
Abstract
Recent studies have shown that the phosphorylation and dephosphorylation of ULK1 and ATG13 are related to autophagy activity. Although ATG16L1 is absolutely required for autophagy induction by affecting the formation of autophagosomes, the post-translational modification of ATG16L1 remains elusive. Here, we explored the regulatory mechanism and role of ATG16L1 phosphorylation for autophagy induction in cardiomyocytes. We showed that ATG16L1 was a phosphoprotein, because phosphorylation of ATG16L1 was detected in rat cardiomyocytes during hypoxia/reoxygenation (H/R). We not only demonstrated that CSNK2 (casein kinase 2) phosphorylated ATG16L1, but also identified the highly conserved Ser139 as the critical phosphorylation residue for CSNK2. We further established that ATG16L1 associated with the ATG12-ATG5 complex in a Ser139 phosphorylation-dependent manner. In agreement with this finding, CSNK2 inhibitor disrupted the ATG12-ATG5-ATG16L1 complex. Importantly, phosphorylation of ATG16L1 on Ser139 was responsible for H/R-induced autophagy in cardiomyocytes, which protects cardiomyocytes from apoptosis. Conversely, we determined that wild-type PPP1 (protein phosphatase 1), but not the inactive mutant, associated with ATG16L1 and antagonized CSNK2-mediated phosphorylation of ATG16L1. Interestingly, one RVxF consensus site for PPP1 binding in the C-terminal tail of ATG16L1 was identified; mutation of this site disrupted its association with ATG16L1. Notably, CSNK2 also associated with PPP1, but ATG16L1 depletion impaired the interaction between CSNK2 and PPP1. Collectively, these data identify ATG16L1 as a bona fide physiological CSNK2 and PPP1 substrate, which reveals a novel molecular link from CSNK2 to activation of the autophagy-specific ATG12-ATG5-ATG16L1 complex and autophagy induction.
Abbreviations
| ACTB | = | actinβ |
| AMPK | = | AMP activated protein kinase |
| ATG | = | autophagy-related |
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BECN1/Beclin 1 | = | autophagy related |
| CD | = | Crohn disease |
| CSNK2 | = | casein kinase 2 |
| GFP | = | green fluorescent protein |
| GNB2L1/RACK1 | = | guanine nucleotide binding protein (G protein)βpolypeptide 2-like 1 |
| GST | = | glutathione S-transferase |
| H/R | = | hypoxia/reoxygenation |
| I/R | = | ischemia/reperfusion |
| LAD | = | left anterior descending |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 β |
| mRFP | = | monomeric red fluorescent protein |
| MTORC1 | = | mechanistic target of rapamycin complex 1 |
| NRVCs | = | neonatal rat ventricular cardiomyocytes |
| OA | = | okadaic acid |
| PE | = | phosphatidylethanolamine |
| PPP1 | = | protein phosphatase 1 |
| PPP2 | = | protein phosphatase 2 |
| PVDF | = | polyvinylidenedifluoride |
| SNP | = | single nucleotide polymorphism |
| SUPT20H/p38IP | = | suppressor of Ty 20 homolog (S. cerevisiae) |
| TBB | = | 4,5,6,7-tetrabromobenzotriazole |
| λPP | = | λ protein phosphatase. |
Introduction
Autophagy plays an important role in many biological processes, such as development, immunity, apoptosis, and cancer.Citation1 Autophagy is a lysosome-dependent bulk degradation pathway, which controls the clearance and recycling of cytoplasmic components for the maintenance of cellular survival.Citation2 Autophagy includes at least 3 pathways: macroautophagy, microautophagy, and chaperone-mediated autophagy.Citation2 Of these, macroautophagy is the most prevalent form of autophagy and has been implicated in cardiovascular diseases.Citation3 In the present study, macroautophagy will be referred to as autophagy.
The autophagic system is tightly regulated by some evolutionarily conserved autophagy-related (ATG) proteins that have been identified from yeasts to mammals.Citation4 Autophagy begins with BECN1/Beclin 1 binding to the class III phosphatidylinositol 3-kinase, PIK3C3, which contributes to the nucleation of the phagophore membrane by recruiting other ATG proteins. During expansion, the cytoplasmic contents are directly enclosed by the double-membrane phagophore, and a complete autophagosome is generated; the autophagosome then fuses with a lysosome to degrade its cargo.Citation5 Autophagosome maturation requires a series of ubiquitin-like conjugation events that mainly involves the LC3-phosphatidylethanolamine (PE) and the ATG12-ATG5-ATG16L1 conjugation systems. These core ATG proteins, including ATG16L1, are indispensable in the generation of the autophagosome. ATG16L1 interacts with the ATG12-ATG5 conjugate to form a dimeric complex, which localizes to phagophores and later promotes MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 β) lipidation by acting as an E3-like ubiquitin ligase.Citation6
Although the function of ATG16L1 during the autophagic process is well documented, less is known about mechanisms governing the regulation of ATG16L1 activity. Given the important role of autophagy, which functions in various biological aspects, it is not unexpected that ATG proteins need to be tightly controlled by post-translational modifications. For instance, acetylation of ULK1 by KAT5/TIP60 induces autophagy, whereas acetylation of ATG5 and ATG12 by EP300/p300 plays an opposite role in the regulation of autophagy.Citation7-8 In addition, direct phosphorylation of ULK1 by AMPK and MTORC1 (mechanistic target of rapamycin complex 1) oppositely regulates the ULK1-ATG13-RB1CC1/FIP200 autophagy-initiating complex.Citation9 However, very little is known about ATG16L1 phospho-regulation and its effects on autophagic flux.
CSNK2 is a highly conserved, constitutively active, and ubiquitously expressed serine-threonine kinase that is typically found as a tetrameric holoenzyme complex of 2 catalytic subunits (CSNK2A1/α and/or CSNK2A2/α') and 2 regulatory (CSNK2B/β) subunits.Citation10 The enzyme is responsible for the phosphorylation of more than 300 substrates and affects a range of biological processes, including chromosome segregation, spindle formation, regulation of apoptosis, the control of cellular proliferation, and circadian rhythm.Citation10 Recent studies also imply a potentially important role for CSNK2 in the regulation of autophagy.Citation11 However, there is no report showing whether or not CSNK2 kinase can directly phosphorylate ATG16L1 to regulate the autophagy machinery.
Protein phosphorylation can be reversed by phosphatases such as PPP1, a ubiquitously expressed eukaryotic serine/threonine protein phosphatase that affects a large variety of cellular functions and processes.Citation12 PPP1 belongs to the phosphoprotein phosphatase family that comprises 4 major subfamilies: PPP1, PPP2CA, PPP2CB, and PPP5.Citation13 Interestingly, like CSNK2, PPP2 (protein phosphatase 2) promotes the pro-autophagic activity of BECN1 via inducing a conformational change in BECN1 in Ras-NIH 3T3 cells.Citation14 However, very little is currently known about the role of PPP1 in autophagy.
In the present study, we show that the serine/threonine kinase CSNK2 phosphorylates ATG16L1 both in vitro and in vivo. Furthermore, we demonstrate that PPP1 dephosphorylates ATG16L1, leading to autophagy inhibition and apoptosis induction. Thus, for the first time, our results uncover a potential role for ATG16L1 in protecting metabolically stressed cardiomyocytes from apoptosis by promoting autophagy through phosphoregulation by the CSNK2-PPP1 complex.
Results
Hypoxia/reoxygenation (H/R) increased autophagy and promoted ATG16L1 phosphorylation in cultured cardiomyocytes
Although phosphorylation of several autophagy-related genes has been previously reported, the signals that stimulate physiological phosphorylation of ATG16L1 remain unknown. To understand the regulatory mechanism underlying ATG16L1-mediated autophagy, we explored whether hypoxia was able to induce phosphorylation of ATG16L1 in cardiomyocytes. Firstly, we assessed autophagy induction in primary neonatal rat ventricular cardiomyocytes (NRVCs) or H9c2 cells (data not shown) that were randomized to control and H/R groups. The H/R group was subjected to 4 h hypoxia followed by reoxygenation for 3 h. In agreement with a previous report,Citation15 the H/R group showed more pronounced autophagy compared with the control group as represented in the western blotting of LC3B-II (). Expression of SQSTM1/p62, a protein sequestered within autophagosomes and subsequently degraded in lysosomes, was markedly decreased (). To independently investigate the accumulation of typical autophagosomes and autolysosomes, we transfected NRVCs with monomeric red fluorescent protein (mRFP)-GFP-LC3 and monitored the change in fluorescent LC3. Monomeric red fluorescent protein (mRFP) does not change its fluorescence even after being exposed to an acidic lysosomal environment where the GFP signal is lost. Therefore, red LC3 puncta indicate autophagosomes and autolysosomes, whereas green LC3 dots only indicate autophagosomes. Thus, when the red puncta merge with the green puncta, the image appears yellow and indicates autophagosomes, while the red only puncta represent autolysosomes.Citation16 As shown in (), H/R significantly increase the numbers of green and red dots. Furthermore, both yellow and red dots were markedly enhanced after H/R, indicating enhanced autophagosomes and autolysosomes (). These data indicated that autophagy was enhanced as a response to H/R in NRVCs.
Figure 1. H/R increased autophagy and promoted ATG16L1 phosphorylation in cultured cardiomyocytes. (A) NRVCs were randomized to control and H/R groups (n = 10). The H/R group was treated with 4 h hypoxia (3% O2, 5% CO2, and 92% N2) followed by 3 h reoxygenation (5% CO2 and 95% O2). Western blotting was performed to detect LC3B-II, SQSTM1, and ACTB. The intensity of the western blot bands was quantified using NIH ImageJ software. (B) Representative images of fluorescent LC3 puncta. NRVCs were transduced with mRFP-GFP-LC3. The numbers of green, yellow and red puncta were significantly higher after H/R in NRVCs (Top panel). H/R significantly increased the number of LC3 dots (lower panel). Error bars represent the mean ± SD of 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. NRVCs were incubated with32P-labeled H3PO4 (5 mCi) with or without H/R. Immunoprecipitation was performed using either anti-ATG16L1 (C) or anti-phosphoserine or anti-phosphothreonine antibody (D) or IgG. SDS/PAGE of the immunoprecipitated proteins was visualized by autoradiography (C). The immunoblotting was analyzed using ATG16L1 antibody (D). (E) His-Tagged WT-ATG16L1, ATG16L1S139A or 3A (S139, S304, T370) mutants were transfected into NRVCs which were incubated with32P-labeled H3PO4 with or without H/R. Immunoprecipitation was performed using anti-His antibody, and the immunoprecipitated proteins were visualized by autoradiography. The representative data comes from 3 independent experiments.
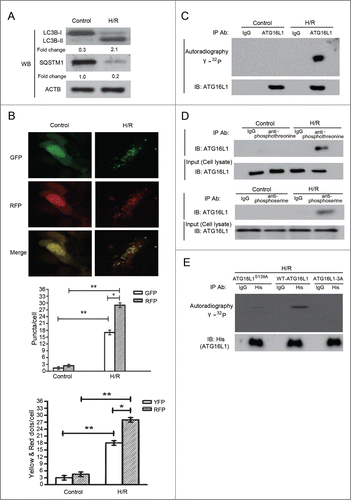
Next, we evaluated whether H/R stimulation of NRVCs is associated with ATG16L1 phosphorylation. Cells were first incubated with H3[32PO4], and then subjected to H/R followed by immunoprecipitation with either an anti-ATG16L1 antibody or nonspecific IgG. Autoradiography confirmed ATG16L1 phosphorylation in NRVCs that were subjected to H/R, as compared to the control incubation (). Phosphorylation of ATG16L1 in NRVCs exposed to H/R was also studied by immunoprecipitation and western blotting with either antiphosphothreonine or antiphosphoserine and anti-ATG16L1 antibodies (). Moreover, calf intestinal alkaline phosphatase treatment resulted in dephosphorylation of ATG16L1, accompanied by a reduction of LC3B-II levels (data not shown). Interestingly, hypoxia alone also increased autophagy and ATG16L1 phosphorylation (Fig. S1A, C). Collectively, these results suggested that H/R treatment induced ATG16L1 phosphorylation, which was strongly related to the induction of autophagy in NRVCs.
CSNK2 was responsible for ATG16L1 phosphorylation in H/R-treated cardiomyocytes
To understand the mechanism underlying the phosphorylation of ATG16L1 and the activation of autophagy, we next sought to identify which protein kinase is responsible for the phosphorylation of this protein. A recent report showed that CK/casein kinase is able to phosphorylate Atg32, a mitophagy receptor in yeast, suggesting that CSNK2 may have a general role to regulate autophagy both in mammalian systems and in yeast.Citation17 Moreover, sequence scanning of ATG16L1 indicated that several serine and threonine residues are highly conserved and surrounded by acidic amino acids (), which is a signature motif (S/TXXE) for CSNK2,Citation18 we therefore reasoned that CSNK2 is a potential kinase responsible for ATG16L1 phosphorylation in H/R-treated H9c2 cells. To test this hypothesis, we treated cardiomyocytes with 4,5,6,7-tetrabromobenzotriazole (TBB), a typical CSNK2 inhibitor,Citation19 and observed a decreased ATG16L1 phosphorylation level in H/R-treated cardiomyocytes (). The mammalian CSNK2 is heterotetrameric form consisting of 2 catalytic subunits (CSNK2A1 and CSNK2A2) and 2 regulatory subunits (CSNK2B). Our in vitro kinase assay showed that both of the CSNK2 catalytic subunits (CSNK2A1 and CSNK2A2), but not the kinase-dead mutants CSNK2A1K68M and CSNK2A2K69M,Citation20 can phosphorylate ATG16L1 (). To further evaluate whether CSNK2 can phosphorylate ATG16L1 under physiological conditions, we performed an immunodepletion analysis of CSNK2 subunits. In H9c2 cell lysates, each endogenous CSNK2 subunit was depleted either individually or in combination with the other subunits via immunoprecipitation with antibodies targeting the CSNK2 subunits. The immunodepleted lysates were then incubated with GST-ATG16L1 to perform an in vitro assay of the kinase activity. We observed that depletion of either of the endogenous CSNK2 subunits in H9c2 cell lysates resulted in a decreased ability to phosphorylate ATG16L1 compared to the IgG control or nontreated lysate (). Moreover, immunodepletion of both CSNK2 catalytic subunits (CSNK2A1 and CSNK2A2) abolished the ability to phosphorylate ATG16L1. These observations supported our hypothesis that CSNK2 is indeed the protein kinase phosphorylating ATG16L1 in H/R-treated cardiomyocytes.
Figure 2. For figure legend, see page 1312.
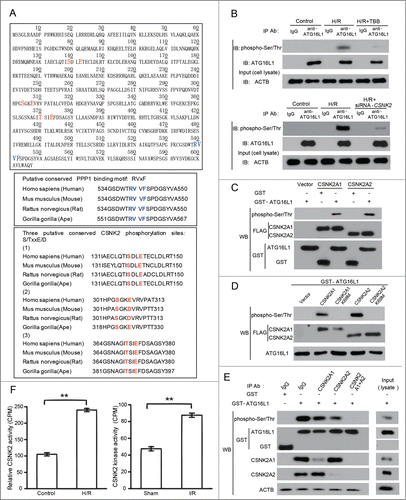
Interestingly, although CSNK2 is a constitutively active kinase, we found that the activity of CSNK2 in cardiomyocytes and mice heart is increased by H/R and I/R, respectively (; Fig. S1B). Importantly, we found that CSNK2 is activated before ATG16L1 phosphorylation and LC3 cleavage (Fig. S1D), which further supports our hypothesis.
Phosphorylation of ATG16L1 on Ser139 selectively enhanced its interaction with the ATG12-ATG5 conjugate
To determine how phosphorylation of ATG16L1 affects autophagy, we examined the known interactors of ATG16L1 including RB1CC1 and the ATG12-ATG5 protein complex.Citation21-22 Given that ATG16L1 is essential for recruiting ATG5 and ATG12 to the phagophore assembly site (PAS) and the ATG12-ATG5-ATG16L1 complex is required for LC3-PE (LC3-II) biogenesis in vivo,Citation22 we hypothesized that phosphorylation of ATG16L1 could regulate the interaction between ATG16L1 and the ATG12-ATG5 complex. To test this hypothesis, we purified His6/V5-tagged ATG16L1 from H/R-treated H9c2 cells by phenyl-Sepharose followed by Ni-NTA resin. Purified ATG16L1 was then dephosphorylated by λ protein phosphatase (λPP) and examined for its binding ability to purified ATG12-ATG5 conjugate in vitro. Dephosphorylation of ATG16L1 by λPP decreased its binding to the ATG12-ATG5 conjugate () and such a phenotype could be rescued by rephosphorylation of ATG16L1 with recombinant CSNK2 ().
Figure 3. Phosphorylation of ATG16L1 on Ser139 selectively enhanced its interaction with the ATG12-ATG5 conjugate. (A) Immunoprecipitation assay showing that dephosphorylation of ATG16L1-V5 purified from H9c2 cells by λPP diminished the ability to bind the ATG12-ATG5 protein complex, which was restored by rephosphorylation by CSNK2. ATG16L1 autoradiography is shown to demonstrate that CSNK2 phosphorylates purified ATG16L1. Bottom histogram shows summary of quantification from independent experiments. Corresponding lane numbers are indicated. (B) In vitro binding assay showing that ATG16L1 purified from E. coli, which lacks endogenous CSNK2, showed increased binding after CSNK2 phosphorylation, which was diminished by dephosphorylation by λPP treatment. Protein input and32P uptake into ATG16L1 are also shown. (C) Affinity isolation assay showing GST-ATG12-ATG5 fusion protein and E. coli-generated ATG16L1 with the indicated ATG16L1 treatment and mutation. CSNK2 phosphorylation and untreated ATG16L1 (3E) or (3A) mutant showed facilitated ATG12-ATG5 conjugate binding compared with untreated wild-type ATG12-ATG5. (D) Immunoprecipitation of FLAG-ATG12-ATG5 co-expressed with glutamate substitutions of CSNK2 phosphorylation sites of ATG16L1. Western blotting was performed with the indicated antibodies. (E) Immunoprecipitation of FLAG-ATG12-ATG5 protein complex co-expressed with alanine substitution of CSNK2 phosphorylation sites. Western blotting was performed with the indicated antibodies. Data are representative of 3 independent experiments and expressed as means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
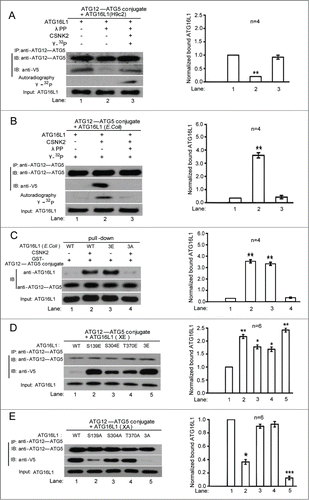
We further validated the roles of CSNK2-mediated phosphorylation of ATG16L1 by repeating the binding assay in E. coli, which lacks CSNK2.Citation23 His6/V5-tagged ATG16L1 expressed in E. coli was purified using Ni-NTA beads followed by CSNK2 treatment, and then analyzed for its association with purified ATG12-ATG5 conjugate. The results indicated that CSNK2 phosphorylation of ATG16L1 promoted its association with the ATG12-ATG5 protein complex, which was abolished by dephosphorylation of ATG16L1 by λPP (). These data indicate that ATG16L1 phosphorylation by CSNK2 is essential for its binding to the ATG12-ATG5 protein complex.
Next, we set out to identify possible phosphorylated sites in ATG16L1 mediated by CSNK2. As shown in (), a sequence analysis performed for ATG16L1 predicted 3 serine and threonine residues as putative CSNK2 phosphorylation acceptor sites: serine 139, serine 304, and threonine 370. We generated a phosphomimetic mutation of ATG16L1 in which all 3 phosphorylation sites were substituted with glutamate, ATG16L1-3E. As expected, ATG16L1-3E purified from E. coli without any treatment showed enhanced binding to the ATG12-ATG5 complex, similar to that of CSNK2-phosphorylated ATG16L1 (). However, a phospho-dead mutation, ATG16L1-3A, purified from E. coli with CSNK2 treatment decreased its binding to the ATG12-ATG5 complex ().
To further identify the critical residue(s) among 3 CSNK2 phosphorylation sites, we co-expressed single residue alanine or glutamate mutants of ATG16L1 with the ATG12-ATG5 conjugate in H9c2 cells, and then explored their interaction via co-immunoprecipitation. We found that any single glutamate mutation increased the association of ATG16L1 with the ATG12-ATG5 complex (). However, co-immunoprecipitation assays using alanine-substituted mutants identified Ser139 as the critical residue (), which showed decreased binding, similar to that of the 3 alanine-substituted mutant, ATG16L1-3A. Additionally, an in vitro kinase assay demonstrated that the highly conserved Ser139 of ATG16L1 was the critical phosphorylated residue for CSNK2 (Fig. S2). Collectively, these results demonstrate that phosphorylation of ATG16L1 on Ser139 positively regulates autophagy through enhancing ATG16L1 binding to the ATG12-ATG5 complex. Notably, this enhancement is specific to the ATG12-ATG5 conjugate and not RB1CC1 (data not shown).
Inhibition of CSNK2 suppressed H/R-induced autophagy and sensitized NRVCs to H/R-induced injury
Because the reversible phosphorylation of ATG16L1 determines its activity, we next explored the functional significance of CSNK2 in H/R-mediated autophagy in cultured NRVCs. Given that inhibition of autophagy promotes apoptosis,Citation24 we hypothesized that the CSNK2 inhibitor (TBB)-induced impairment of autophagy might enhance the apoptotic rates in cultured NRVCs. To confirm this hypothesis, NRVCs were assigned to 4 groups: Control, NRVCs were maintained in normoxic conditions without TBB treatment; TBB, NRVCs were treated with TBB and incubated in normoxic condition; H/R, NRVCs. were treated with hypoxia (4 h) and subsequent reoxygenation (3 h); H/R+TBB, NRVCs pretreated with TBB were subjected to treatment as described for the H/R group.
We found that the expression levels of LC3B-II in the H/R+TBB group were significantly reduced with significantly increased levels of SQSTM1 (; Fig. S3A). To quantify the GFP-LC3 dots, we transfected NRVCs with GFP-LC3 and examined the abundance of GFP-LC3 puncta in every group. Our results demonstrated that the GFP-LC3 dots were no different in control and TBB groups (). However, the GFP-LC3 dots were increased in NRVCs subjected to H/R. After pretreatment with TBB, the GFP-LC3 dots were significantly decreased in NRVCs exposed to H/R. The data indicated that TBB decreased GFP-LC3 fusion proteins in NRVCs subjected to H/R. Because both induction of the autophagic process and a block in the autophagosome maturation increase the ratio of LC3B-II/LC3B-I, by employing the autophagy inhibitor chloroquine (CQ), we monitored autophagic flux to distinguish between these 2 events (Fig. S4); these data indicated that TBB resulted in a block in autophagy.
Figure 4. Inhibition of CSNK2 suppressed H/R-induced autophagy and sensitized NRVCs to H/R-induced injury. NRVCs were assigned to 4 groups: Control, NRVCs were maintained in normoxic conditions without the CSNK2 inhibitor TBB treatment; TBB, NRVCs were treated with TBB (5 μM) and incubated in normoxic conditions; H/R, NRVCs were subjected to 4 h of hypoxia followed by 3 h of reoxygenation; H/R+TBB, NRVCs pretreated with TBB for 60 min were subjected to 4 h of hypoxia followed by 3 h of reoxygenation. NRVCs transfected with scramble or siRNA-CSNK2 were also divided into 4 groups as described above: Control, siRNA-CSNK2, H/R, and H/R+siRNA-CSNK2. (A) The cells in every group were then lysed and subjected to western blotting for the expression of LC3B-II and SQSTM1. (B) Representative punctate images of GFP-LC3 in NRVCs. NRVCs were transfected with the GFP-LC3 plasmid for 24 h and then treated with or without H/R or TBB or siRNA (upper panel). GFP-LC3 aggregates were quantified using ImageJ (mean ± SD; n = 100 cells from 3 independent experiments, lower panel). (C) NRVCs were transfected with empty vector or plasmid containing WT-ATG16L1 or the S139E mutant, as indicated, for 24 h and then treated as described above in (A). Western blotting was performed to detect LC3B-II and SQSTM1 protein levels. (D) Quantitative assessment of total cell death by ANXA5/PI staining. Flow cytometry results were displayed as quantitative bar graphs. Data are representative of 3 independent experiments and expressed as means ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
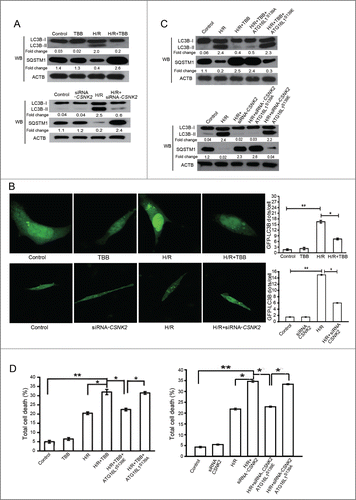
To confirm that the role of TBB in autophagy is mediated through the dephosphorylation of ATG16L1 at Ser139, a rescue experiment was performed using ATG16L1S139A or ATG16L1S139E mutant plasmids transfected into NRVCs pretreated with TBB. As expected, the S139E mutant, but not ATG16L1S139A, restored the reduced levels of LC3B-II in NRVCs treated with H/R+TBB (). These results demonstrated that CSNK2 specifically activated autophagy in NRVCs through the phosphorylation of ATG16L1 at Ser139.
Given that 2 processes (autophagy and apoptosis) often regulate each other in opposite directions, we assessed the total cell death with ANXA5/annexin A5-propidium iodide (PI) staining in the aforementioned group. The cell death rates in the H/R group were markedly increased (P < 0.05 vs. control) whereas pre-treatment with TBB significantly further elevated H/R-induced total cell death (P < 0.05 vs. H/R) (; Fig. S3B). As expected, the ATG16L1S139E mutant, but not ATG16L1S139A, rescued the elevated total cell death rates in NRVCs.
SiRNA-mediated CSNK2 knockdown experiments further strengthened the results that ATG16L1 is phosphorylated by CSNK2 () and that this response is responsible for the development of autophagy and cellular protection against H/R ().
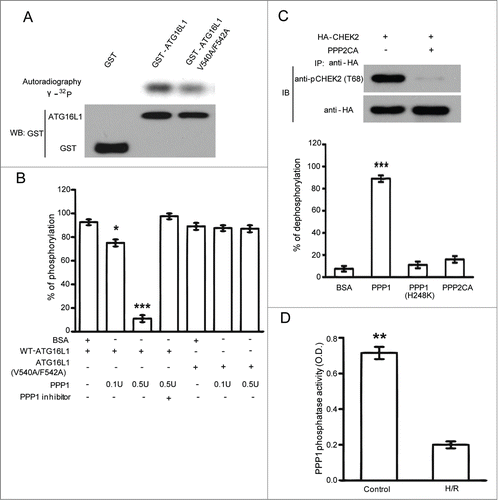
PPP1 dephosphorylated ATG16L1 in vitro
Although control of protein activities by phosphorylation is a widespread regulatory mechanism, these modifications are often fine-tuned through the coordinated activities of phosphatases. We next sought to identify which phosphatase is responsible for the dephosphorylation of ATG16L1. Given that PPP1 antagonizes the roles of CSNK2 kinase in various events,Citation25 one candidate enzyme is PPP1 whose role in ATG16L1 signaling is unclear. PPP1 is ubiquitously expressed and a major serine/threonine phosphatase.Citation26 Through dephosphorylating different substrates, PPP1 is shown to be essential for cell division, and participates in the regulation of diverse biological processes, including metabolism, Parkinson disease, muscle contraction, drug addiction, cancer, protein synthesis, neuronal signaling, apoptosis, and autophagy which is of specific interest for the current study.Citation27 Some PPP1 substrates and most of PPP1 regulators bind PPP1 via an evolutionarily conserved PPP1-binding motif, the (R/K)Vx(F/W) (x denotes any residue) consensus, which functions as a docking site for PPP1.Citation28 Interestingly, ATG16L1 contains a single RVxF (539RVVF542) motif () that can behave as a putative PPP1 binding site. To test whether or not ATG16L1 is a substrate for PPP1 dephosphorylation, we used CSNK2-phosphorylated GST-ATG16L1 as substrate and performed in vitro dephosphorylation assays (). CSNK2-phosphorylated ATG16L1 was greatly dephosphorylated with increasing amounts of purified PPP1 (). As expected, the phosphatase activity of PPP1 was completely inhibited by the protein phosphatase inhibitor-2 (I-2) (). Interestingly, we found that the dephosphorylation rate was markedly impaired for ATG16L1 with mutations in the potential PPP1 docking site (V540A/F542A), which blocked the binding of PPP1 to its substrates (). The V540A/F542A mutant did not influence the phosphorylation by CSNK2 (). Strikingly, PPP2CA could not dephosphorylate CSNK2-phosphorylated ATG16L1 ( lower panel), although the same amount of PPP2CA could efficiently dephosphorylate CHEK2 at Thr68 which is a known substrate for PPP2CA ().Citation29 Moreover, the catalytically inactive PPP1 mutant (H248K),Citation30 could not decrease the phosphorylation of ATG16L1. These data suggest that PPP1, but not PPP2CA, dephosphorylates CSNK2-phosphorylated ATG16L1 in vitro, which indicates that PPP1 may be the major phosphatase responsible for the dephosphorylation of ATG16L1. Interestingly, we also found that H/R inhibited PPP1 activity in cardiomyocytes ().
Figure 5. PPP1 dephosphorylated ATG16L1 in vitro. (A) In vitro kinase assays used CSNK2 kinase to phosphorylate GST-ATG16L1 with or without V540A/F542A mutations. (B) In vitro dephosphorylation assays using CSNK2-phosphorylated GST-ATG16L1 as substrate as described under Materials and Methods. Results from 3 independent experiments are presented. (C) In vitro dephosphorylation assays using phosphatases PPP1, catalytically inactive PPP1 (H248K), and PPP2CA. The graph indicates the percentage of dephosphorylation from 3 independent experiments. HEK293T cells were transfected with HA-CHEK2. Cells were lysed and immunoprecipitated with anti-HA antibody after cisplatin exposure. The same amount of PPP2CA was used to dephosphorylate immunoprecipitated HA-CHEK2. (D) H/R-mediated inhibition of PPP1 activity. Cardiomyocytes exposed to normoxia or H/R were immunoprecipitated with an anti-PPP1 antibody and serine/threonine phosphatase activity was measured in immunoprecipitates. The data are presented as the means ± SD of 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
PPP1 interacted with and dephosphorylated ATG16L1 in vivo
Next, we further investigated whether PPP1 could dephosphorylate ATG16L1 in vivo. To this end, we knocked down PPP1 expression using specific siRNA against PPP1 in NRVCs and H9c2 cells, or treated NRVCs and H9c2 cells with okadaic acid (OA), an inhibitor of Ser/Thr-protein phosphatases, at various concentrations. OA inhibits PPP2CA at nanomolar concentrations, whereas inhibition of PPP1 requires much higher OA concentrations.Citation31 PPP1 knockdown or OA treatment (1 μM for 30 min) significantly enhanced the phosphorylation of ATG16L1 (). However, the catalytically inactive PPP1 mutant (H248K), could not decrease the phosphorylation of ATG16L1 in transfected cells (). In line with our in vitro dephosphorylation results (), the ATG16L1V540A/F542A mutant was less efficiently dephosphorylated when compared with wild-type ATG16L1 (). These findings prompted us to test whether ATG16L1 forms a complex with PPP1. To address this question, we performed immunoprecipitation with transfected vectors and found that PPP1 WT, but not the inactive mutant PPP1H248K, associated with ATG16L1, suggesting PPP1 catalytically binds to ATG16L1 (). Moreover, the PPP1 inactive mutant did not interact with ATG16L1V540A/F542A either (data not shown). Interestingly, the ATG16L1S139A mutant showed enhanced interaction with PPP1 when compared with WT ATG16L1 in transfected cells (). Thus, our data indicate that PPP1 tends to associate with the nonphosphorylated form of ATG16L1, whereas CSNK2 prefers to bind with Ser139-phosphorylated ATG16L1.
Figure 6. For figure legend, see page 1318.
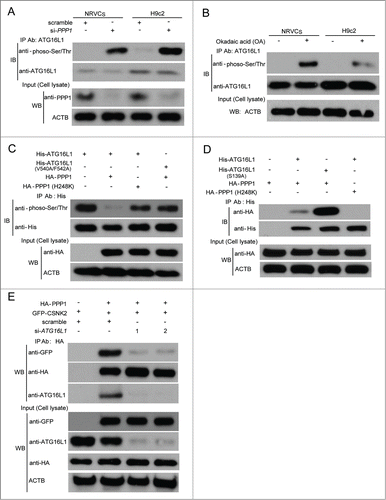
Surprisingly, we found that CSNK2 also interacted with PPP1 in NRVCs (). However, none of CSNK2 subunits has a consensus PPP1-binding motif (R/KV/IXF/W) or FXXBXB motif.Citation32 We therefore hypothesized that an adaptor protein mediates the association of CSNK2 and PPP1. To test whether ATG16L1 functions as an adaptor protein to regulate the interaction between CSNK2 and PPP1, we compared CSNK2-PPP1 association with or without ATG16L1 knockdown () and observed decreased CSNK2-PPP1 interaction following ATG16L1 knockdown. Such an observation further confirms that ATG16L1 is required for the CSNK2-PPP1 association.
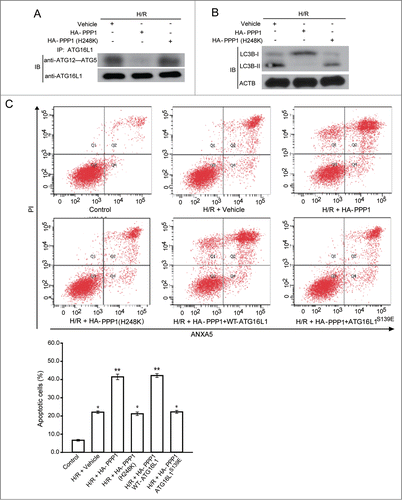
PPP1 overexpression decreased H/R-induced autophagy and increased apoptosis in cardiomyocytes
Finally, we determined that PPP1-mediated dephosphorylation of ATG16L1 at Ser139 is of functional importance for the inhibition of autophagy. Strikingly, we found that, in contrast to treatment with vehicle, overexpression of wild-type PPP1, but not the phosphatase-dead mutant PPP1H248K, inhibited the interaction between ATG16L1 and the ATG12-ATG5 complex, decreased H/R-induced autophagy, and thus increased NRVC death rate (). To confirm that the role of PPP1 in autophagy is mediated through the dephosphorylation of ATG16L1 at Ser139, a rescue experiment was performed in which wild-type ATG16L1 and the S139E mutant plasmids were transfected into PPP1-transfected cells. As expected, S139E mutant, but not wild-type ATG16L1, reversed the increased NRVC death rate in PPP1-transfected cells (). These results demonstrated that PPP1 specifically regulates autophagy through the dephosphorylation of ATG16L1 at Ser139, and further reinforce our hypothesis that the reversible phosphorylation of ATG16L1 at Ser139 by PPP1 and CSNK2 dictates the interaction between ATG16L1 and the ATG12-ATG5 conjugate associated with autophagy.
Figure 7. PPP1 overexpression decreased H/R-induced autophagy and increased apoptosis in cardiomyocytes. (A) H9c2 cells were transfected with plasmids containing vehicle, or WT HA-PPP1, or its inactive mutant HA-PPP1 (H248K), and then exposed to H/R. Immunoprecipitation (IP) was performed with an anti-ATG16L1 antibody. Co-IP of ATG12-ATG5 was detected through western blotting. (B) Cell lysates from (A) were analyzed via western blotting by probing with LC3B antibody. (C) H9c2 cells were transfected with the indicated plasmids and then exposed to H/R. Apoptosis was measured by ANXA5/PtdIns staining. Experiments were repeated at least 3 times. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Although autophagy-regulating signaling pathways have been remarkably well characterized in recent years, molecular mechanisms regulating this process are not fully studied. It is widely thought that kinases and phosphatases cooperatively control protein activity. However, previous studies were mostly focused on several positive or negative regulators of autophagy, such as AMPK, MTORC1, ULK1, and the class III phosphatidylinositol 3-kinase complex, which affect autophagy induction in the early phase.Citation33 Here, for the first time, we (i) reported that ATG16L1 is a phosphoprotein and phosphorylation of ATG16L1 promotes autophagosome maturation; (ii) further identified CSNK2 as an important kinase responsible for phosphorylation of ATG16L1 and serine 139 as its major substrate residue; (iii) demonstrated that ATG16L1 associates with the ATG12-ATG5 complex in a Ser139 phosphorylation-dependent manner, whereas CSNK2 inhibitor treatment disrupted the ATG12-ATG5-ATG16L1 complex; (iv) provided evidence that wild-type PPP1, but not the inactive mutant, interacts with ATG16L1, which dephosphorylates CSNK2-phosphorylated ATG16L1; (v) further identified the539RVVF542 consensus site for PPP1 binding in the C-terminal tail of ATG16L1; importantly, (vi) demonstrated that phosphorylation of ATG16L1 on Ser139 was responsible for H/R-induced autophagy in cardiomyocytes, which protects cardiomyocytes from apoptosis. Collectively, we provided a novel molecular mechanism suggesting roles for CSNK2 and PPP1 in the regulation of autophagy by phosphorylation of ATG16L1.
CSNK2 has previously been indicated as being involved in the autophagic process. CSNK2 dissociates BECN1 from BCL2 and initiates autophagy by phosphorylating BCL2 at serine and threonine residues.Citation34 CSNK2 also phosphorylates Ser403 of SQSTM1 directly and promotes selective autophagic clearance of ubiquitinated proteins.Citation35 A recent report shows that the yeast homolog of CSNK2 is able to phosphorylate Atg32, a mitophagy receptor in yeast, suggesting that CSNK2 may have a general role to regulate mitophagy both in mammalian systems and in yeast.Citation17 However, the effect of CSNK2 on autophagy is controversial, because CSNK2 significantly inhibits autophagy induction in human glioblastoma cells (M059K and T98G).Citation36 In this study, we provide both in vitro and in vivo evidence demonstrating that ATG16L1 is a novel substrate of CSNK2, and phosphorylation of ATG16L1 is induced by H/R in a CSNK2-dependent manner. Phosphorylation of ATG16L1 on Ser139 has not been observed previously. It is worth noting that several other residues (such as Ser213, The265 and Ser287) of ATG16L1 are predicted to be phosphorylated; however, none of these phosphorylation sites have been confirmed, and their relevant kinases have not yet been identified. Additionally, phosphorylation of other autophagic components (such as autophagy-related proteins) by other kinases has already been documented. For example, MAPK8/JNK1 plays a critical role in the early steps of autophagy whereas the MEKK4-MAPK/p38 axis-induced phosphorylation of ATG5 blocks autophagic flux in a late stage.Citation5,37 Recently, SUPT20H/p38IP was shown to bind to ATG9 and to be essential for starvation-induced autophagy.Citation38 We think that identification of the remaining phosphorylation site(s) and the relevant kinase(s) should provide useful information for a better understanding of the cellular function of ATG16L1.
Contrary to current rapidly increasing knowledge of autophagy-related kinases, our understanding of phosphatase function, regulation, and substrate interaction involved in this process is still quite limited. Given that a delicate balance between kinase and phosphatase activities determines appropriate regulation of different cellular processes, dysregulation of a single key phosphatase orchestrating these pathways may have important effects on cellular functions, especially on induction of autophagy.Citation39 Indeed, phosphatases have recently been linked to the autophagy pathway, such as several well-known phosphatases (PTEN, PPM1D/WIP1, PTPRS/PTPσ, and PPP2CA), and newly identified phosphatases (PGAM5, SGPP1 [sphingosine-1-phosphate phosphatase 1] and calcineurin).Citation20,40-43 However, none of these studies have directly linked the activity of phosphatases to ATG16L1 function. This study not only identified PPP1 as a phosphatase that directly dephosphorylates CSNK2-phosphorylated ATG16Ll, but also indicated that ATG16Ll binds to PPP1 through a RVxF docking motif, further supporting the opposing roles of CSNK2 kinase and PPP1 during autophagy flux.
What might be the significance of autophagy regulation by CSNK2 and PPP1 under physiological conditions? Autophagy is an important player in cardiovascular disease development such as atherosclerosis, cardiac ischemia/reperfusion (I/R), cardiomyopathy, heart failure, and hypertension.Citation44-45 However, the pathophysiological role of autophagy in cardiovascular diseases is controversial. Apoptosis invariably contributes to cell death, but autophagy plays dual roles in cell death or survival, which might depend on the cellular context.Citation46-47 Some studies have indicated that upregulation of autophagy is protective during myocardium ischemia.Citation48 For example, inhibition of PPP1 promotes phosphorylation of EIF2S1/eIF2α to induce autophagy which protects cardiomyocytes against tunicamycin-induced apoptosis and I/R injury.Citation49 In the present study, we uncovered a new mechanism involving CSNK2-PPP1 in protecting cardiomyocytes from H/R-mediated apoptosis via the regulation of ATG16L1 activity. Because dysregulation of autophagy has also been linked to cancer and other diseases, we think that our results will be useful for shedding light on the molecular mechanisms underlying these diseases and offer multiple targets for therapeutic intervention.
ATG16L1, a bona fide autophagy protein interacting with the ATG12-ATG5 conjugate, is required for mediating the conjugation of PE to LC3 and the formation of the autophagosome in both yeast and mammals.Citation50 Recently, ATG16L1 has drawn special attention because of its identification as a critical risk gene for Crohn disease (CD). Genetic and functional findings suggest that dysregulation of ATG16L1 stimulates inflammatory reactions and is closely related to the aberrant Paneth cell function.Citation51 It was reported that autophagy may act as an important mechanism in linking host genetic susceptibility to immune dysregulation that leads to the pathogenesis of CD.Citation52 ATG16L1 has 7 WD-repeats in its C-terminal domain, suggesting that these may provide a platform for further protein-protein interactions, highly reminiscent of GNB2L1/Rack1 (guanine nucleotide binding protein [G protein], β polypeptide 2-like 1) which also has 7 WD-repeats and can interact with PPP2CA.Citation53 Phosphorylation is well known to represent a post-translational mechanism for the rapid regulation of protein activity. Herein, we provided the first evidence of the post-translational modification of ATG16L1, suggesting that regulation of ATG16L1 phosphorylation is a critical step in autophagy induction in hypoxic cardiomyocytes. Although there were many studies about the effects of autophagy on cardiovascular diseases, very little was known about ATG16L1's role in the process, which definitely needs to be further investigated to provide additional insights into understanding the physiological function of ATG16L1. Given the crosstalk between H/R and inflammation, investigating the pathway of ATG16L1-mediated autophagy and its regulation in the context of cardiomyocytes can expand our future understanding of CD pathogenesis if a similar observation holds true, albeit in a different cell system.
In conclusion, for the first time, our results demonstrated that reversible phosphorylation of ATG16L1 plays a crucial role in fine-tuning autophagy activity in response to cardiomyocyte H/R, showing a selective regulation of ATG16L1 activity via CSNK2-PPP1, which oppositely control autophagosome maturation.
Materials and Methods
Plasmids
The human ATG16L1 constructs and V5 epitope-tagged PPP1 construct have been described previously.Citation54-55 Other plasmids used were as follows: HA-tagged CSNK2 (Addgene, 27086), CSNK2A1 (Addgene, 23678), CSNK2A2 (Addgene, 27087), ATG5 (Addgene, 22948), ATG12 (Addgene, 27049) and FLAG-ATG16L1 constructs (Addgene, 24302). All PCR-derived constructs were verified by sequencing. The GFP-LC3 expression vector and LC3 tagged with GFP and RFP in tandem (mRFP-GFP-LC3) were kindly provided by Dr. Xiao Xie (Rutgers University). The human PPP1 clone (OpenBiosystems, 3956353) was subcloned into the pcDNA-HA vector.Citation56 Deletion constructs were generated using PCR and verified by sequencing and restriction enzyme digestion. All point mutations were generated by the QuikChange site-directed PCR mutagenesis kit (Stratagene, 200523) and verified by sequencing. The following primers for the RVxF motif were used to introduce the alanine point mutations (V540A/F542A) into full-length human ATG16L1 plasmid, and mutated residues are underlined: 5′-GCGGCTCTGACTGGACCAGAGCTGTCGCCAGCCCTGATGG-3′ (forward) and 3′-CCATCAGGGCTGGCGACAGCTCTGGTCCAGTCAGAGCCGC-5′ (reverse). Other point mutations were also introduced into the nonphosphorylatable mutant and mutated residues are underlined. These are as follows: S304A, 5′-GCTCATCCTGGTGCTGGTAAAGAAGTG AGG-3′ (forward) and 3′-CCTCACTTCTTTACCAGCACCAGGATGAGC-5′ (reverse). S370A, 5′-TGCAGGAATTGCAAGCATTGAATTTGATAG-3′ (forward) and 3′-CTATCAAATTCAATGCTTGCAATTCCTGCA-5′ (reverse). S139A, 5′-GTTTGCAGACTATCGCTGACCTGGAGACGG-3′ (forward) and 3′-CCGTCTCCAGGTCAGCGATAGTCTGCAAAC-5′ (reverse). Plasmids containing the ATG16L1 phosphomimetic mutant (S/T to E) were generated using primers as follows: S304E, 5′-GCTCATCCTGGTGAAGGTAAAGAAGTGAGG-3′ (forward) and 3′-CCTCAC TTCTTTACCTTCACCAGGATGAGC-5′ (reverse). S370E, 5′-TGCAGGAATTGAAAGCAT TGAATTTGATAG-3′ (forward) and 3′-CTATCAAATTCAATGCTTTCAATTCCTGCA-5′ (reverse). S139E, 5′-GTTTGCAGACTATCGAGGACCTGGAGACGG-3′ (forward) and 3′-CCG TCTCCAGGTCCTCGATAGTCTGCAAAC-5′ (reverse).
Reagents, siRNA, and antibodies
Reagents used were as follows: purified protein kinase CSNK2 (New England Biolabs, P6010S), λPP (New England Biolabs, P5501-01), PPP1 (New England Biolabs, P0754S). Protein kinase CSNK2 inhibitor 4,5,6,7-tetrabromobenzotriazole, TBB (Sigma-Aldrich, T0826). Okadaic acid, (LC Laboratories, O-2220) was dissolved in methanol to 1 mM, PPP1 inhibitors (Santa Cruz Biotechnology, SC-45044). Cisplatin (Sigma-Aldrich, 479306). siRNA for CSNK2 (Santa Cruz Biotechnology, SC-29919), siRNA for human PPP1, targeting all 3 isoforms (Santa Cruz Biotechnology, SC-43545). Transfection was done with Oligofectamine (Invitrogen Life Technologies, 12252-011) as recommended by the manufacturer. Anitbodies used were as follows: anti-PPP1 monoclonal antibody (Santa Cruz Biotechnology, SC-7482), anti-ACTB (Sigma-Aldrich, A1978), anti-V5 antibody (Invitrogen, R960-25), anti-FLAG (Sigma-Aldrich, F3165), anti-HA (Sigma-Aldrich, 9658-100), anti-SQSTM1 (Sigma-Aldrich, P0068), rabbit polyclonal anti-LC3B antibody (Sigma-Aldrich, L7543), glutathione S-transferase (GST) antibodies (Sigma-Aldrich, A7340-5 mL), anti-His (Bethyl Laboratories, A190-112A), rabbit anti-ATG12-ATG5 complex antibody (Biocompare, CPBT-66438RA), rabbit polyclonal antibodies to phosphoserine (Abcam, ab9332), phosphothreonine (Abcam, ab9337), phosphoserine/threonine (Abcam, ab17464) and anti-CHEK2 phospho T68 (Abcam, ab32148).
GFP-LC3 puncta assay
The method used to evaluate fluorescent LC3 puncta in vivo has been described previously.Citation16 Briefly, H9c2 cells or NRVCs were plated into a Φ20 mm dish (NEST, 801001) and transiently transfected with GFP-LC3 or mRFP-GFP-LC3 plasmid for 24 h. The cells were then fixed in 3.7% paraformaldehyde. The number of GFP-LC3 puncta per cell in GFP-LC3-, or mRFP-GFP-LC3-positive cells was counted under a laser-scanning confocal microscope (LEICA TCS SP2, Germany).
λ-Phosphatase treatment
Indicated cells were lysed in Nonidet P-40 buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Nonidet P-40 [Roche, 11754599001]). The lysates were treated or not with 400 units (1 μL) of λPP (New England Biolabs, P0753) in the presence of 1 mM MnCl2 at 30°C for 30 min. A mixed solution of 10 mM sodium orthovanadate and 50 mM sodium fluoride was used as a λPP inhibitor. The reaction was stopped by the addition of SDS sample buffer followed by 5 min of heating at 95°C.
Flow cytometry
The assay was performed according to the manufacturer's instructions. Briefly, cells were harvested, washed with phosphate-buffered saline (PBS; Sigma-Aldrich, P4417), suspended in ANXA5 binding buffer (10 mM HEPES, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2), stained with Alexa Fluor 488 ANXA5-PtdIns (Invitrogen, V13241), and analyzed by flow cytometry. For each experiment, 10,000 cells were analyzed using the BD LSRII (BD Biosciences, USA) and FACS Diva Software (BD Biosciences, USA).
Immunoprecipitation
After cells were lysed in lysis buffer (2% Nonidet P-40 buffer in PBS supplemented with protease inhibitors [Roche, 11836145001]) for 20 min, the nuclear and cellular debris were cleared by centrifugation. Immunoprecipitation was performed using rabbit polyclonal anti-GFP (Life Technologies, K481001), mouse monoclonal anti-FLAG, M2 (Sigma-Aldrich, F-3165) or anti-hemagglutinin (HA) antibody (Life Technologies, A-21287), and protein A/Sepharose (Amersham Biosciences, 17-0780-01) or protein G/Sepharose (Amersham Biosciences, 17-0885-01). Immunoprecipitates were washed 6 times in lysis buffer and eluted in SDS sample buffer. Proteins were separated by SDS-PAGE and analyzed by immunoblotting.
Hypoxia/reoxygenation
Hypoxia/reoxygenation (H/R) was performed as previously described.Citation57 H/R conditions were simulated by culturing cells in a hypoxia chamber (MIC-101, Billups-Rothenberg, Inc., Del Mar, CA). H9c2 cells were cultured in nutrient-rich medium for 3 d and washed with PBS. Hypoxia was achieved by placing the cells in a hypoxia chamber filled with 5% CO2 and 95% N2 at 37°C for 4 h. After hypoxia exposure, the cells were reoxygenated with 5% CO2 and 95% O2 for 3 h in DMEM with 10% fetal bovine serum (FBS; GE Healthcare HyClone, SH3007102).
Cell culture and transfection
HEK293T cells (Thermo Scientific, HCL4517) were maintained in DMEM (Invitrogen, 12430047) containing 10% FBS and L-glutamine (Invitrogen, 25030-081) plus 100 units/mL of penicillin and 100 μg/mL streptomycin (Invitrogen, 15140-122) at 37°C in a humidified atmosphere containing 5% CO2.
Isolation and culture of primary rat neonatal cardiomyocytes was performed as described previously.Citation58 The animal procedures were in compliance with IACUC approved protocols. Briefly, whole hearts from 2- to 5-d-old rats were isolated, minced, and rinsed in a balanced salt solution (1.26 mM CaCl2, 137 mM NaCl, 0.338 mM NaH2PO4, 5.5 mM MgSO4, 5.3 mM KCl, 0.8 mM MgSO4, 0.44 mM KH2PO4, 4.17 mM NaHCO3, pH 7.3-7.4). Five cycles of digestion using collagenase (95 U/mL; Sigma-Aldrich, C0130) and 0.6 U/mL of pancreatin (Sigma-Aldrich, P7545) were performed. The tissue pieces were incubated and shaken gently for 20 min at 37°C in the balanced salt solution. At the end of each cycle, the suspension was centrifuged (×500 g, 10 min) and the cell pellet resuspended in 4 mL of the digestion solution. Supernatant fractions from the 5 cycles were centrifuged (500 × g, 10 min) and resuspended in the cardiac medium containing DMEM and M199 (Sigma-Aldrich, M-4530) (4:1) supplemented with horse serum (10%; Sigma-Aldrich, H1270), fetal calf serum (5%; Sigma-Aldrich, F7524), streptomycin (100 μg/mL) and penicillin (100 U/mL). After cell plating on noncoated culture dishes for 1.5 h to allow attachment of nonmyocyte cells, the remaining cell suspension containing neonatal cardiomyocytes was collected and seeded at 5 × 104 cells/cm2 in collagen I-coated culture dishes (Sigma-Aldrich, S3315) in the cardiac medium and incubated in 5% CO2 at 37°C. The medium was replaced after 72 h and cardiomyocytes were allowed to reach confluence before use.
H9c2 cardiomyocytes derived from rat myocardium were obtained (ATCC, CRL-1446). H9c2 cells were cultured in DMEM with 10% FBS without penicillin/streptomycin in a humidified atmosphere of 5% CO2 at 37°C. The medium was changed every 2 d. TBB was added 1 h prior to H/R. H9c2 cells were assigned to 4 groups: (1) Control, H9c2 cells were maintained in normoxic conditions without TBB treatment; (2) TBB, H9c2 cells were treated with TBB and incubated in normoxic conditions; (3) H/R, H9c2 cells were subjected to 4 h of hypoxia followed by 3 h of reoxygenation; (4) H/R+TBB, H9c2 cells pretreated with TBB were subjected to 4 h of hypoxia followed by 3 h of reoxygenation. H9c2 cells were treated with TBB (100 μM) for the indicated times as described previously.Citation3 All the transient overexpression transfections were performed using Attractene (Qiagen, 301005) following the manufacturer's instructions.
Western blotting
Whole cell extracts were prepared using RIPA buffer (Sigma-Aldrich, R0278). The proteins were monitored with a western blot analysis. The samples were denatured by boiling for 5 min in loading buffer, subjected to 10% SDS-PAGE, and then electroblotted onto polyvinylidenedifluoride (PVDF) membrane (Invitrogen, LC2005). Next, the membranes were incubated with the indicated primary antibodies, followed by washing with Tris-buffered saline-Tween 20 [T-BST; Tris-Buffered Saline (Bio-Rad, 170-6435); 0.1% Tween-20 (Bio-Rad, 170-6531)] for 30 min and then incubated with a horseradish peroxidase-conjugated secondary antibody (Kirkegaard & Perry Labs, 14-12-06) at room temperature for 1 h. After being washed again with T-BST buffer for 30 min, immunolabeled bands were detected using an enhanced chemiluminescence (ECL) kit (SuperSignal; Pierce Biotechnology, 32106). The primary antibodies were used at a dilution of 1:1000, and secondary antibodies were used at a dilution of 1:2000. The anti-ACTB was used as the loading control. The blots were scanned using an Epson Perfection PhotoScanner (Epson, Japan). Finally, data analysis was performed with ImageJ (National Institutes of Health, Bethesda, MD) by measuring the densities of immunoreactive bands.
Recombinant protein expression and purification
All proteins were expressed in E. coli Rosetta pLySS cells.Citation59 GST-tagged ATG16L1 was expressed from pOPTG.Citation60 Briefly, GST-tagged ATG16L1 was expressed in E. coli Rosetta pLySS from pOPTG-ATG16L1. Cells were grown at 37°C to an OD600 = 0.8, induced with 1 mM IPTG (Sigma-Aldrich, I6758) and grown for another 4 h. Cells were disrupted by freeze thawing and the cleared lysate was incubated with glutathione beads (glutathione Sepharose 4B; GE Healthcare, 17-0756-01). The protein was cleaved off from the beads with TEV protease (Sigma-Aldrich, T4455), concentrated and applied to a 16/60 S75 gel exclusion column (GE Healthcare, 17-1068-01). The protein was eluted with 150 mM NaCl, 50 mM HEPES, pH 7.5, 1 mM DTT. The ATG12-ATG5 conjugate was produced in E. coli Rosetta pLySS by co-expression of ATG12, and hexahistidine-tagged ATG5. Cells were grown at 37°C to an OD600 = 0.8, induced with 1 mM IPTG and grown for another 4 h at 37°C. Harvested cells were resuspended in the resuspension buffer [300 mM NaCl, 50 mM HEPES, pH 7.5, 10 mM Imidazole, 2.5 mM Pefablock (Roth, 11429868001), 1 mM MgCl2, 2 mM 2-mercaptoethanol, DNase (Sigma-Aldrich, D5025)], and disrupted by the freeze-thaw method and sonication.Citation61
The cleared lysate was applied to a HisTrap column (GE Healthcare, 17-5247-01) and the proteins were eluted by a step-wise (0-250 mM) imidazole (Sigma-Aldrich, CI2399-500G) gradient. The eluate was concentrated using Amicon Ultra centrifugal filter (MW cut-off 30 kDa; Sigma-Aldrich, Z717185) and further purified using a 16/60 S200 size exclusion column (GE Healthcare, 17-1069-01).
GST affinity isolation assay
GST-ATG16L1 proteins were purified from E. coli BL21 using glutathione-sepharose beads according to a standard procedure.Citation62 After washes with PBS, purified ATG16L1 beads in HSE buffer (20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1% Triton ×-100) were mixed with purified CSNK2 or PPP1 and incubated for 1 h at 4°C. After washes with HSE buffer, bound CSNK2 or PPP1 was analyzed by SDS-PAGE and immunoblotting. Briefly, GST-ATG16L1 or GST control proteins prebound to glutathione-sepharose beads were mixed with 100 ng of pure PPP1 protein, incubated for 90 min, washed with 0.1% Triton X-100 (Fisher, BP151-100) in phosphate-buffered Ca2+/Mg2+-free saline (Sigma-Aldrich, D5773), and then eluted by heating in SDS sample buffer. HEK293T cell lysates were obtained by washing near confluent 10-cm plates with PBS and lysing in buffer (1% Triton X-100, 1× Complete protease inhibitor [Roche, 11836153001], 50 mM NaF) on ice, followed by incubation on ice with mixing for 30 min. After centrifugation (16,000 × g) for 20 min, the supernatant fraction was added to glutathione beads pre-coated with wild-type and mutant GST-ATG16L1. After a 3-h incubation with mixing at 4°C, beads were centrifuged and washed with lysis buffer, and bound proteins were eluted in SDS sample buffer.
In vitro kinase assay
1-2 μg of GST-ATG16L1 was incubated with 0.5-1 μg of active recombinant CSNK2 kinase in kinase buffer (50 mM Tris-HCl, pH 7.4, 50 mM NaCl, 10 mM MgCl2, 10 mM β-glycerophosphate, 1 mM dithiothreitol [DTT], 100 μM ATP [Sigma-Aldrich, A26209]) in the presence of 5 μCi of [γ-32P] ATP (3000 Ci/mM; PerkinElmer Life Sciences). The reaction was carried out at 30°C for 30 min and stopped by the addition of SDS loading buffer. The samples were resolved by SDS-PAGE, transferred onto PVDF (Millipore, UFC30GVNB), and visualized by autoradiography followed by western blot analysis.
In vitro dephosphorylation/phosphatase assay
GST-ATG16L1 was first phosphorylated by CSNK2A in vitro as described above. Phosphorylated GST-ATG16L1 was affinity isolated by glutathione-agarose (Santa Cruz Biotechnology, SC-2009) and extensively washed with Nonidet P-40 buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 1% Nonidet P-40). The washed beads were then resuspended in Nonidet P-40 buffer and split into tubes and appropriate protein phosphatases (PPP1 [New England Biolabs, P0754], or PPP2CA [Calbiochem, 539508]) or BSA as a negative control. For inhibitor-2 (New England Biolabs, P0755) treatment, 2.5 units of inhibitor-2 were added. The reaction was incubated at 30°C for 30 min. The beads and supernatant fraction were carefully separated by centrifugation. The relative amounts of32P released into the supernatant fraction and the32P bound to GST-ATG16L1 were quantified by liquid scintillation counting (Beckman, 500603).
CSNK2 kinase activity assay
Cells were lysed by sonication and supernatant fractions were collected. Aliquots of cell lysates containing 50 μg of protein were assayed for CSNK2 kinase activity by the Casein Kinase 2 Assay Kit (Upstate Cell Signaling Solutions, 17-312) following the manufacturer's instructions. Using a scintillation counter, the phosphotransferase activity of CSNK2 was quantified. Triplicate reactions were run in the presence or absence of CSNK2 inhibitor. The nonspecific activity measured in the reactions containing inhibitor was subtracted from activity measured in reactions without inhibitor. Briefly, samples were divided into 2 groups with and without TBB treatment. Specific CSNK2 activity was calculated using the equation: CSNK2 activity = cpm value 1 (the group without TBB) - cpm value 2 (the group with TBB treatment). Data were expressed as means ± SD (n = 3).
PPP1 phosphatase activity assay
PPP1 phosphatase activity was measured using a serine/threonine phosphatase assay kit according to the manufacturer's instructions (Upstate Biotechnology, 17-127). Briefly, cells were put into ice cold PBS and lysed by sonication. The pellet was resuspended in Buffer C (420 mM NaCl, 20 mM HEPES, pH 7.9, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM phenylmethanesulfonyl fluoride [Sigma-Aldrich, P7626], 1 mM DTT, 1 mg/ml each of leupeptin [Sigma-Aldrich, L2884] and aprotinin [Sigma-Aldrich, A6103]). PPP1 was immunoprecipitated from supernatant fractions using 1 mg/ml of anti-PPP1 (Upstate Biotechnology, 07-273) and protein A/G Plus-Agarose beads (Santa Cruz Biotechnology, SC-2003). On-bead phosphatase activity was determined by co-incubation of the beads with a known PPP1 substrate phosphopeptideCitation63 and measurement of phosphate liberation.
In vivo myocardial ischemia/reperfusion (I/R) model
Wild-type mouse (C57BL6/J) hearts were subjected to global ischemia/reperfusion (I/R) during Langendorff perfusion.Citation64 The animal study was approved by the Institutional Animal Care. Surgical induction of myocardial I/R was performed as follows: Briefly, mice were anesthetized by an intraperitoneal injection with pentobarbital sodium (50 mg/kg; Sigma-Aldrich, P3761). After performing a left thoracotomy, the left anterior descending (LAD) coronary artery was ligated using a 6-0 silk suture around fine PE-10 tubing with a slip knot. To induce I/R injury, mice were subjected to 30 min of LAD ischemia followed by 60 min of reperfusion, respectively. For the sham group, a suture was passed under the LAD but not tied. After experiments, mice were euthanized with pentobarbital sodium (200 mg/kg) by an intraperitoneal injection, and the ischemic-reperfused tissue was isolated and used for further assays.
Statistical analysis
All data are presented as the means ± SD and were statistically analyzed using SPSS software (IBM Inc., USA), version 13.0. The data were analyzed using one-way ANOVAs and a 2-tailed, unpaired Student t test to determine statistical significance and were further evaluated using the Bonferroni post hoc tests. A value of P < 0.05 was considered to be statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1060386_supplemental_files.doc
Download MS Word (1.3 MB)Funding
This study was supported by grants from the National Natural Science Foundation of China (81070176, 81270282, 81470389, 81430055 and 81172139).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Mizushima N. Autophagy: process and function. Genes Dev 2007; 21:2861-73; PMID:18006683; http://dx.doi.org/10.1101/gad.1599207
- Joubert PE, Werneke SW, de la Calle C, Guivel-Benhassine F, Giodini A, Peduto L, Levine B, Schwartz O, Lenschow DJ, Albert ML. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J Exp Med 2012; 209:1029-47; PMID:22508836; http://dx.doi.org/10.1084/jem.20110996
- Wirawan E, Vanden Berghe T, Lippens S, Agostinis P, Vandenabeele P. Autophagy: for better or for worse. Cell Res 2012; 22:43-61; PMID:21912435; http://dx.doi.org/10.1038/cr.2011.152
- Chen Y, Klionsky DJ. The regulation of autophagy-unanswered questions. J Cell Sci 2011; 124:161-70; PMID:21187343; http://dx.doi.org/10.1242/jcs.064576
- Keil E, Hocker R, Schuster M, Essmann F, Ueffing N, Hoffman B, Liebermann DA, Pfeffer K, Schulze-Osthoff K, Schmitz I. Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ 2013; 20:321-32; PMID:23059785; http://dx.doi.org/10.1038/cdd.2012.129
- Reggiori F, Komatsu M, Finley K, Simonsen A. Autophagy: more than a nonselective pathway. Intl J Cell Biol 2012; 2012:219625; PMID:22666256; http://dx.doi.org/10.1155/2012/219625
- Lee IH, Finkel T. Regulation of autophagy by the p300 acetyltransferase. J Biol Chem 2009; 284:6322-8; PMID:19124466; http://dx.doi.org/10.1074/jbc.M807135200
- Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, Zhang SM, Lian G, Liu Q, Ruan K, et al. Protein phosphorylation-acetylation cascade connects growth factor deprivation to autophagy. Autophagy 2012; 8:1385-6; PMID:22717509; http://dx.doi.org/10.4161/auto.20959
- Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/10.1128/MCB.06159-11
- Singh NN, Ramji DP. Protein kinase CK2, an important regulator of the inflammatory response? J Mol Med 2008; 86:887-97; PMID:18437331; http://dx.doi.org/10.1007/s00109-008-0352-0
- Al Quobaili F, Montenarh M. CK2 and the regulation of the carbohydrate metabolism. Metabolism 2012; 61:1512-7; PMID:22917893; http://dx.doi.org/10.1016/j.metabol.2012.07.011
- Cohen PT. Protein phosphatase 1-targeted in many directions. J Cell Sci 2002; 115:241-56; PMID:11839776
- Peti W, Nairn AC, Page R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J 2013; 280:596-611; PMID:22284538; http://dx.doi.org/10.1111/j.1742-4658.2012.08509.x
- Kim YK, Ahn JH, Lee M. Src Family Kinase Inhibitor PP2 Induces LC3 Conversion in a Manner That is Uncoupled from Autophagy and Increases Apoptosis in Multidrug-Resistant Cells. Biomol Ther (Seoul) 2012; 20:393-8; PMID:22284538; http://dx.doi.org/10.4062/biomolther.2012.20.4.393
- Ma H, Guo R, Yu L, Zhang Y, Ren J. Aldehyde dehydrogenase 2 (ALDH2) rescues myocardial ischaemia/reperfusion injury: role of autophagy paradox and toxic aldehyde. Eur Heart J 2011; 32:1025-38; PMID:20705694; http://dx.doi.org/10.1093/eurheartj/ehq253
- Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of foxO by sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res 2010; 107:1470-82; PMID:20947830; http://dx.doi.org/10.1161/CIRCRESAHA.110.227371
- Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res 2014; 24:787-95; PMID:24903109; http://dx.doi.org/10.1038/cr.2014.75
- Xavier CP, Rastetter RH, Blomacher M, Stumpf M, Himmel M, Morgan RO, Fernandez MP, Wang C, Osman A, Miyata Y, et al. Phosphorylation of CRN2 by CK2 regulates F-actin and Arp2/3 interaction and inhibits cell migration. Sci Rep 2012; 2:241; PMID:22355754; http://dx.doi.org/10.1038/srep00241
- Pagano MA, Bain J, Kazimierczuk Z, Sarno S, Ruzzene M, Di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F, et al. The selectivity of inhibitors of protein kinase CK2: an update. Biochem J 2008; 415:353-65; PMID:18588507; http://dx.doi.org/10.1042/BJ20080309
- Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell 2014; 54:362-77; PMID:24746696; http://dx.doi.org/10.1016/j.molcel.2014.02.034
- Nishimura T, Kaizuka T, Cadwell K, Sahani MH, Saitoh T, Akira S, Virgin HW, Mizushima N. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep 2013; 14:284-91; PMID:23392225; http://dx.doi.org/10.1038/embor.2013.6
- Fujita N, Morita E, Itoh T, Tanaka A, Nakaoka M, Osada Y, Umemoto T, Saitoh T, Nakatogawa H, Kobayashi S, et al. Recruitment of the autophagic machinery to endosomes during infection is mediated by ubiquitin. J Cell Biol 2013; 203:115-28; PMID:24100292; http://dx.doi.org/10.1083/jcb.201304188
- Chou MF, Prisic S, Lubner JM, Church GM, Husson RN, Schwartz D. Using bacteria to determine protein kinase specificity and predict target substrates. PLoS One 2012; 7:e52747; http://dx.doi.org/10.1371/journal.pone.0052747
- Emdad L, Qadeer ZA, Bederson LB, Kothari HP, Uzzaman M, Germano IM. Is there a common upstream link for autophagic and apoptotic cell death in human high-grade gliomas? Neuro-oncology 2011; 13:725-35; PMID:21727211; http://dx.doi.org/10.1093/neuonc/nor053
- Lee HM, Chen R, Kim H, Etchegaray JP, Weaver DR, Lee C. The period of the circadian oscillator is primarily determined by the balance between casein kinase 1 and protein phosphatase 1. Proc Natl Acad Sci U S A 2011; 108:16451-6; PMID:21930935; http://dx.doi.org/10.1073/pnas.1107178108
- Ragusa MJ, Dancheck B, Critton DA, Nairn AC, Page R, Peti W. Spinophilin directs protein phosphatase 1 specificity by blocking substrate binding sites. Nat Struct Mol Biol 2010; 17:459-64; PMID:20305656; http://dx.doi.org/10.1038/nsmb.1786
- Peti W, Nairn A, Page R. Structural basis for protein phosphatase 1 regulation and specificity. FEBS J 2013; 280:596-611; http://dx.doi.org/10.1111/j.1742-4658.2012.08509.x
- Wakula P, Beullens M, van Eynde A, Ceulemans H, Stalmans W, Bollen M. The translation initiation factor eIF2beta is an interactor of protein phosphatase-1. Biochem J 2006; 400:377-83; PMID:16987104; http://dx.doi.org/10.1042/BJ20060758
- Liang X, Reed E, Yu JJ. Protein phosphatase 2A interacts with Chk2 and regulates phosphorylation at Thr-68 after cisplatin treatment of human ovarian cancer cells. Int J Mol Med 2006; 17:703-8; PMID:16596250
- Peirce MJ, Cox SE, Munday MR, Peachell PT. Preliminary characterization of the role of protein serine/threonine phosphatases in the regulation of human lung mast cell function. Br J Pharmacol 1997; 120:239-46; PMID:9117116; http://dx.doi.org/10.1038/sj.bjp.0700915
- Traweger A, Wiggin G, Taylor L, Tate SA, Metalnikov P, Pawson T. Protein phosphatase 1 regulates the phosphorylation state of the polarity scaffold Par-3. Proc Natl Acad Sci U S A 2008; 105:10402-7; PMID:18641122; http://dx.doi.org/10.1073/pnas.0804102105
- Parnell SC, Puri S, Wallace DP, Calvet JP. Protein phosphatase-1alpha interacts with and dephosphorylates polycystin-1. PloS One 2012; 7:e36798; PMID:22675472; http://dx.doi.org/10.1371/journal.pone.0036798
- Kroemer G, Marino G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280-93; PMID:20965422; http://dx.doi.org/10.1016/j.molcel.2010.09.023
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571-80; PMID:21311563; http://dx.doi.org/10.1038/cdd.2010.191
- Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell 2011; 44:279-89; PMID:22017874; http://dx.doi.org/10.1016/j.molcel.2011.07.039
- Olsen BB, Svenstrup TH, Guerra B. Downregulation of protein kinase CK2 induces autophagic cell death through modulation of the mTOR and MAPK signaling pathways in human glioblastoma cells. Int J Oncol 2012; 41:1967-76; PMID:23007634; http://dx.doi.org/10.3892/ijo.2012.1635
- Wong CH, Iskandar KB, Yadav SK, Hirpara JL, Loh T, Pervaiz S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PloS One 2010; 5:e9996; PMID:20368806; http://dx.doi.org/10.1371/journal.pone.0009996
- Webber JL, Tooze SA. Coordinated regulation of autophagy by p38alpha MAPK through mAtg9 and p38IP. EMBO J 2010; 29:27-40; PMID:19893488; http://dx.doi.org/10.1038/emboj.2009.321
- Puustinen P, Rytter A, Mortensen M, Kohonen P, Moreira JM, Jaattela M. CIP2A oncoprotein controls cell growth and autophagy through mTORC1 activation. J Cell Biol 2014; 204:713-27; PMID:24590173; http://dx.doi.org/10.1083/jcb.201304012
- Errafiy R, Aguado C, Ghislat G, Esteve JM, Gil A, Loutfi M, Knecht E. PTEN increases autophagy and inhibits the ubiquitin-proteasome pathway in glioma cells independently of its lipid phosphatase activity. PloS One 2013; 8:e83318; PMID:24349488; http://dx.doi.org/10.1371/journal.pone.0083318
- Martin KR, Xu Y, Looyenga BD, Davis RJ, Wu CL, Tremblay ML, Xu HE, MacKeigan JP. Identification of PTPsigma as an autophagic phosphatase. J Cell Sci 2011; 124:812-9; PMID:21303930; http://dx.doi.org/10.1242/jcs.080341
- Lepine S, Allegood JC, Park M, Dent P, Milstien S, Spiegel S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell Death Differ 2011; 18:350-61; PMID:20798685; http://dx.doi.org/10.1038/cdd.2010.104
- Dwivedi M, Song HO, Ahnn J. Autophagy genes mediate the effect of calcineurin on life span in C. elegans. Autophagy 2009; 5:604-7; PMID:19279398
- Hamacher-Brady A, Brady NR, Gottlieb RA. Enhancing macroautophagy protects against ischemia/reperfusion injury in cardiac myocytes. J Biol Chem 2006; 281:29776-87; PMID:16882669; http://dx.doi.org/10.1074/jbc.M603783200
- Pu J, Yuan A, Shan P, Gao E, Wang X, Wang Y, Lau WB, Koch W, Ma XL, He B. Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur Heart J 2013; 34:1834-45; PMID:22307460; http://dx.doi.org/10.1093/eurheartj/ehs011
- Pu J, Mintz GS, Biro S, Lee JB, Sum SY, Madden SP, Burke AP, Zhang P, He B, Goldstein JA, et al. Insights into echo-attenuated plaques, echolucent plaques, and plaques with spotty calcification: novel findings from comparisons among intravascular ultrasound, near-infrared spectroscopy, and pathological histology in 2,294 human coronary artery segments. J Am Coll Cardiol 2014; 63:2220-33; http://dx.doi.org/10.1016/j.jacc.2014.02.576
- Wohlgemuth SE, Calvani R, Marzetti E. The interplay between autophagy and mitochondrial dysfunction in oxidative stress-induced cardiac aging and pathology. J Mol Cell Cardiol 2014; 71:62-70; PMID:24650874; http://dx.doi.org/10.1016/j.yjmcc.2014.03.007
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007; 100:914-22; PMID:17332429; http://dx.doi.org/10.1161/01.RES.0000261924.76669.36
- Liu CL, He YY, Li X, Li RJ, He KL, Wang LL. Inhibition of serine/threonine protein phosphatase PP1 protects cardiomyocytes from tunicamycin-induced apoptosis and I/R through the upregulation of p-eIF2alpha. Int J Mol Med 2014; 33:499-506; PMID:24366244; http://dx.doi.org/10.3892/ijmm.2013.1603
- Dooley HC, Razi M, Polson HE, Girardin SE, Wilson MI, Tooze SA. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell 2014; 55:238-52; PMID:24954904; http://dx.doi.org/10.1016/j.molcel.2014.05.021
- Zhai Z, Wu F, Dong F, Chuang AY, Messer JS, Boone DL, Kwon JH. Human autophagy gene ATG16L1 is post-transcriptionally regulated by MIR142-3p. Autophagy 2014; 10:468-79; PMID:24401604; http://dx.doi.org/10.4161/auto.27553
- Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011; 474:298-306; PMID:21677746; http://dx.doi.org/10.1038/nature10208
- Kiely PA, Baillie GS, Lynch MJ, Houslay MD, O'Connor R. Tyrosine 302 in RACK1 is essential for insulin-like growth factor-I-mediated competitive binding of PP2A and beta1 integrin and for tumor cell proliferation and migration. J Biol Chem 2008; 283:22952-61; PMID:18567578; http://dx.doi.org/10.1074/jbc.M800802200
- Murthy A, Li Y, Peng I, Reichelt M, Katakam AK, Noubade R, Roose-Girma M, DeVoss J, Diehl L, Graham RR, et al. A Crohn's disease variant in Atg16l1 enhances its degradation by caspase 3. Nature 2014; 506:456-62; PMID:24553140; http://dx.doi.org/10.1038/nature13044
- Fang Y, Sathyanarayanan S, Sehgal A. Post-translational regulation of the Drosophila circadian clock requires protein phosphatase 1 (PP1). Genes Dev 2007; 21:1506-18; PMID:17575052; http://dx.doi.org/10.1101/gad.1541607
- Xiao L, Chen Y, Ji M, Dong J. KIBRA regulates Hippo signaling activity via interactions with large tumor suppressor kinases. J Biol Chem 2011; 286:7788-96; PMID:21233212; http://dx.doi.org/10.1074/jbc.M110.173468
- Cheng Y, Zhu P, Yang J, Liu X, Dong S, Wang X, Chun B, Zhuang J, Zhang C. Ischaemic preconditioning-regulated miR-21 protects heart against ischaemia/reperfusion injury via anti-apoptosis through its target PDCD4. Cardiovasc Res 2010; 87:431-9; PMID:20219857; http://dx.doi.org/10.1093/cvr/cvq082
- Hadad I, Veithen A, Springael JY, Sotiropoulou PA, Mendes Da Costa A, Miot F, Naeije R, De Deken X, Entee KM. Stroma cell-derived factor-1α signaling enhances calcium transients and beating frequency in rat neonatal cardiomyocytes. PLoS One 2013; 8:e56007; http://dx.doi.org/10.1371/journal.pone.0056007
- Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, Kraft C, Martens S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J 2012; 31:4304-17; PMID:23064152; http://dx.doi.org/10.1038/emboj.2012.278
- Gonzalez B, Garrido F, Ortega R, Martinez-Julvez M, Revilla-Guarinos A, Perez-Pertejo Y, Pérez-Pertejo Y, Velázquez-Campoy A, Sanz-Aparicio J, Pajares MA. NADP+ binding to the regulatory subunit of methionine adenosyltransferase II increases intersubunit binding affinity in the hetero-trimer. PloS One 2012; 7:e50329; PMID:23189196; http://dx.doi.org/10.1371/journal.pone.0050329
- Romanov J, Walczak M, Ibiricu I, Schuchner S, Ogris E, Kraft C, Martens S. Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation. EMBO J 2012; 31:4304-17; PMID:23064152; http://dx.doi.org/10.1038/emboj.2012.278
- Matsushita M, Suzuki NN, Obara K, Fujioka Y, Ohsumi Y, Inagaki F. Structure of Atg5.Atg16, a complex essential for autophagy. J Biol Chem 2007; 282:6763-72; PMID:17192262; http://dx.doi.org/10.1074/jbc.M609876200
- Jideama NM, Crawford BH, Hussain AK, Raynor RL. Dephosphorylation specificities of protein phosphatase for cardiac troponin I, troponin T, and sites within troponin T. Int J Biol Sci 2006; 2:1-9; PMID:16585947
- Wan TC, Tosh DK, Du L, Gizewski ET, Jacobson KA, Auchampach JA. Polyamidoamine (PAMAM) dendrimer conjugate specifically activates the A3 adenosine receptor to improve post-ischemic/reperfusion function in isolated mouse hearts. BMC Pharmacol 2011; 11:11; PMID:22039965; http://dx.doi.org/10.1186/1471-2210-11-11