
Abstract
Currently, there is limited understanding about hormonal regulation of mitochondrial turnover. Thyroid hormone (T3) increases oxidative phosphorylation (OXPHOS), which generates reactive oxygen species (ROS) that damage mitochondria. However, the mechanism for maintenance of mitochondrial activity and quality control by this hormone is not known. Here, we used both in vitro and in vivo hepatic cell models to demonstrate that induction of mitophagy by T3 is coupled to oxidative phosphorylation and ROS production. We show that T3 induction of ROS activates CAMKK2 (calcium/calmodulin-dependent protein kinase kinase 2, β) mediated phosphorylation of PRKAA1/AMPK (5′ AMP-activated protein kinase), which in turn phosphorylates ULK1 (unc-51 like autophagy activating kinase 1) leading to its mitochondrial recruitment and initiation of mitophagy. Furthermore, loss of ULK1 in T3-treated cells impairs both mitophagy as well as OXPHOS without affecting T3 induced general autophagy/lipophagy. These findings demonstrate a novel ROS-AMPK-ULK1 mechanism that couples T3-induced mitochondrial turnover with activity, wherein mitophagy is necessary not only for removing damaged mitochondria but also for sustaining efficient OXPHOS.
Abbreviations
| Baf | = | bafilomycin A1 |
| B.W | = | body weight |
| BECN1 | = | Beclin 1, autophagy related |
| BNIP3L/NIX | = | BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like |
| CAMKK2 | = | calcium/calmodulin-dependent protein kinase kinase 2, β |
| CQ | = | chloroquine |
| KD | = | knockdown |
| DNM1L/Drp1 | = | dynamin 1-like |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| MTOR | = | mechanistic target of rapamycin |
| OCR | = | oxygen consumption rate |
| OXPHOS | = | oxidative phosphorylation |
| PPARGC1A | = | peroxisome proliferator-activated receptor gamma, coactivator 1 α |
| PRKAA1/AMPK | = | protein kinase, AMP-activated, α 1 catalytic subunit |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| RPS6KB | = | ribosomal protein S6 kinase |
| ROS | = | reactive oxygen species |
| SQSTM1/p62 | = | sequestosome 1 |
| TEM | = | transmission electron microscopy |
| THRB | = | thyroid hormone receptor β |
| T3 | = | thyroid hormone (triiodothyronine). |
Introduction
Mitochondria form a hub for hormonal regulation of lipid metabolism and oxidative phosphorylation (OXPHOS) to meet cellular energy demands. However, increased OXPHOS generates reactive oxygen species (ROS) that can damage mitochondria and lead to mitochondrial fatigue within the cell.Citation1 In response, challenged cells maintain mitochondrial quality control through mitochondrial turnover that is comprised of repair and/or removal of damaged mitochondria as well as concomitant synthesis of new mitochondria. Thus, mitochondrial turnover is needed for cell survival whenever there is increased oxidative phosphorylation and secondary ROS productionCitation2,3 In this connection, both yeast and mammalian cells have the ability to selectively remove damaged mitochondria through autophagy (mitophagy),Citation4-6 a process that primarily involves engulfment of mitochondria by autophagosomes, followed by their digestion after autophagosome/lysosome fusion.Citation7
Although mitophagy has been increasingly recognized as a major mechanism for bulk and selective degradation of mitochondria,Citation8 it has been largely regarded as either a last resort measure to remove severely damaged mitochondria during cell stressCitation9 or excess mitochondria during development.Citation10 In addition, most of the mechanistic details about the induction of mitophagy have been derived from pharmacological and genetic studies performed in vitro.Citation11 Recently, several studies have shown a direct correlation between mitophagy and mitochondrial activity under physiological conditions,Citation12,13 suggesting that steady-state mitophagy may commonly occurs in metabolically active cells in order to prevent, rather than repair mitochondrial damage. However, it is not known whether this coupling of mitophagy and OXPHOS occur during hormone-regulated metabolism under physiological conditions. The study of this potential relationship is particularly germane given the increasing incidence of metabolic disorders worldwide, and our awareness that hormonal and mitochondrial dysfunction can contribute to metabolic disorders.Citation14,15
Thyroid hormone (T3) is the major endocrine regulator of metabolic rate, and its hypermetabolic effects have been well-characterized.Citation16 T3 also has profound impacts on mitochondrial function and turnover by regulating processes such as mitogenesis, proton leak, OXPHOS, and ROS generation.Citation17 Thyroid hormone (T3) stimulates mitochondrial biogenesisCitation18 by increasing PPARGC1A (peroxisome proliferator-activated receptor gamma, coactivator 1 α) gene expression. It also promotes β-oxidation of fatty acids by increasing substrate availability/selectivity in hepatic mitochondria through induction of CPT1A (carnitine palmitoyltransferase 1A [liver]) and PDK4 (pyruvate dehydrogenase kinase, isozyme 4),Citation19 and stimulation of lipophagyCitation20 These, in turn, lead to increased oxidative phosphorylation and ROS production. Indeed, previous reports have shown that severe hyperthyroidism is associated with increased ROS production and cellular damage.Citation21-25 Surprisingly, although T3 has been reported to concurrently induce mitochondrial activity and turnover,Citation26 the underlying mechanism for their interrelationship is not well understood.
Recently, we and othersCitation20,27 have shown that T3 is a potent inducer of autophagy, and this process is critical for β-oxidation of fatty acids and oxidative phosphorylation in mitochondria. However, it is not known whether T3-mediated autophagy participates in mitochondrial turnover. Accordingly, we examined whether T3-mediated induction of mitochondrial activity is associated with mitophagy. Using both in vitro and in vivo models, we found that stimulation of autophagy by T3 was regulated by mitochondrial activity via production of ROS and activation of CAMKK2 and PRKAA1/AMPK signaling in hepatic cells. We also observed that phosphorylation of a PRKAA1/AMPK substrate, ULK1, was a prerequisite for mitochondrial targeting by autophagic machinery. Perturbation of ULK1-dependent mitophagy severely impaired mitochondrial function. Our results thus provide direct evidence for hormonal regulation of the homeostatic and metabolic coupling of mitophagy with mitochondrial activity, and may help explain how T3 can sustain its prolonged calorigenic action in metabolically active tissues such as the liver.
Results
T3 stimulates mitochondrial activity and ROS generation in THRB-HepG2 cells
To study the effect of T3 on mitochondrial function and autophagy in a cell-autonomous manner, we used previously characterized THRB (thyroid hormone receptor, β)-expressing HepG2 cells.Citation28 T3 increased basal respiration as well as the maximal and spare respiratory capacity in these cells in a dose- and time-dependent manner suggesting a net increase in mitochondrial activity (). Since circulating levels of T3 are in the nM range, these findings show that its ability to increase mitochondrial function occur at physiological doses. Since increased cellular respiration is accompanied by an elevated mitochondrial membrane potential (Δψm), we stained control and T3-treated cells with tetramethylrhodamine, ethyl ester (TMRE) and observed a significant increase in Δψm in T3-treated cells (; Fig. S1A), further confirms increased mitochondrial activity. Furthermore, this time-dependent increase in mitochondrial respiration by T3 was associated with its transcriptional induction of target genes such as CPT1A in THRB-HepG2 cells () suggesting a genomic contribution to mitochondrial stimulation by T3 under these conditions.
Figure 1. T3 increases mitochondrial activity in hepatic cells. (A and B) Seahorse analysis of oxygen consumption rate (OCR) in T3-treated THRB-HepG2 cells at different dose for 48 h. (C and D) Seahorse analysis of OCR in 100 nM T3-treated THRB-HepG2 cells at variable time periods. OCR was measured continuously throughout the experimental period at baseline and in the presence of the indicated drugs. Bars represent the mean of the respective individual ratios ±SEM (n = 5). The asterisk indicates P < 0.05. (E) Mitochondrial membrane potential in T3 (100 nM/48 h)-treated THRB-HepG2 cells as assessed by TMRE (a mitochondrial membrane potential indicator) mean fluorescence intensity (MFI). (F) qPCR data showing CPT1A mRNA levels in THRB-HepG2 cells after T3 treatment (100 nM) at different time points. Values are means ±SD (n = 5). The asterisk indicates P < 0.05.
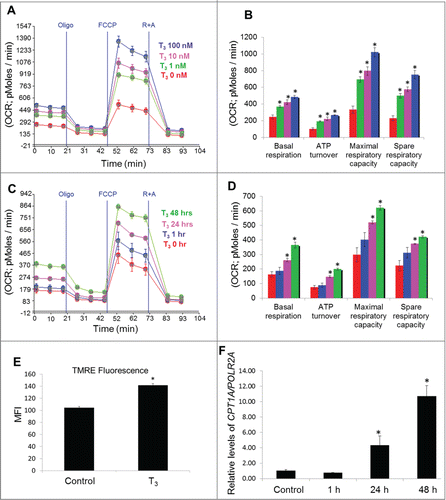
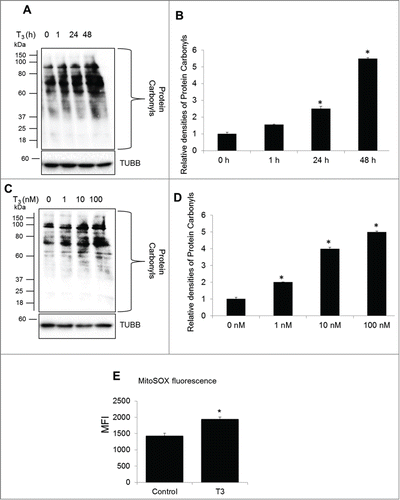
Electron leak during the passage of electrons via the electron transport chain produces superoxide anions (O2.−) that are converted to ROS.Citation29 To assess the oxidative stress induced by T3 in cells undergoing increased mitochondrial activity, we employed immunoblot-based detection of carbonyl groups introduced into proteins by oxidative reactions. Our results showed that T3 increased oxidative stress in THRB-HepG2 cells in both a dose- and time-dependent manner, coincident with its stimulation of mitochondrial activity (). We also used MitoSOX, a fluorophore that is activated by mitochondrial superoxide oxidation to assess ROS formation, and confirmed that T3 treatment led to mitochondrial production of ROS ( and Fig. S1B).
Figure 2. T3 increases oxidative stress in hepatic cells. (A) Protein oxidation analysis using the oxyblot method in THRB-HepG2 cells at different doses of T3 for 48 h. (B) Quantification of Oxyblot assays (bars represent the mean of the respective individual ratios ±SD , n = 3,*P < 0 .05). (C) Protein oxidation analysis using the oxyblot method in THRB-HepG2 cells with 100 nM T3 for different time periods. (D) Quantification of Oxyblot assays (n = 3, *P < 0 .05). (E) Mitochondrial superoxide generation in T3 (100 nM/48 h)-treated cells measured by MitoSOX. Bars represent the mean of the respective fluorescence intensity ±SD (n = 5, *P < 0.05). TUBB, β-tubulin; MFI, mean fluorescence intensity.
T3 induces mitophagy in THRB-HepG2 cells
As we observed previously,Citation20 T3 induced autophagosome formation and autophagic flux when LC3-II was detected in a dose- and time-dependent fashion (; Fig. S2). To determine the occurrence of mitochondrial autophagy or “mitophagy,” we used a tandem-tagged RFP-EGFP chimeric plasmid, pAT016, that encoded a mitochondrial-targeting signal sequence fused in-frame with RFP and EGFP genes (tandem-tagged mt-RFP-EGFP).Citation30 The use of this plasmid benefits from the different stabilities of RFP and GFP in an acidic environment. The GFP signal is quenched at lower pH whereas RFP can be visualized in acidic autolysosomes; thus, increased RFP/red-only fluorescence in the lysosomes indicates completion of the mitophagic process. Using this assay, we observed that T3 increased autolysosome-resident mitochondria (red fluorescent dots without any green fluorescence) at concentrations as low as 1 nM and as early as 24 h ( and ). Validation of the RFP-only signal as lysosomal resident was shown previously for this technique,Citation31 using lysosomal markers and also confirmed by us using the lysosomal acidification inhibitor, bafilomycin A1 (Baf) (Fig. S3).
Figure 3. T3 increases autophagy and mitophagy in hepatic cells. (A and B) Representative immunoblot and quantitation showing LC3-II levels in THRB-HepG2 cells treated with indicated doses of T3 for 48 h. Bars represent the mean of the respective individual ratios ±SD (n = 3, *P < 0 .05). (C and D) Representative Immunoblot and quantification showing LC3-II levels in THRB-HepG2 cells treated with 100 nM T3 for indicated time periods. Bars represent the mean of the respective individual ratios ±SD (n = 3, *P < 0 .05). (E) Monitoring mitophagic flux using dual fluorescence Mito-mRFP-EGFP reporter (pAT016). Lysosomal delivery of the tandem fusion protein Mito-mRFP-EGFP along with entire mitochondria results in differential quenching and degradation of the 2 individual fluorochromes, thus allowing for visual analysis of mitophagic flux. THRB-HepG2 cells transiently expressing Mito-mRFP-EGFP were treated with 1 nM or 100 nM T3 for 48 h followed by visualization using confocal microscopy (40X magnification). Nuclei were stained with DAPI (blue). In the images, fluorescence signals indicates the expression of Mito-mRFP-EGFP targeting mitochondria: yellow color, no mitophagy or normal cytosolic mitochondria; red color, mitophagy or mitochondria inside lysosomes. (F) Quantitative analysis of the RFP (red-only) fluorescence to denote % mitophagy was done. Quantification of images (at least 20 transfected cells per each sample in 3 different fields) was conducted with ImageJ software. Bars represent the mean of the respective individual ratios ±SD (*, P < 0 .05).
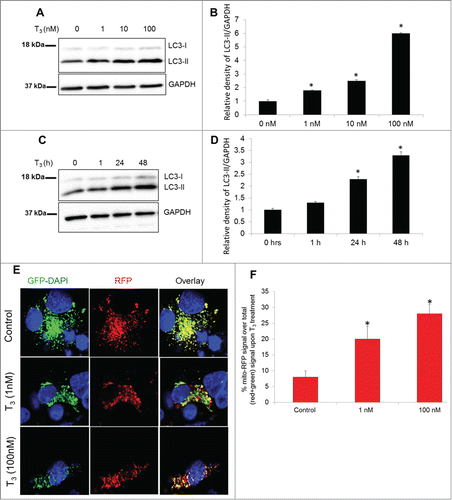
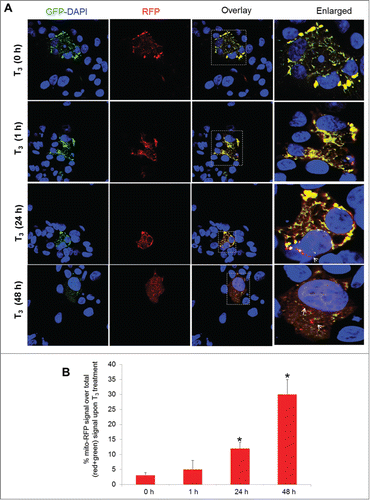
Figure 4. T3 increases mitophagic flux time dependently in hepatic cells. (A) THRB-HepG2 cells transiently expressing Mito-mRFP-EGFP were treated with 100 nM T3 for different time periods followed by visualization using confocal microscopy (40X magnification). Nuclei were stained with DAPI (blue). In the images, fluorescence signals indicate the expression of Mito-mRFP-EGFP targeting mitochondria: yellow color, no mitophagy or normal mitochondria; red color, mitophagy or mitochondria inside lysosomes. (B) Quantitative analysis of the RFP (red) fluorescence to denote % mitophagy. Quantification of images (at least 10 transfected cells per each sample in 3 different fields) was conducted with ImageJ software. Bars represent the mean of the respective individual ratios ±SD (*, P < 0 .05).
To further validate the ability of T3 to increase mitochondrial turnover by mitophagy, we used a genetically encoded pH-sensitive mitochondrial probe called mito-Keima.Citation32 pMT-mKeima is a DsRed variant that targets the mitochondrial matrix, and is excited preferentially by a 440 nm laser line at a pH of 7.4 and a 550 nm laser line at pH of 4.5 (the pH of lysosomes). Mitochondria that have been delivered to acidic lysosomes are identified by comparing the intensity of mito-Keima emissions at 610 nm after sequential laser excitation at 440 and 550 nm. Using this strategy, we found a significant increase in the lysosome-resident mitochondria (red) in T3-treated cells compared to untreated control cells. ().
Figure 5. T3 increases autophagosomal delivery of mitochondria in hepatic cells. (A) pMT-mKeima-Red plasmid transfected cells were treated with +/− T3 (100 nM/48 h) followed by cell imaging (40X magnification). Nuclei appear blue. The elongated cytosolic mitochondria are at neutral pH shown as green (pseudo color) excitation wavelength of 440 nm and emission wavelength 610 nm, whereas lysosomal resident mitochondria appear as rounded shown as red (pseudo color) with excitation wavelength of 550 nm and emission wavelength 610 nm. (B) Quantitation of live cell fluorescent imaging of autophagy. Cells with ≥5 puncta preferentially excited at 550 nm relative to 440 nm were scored for at least 50 cells transfected per condition (n = 3). Asterisk indicates P < 0.05. (C) Electron micrograph of untreated control and T3 (100 nM/48 h)-treated THRB-HepG2 cells. Note the increased number of autophagic vesicle resident mitochondria (denoted with arrows) in T3-treated cells. Scale Bar = 0.2 µm. (D) Bar graphs showing % of autophagosomes (AVs) containing mitochondria in control and T3-treated THRB-HepG2 cells based on EM micrograph images. Scoring was done by counting 5 to 7 different cells in 5 random fields per condition (n=3). Asterisk indicates P < 0.05. (E, F) Representative immunoblot and densitometric analysis showing levels of mitochondrial proteins COX4I1 and PDHA in control and T3 (100 nM/48 h)-treated THRB-HepG2 whole cell lysates with or without Baf (10 nM/24 h) (SD, n = 3). Asterisk indicates P < 0.05 when compared between T3+Baf vs. Baf alone.
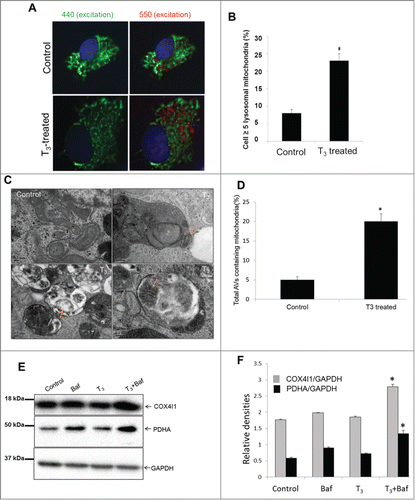
Next, we used electron microscopy to demonstrate increased numbers of mitochondria in autolysosomes (fused autophagosomes and lysosomes) after T3 treatment ().
Finally, we used Baf to inhibit autophagy and found that the levels of mitochondrial proteins such as PDHA (pyruvate dehydrogenase [lipoamide] α) and COX4I1/COX IV (cytochrome c oxidase subunit IV isoform 1) significantly accumulated in cells treated with T3+Baf compared to cells treated with Baf alone. These findings provide further evidence for the degradation of mitochondria by autophagy ().
We also examined the expression of key mitophagic genes and found that T3 significantly increased BNIP3, BNIP3L/NIX, ULK1, LC3, and SQSTM1/p62 mRNA expression; however, it did not change the expression of several other genes such as PINK1, PARK2/PARKIN, and RHEB that previously have been implicated in mitophagy,Citation33 (Fig. S4A). In addition to the induction of autophagic/mitophagic gene expression, we also observed a concomitant increase in the expression of mitochondrial biogenesis regulators such as PPARGC1A and TFAM as well as mitochondrial genes such as, COX4I1, which could explain the small difference in the mitochondrial protein seen after T3 treatment (Fig. S4B).
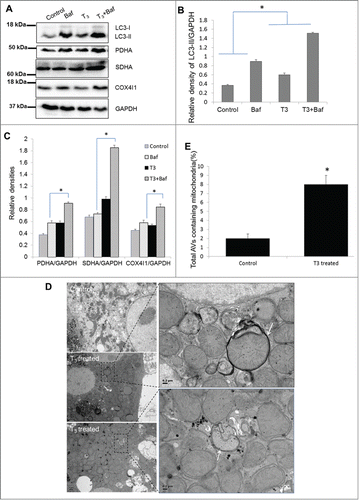
In order to demonstrate that the induction of mitophagy in THRB-expressing HepG2 cells was not due to artifacts such as THRB overexpression or cell line-specific effects, we conducted studies of autophagy in primary mouse hepatocytes. T3 induced autophagic flux () and mitophagy in primary mouse hepatocytes; as evident by mitochondrial protein accumulation and autophagosome resident mitochondria (); thus demonstrating that these cell-autonomous effects occur in primary hepatic cells with normal THRB expression.
Figure 6. T3 increases mitophagy in primary hepatocytes. (A-C) Representative blots and densitometric analysis showing the levels of LC3-II and mitochondrial markers PDHA, SDHA, and COX4I1 with and without Baf (40 nM/8 h) in primary mouse hepatocytes treated with 100 nM T3 for 24 h (n = 3). Asterisk indicates P < 0.05 when indicated relative densities of proteins/GAPDH between T3+Baf/T3 is compared to that in Baf/control. (D) Electron micrograph of primary mouse hepatocytes treated with T3. EM of untreated control and T3-treated (100 nM/24 h) mouse hepatocytes showing increased mitophagy (denoted by arrows showing autophagosomes containing mitochondria) under T3 treatment. Scale bar =1 µm and in enlarged figures are 0.2 µm. (E) Bar graphs showing % of autophagosomes (AVs) containing mitochondria in control and T3-treated primary mouse hepatocytes based on EM micrograph images. Scoring was done by counting 10 to 15 different autophagic vesicles in 5 random fields per condition (n = 3, *p < 0.05).
T3 induces autophagy via the ROS-PRKAA1/AMPK pathway
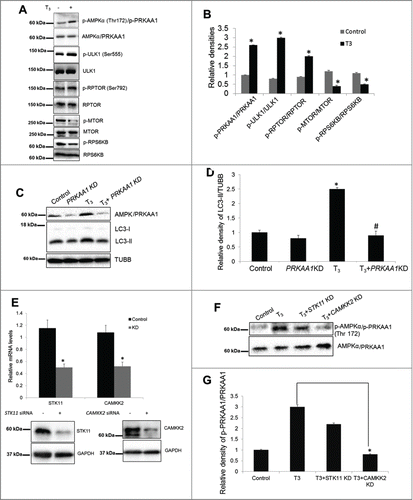
Recently, ROS signaling has been implicated in activation of AMP-activated protein kinase (AMPK) and autophagy.Citation34,35 Additionally, T3 can activate PRKAA1/AMPK in the liver.Citation36 Thus, we examined further the relationships between oxidative stress, PRKAA1/AMPK phosphorylation, and autophagy since oxidative stress classically has been associated with T3-induced mitochondrial activity.Citation22 Our results showed that PRKAA1/AMPK signaling is indeed activated by T3 in THRB-HepG2 cells (). Immunoblots showed a significant increase in the phosphorylation of PRKAA1/AMPKα1 (Thr172) and its downstream target, ULK1 (Ser555), in T3-treated cells () Since PRKAA1/AMPK inhibits MTOR activity by phosphorylating RPTOR,Citation37 we also checked phosphorylation at the PRKAA1/AMPK-regulated site on RPTOR and found that its phosphorylation was increased by T3 (.). Furthermore, phosphorylation of MTOR and its downstream target, RPS6KB, was decreased () after T3 treatment. We then confirmed the critical role of PRKAA1/AMPK in T3-induced autophagy by using PRKAA1 siRNA, and observed that induction of autophagy by T3 is PRKAA1/AMPK mediated () Both STK11/LKB1 (serine/threonine kinase 11) and CAMKK2 have been implicated in the regulation of PRKAA1/AMPK activityCitation38 Therefore, we knocked down both STK11 and CAMKK2 () to assess the effect of these upstream kinases on T3 induced PRKAA1/AMPK activation. Loss of CAMKK2 significantly impaired T3 induction of PRKAA1/AMPK whereas STK11 ablation had only minor effect ().
Figure 7. T3-induced autophagy is CAMKK2-AMPK-mediated. (A and B) Representative blots and densitometric analysis showing the phosphorylated and total protein levels of PRKAA1/AMPKα1, ULK1, RPTOR, MTOR, and RPS6KB in THRB-HepG2 cells treated with T3 (100 nM/48 h). Bars represent the mean of the respective individual ratios ±SD (n = 5, *P < 0 .05). (C and D) Representative blots and densitometric analysis showing the effect of PRKAA1 knockdown (KD) on LC3-II levels in THRB-HepG2 cells treated with T3 (100 nM/48 h). Bars represent the mean of the respective individual ratios ±SD (n = 5, *P < 0 .05 vs control and #P < 0 .05 vs T3 alone). (E) Efficacy of STK11 and CAMMK2 Knock down (KD) at RNA and protein levels. (F and G) Representative blots and densitometric analysis showing the effect of STK11 and CAMKK2 KD on PRKAA1/AMPK activation in THRB-HepG2 cells treated with T3 (100 nM/48 h). Bars represent the mean of the respective individual ratios ±SD (n = 3, *P < 0 .05).
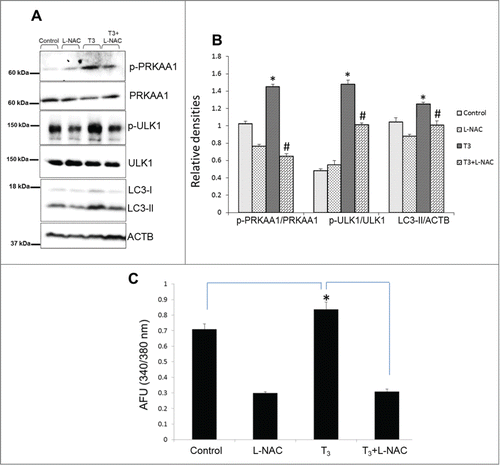
We then determined whether T3-induced ROS occurred upstream or downstream of PRKAA1/AMPK activation. Using the antioxidant, N-acetyl-L-cysteine (L-NAC), we found that quenching ROS (Fig. S5A) production abrogated T3–induced PRKAA1/AMPK and ULK1 phosphorylation as well as autophagy (). These findings clearly demonstrate that ROS production occurs upstream of PRKAA1/AMPK activation and is necessary for induction of autophagy by T3. Previously, it has been shown that ROS-dependent increases in cytosolic calcium activates PRKAA1/AMPK via CAMKK2.Citation39 Since T3-mediated activation of PRKAA1/AMPK was dependent upon CAMKK2 (), we examined whether T3 induction of oxidative phosphorylation and ROS production led to increased intracellular Ca2+ and activation of CAMKK2. Using a Ca2+ sensing probe, Fura-2AM, we found that T3 treatment increased intracellular Ca2+. Additionally, both basal and T3-induced intracellular Ca2+ levels were suppressed by L-NAC (). Taken together, the foregoing results showed that increased ROS production by T3 likely triggers autophagy and mitophagy through increased intracellular Ca2+ and activation of CAMKK2-PRKAA1/AMPK signaling. Interestingly, in contrast to its ability to abolish T3-induced autophagy, L-NAC had only modest effects on the transcription of autophagic genes by T3 (Fig. S5B). These findings suggest that transcriptional regulation of autophagy gene expression by T3 likely occurs independently of this ROS-mediated signaling pathway.
Figure 8. T3-induced oxidative stress mediates PRKAA1/AMPK-ULK1 activation. (A and B) Representative blots showing total or phosphorylated PRKAA1/AMPK, LC3-II and ULK1 protein levels in whole cell lysates after addition of ROS inhibitor (L-NAC; 5 mM) in THRB-HepG2 treated with T3(100 nM/48 h). Bars represent the mean of the respective individual ratios ±SD (n = 3). The asterisk (* P < 0 .05) indicates a difference between T3 alone vs. control and (#P < 0 .05) indicates difference between T3+L-NAC vs. T3-alone treated cells. (C) Cells were treated with T3 (100 nM/24 h +/− LNAC (5 mM), and then loaded with Fura-2AM (1 µM). Intracellular Ca2+ was measured by fluorescence analysis. The data were presented as mean of arbitrary fluorescence units (AFU) ± SD (n = 6, *P < 0 .05).
T3 increases mitochondrial translocation and recruitment of autophagic proteins
Since mitochondrial localization of the autophagic machinery is required for mitophagy priming,Citation40,41 we measured the levels of autophagic proteins in purified mitochondrial fractions, which were verified to be free from cytosolic and lysosomal contamination (). Our results showed that T3 treatment increased the localization of ULK1, SQSTM1, and LC3 within the mitochondrial fraction of HepG2 cells (). These findings were consistent with earlier findings that showed both ULK1 and SQSTM1 were involved in selective mitochondrial recognition during mitophagy.Citation42 Additionally, we observed that DNM1L/Drp1 (dynamin 1-like), a protein associated with mitochondrial fission and mitophagy,Citation43 was preferentially recruited to mitochondria in response to T3 treatment (). Interestingly, we also observed increased mitochondrial protein ubiquitination in T3-treated cells (). Since mitochondrial ubiquitination precedes various types of mitophagy,Citation44 it is likely that T3-induced mitophagy involves ubiquitination of mitochondrial proteins as part of the priming process. Confocal imaging of FLAG-tagged ULK1 with the mitochondrial marker TOMM20 showed that T3 increased translocation of ULK1 into hepatic mitochondria, providing further evidence for mitophagy priming by autophagy proteins ().
Figure 9. Mitophagy proteins translocate to mitochondria and are necessary for T3 stimulation of mitophagy. (A) Immunoblot showing mitochondrial protein ubiquitination and localization of ULK1, SQSTM1, LC3B-II, and DNM1L proteins in isolated mitochondrial fraction from T3 (100 nM/48 h)-treated THRB-HepG2 cells. Purity/enrichment of the mitochondrial fraction (Mito) was verified by the absence of TUBB/β-tubulin (cytosolic) and LAMP1 (lysosomal) relative to its level in the whole cell lysate (WCL) for the same amount of VDAC levels. (B) THRB-HepG2 cells transfected with Flag-ULK1 construct were treated with T3 (100 nM/48 h) and subsequently, immunostained with anti-TOMM20 (green) and anti-Flag (red) antibodies (40X magnification). Nuclei were stained with DAPI. In the fluorescent images, yellow color indicates the colocalization of ULK1 to the mitochondria. (C) Quantification of colocalization of Flag-ULK1 in the mitochondria (at least 15 transfected cells per each sample in 3 different fields) was conducted with ImageJ software. Bars represent the mean of the respective individual ratios ±SD (*P < 0 .05). (D) THRB-HepG2 cells transiently expressing Mito-mRFP-EGFP were treated with 100 nM T3 for 48 h with or without ULK1 KD followed by visualization using confocal microscopy (40X magnification). Nuclei were stained with DAPI (blue). In the images, fluorescence signals indicate the expression of Mito-mRFP-EGFP targeting mitochondria: yellow color, no mitophagy; red color, mitophagy. (E) Quantitative analysis of the RFP (red) fluorescence to denote % mitophagy. Quantification of images (at least 10 transfected cells per each sample in 3 different fields) was conducted with ImageJ software. Bars represent the mean of the respective individual ratios ±SD (*P < 0 .05).
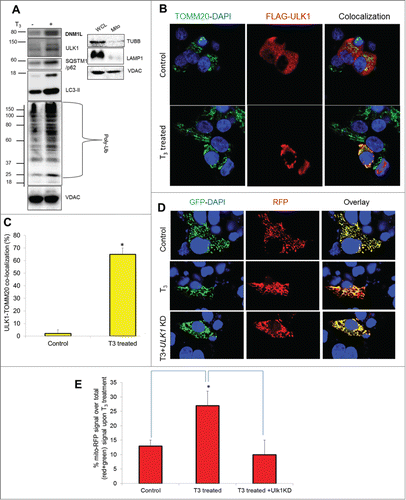
We next demonstrated the importance of ULK1 in the priming process for mitophagy by measuring mitophagic flux in ULK1 knockdown (KD) cells treated with T3. Similar studies also were conducted on other known autophagic/mitophagic proteins (Fig. S6). Our results showed that both ULK1 and SQSTM1 KDs reduced T3-induced mitophagy in THRB-HepG2 cells (; Fig. S6) Although ULK1 and SQSTM1 expression were critical for mitophagy, neither ULK1 nor SQSTM1, were necessary for basal and T3-induced general autophagy in our cell system (Fig. S7A-C). Similarly, knockdown of the mitophagy regulator, BNIP3L, impaired T3-induced mitophagy but had no effect on T3-induced general autophagy. In contrast, BECN1 was essential for both mitophagy and general autophagy (Fig. S7D, E).
Mitophagy is essential for maintaining T3-induced mitochondrial activity and regulating oxidative stress
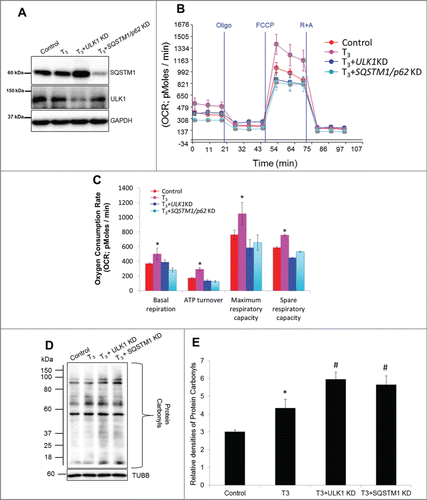
We next determined whether mitophagy inhibition has a negative impact on T3-stimulated mitochondrial function. Accordingly, we knocked down ULK1 and SQSTM1 in HepG2 cells by siRNA (), and studied their effects on T3-induced respiration. Our results showed that T3-induction of mitochondrial respiration was reduced when either ULK1 or SQSTM1 were knocked down (). Interestingly, neither ULK1 KD nor SQSTM1 KD affected mitochondrial function under basal conditions (Fig. S8A), suggesting that their critical roles are distinct features of T3-mediated mitochondrial stimulation. Similar dampening of T3-stimulated mitochondrial respiration occurred with lysosomal inhibition (Fig. S8B) and ROS inhibition with L-NAC (Fig. S8B). Taken together, these data strongly support the notion that mitophagy is critical for maintaining high mitochondrial activity by T3. In line with these findings, we also found that loss of both ULK1 and SQSTM1 aggravated cellular oxidative stress induced by T3 ().
Figure 10. Inhibition of T3-induced mitophagy impairs mitochondrial activity and increases oxidative stress. (A) Immunoblot showing knockdown (KD) efficiency of ULK1 and SQSTM1 in THRB-HepG2 cells. (B) Seahorse analysis of oxygen consumption in T3 (100 nM/48 h)-treated THRB-HepG2 cells +/− ULK1 and SQSTM1 KD. OCR was measured as described in . (C) Representative plot basal/maximal respiration as well as spare respiratory capacity in T3 (100 nM/48 h)-treated THRB-HepG2 cells. Bars represent the mean of the respective individual ratios ±SD (n = 5). The asterisk (*P < 0 .05) indicates difference compared between T3 alone vs. control and (#P < 0 .05) indicates difference between T3+ULK1 or SQSTM1 KD vs. T3 alone treated cells. (D) Protein oxidation analysis using the oxyblot method in T3 (100 nM/48 h)-treated THRB-HepG2 cells +/− ULK1 and SQSTM1 KD (E) Quantification of Oxyblots (bars represent the mean of the respective individual ratios ±SD , n = 3). The asterisk (*P < 0 .05) indicates difference compared between T3 alone vs. control and (#P < 0 .05) indicates difference between T3 +ULK1 or SQSTM1 KD vs. T3-alone treated cells.
T3 induces hepatic oxidative stress and mitophagy in vivo
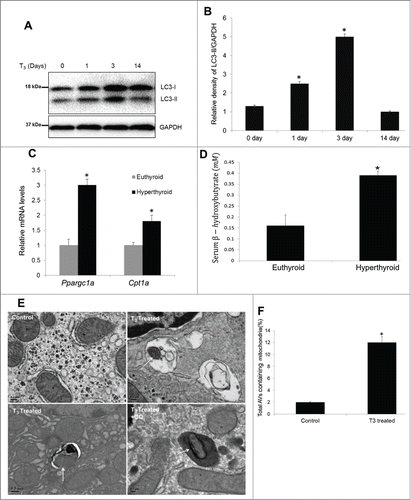
Our results showed that T3 induction of mitochondrial activity and oxidative stress was associated with mitophagy in vitro; therefore, we next studied these phenomena in vivo. Accordingly, we injected mice with T3 for 1, 3, and 14 d, and measured LC3-II levels in livers from mice sacrificed on indicated time periods. Our results suggested that hepatic autophagy was induced acutely from d 1 to 3 and then decreased after chronic T3 treatment for 14 d (). The increase in hepatic autophagy in hyperthyroid mice was associated with increased expression of metabolic and mitochondrial biogenesis genes such as Cpt1a and Ppargc1a as well as increased β-oxidation ().
Figure 11. T3 concomitantly increases autophagy, mitochondrial biogenesis, and activity in vivo. (A, B) Immunoblot and densitometric analysis of LC3-II levels in T3-treated (hyperthyroid) mice at a dose of 10 µg/100 g body weight (B.W.) daily for different time periods (n = 5,*P < 0 .05). (C) qPCR data showing mRNA levels of Cpt1a and Ppargc1a from livers of T3-treated mice (10 µg T3/100 g B.W. for 3 d). Values are means ±SD (n = 5). (D) Serum β-hydroxybutyrate in the above described animal groups was measured using manufacturer's guidelines. Values are means ±SEM (n = 5). The asterisk indicates *P < 0 .05. (E) Electron micrograph and quantitation of livers from untreated control (euthyroid) and T3-treated (hyperthyroid, injected with 10 µg T3/100 g B.W. for 3 d) mice. Note the increased number of autophagic vesicle resident mitochondria in T3-treated animals (shown by arrows). Liver section from hyperthyroid mice cotreated with CQ clearly showed mitochondria inside lysosomal compartment. (F) Bar graphs showing % of autophagosomes/ autophagic vesicles containing mitochondria in control and T3-treated mice based on EM micrograph images. Scoring was done by counting 5 to 10 different cells containing autophagosomes (AVs) in 5 random fields per condition (n = 5, *P < 0.05).
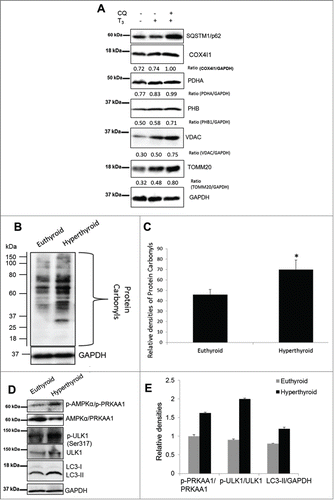
We next examined hepatic mitophagy by EM in mice treated with T3 for 3 d. Similar to our in vitro data, we found increased localization of mitochondria within autophagosomes (autophagic vesicles) in livers from T3-treated (hyperthyroid) mice () In these experiments, we co-treated hyperthyroid mice with the lysosomal inhibitor, chloroquine (CQ) to confirm delivery of mitochondria into lysosomes since it is difficult to identify digested mitochondria in lysosomes. We found further evidence for autophagy-mediated degradation of mitochondria when we measured the expression levels of specific mitochondrial proteins in livers from control, hyperthyroid (T3-treated), and hyperthyroid mice co-treated with CQ. Our results showed that T3 increased hepatic mitochondrial protein levels relative to the loading control (GAPDH) in agreement with earlier reports that described induction of mitochondrial biogenesis by T3.Citation18 However, we observed a further increase in mitochondrial protein levels in hyperthyroid mice co-treated with CQ (), suggesting that T3 also increased lysosomal degradation of mitochondria. In tandem with the increase in mitochondrial activity, T3 induced hepatic ROS production () as well as PRKAA1/AMPK and ULK1 activation in vivo (). Taken together, our findings suggested that T3 regulates mitochondrial turnover and function by stimulating both mitochondrial synthesis and mitophagy in vivo.
Figure 12. T3 increases oxidative stress and mitophagy in vivo. (A) A representative immunoblot using whole cell lysates showing hepatic levels of mitochondrial proteins in vehicle (PBS), T3 (10 µg/100 g B.W. for 3 d) - and T3+CQ (i.p. administration CQ at 60 mg/kg B.W. for 3 d) - treated mice. (B) Protein oxidation analysis using the Oxyblot method on liver tissues from euthyroid and hyperthyroid (injected with 10 µg T3/100 g B.W. for 3 d) mice. (C) Quantification of Oxyblot assays (n = 5). (D and E) Representative blots and densitometric analysis showing total and phosphorylated PRKAA1/AMPK, ULK1, and LC3-II in whole cell lysates after T3 treatment (10 µg T3/100 g B.W. for 3 days) in vivo. Bars represent the mean of the respective individual ratios ±SD (n = 5). Asterisk indicates P < 0.05.
Discussion
In this manuscript, we have provided several lines of evidence showing that oxidative phosphorylation, ROS production, and mitophagy are induced by T3 in hepatic cells in an interdependent manner. First, we demonstrated that induction of mitochondrial respiration by T3 is accompanied by increased generation of ROS and autophagy. Second, we used multiple cellular imaging methods to demonstrate that the increase in mitochondrial activity was accompanied by an increase in mitophagy. Third, we showed that T3-induced ROS initiates autophagy by a novel pathway involving an increase in intracellular Ca2+, level, activation of CAMKK and PRKAA1/AMPK activities, and the phosphorylation and mitochondrial translocation of autophagic proteins such as ULK1. Finally, we showed that inhibition of mitophagy markedly impaired T3-induced mitochondrial function and increased oxidative stress.
It is important to note that both induction of mitochondrial activity and mitophagy by T3 occurred at 1 nM and increased in a concentration-dependent manner. Thus, T3 was able to induce mitophagy at both physiological and pharmacological concentrations. Since both T3-mediated mitophagy and mitochondrial activity occurred after 24 h of treatment, it is likely that they involve changes in gene expression. In support of this notion, we previously showed that T3-mediated autophagy was THRB-dependent.Citation20 Nonetheless, we cannot completely eliminate the possibilities of nongenomic effects that are THRB-dependent or the involvement of other thyroid hormone metabolites that may act directly on the mitochondriaCitation17 or activate other signaling pathways.
We also discovered a novel ROS-dependent signaling cascade that mediated the induction of mitophagy by T3. First, we found that ROS generation was temporally-coupled with increased mitochondrial function in T3-stimulated cells. In this connection, ROS have been shown to stimulate autophagy and mitophagy in yeast and mammals.Citation3,5,45 Next, we found that T3 activated PRKAA1/AMPK and its target, ULK1, and induced autophagy, in a ROS-dependent manner. Indeed, the chemical antioxidant, L-NAC, abrogated T3s effects on these processes. Interestingly, we then found an essential role for T3 to increase intracellular Ca2+ level and activate CAMKK2 at the beginning this signaling cascade. Furthermore, we showed that this kinase was indispensable for T3-induced PRKAA1/AMPK activation. Thus, our findings are consistent with the notion that ROS induction of intracellular Ca2+ release activates CAMKK2, an upstream activator of PRKAA1/AMPK to stimulate ULK1 phosphorylation. Since ROS activates CAMKK2 via intracellular Ca2+ release,Citation39 it seems likely that CAMKK2 activation serves as an essential link between metabolic stress and mitophagy. Thus, it is tempting to speculate that other extracellular stimuli or metabolic conditions that generate hepatic ROS may stimulate PRKAA1/AMPK and autophagy by using this same pathway.
Translocation of general autophagy machinery to mitochondria is essential for the sorting and selective degradation of damaged mitochondria. Based on our results, ULK1 translocation to mitochondria likely marks mitochondria for T3-stimulated autophagy. Our results are supported by recent findings which show that translocation of ULK1 to mitochondria triggers selective mitochondrial autophagy.Citation40,41 Collectively, our results suggest that ROS-activated PRKAA1/AMPK signaling is essential for priming mitochondrial degradation through its activation of ULK1. In order to determine the specific contribution of ULK1 and SQSTM1 to mitophagy vs. general autophagy, we performed siRNA knockdown of these genes and assessed their effects on these 2 processes. Surprisingly, although both ULK1 and SQSTM1 were essential for T3-induced mitophagy, their loss had no significant impact on autophagosome formation in THRB-HepG2 cells in either basal conditions or after T3 treatment. These findings suggest that ULK1 and SQSTM1 are specifically required for mitophagy and not for general autophagy induced by T3. Of note, we found that T3 also induced the translocation of mitochondrial fission protein, DNM1L, an event that precedes mitophagy.Citation43 Interestingly, it recently has been shown that hormonal stimulation of mitochondrial fission is employed by brown adipocytes to amplify energy expenditure.Citation46
We found that T3 induced the expression of mitophagy-related genes such as BNIP3L and BNIP3 but does not affect the expression of other mitophagy genes such as PINK1 and PARK2. Although the latter proteins are commonly implicated in mitophagy, recent studies show that initiation of mitophagy can include alternative mechanisms such those involving BNIP3L.Citation8 Loss of BNIP3L impaired T3-induced mitophagy in our system; however, further studies are needed to elucidate the precise roles of BNIP3L and/or other related proteins in T3-induced mitophagy.
Here we show a functional dependence of T3-stimulated mitochondrial activity on mitophagy since mitophagy was required for T3-induction of oxidative phosphorylation. This finding was surprising since mitophagy previously had been thought to be associated with mitochondrial dysfunction.Citation47 However, our data suggest that the co-occurrence of mitophagy, mitochondrial biogenesis, and increased mitochondrial activity in T3-treated cells may be necessary in order to sustain hepatic mitochondrial turnover without major ROS-induced mitochondrial and cellular damage. Our findings are consistent with recent studies in which mitophagy increases mitochondrial biogenesis,Citation48 prevents loss of mitochondrial function and mitochondrial aging,Citation12 and also reduces mitochondrial oxidative stress.Citation49,50 Thus, ROS-stimulated mitophagy may be part of a physiological homeostatic response to maintain hepatic cell viability and function in hypermetabolic states.
We and others have shown that T3 can activate another type of autophagy, lipophagy, in hepatic cells.Citation20,27 We observed the co-occurrence of lipophagy and mitophagy at all the T3 doses used in this study (1 to 100 nM) (Sinha and Yen, unpublished data); however, lipophagy was best visualized when cells were preloaded with exogenous fatty acids. Of note, we found that it was not necessary to preload cells with fatty acids to observe mitophagy, and since we used higher magnification images to visualize mitophagy events, endogenous lipid droplets in autophagosomes do not appear prominently in the EM images of this current study. At this point, we cannot determine conclusively whether mitophagy, lipophagy, or both, are the major drivers for the beneficial effect of T3 on hepatic lipid metabolism. We believe that both processes are coordinated such that lipophagy helps to deliver fuel to the mitochondria and mitophagy helps to maintain a healthy population of mitochondria that can effectively utilize the delivered fuel (). Furthermore, our study illustrates the point that mitochondria may act as both the source as well as the target of hormone-induced autophagy.
Figure 13. Model of T3-induced hepatic mitophagy. T3 increases mitochondrial activity by either increasing mitochondrial fuel delivery via transcription of genes like CPT1A and PDK4 and by increasing mitochondrial biogenesis via PPARGC1A. Increased mitochondrial function leads to increased respiration which in turn causes ROS generation. ROS increases activation of PRKAA1/AMPK via Ca2+-CAMKK2 signaling. PRKAA1/AMPK activation leads to 2 distinct bifurcated pathways with one increasing mitophagy through ULK1 and the other increasing general autophagy such as lipophagy via MTOR suppression. T3 also augment the transcription of key autophagy genes via a still uncharacterized nuclear pathway which may include THR's or other transcription factors. A concurrent increase in both mitophagy and lipophagy may help sustain mitochondrial respiration with lipophagy providing fuel for oxidation and mitophagy helping to maintain homeostatic mitochondrial turnover.
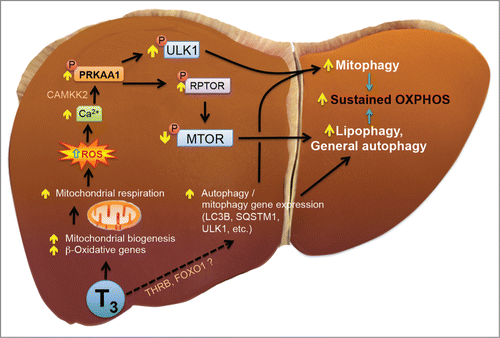
Taken together, our data demonstrate that T3 maintains increased hepatic metabolic activity by promoting turnover of the intracellular pool of mitochondria through increased rates of mitophagy and mitochondrial synthesis. Thus, induction of mitophagy by intracellular ROS derived from increased mitochondrial energy production can help prevent the accumulation of damaged mitochondria as well as promote cellular health and function in hypermetabolic states. Mitochondrial function and its quality control are important factors in metabolic diseases such as diabetes and nonalcoholic fatty liver disease as well as aging.Citation51,52 Our studies thus suggest that T3 or its analogs may be beneficial for treating these conditions by promoting mitochondrial turnover.Citation53 Lastly, our finding that T3 stimulation of mitophagy occurs via a pathway involving increased ROS production; intracellular Ca2+ increase; CAMKK2, PRKAA1/AMPK, and ULK1 activation now provides several new therapeutic targets for maintaining mitochondrial number and function that could lead to novel treatments for metabolic and aging-related diseases.Citation54
Materials and Methods
Reagents
Triiodothyronine (T3) (IRMM469–1EA), N-acetyl-L-cysteine (L-NAC; A7250), bafilomycin A1 (Baf; B1793) and chloroquine (CQ; C6628) were purchased from Sigma-Aldrich. Antibodies were procured from Cell Signaling Technology (LC3B, 2775; SQSTM1/p62, 5114; PRKAA1/AMPKα1, 2795; phospho-PRKAA1/AMPKα (Thr172) (D79.5E) rabbit mAb, 4188, ULK1 (D8H5) rabbit mAb, 8054; phospho-ULK1 (Ser555) (D1H4) rabbit mAb, 5869, MTOR (7C10) rabbit mAb, 2983; phospho-MTOR (Ser2448) (D9C2) XP® rabbit mAb, 5536, RPS6KB/p70S6 kinase (49D7) rabbit mAb, 2708; phospho-RPS6KB/p70S6 kinase (Thr389) (108D2) rabbit mAb, 9234; RPTOR/Raptor (24C12) rabbit mAb, 2280; phospho-RPTOR/Raptor (Ser792), 2083; DNM1L/DRP1 (D6C7) rabbit mAb, 8570; GAPDH (D16H11) XP® rabbit mAb, 5174; Mitochondrial Marker Antibody Sampler Kit 8674; TUBB/β-tubulin, 2146; BECN1/Beclin-1, 3738) and Santa Cruz Biotechnology (ACTB/β-actin (ACTBD11B7), sc-81178; STK11/LKB1 (E-9), sc-374334; CAMKK2/CaMKKβ (H-95), sc-50341; BNIP3L/Nix (N-19), sc-5860. Culture media, MitoTracker Red (Molecular Probes, M-7512), MitoSOX (Molecular Probes, M-36008), DCFH-DA (Molecular Probes, TD-399), TMRE (Molecular Probes, T-669), Fura-2AM (Molecular Probes, F-1201), and transfection reagents were from Invitrogen,USA. Silencer® Select siRNAs from Ambion® were used at a concentration of 10 nM (ULK1 s15963, PRKAA1 s100, SQSTM1 s16960, CAMKK2 s20927, STK11 s13579, BECN1 s16537, BNIP3L s2062). CoralHue® Mitochondria-targeted mKeima-Red expression plasmid (pMT-mKeima-Red) was purchased from MBL International Corporation (AM-V0251), and eGFP-LC3 (Addgene plasmid 21073) plasmids were gifts from Prof. T. Yoshimori (Osaka University, Osaka, Japan). Mito-RFP-EGFP plasmid was a kind gift from Dr. Andreas Till (Institute of Clinical Molecular Biology; Christian-Albrechts-University of Kiel; Kiel, Germany) and described in Ref.Citation30
Animals
Male C57BL/6 mice (8- to 10-wk-old) were purchased and housed in hanging polycarbonate cages under a 12 h/12 h light/dark schedule. Animals were euthanized in CO2 chambers, and blood was drawn by cardiac puncture. All mice were maintained according to the Guide for the Care and Use of Laboratory Animals (NIH publication no. One.0.0. Revised 2011), and experiments were approved by the IACUCs at Duke-NUS Graduate Medical School.
Cell culture
HepG2 cells with stably transfected human THRB were maintained at 37°C in DMEM supplemented with 10% fetal bovine serum as described previously.Citation20 For T3 treatment, cells were preconditioned in TH depleted media (DMEM+Dowex stripped 10% fetal bovine serum,Citation20 for 3 d before adding T3. Primary mouse hepatocytes were isolated and cultured using a standard 2-step collagenase perfusion protocol. For knockdown studies, HepG2 cells were treated with specific siRNA (10 nM) for 24 h before adding T3 for the next 48 h.
RNA isolation and real-time PCR
Total RNA was isolated and qPCR performed using the QuantiTect SYBR Green PCR Kit (Qiagen, 204141) in accordance to the manufacturer's instructions. Primer sequence provided as (Fig. S9).
Western blotting
Cells or tissue samples were lysed using CelLytic™ M Cell Lysis Reagent (Sigma, C2978) and immunobloting was performed as described previously.Citation20 Densitometry analysis was performed using Image J software (NIH, Bethesda, MD, USA).
Immunofluorescence studies and transmission electron microscopy
Immunofluorescences experiments were performed as described previouslyCitation55 and cell imaging was performed using an Operetta® High Content Imaging System (PerkinElmer, Massachusetts, USA) LSM710 Carl Zeiss (Carl Zeiss Microscopy GmbH, Germany) confocal microscopy images were obtained from a z confocal plane and quantification was done in single planes. Transmission electron microscopy was performed as described previously.Citation55 Imaging was performed on an Olympus EM208S transmission electron microscope; Japan.
Mitochondrial membrane potential and superoxide detection
Cells were treated with T3 or the vehicle for a period of 48 h. Fluorochromes TMRE (to detect membrane potential) or MitoSOX (to detect mitochondrial superoxide radical), were used as per manufacturer's instructions and analyzed using flow cytometry (MACSQuant Analyzer 10, Miltenyi Biotec,Germany).
Measurement of intracellular Ca2+
Intracellular Ca2+ was monitored using the Ca2+-sensitive fluorescent dye, Fura 2-acetoxymethyl ester (Fura-2AM; 1 μmol/L) as per the manufacturer's instructions.
Mitochondrial fraction preparation
Mitochondrial fraction was prepared using the Mitochondria Isolation Kit for Cultured Cells (Thermo Scientific™ Pierce™, 89874) as per the manufacturer's instructions.
In vivo protein oxidation assay
Protein carbonyl formation as a measure of oxidative stress was measured using an Oxyblot system (Millipore, S7150) according to the manufacturer's instructions.
Seahorse XF-24 metabolic flux analysis
Oxygen consumption was measured at 37°C using an XF24 extracellular analyzer (Seahorse Bioscience,USA). Thirty thousand HepG2 cells were seeded in 24-well XF24 culture plates and treated with or without T3 prior to the assay. Optimization of reagents was performed using the Mito stress test kit from Seahorse Bioscience (103015-100) using the protocol and algorithm program in the XF24 analyzer. Basal OCR is [OCR with substrates - OCR with rotenone and antimycin A]. ATP turnover corresponds to the OCR used for mitochondrial ATP synthesis, and reflects the OCR inhibited by oligomycin. However, ATP could be generated in glycolysis, and oligomycin does not inhibit that. Therefore, here we use the term “ATP turnover” strictly for ATP generated via oxidative phosphorylation due to the ATP synthase. Maximal OCR or respiratory capacity is [OCR with FCCP - OCR with rotenone and antimycin A. The spare respiratory capacity (SRC) is OCR with FCCP - Basal OCR].
Calculations and statistics
Results were expressed as either SD or mean ± standard error of the mean (SEM). The statistical significance of differences (*, P < 0.05) was assessed by Student t test. Two-way ANOVA analyses followed by Tukey's post-hoc were performed when comparing different groups.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
supplemental_files.zip
Download Zip (23.1 MB)Acknowledgments
We would like to thank Drs. C.B. Newgard (Duke University Medical Center, Durham, NC), Sherwin Xie, Andrea Lim, Li Ying, Scott A. Summers, Karl Tryggvason, and Kristmundur Sigmundsson (Duke-NUS, Singapore), Dr. Martin L. Privalsky (Department of Microbiology, University of California, Davis, Davis, California) and Dr. Andreas Till (Institute of Clinical Molecular Biology; Christian-Albrechts-University of Kiel; Kiel, Germany) for their helpful advice and technical support and reagents.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported by A*STAR Translational Clinical Research Partnership Award 13/1/96/19/692, NMRC/CSA/0054/2013 (PMY), NMRC/CIRG/1340/2012 (PMY), and NMRC/BNIG/2025/2014 (RAS) from the MInistry of Health, Singapore.
Related Research Data
References
- Kakkar P, Singh BK. Mitochondria: a hub of redox activities and cellular distress control. Mole Cell Biochem 2007; 305:235-53; PMID:17562131; http://dx.doi.org/10.1007/s11010-007-9520-8
- Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res 2009; 37:2539-48; PMID:19264794; http://dx.doi.org/; http://dx.doi.org/10.1093/nar/gkp100
- Scherz-Shouval R, Elazar Z. Regulation of autophagy by ROS: physiology and pathology. Trends Biochem Sci 2011; 36:30-8; PMID:20728362; http://dx.doi.org/10.1016/j.tibs.2010.07.007
- Tolkovsky AM. Mitophagy. Biochimica et Biophysica Acta 2009; 1793:1508-15; PMID:19289147; http://dx.doi.org/10.1016/j.bbamcr.2009.03.002
- Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, Kang D. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. J Biol Chem 2012; 287:3265-72; PMID:22157017; http://dx.doi.org/10.1074/jbc.M111.280156
- Dodson M, Darley-Usmar V, Zhang J. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med 2013; 63:207-21; PMID:23702245http://dx.doi.org/10.1016/j.freeradbiomed.2013.05.014
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death differ 2013; 20:31-42; PMID:22743996; http://dx.doi.org/10.1038/cdd.2012.81;
- Redmann M, Dodson M, Boyer-Guittaut M, Darley-Usmar V, Zhang J. Mitophagy mechanisms and role in human diseases. Int J Biochem Cell Biol 2014; 53:127-33; PMID:24842106; http://dx.doi.org/10.1016/j.biocel.2014.05.010
- Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 2012; 125:795-9; PMID:22448035; http://dx.doi.org/10.1242/jcs.093849
- Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 2008; 454:232-5; PMID:18454133; http://dx.doi.org/10.1038/nature07006
- Liu L, Sakakibara K, Chen Q, Okamoto K. Receptor-mediated mitophagy in yeast and mammalian systems. Cell Res 2014; 24:787-95; PMID:24903109; http://dx.doi.org/10.1038/cr.2014.75
- Melser S, Chatelain EH, Lavie J, Mahfouf W, Jose C, Obre E, Goorden S, Priault M, Elgersma Y, Rezvani HR, et al. Rheb regulates mitophagy induced by mitochondrial energetic status. Cell Metab 2013; 17:719-30; PMID:23602449; http://dx.doi.org/10.1016/j.cmet.2013.03.014
- Graef M, Nunnari J. Mitochondria regulate autophagy by conserved signalling pathways. EMBO J 2011; 30:2101-14; PMID:21468027; http://dx.doi.org/10.1038/emboj.2011.104
- Soleimanpour SA, Gupta A, Bakay M, Ferrari AM, Groff DN, Fadista J, Spruce LA, Kushner JA, Groop L, Seeholzer SH, et al. The diabetes susceptibility gene Clec16a regulates mitophagy. Cell 2014; 157:1577-90; PMID:24949970; http://dx.doi.org/10.1016/j.cell.2014.05.016
- Ryter SW, Koo JK, Choi AM. Molecular regulation of autophagy and its implications for metabolic diseases. Curr Opin Clin Nutr Metab Care 2014; 17:329-37; PMID:24848530; http://dx.doi.org/10.1097/MCO.0000000000000068
- Sinha RA, Singh BK, Yen PM. Thyroid hormone regulation of hepatic lipid and carbohydrate metabolism. Trends Endocrinol Metab: TEM 2014; 25(10):538-45; PMID:25127738.
- Cioffi F, Senese R, Lanni A, Goglia F. Thyroid hormones and mitochondria: with a brief look at derivatives and analogues. Mol Cell Endocrinol 2013; 379:51-61; PMID:23769708; http://dx.doi.org/10.1016/j.mce.2013.06.006
- Weitzel JM, Iwen KA. Coordination of mitochondrial biogenesis by thyroid hormone. Mol Cell Endocrinol 2011; 342:1-7; PMID:21664416; http://dx.doi.org/10.1016/j.mce.2011.05.009
- Thakran S, Sharma P, Attia RR, Hori RT, Deng X, Elam MB, Park EA. Role of sirtuin 1 in the regulation of hepatic gene expression by thyroid hormone. J Biol Chem 2013; 288:807-18; PMID:23209300; http://dx.doi.org/10.1074/jbc.M112.437970
- Sinha RA, You SH, Zhou J, Siddique MM, Bay BH, Zhu X, Privalsky ML, Cheng SY, Stevens RD, Summers SA, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest 2012; 122:2428-38; PMID:22684107; http://dx.doi.org/10.1172/JCI60580
- Fernandez V, Tapia G, Varela P, Romanque P, Cartier-Ugarte D, Videla LA. Thyroid hormone-induced oxidative stress in rodents and humans: a comparative view and relation to redox regulation of gene expression. Comp Biochem Physiol Toxicol Pharmacol 2006; 142:231-9; PMID:16298169; http://dx.doi.org/10.1016/j.cbpc.2005.10.007
- Venditti P, Di Meo S. Thyroid hormone-induced oxidative stress. Cell Mol Life Sci: CMLS 2006; 63:414-34; PMID:16389448; http://dx.doi.org/10.1007/s00018-005-5457-9
- Videla LA, Fernandez V, Tapia G, Varela P. Thyroid hormone calorigenesis and mitochondrial redox signaling: upregulation of gene expression. Front Biosci : J virtual library 2007; 12:1220-8; PMID:17127375; http://dx.doi.org/10.2741/2140
- Videla LA. Hormetic responses of thyroid hormone calorigenesis in the liver: Association with oxidative stress. IUBMB Life 2010; 62:460-6; PMID:20503439.
- Zambrano A, Garcia-Carpizo V, Gallardo ME, Villamuera R, Gomez-Ferreria MA, Pascual A, Buisine N, Sachs LM, Garesse R, Aranda A. The thyroid hormone receptor β induces DNA damage and premature senescence. J Cell Biol 2014; 204:129-46; PMID:24395638; http://dx.doi.org/10.1083/jcb.201305084
- Gross NJ. Control of mitochondrial turnover under the influence of thyroid hormone. J Cell Biol 1971; 48:29-40; PMID:5545111; http://dx.doi.org/10.1083/jcb.48.1.29
- Tseng YH, Ke PY, Liao CJ, Wu SM, Chi HC, Tsai CY, Tseng YH, Ke PY, Liao CJ, Wu SM, Chi HC, Tsai CY. Chromosome 19 open reading frame 80 is upregulated by thyroid hormone and modulates autophagy and lipid metabolism. Autophagy 2014; 10:20-31; PMID:24262987; http://dx.doi.org/10.4161/auto.26126
- Chan IH, Privalsky ML. Isoform-specific transcriptional activity of overlapping target genes that respond to thyroid hormone receptors alpha1 and beta1. Mol Endocrinol (Baltimore, Md) 2009; 23:1758-75; PMID:19628582; http://dx.doi.org/10.1210/me.2009-0025
- Turrens JF. Mitochondrial formation of reactive oxygen species. J physiol 2003; 552:335-44; PMID:14561818; http://dx.doi.org/10.1113/jphysiol.2003.049478
- Kim SJ, Khan M, Quan J, Till A, Subramani S, Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathogens 2013; 9:e1003722; PMID:24339771; http://dx.doi.org/10.1371/journal.ppat.1003722
- Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG, Siddiqui A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc Natl Acad Sci USA 2014; 111:6413-8; PMID:24733894; http://dx.doi.org/10.1073/pnas.1321114111
- Katayama H, Kogure T, Mizushima N, Yoshimori T, Miyawaki A. A sensitive and quantitative technique for detecting autophagic events based on lysosomal delivery. Chem Biol 2011; 18:1042-52; PMID:21867919; http://dx.doi.org/10.1016/j.chembiol.2011.05.013
- Ding WX, Yin XM. Mitophagy: mechanisms, pathophysiological roles, and analysis. Biol Chem 2012; 393:547-64; PMID:22944659.
- Li L, Chen Y, Gibson SB. Starvation-induced autophagy is regulated by mitochondrial reactive oxygen species leading to AMPK activation. Cell Signal 2013; 25:50-65; PMID:23000343; http://dx.doi.org/10.1016/j.cellsig.2012.09.020
- Rahman M, Mofarrahi M, Kristof AS, Nkengfac B, Harel S, Hussain SN. Reactive oxygen species regulation of autophagy in skeletal muscles. Antioxid Redox Signal 2014; 20:443-59; PMID:24180497; http://dx.doi.org/10.1089/ars.2013.5410
- Vargas R, Ortega Y, Bozo V, Andrade M, Minuzzi G, Cornejo P, Fernandez V, Videla LA. Thyroid hormone activates rat liver adenosine 5,-monophosphate-activated protein kinase: relation to CaMKKb, TAK1 and LKB1 expression and energy status. J Biol Regul Homeost Agents 2013; 27:989-99; PMID:24382180.
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214-26; PMID:18439900; http://dx.doi.org/10.1016/j.molcel.2008.03.003
- Ruderman NB, Carling D, Prentki M, Cacicedo JM. AMPK, insulin resistance, and the metabolic syndrome. J Clin Invest 2013; 123:2764-72; PMID:23863634; http://dx.doi.org/10.1172/JCI67227
- Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers AMPK activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Mol Cell Biol 2011; 31:3531-45; PMID:21670147; http://dx.doi.org/10.1128/MCB.05124-11
- Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 2012; 125:1488-99; PMID:22275429; http://dx.doi.org/10.1242/jcs.094110
- Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, et al. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 2014; 15(5):566-75; PMID:24671035
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61.; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochim Biophys Acta 2012; 1823:2297-310; PMID:22917578; http://dx.doi.org/10.1016/j.bbamcr.2012.08.007
- Springer W, Kahle PJ. Regulation of PINK1-Parkin-mediated mitophagy. Autophagy 2011; 7:266-78; PMID:21187721; http://dx.doi.org/10.4161/auto.7.3.14348
- Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res 2005; 8:3-5; PMID:15798367; http://dx.doi.org/10.1089/rej.2005.8.3
- Wikstrom JD, Mahdaviani K, Liesa M, Sereda SB, Si Y, Las G, Twig G, Petrovic N, Zingaretti C, Graham A, et al. Hormone-induced mitochondrial fission is utilized by brown adipocytes as an amplification pathway for energy expenditure. EMBO J 2014; 33:418-36; PMID:24431221
- Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J, Dorn GW 2nd, Brady MJ, Macleod KF. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol 2012; 32:2570-84; PMID:22547685; http://dx.doi.org/10.1128/MCB.00167-12
- Carchman EH, Whelan S, Loughran P, Mollen K, Stratamirovic S, Shiva S, Rosengart MR, Zuckerbraun BS. Experimental sepsis-induced mitochondrial biogenesis is dependent on autophagy, TLR4, and TLR9 signaling in liver. FASEB J 2013; 27:4703-11; PMID:23982147; http://dx.doi.org/10.1096/fj.13-229476
- Bin-Umer MA, McLaughlin JE, Butterly MS, McCormick S, Tumer NE. Elimination of damaged mitochondria through mitophagy reduces mitochondrial oxidative stress and increases tolerance to trichothecenes. Proc Natl Acad Sci USA 2014; 111(32):11798-803; PMID:25071194
- Zhu H, Foretz M, Xie Z, Zhang M, Zhu Z, Xing J, Leclerc J, Gaudry M, Viollet B, Zou MH. PRKAA1/AMPKalpha1 is required for autophagy-dependent mitochondrial clearance during erythrocyte maturation. Autophagy 2014; 10:1522-34; PMID:24988326
- Czaja MJ, Ding WX, Donohue TM, Jr., Friedman SL, Kim JS, Komatsu M, Lemasters JJ, Lemoine A, Lin JD, Ou JH, et al. Functions of autophagy in normal and diseased liver. Autophagy 2013; 9:1131-58; PMID:23774882; http://dx.doi.org/10.4161/auto.25063
- Blake R, Trounce IA. Mitochondrial dysfunction and complications associated with diabetes. Biochim Biophys Acta 2014; 1840:1404-12; PMID:24246956; http://dx.doi.org/10.1016/j.bbagen.2013.11.007
- Cable EE, Finn PD, Stebbins JW, Hou J, Ito BR, van Poelje PD, Linemeyer DL, Erion MD. Reduction of hepatic steatosis in rats and mice after treatment with a liver-targeted thyroid hormone receptor agonist. Hepatology 2009; 49:407-17; PMID:19072834; http://dx.doi.org/10.1002/hep.22572
- Andreux PA, Houtkooper RH, Auwerx J. Pharmacological approaches to restore mitochondrial function. Nat Rev Drug Discov 2013; 12:465-83; PMID:23666487; http://dx.doi.org/10.1038/nrd4023
- Sinha RA, Farah BL, Singh BK, Siddique MM, Li Y, Wu Y, Ilkayeva OR, Gooding J, Ching J, Zhou J, et al. Caffeine stimulates hepatic lipid metabolism by the autophagy-lysosomal pathway in mice. Hepatology 2014; 59:1366-80; PMID:23929677; http://dx.doi.org/10.1002/hep.26667