; PMID:24970805
- Manov I, Pollak Y, Broneshter R, Iancu TC. Inhibition of doxorubicin-induced autophagy in hepatocellular carcinoma Hep3B cells by sorafenib–the role of extracellular signal-regulated kinase counteraction. FEBS J 2011; 278:3494-507; PMID:21790999; http://dx.doi.org/10.1111/j.1742-4658.2011.08271.x
- Cereghetti GM, Stangherlin A, Martins de Brito O, Chang CR, Blackstone C, Bernardi P, Scorrano L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc Natl Acad Sci U S A 2008; 105:15803-8; PMID:18838687; http://dx.doi.org/10.1073/pnas.0808249105
- Gao N, Kramer L, Rahmani M, Dent P, Grant S. The three-substituted indolinone cyclin-dependent kinase 2 inhibitor 3-[1-(3H-imidazol-4-yl)-meth-(Z)-ylidene]-5-methoxy-1,3-dihydro-indol-2-one (SU9516) kills human leukemia cells via down-regulation of Mcl−1 through a transcriptional mechanism. Mol Pharmacol 2006; 70:645-55; PMID:16672643; http://dx.doi.org/10.1124/mol.106.024505
- Wang Z, Zhang J, Wang Y, Xing R, Yi C, Zhu H, Chen X, Guo J, Guo W, Li W, et al. Matrine, a novel autophagy inhibitor, blocks trafficking and the proteolytic activation of lysosomal proteases. Carcinogenesis 2013; 34:128-38; PMID:23002236; http://dx.doi.org/10.1093/carcin/bgs295
- Li G, Zhou T, Liu L, Chen J, Zhao Z, Peng Y, Li P, Gao N. Ezrin dephosphorylation/downregulation contributes to ursolic acid-mediated cell death in human leukemia cells. Blood Cancer J 2013; 3:e108; PMID:23584398; http://dx.doi.org/10.1038/bcj.2013.7