
Abstract
Mutations in PINK1 and PARK2/Parkin are a main risk factor for familial Parkinson disease. While the physiological mechanism of their activation is unclear, these proteins have been shown in tissue culture cells to serve as a key trigger for autophagy of depolarized mitochondria. Here we show that ablation of the mitochondrial rhomboid protease PARL leads to retrograde translocation of an intermembrane space-bridging PINK1 import intermediate. Subsequently, it is rerouted to the outer membrane in order to recruit PARK2, which phenocopies mitophagy induction by uncoupling agents. Consistent with a role of this retrograde translocation mechanism in neurodegenerative disease, we show that pathogenic PINK1 mutants which are not cleaved by PARL affect PINK1 kinase activity and the ability to induce PARK2-mediated mitophagy. Altogether we suggest that PARL is an important intrinsic player in mitochondrial quality control, a system substantially impaired in Parkinson disease as indicated by reduced removal of damaged mitochondria in affected patients.
Abbreviations
| AIFM | = | apoptosis-inducing factor, mitochondrion-associated |
| CCCP | = | carbonyl cyanide m-chlorophenyl hydrazone |
| CTD | = | C-terminal domain |
| Δψm | = | inner mitochondrial membrane potential |
| FPP | = | fluorescence protease protection assay |
| FSC | = | forward scatter |
| IMS | = | intermembrane space |
| IRES | = | internal ribosome entry site |
| LETM1 | = | leucine zipper-EF-hand-containing transmembrane protein 1 |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 |
| nt | = | nontargeting |
| NTD | = | N-terminal domain |
| PARK2/Parkin | = | parkin RBR E3 ubiquitin protein ligase |
| PD | = | Parkinson disease |
| PINK1 | = | PTEN induced kinase 1 |
| PARL | = | presenilin associated, rhomboid-like |
| shRNA | = | small hairpin RNA |
| siRNA | = | small interfering RNA |
| SSC | = | side scatter |
| TIMM | = | translocase of inner mitochondrial membrane |
| TM | = | transmembrane |
| TOMM | = | translocase of outer mitochondrial membrane |
| VDAC | = | voltage-dependent anion channel |
| WT | = | wild type. |
Introduction
Parkinson disease (PD) is one of the most frequent neurodegenerative disorders. While the cause of sporadic, age-onset PD is unclear, several risk factors underlying inherited, juvenile-onset parkinsonism are known.Citation1 Recent insights into the function and dysfunction of the 2 most commonly mutated genes in autosomal-recessive PD, namely PINK1 (PTEN induced kinase 1) and PARK2 (parkin RBR E3 ubiquitin protein ligase),Citation2,3 have demonstrated their central role in mitochondrial quality control.Citation4 Taken together with compelling evidence that mitochondrial dysfunction is central in the etiology of PD,Citation5 this indicates that insufficient removal of damaged mitochondria and subsequent accumulation of toxic stimuli serve as one of the triggers of neurodegeneration in PD.Citation6,7 Although studies in knockout mice indicate that PINK1 has additional functions and compensatory mechanisms for PINK1 deficiency may exist,Citation8-11 in cultured cells treated with agents uncoupling the mitochondrial membrane potential PINK1 has been shown to recruit the cytosolic E3 ubiquitin protein ligase PARK2 to mitochondria.Citation12 Thereby, PINK1-catalyzed phosphorylation of PARK2 and ubiquitin is thought to mediate the E3 ubiquitin protein ligase activity and ubiquitination of outer mitochondrial membrane proteins.Citation13-18 The effects of mitochondrial PARK2 activation range from proteasomal degradation of certain outer membrane proteinsCitation19 to lysosomal destruction of entire damaged mitochondria by macroautophagy, a process known as mitophagy.Citation12,20-22
PINK1 is a highly conserved, ubiquitously expressed serine/threonine kinaseCitation3,23 with an N-terminal matrix targeting sequence followed by a transmembrane (TM) domain that serves as signal anchor.Citation24,25 In a mechanism that has not been fully resolved yet, its 66-kDa PINK1 precursor (PINK1–66) shows dual targeting to the inner and outer mitochondrial membrane.Citation12,26,27 Recently, we and others have shown that PINK1–66 targeted to the inner mitochondrial membrane by the canonical import pathway is processed within its N-terminal signal anchor by the rhomboid intramembrane protease PARL (presenilin associated, rhomboid-like), leading to release of a 55-kDa C-terminal fragment (PINK1–55) either into the intermembrane space (IMS) or the cytoplasm.Citation28-31 Since fully imported proteins cannot pass the outer membrane of healthy mitochondria unassisted, we have concluded that PARL cleaves a PINK1–66 import intermediate that has been released from the translocase of inner mitochondrial membrane (TIMM) into the lipid bilayer, but has only partially passed the translocase of outer mitochondrial membrane (TOMM) with its C terminus.Citation29 Processed PINK1–55, which lacks any bona fide targeting signal, can slide back through TOMM into the cytoplasm, where it is targeted for proteasomal degradation via the N-end rule pathway.Citation32-34 However, upon disruption of the mitochondrial membrane potential by the ionophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP), PINK1–66 cannot be integrated into the inner membrane and consequently it does not become a substrate for PARL.Citation28-31 In this situation, stabilization and prolonged interaction with the TOMM complex is observed, leading to PINK1–66 integration into the outer membrane by an ill-defined pathway.Citation35-37 Furthermore it has been shown that also the accumulation of misfolded proteins in the mitochondrial matrix can cause PINK1–66 accumulation and PARK2 activation.Citation38 Since the molecular mechanism of PINK1–66 targeting to the outer mitochondrial membrane is unclear and alternative roles of PARL in the control of PINK1 have been suggested,Citation12,31,39,40 the relevance of the PINK1-PARK2 pathway in mitochondrial quality control and its relationship to PARL are currently still under debate.
Here we show that loss of PARL activity leads to accumulation of PINK1–66, mitochondrial PARK2 recruitment and subsequently to mitophagy, mimicking the situation in depolarized organelles. We find that PINK1–66 anchored in the inner membrane is not sufficient for activation of the mitophagy pathway; its IMS-spanning import intermediate has to be rerouted to the outer membrane in order to become active. Moreover, we demonstrate that this retrograde translocation depends on specific PINK1 TM domain features, indicating that mutations within this region interfere with removal of damaged mitochondria by autophagy. Impairment of this recently recognized quality control pathway and subsequent accumulation of dysfunctional mitochondria may be critical determinants during the development of neurodegenerative diseases.
Results
Impaired PINK1–66 processing leads to mitochondrial PARK2 recruitment
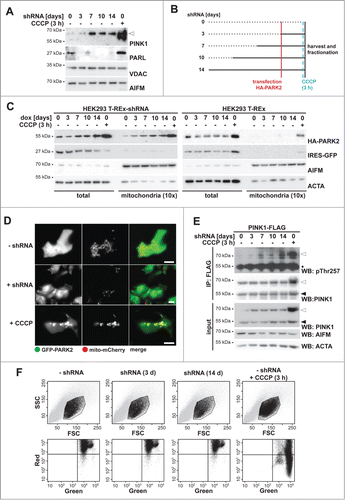
Recent evidence indicates that PARL knockdown in HEK293T cells leads to accumulation of ectopically expressed PINK1 at the mitochondrial surface.Citation29 However, it remained unclear whether this triggers PARK2 activation and whether it corresponds to a physiological response. To investigate this further, we used a cell line stably expressing a doxycycline-inducible PARL-specific shRNACitation29 and studied endogenous PINK1. We tested whether PARL knockdown and subsequent PINK1 accumulation affect PARK2 recruitment. Western blotting of an enriched mitochondrial fraction revealed that PARL knockdown leads to accumulation of some PINK1 within 3 d, and robust levels were observed after 7 d, whereas no effect was observed in control cells lacking the shRNA ( and Fig. S1A). Next, we used the PARL knockdown cells and transfected HA-tagged PARK2 at different times after induction of the shRNA (as outlined in ). Western blot analysis of isolated mitochondria revealed a shift of cytosolic HA-PARK2 into the mitochondrial fraction already after 3 d of PARL ablation (). Although less pronounced than observed upon CCCP treatment, this result mimics activation of the PINK1-PARK2 pathway even in absence of chemical stressors. Doxycycline treatment of HEK293 T-REx cells lacking the PARL knockdown construct, however, did not show any mitochondrial HA-PARK2 recruitment unless CCCP was added, demonstrating that the effect caused by the knockdown is specific (). A consistent effect of PARL knockdown was observed for endogenous PARK2 (Fig. S1B), highlighting the physiological relevance. The effect on endogenous as well as ectopically expressed PARK2 became more pronounced upon prolonged induction of the shRNA (for 7 d and longer) ( and Fig. S1B). Interestingly, under PARL knockdown conditions the steady-state level of ectopically expressed HA-PARK2 increased (). Since we observed identical HA-PARK2 levels upon acute CCCP treatment irrespective of how long PARL has initially been knocked down (Fig. S1C), this indicates that under the overexpression conditions PARK2 is stabilized by its PINK1-mediated recruitment to mitochondria. Consistent with mitochondrial recruitment, upon PARL knockdown bright spots of GFP-tagged PARK2 that colocalized with mito-mCherry were observed by fluorescence microscopy in approximately 15% of the cells ( and Fig. S1D). Noninduced cells, however, showed a homogenous diffuse cytosolic GFP-PARK2 distribution in all cells and GFP-PARK2 was only recruited upon CCCP treatment ( and Fig. S1D). Interestingly, acute treatment with CCCP caused formation of much more and brighter GFP-PARK2 structures, indicating that the entire mitochondrial network was affected. The more-subtle GFP-PARK2-relocalization observed upon PARL knockdown, however, indicates that the PINK1 pathway is activated only in a fraction of the mitochondrial network. Consistent with PINK1 activation, knockdown of PARL leads to autocatalyzed PINK1 phosphorylation at threonine 257 (pThr257) (), which previously has been reported to correlate with its activity.Citation14 PARL knockdown, however, did not affect the mitochondrial membrane potential as determined by detection of the fluorescent dye JC-1, which stains healthy mitochondria orange, whereas CCCP treatment shifted the color to green (). Taken together, these results strongly suggest that reducing the PARL gene dose leads to PINK1 activation and subsequent PARK2 recruitment without affecting the mitochondrial membrane potential. We note, however, that PARL also may cleave additional unknown substrates that may influence PARK2 activity.
Figure 1 (See previous page). PARL ablation leads to mitochondrial PARK2 recruitment. (A) Endogenous PINK1–66 (white triangle) accumulated upon PARL knockdown. In order to detect robust PINK1 levels, mitochondrial membrane proteins were enriched by subcellular fractionation and sodium carbonate extraction. Mitochondrial reference proteins such as VDAC (voltage-dependent anion channel) and AIFM (apoptosis-inducing factor, mitochondrion-associated) were not affected by PARL knockdown. As control, PINK1–66 accumulation was induced by CCCP for 3 h. (B) Experimental outline to analyze the influence of PARL ablation on mitochondrial PARK2 recruitment. PARL knockdown in HEK293 T-REx-shRNA cells was induced by doxycycline as indicated. Cells were transfected with HA-PARK2 36 h before harvesting. As control, cells were treated with CCCP for 3 h. (C) PARK2 accumulated in total cell extracts and was recruited to mitochondria upon doxycycline (dox)-induced PARL knockdown (left panel). This effect was specific, since PARK2 did not accumulate in non-shRNA-expressing parental HEK293 T-REx cells treated with doxycycline for the indicated time (right panel). As control, PARK2 recruitment was induced by CCCP. IRES-GFP and the cellular markers AIFM and ACTA/actin were used as transfection and loading control, respectively. (D) Upon PARL knockdown for 14 d (+ shRNA), GFP-PARK2 colocalized with the mitochondrial marker mito-mCherry similarly, but less pronounced as observed in cells treated with CCCP for 3 h. Scale bars: 10 µm. (E) PINK1 was activated upon PARL knockdown leading to its autophosphorylation at threonine 257 (pThr257), as assessed by immunoprecipitation (IP) and western blot (WB) analysis of ectopically expressed PINK1-FLAG from the detergent solubilized mitochondrial membrane fraction. Asterisks, cross-reacting immunoglobulin heavy chain. (F) FACS-based JC-1 assay shows that doxycycline-induced PARL knockdown for 3 and 14 d (d) did not influence the mitochondrial membrane potential in HEK293 T-REx-shRNA cells. Healthy cells showed red and green stained mitochondria, whereas cells with dissipated membrane potential caused by CCCP were predominantly green. FSC, forward scatter; SSC, side scatter.
Blocking PARL-catalyzed PINK1 processing induces mitophagy
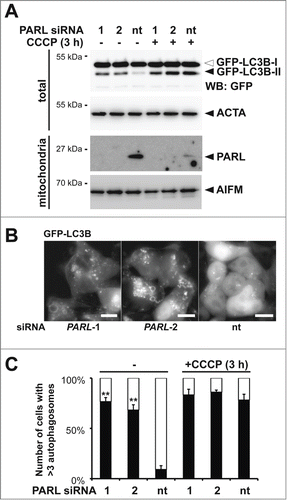
Since PINK1 and PARK2 have been shown to trigger several different stress response pathways and the molecular mechanism of PINK1 activation is unclear,Citation30,31,39 we thought to validate the effect of PARL knockdown on mitophagy induction. Therefore, we tested whether siRNA-mediated PARL knockdown in HEK293 T-REx cells stably expressing the GFP-tagged autophagy marker MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3) leads to formation of the activated lipidated GFP-LC3B-II, which exhibits a faster electrophoretic mobility on SDS-PAGE compared to cytosolic, unlipidated GFP-LC3B-I.Citation41 Knockdown of PARL by using 2 independent targeting sequences results in LC3B activation, whereas in cells transfected with a control siRNA significant GFP-LC3B-II levels were only detected upon CCCP treatment (). Consistent with this, we observed GFP-LC3B positive bright structures in cells transfected with PARL-specific siRNAs (). To discriminate between mitophagy and general macroautophagy we cotransfected mCherry-PARK2. Fluorescence microscopy showed that GFP-LC3B and mCherry-PARK2 colocalized in PARL-ablated cells, however, less pronounced than chemical uncoupling (Fig. S2A). This result indicates that PARL knockdown triggers recruitment of certain PARK2-decorated mitochondria to phagophores. Although compared to CCCP treatment, upon PARL knockdown fewer mitochondria per cell were recruited with approximately 75% of the PARL-ablated cells showing more than 3 GFP-LC3B and mCherry-PARK2 double positive structures, the effect was nearly as penetrant as observed upon uncoupling of the membrane potential (). Taken together with the intermediate effect on GFP-PARK2 recruitment ( and Fig. S1D), this result indicates that in our inducible knockdown system, local recruitment of the autophagy machinery is observed. Since ectopic expression of GFP-LC3B alone may trigger formation of autophagosomes, recruitment of mCherry-PARK2 was also followed in noninduced cells. In these cells PARK2 stayed exclusively cytosolic (Fig. S2B), confirming that the effect observed upon induction was specifically caused by PARL knockdown.
Figure 2. Blocking PARL-catalyzed PINK1 processing induces PARK2-mediated mitophagy. (A) PARL knockdown by 2 independent siRNAs (1 and 2) for 10 d in HEK293 T-REx cells expressing the autophagosomal marker GFP-LC3B induced conversion of soluble, cytoplasmic GFP-LC3B-I to lipidated, membrane-bound, activated GFP-LC3B-II. In cells transfected with the nontargeting siRNA (nt) autophagy was only induced upon treatment with CCCP. The cellular markers AIFM and ACTA/actin were used as loading control. (B) siRNA-mediated PARL knockdown in stable GFP-LC3B expressing cells triggered formation of autophagosomes, whereas GFP-LC3B stayed cytosolic in control cells. Scale bars: 10 μm. (C) PARL knockdown by 2 independent siRNAs (1 and 2) triggered mCherry-PARK2 translocation and autophagosome formation whereas cells transfected with a nontargeting siRNA (nt) responded only in presence of CCCP. See Figure S2A for representative picture. Cells showing more than 3 GFF-LC3B and mCherry-PARK2 positive structures were defined as autophagy-positive. Per sample 100 cells were analyzed (means ±SEM, n=4 ). Significant changes of PARL ablation in absence of CCCP vs. nt control are indicated (**P<0.01; Student t test).
PARL generates soluble PINK1–55 that cannot associate with the outer mitochondrial membrane
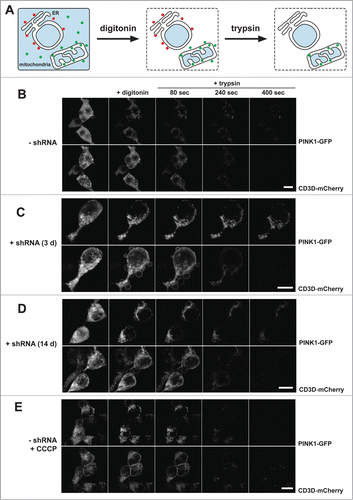
We previously showed the increase of the outer membrane pool of PINK1 upon PARL knockdown by applying biochemical protease protection assays of isolated mitochondria.Citation29 To further investigate this effect in cells we performed quantitative fluorescence protease protection (FPP) assaysCitation42 with inducible PARL HEK293 T-REx-shRNA cells (). We imaged cells coexpressing PINK1-GFP and the type I membrane protein CD3D-mCherry (with C-terminal mCherry tag), before and after selective permeabilization of the plasma membrane with digitonin. This approach allows for the quantification of the bound portion versus the unbound, freely diffusible cytosolic portion of a molecule. As it had been observed before in a similar setup, prior permeabilization, both a cytosolic as well as a mitochondrial pool of PINK1-GFP was detectable.Citation29 Upon digitonin treatment, cytosolic PINK1-GFP vanished from the cells by diffusion, while membrane-bound mitochondrial PINK1-GFP remained inside the cells ( and Fig. S3A). The bound portion of PINK1-GFP was further investigated by exogenous protease application. Only exposed proteins are accessible by the protease and can thus be digested. Proteins protected by surrounding membranes remain intact. Mitochondrial PINK1-GFP showed partial protection from exogenously added trypsin, whereas the reference protein CD3D-mCherry was nearly completely degraded by the protease ( and Fig. S3B). Taken together with our previous analyses of submitochondrial PINK1 localization,Citation29 this corroborated our results that PINK1–66 undergoes dual targeting to the inner and outer mitochondrial membranes. In cells expressing PARL-specific shRNA for 3 d, we observed an increase of the mitochondrial PINK1-GFP signal from 29% to 47% (P < 0.05) (Fig. S3A). Upon trypsin application, the amount of protected PINK1-GFP signal was comparable to that observed in noninduced cells ( and Fig. S3B). This indicates that due to suppression of PARL-triggered turnover, PINK1–66-GFP accumulated at the inner mitochondrial membrane. However, since a fraction of PINK1-GFP was accessible to trypsin, we conclude that a certain fraction of the stabilized PINK1–66 becomes associated with the outer membrane. Upon prolonged PARL knockdown, mitochondrial accumulation and outer membrane association increased by 17% (P < 0.05) and 10% (P = 0.01), respectively ( and Fig. S3A). In contrast, import of the inner mitochondrial protein LETM1 (leucine zipper-EF-hand-containing transmembrane protein 1) was not affected by PARL knockdown (result not shown). This specific effect on PINK1 implies that prolonged PARL knockdown leads to an increase of the outer membrane-attached pool of PINK1–66. Although less pronounced as observed upon chemical uncoupling in CCCP-treated cells ( and Fig. S3B), increase of the surface accessible pool suggests that in both cases PINK1–66 adopts the topology of a bona fide outer membrane protein. Consistent results were obtained by a classical protease protection assay with isolated mitochondria (Fig. S3C). Taken together, these results show that (upon loss of PARL activity) unprocessed PINK1–66 reaches the outer mitochondrial membrane even when an intact inner membrane potential allows its protein import through TIMM. Processed PINK1–55, however, cannot associate with the outer membrane and, consequently, gets dislocated from the mitochondria to become a substrate for proteasomal degradation.
Figure 3 (See previous page). PINK1–66 shows dual targeting to the inner and outer mitochondrial membrane. (A) Outline of the FPP assay analyzing localization and topology of ectopically expressed PINK1-GFP. (B–E) PINK1-GFP was coexpressed with the endoplasmic reticulum (ER)-resident type I membrane protein CD3D-mCherry in HEK293 T-REx cells expressing a PARL-specific shRNA as indicated. Upon expression for 30 h, cells were permeabilized with digitonin to release cytosolic content and subsequently treated with the protease trypsin. PARL knockdown for 3 d (C) and more pronounced for 14 d (D) stabilized PINK1–66 leading to a subpopulation that was susceptible for exogenously added trypsin, as it had been observed upon CCCP treatment (E). Scale bars: 10 μm. See Figure S3 for quantification.
PINK1–66 spanning both membranes slides back to the mitochondrial surface to induce PARK2 recruitment
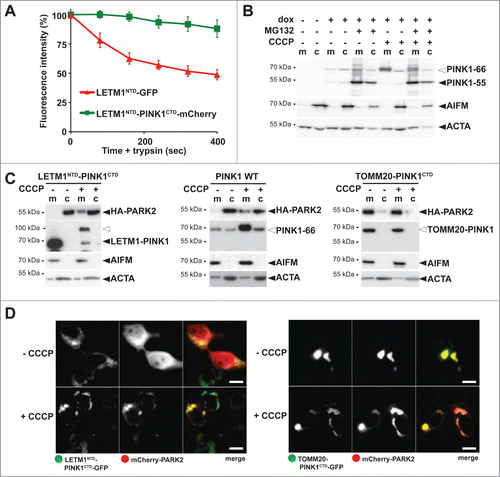
Since PARL-catalyzed cleavage of PINK1–66 in the inner mitochondrial membrane leads to the release of PINK1–55 into the cytosol, we previously reasoned that PARL cleaves an import intermediate of PINK1 that has only partially passed the TOMM complex.Citation29 To test whether this form exists and whether it is sufficient to recruit PARK2, we fused the C-terminal kinase domain (CTD) of PINK1 to the N-terminal TM domain (NTD) of the inner membrane protein LETM1, which is not cleaved by PARL (Fig. S4A). To determine topology of this chimera, we tagged LETM1NTD-PINK1CTD C-terminally with mCherry and compared it with a LETM1 signal anchor-GFP fusion (LETM1NTD-GFP) by performing quantitative FPP assays ( and Fig. S4B). Consistent with the delayed passage of PINK1CTD through the TOMM complexCitation25 and its 2-membrane-spanning form facing the cytosol with its C terminus, LETM1NTD-PINK1CTD-mCherry was partially accessible to trypsin, whereas LETM1NTD-GFP was fully protected ( and Fig. S4B). Similarly, the LETM1-PINK1 chimera was partially surface accessible in isolated mitochondria (Fig. S4C). In order to establish modest uniform expression of this chimera and to allow comparison with PINK1 wild type (WT), we generated inducible stable cells lines using the Flp-In T-REx system and tested them for mitochondrial PARK2 recruitment by cellular fractionation. Importantly, like for endogenous PINK1, under these mild overexpression conditions also for PINK1 WT no 55-kDa form was detected unless the proteasome was inhibited by MG132 (). For both constructs investigated, PINK1 WT and the LETM1NTD-PINK1CTD chimera, no recruitment of PARK2 was observed unless import was blocked by CCCP (). Similarly, by using stable cell lines expressing GFP-tagged PINK1 we did not detect any recruitment of mCherry-PARK2 unless CCCP was added (Fig. S4D). However, consistent with previous results of outer membrane targeted PINK1, its kinase domain fused to the outer membrane protein TOMM20 constantly recruited PARK2 ().Citation12 In cells coexpressing the TOMM20-PINK1CTD-GFP chimera with mCherry-PARK2, no cytosolic PARK2 was ascertained and the mitochondrial network was clustered to a few unphysiological large aggregates (). Taken together, our results show that PINK1 anchored to the inner membrane cannot recruit PARK2. Instead, PINK1 has to be fully targeted to the outer mitochondrial membrane in order to productively interact with PARK2.Citation12
Figure 4. PINK1 stably anchored to the inner membrane cannot recruit PARK2. (A) The C terminus comprising the PINK1 kinase domain delays mitochondrial import as shown by quantitative FPP assay of HEK293T cells coexpressing LETM1NTD-GFP and LETM1NTD-PINK1CTD-mCherry (>60 cells were analyzed, n = 3, means ±SEM ). See for experimental outline and Figure S4B for representative picture. (B) PARL-catalyzed cleavage of ectopically expressed PINK1–66 in HEK293 T-REx cells led to proteasomal degradation of PINK1–55 as shown by sensitivity to MG132 (2 µM) compared to vehicle control. (C) PINK1 anchored in the inner membrane (LETM1NTD-PINK1CTD) interacted with PARK2 only in CCCP-treated cells, whereas the outer membrane targeted TOMM20-PINK1CTD chimera recruited PARK2 independently of uncoupling of the mitochondrial membrane potential. Cells were fractionized and recruitment of ectopically expressed HA-PARK2 from the soluble nonmitochondrial fraction (c) to mitochondria (m) was analyzed. The cellular markers AIFM and ACTA/actin were used as fractionation control. White triangle indicates unprocessed pre form of LETM1-PINK1 observed upon block of mitochondrial import (D) Whereas expression of LETM1NTD-PINK1CTD-GFP did not lead to recruitment of mCherry-PARK2 to mitochondria, coexpression of TOMM20-PINK1CTD-GFP was sufficient to stabilize PARK2 on the mitochondrial surface even in absence of dissipation of the membrane potential by CCCP. Scale bars: 10 μm.
Inner membrane-trapped PINK1 blocks CCCP-triggered PARK2 recruitment
Next, we tested whether the catalytic serine-to-alanine mutant of human PARL (S277A) traps and stabilizes ectopically expressed PINK1, as had been observed previously for other rhomboid proteases.Citation43,44 This dominant-negative effect was particularly pronounced using C-terminal tagged PARLS277A, as shown by immunoprecipitation (). Interestingly, also coexpression of C-terminally tagged PARL WT copurified traces of PINK1–66 and caused a slight decrease of PINK1–55 generation ( and Fig. S5A). Since tagging of PARL did not affect mitochondrial targeting and insertion into the inner membrane (), this result suggests that blocking PARL at its C terminus interferes with its activity, as it has been observed previously for other rhomboid family proteins.Citation45-48 Although the exact role of the PARL C terminus in the rhomboid reaction cycle remains to be addressed, we used the GFP-tagged substrate-trapping SA-mutant as a tool to investigate how prolonged stabilization of PINK1–66 WT at the inner membrane affects PARK2 recruitment. In order to ensure uniform expression and to minimize toxicity of this construct, we generated an inducible mouse PARLS275A-GFP expressing T-REx cell line. Induction of PARLS275A-GFP stabilized endogenous PINK1, but we did not observe any significant mitochondrial PARK2 recruitment as assessed by cellular fractionation () and fluorescence microscopy (). These results indicate that the submitochondrial PINK1 localization, and not simply its quantity, determines whether PARK2 is recruited.
Figure 5. Catalytic PARL mutant traps PINK1–66, thereby blocking the CCCP-triggered PARK2 recruitment. (A) GFP-tagged catalytic human PARLS277A mutant (SA) traps ectopically expressed PINK1–66. Mitochondrial membranes were solubilized with Triton X-100 and GFP-tagged PARL constructs were immunoprecipitated (IP) and analyzed by western blotting (WB) using the indicated antibodies. (B) Tagging of mouse PARLS275A at its C terminus with GFP did not affect its targeting to mitochondria. Scale bars: 10 μm. (C) Trapping of endogenous PINK1–66 by inducibly expressed mouse PARLS275A-GFP in the inner membrane did not lead to mitochondria-associated HA-PARK2 irrespective of dissipation of the membrane potential, whereas treatment of noninduced cells with CCCP resulted in HA-PARK2 recruitment from the cytosol (c) to the mitochondrial fraction (m). For detection of endogenous PINK1–66 (white triangle), mitochondrial membrane proteins were enriched in the pellet fraction (p) of a sodium carbonate extraction. s, extracted supernatant. (D) Dominant-negative mouse PARLS275A-GFP mutant prevented recruitment of mCherry-PARK2 to mitochondria even when the mitochondrial membrane potential was disrupted by CCCP. Scale bars: 10 μm. (E) Quantification of experiments shown in (d) and Figure S5B. Quantification of cells with mitochondria-located mCherry-PARK2 compared to cytosolic PARK2 in dependence of mouse PARLS275A-GFP expression and CCCP is shown. Stable expression of PARLS275A-GFP was induced with doxycycline. Per sample 100 cells were analyzed (means ±SEM, n = 4 ). Significant change between CCCP treatment in presence and absence of PARLSA-GFP is indicated (***P<0.001; Student t test).
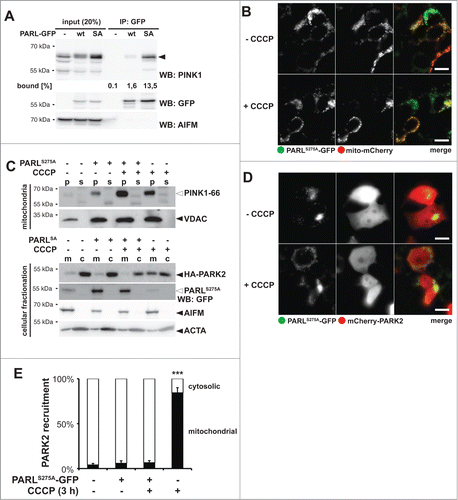
Since we previously had observed that ectopic expression of PARL can boost PINK1 processing,Citation29 we asked whether a putative mitochondrial pool of unprocessed PINK1–66 influences PARK2 recruitment or whether only newly synthesized PINK1–66 inserted into the TOMM complex is bioactive. We used the PARLS275A-GFP substrate trap to stabilize endogenous PINK1–66 at the inner membrane and asked whether this would interfere with CCCP-induced PARK2 recruitment, which is predicted to affect newly synthesized proteins. The rationale was that a preexisting pool of PINK1–66 fully imported into the IMS is predicted to have no influence on CCCP-mediated PARK2 recruitment at the outer membrane, whereas a putative 2-membrane-spanning form may interfere. Interestingly, in presence of PARLS275A-GFP, treatment of cells for 3 h with CCCP did not cause any recruitment of ectopically expressed PARK2 into the mitochondrial fraction (). Consistent with this dominant-negative effect, coexpression of PARLS275A-GFP with mCherry-PARK2 did not show any colocalization irrespective of treatment with CCCP ( and Fig. 5E). This block of PARK2 translocation was specifically caused by trapping of endogenous PARL substrates, since CCCP treatment of the stable cells without PARLS275A-GFP induction showed mitochondrial PARK2 recruitment ( and Fig. S5B). Taken together, these results indicate that the fate of PINK1–66 at the inner and outer mitochondrial membrane is functionally coupled and PINK1–66 trapped by PARLS275A-GFP blocks newly synthesized PINK1–66 to recruit PARK2 probably by sequestering a limiting factor in mitochondrial PINK1–66 import. Although the exact molecular mechanism of how PARL is linked to PINK1s outer membrane targeting and how it affects mitochondrial PARK2 recruitment remains to be determined, this result shows that PARL can play a central role in mitochondrial quality control.
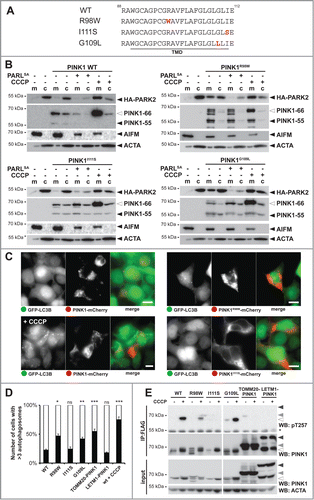
Retrograde translocation depends on specific PINK1 TM features
Our model that PARL tunes activation of the PINK1-PARK2 pathway predicts that mutations interfering with PINK1 turnover may affect the rate of mitophagy, with a predicted impact on mitochondrial disorders. Consistent with this hypothesis, we recently showed that 2 PD-associated heterozygous variants in the PINK1 TM domain, namely R98W and I111S, or mutation of a conserved glycine residue block PARL-catalyzed processing ().Citation29 In order to address the relevance of this effect, we generated inducible stable cell lines expressing these mutants and analyzed their ability to recruit PARK2. As it had been observed upon transient transfection,Citation29 all 3 PINK1 TM domain mutants were processed in mitochondria by so far uncharacterized PARL-independent proteases (). Despite that alternative processing, PINK1R98W efficiently triggered translocation of ectopically expressed PARK2 into the mitochondrial fraction even in absence of CCCP ( and Fig. S6A). Since coexpression of the PARLS277A substrate-trapping mutant reverted this constitutive PARK2 recruitment, we conclude that the R98W mutation does neither affect insertion into the inner membrane nor its transient interaction with PARL. Interestingly, the second PD-associated TM mutant, PINK1I111S, did not show any constitutive activation and triggered mitochondrial PARK2-recruitment only in the presence of CCCP similar as it has been observed for the WT construct ( and Fig. S6A). This indicates that the I111S mutant can be targeted to the outer membrane when it got stuck in the TOMM complex mediated by decreased mitochondrial membrane potential, but as soon it reaches the inner membrane it becomes a membrane protein that cannot slide back to the mitochondrial surface. In contrast, blocking PARL-catalyzed cleavage by mutating the invariant glycine-109 to leucineCitation29 triggers PARK2 recruitment in absence of uncoupler (). Consistent with alternative fates of the PINK1 mutants tested, upon coexpression with GFP-LC3B, PINK1R98W or PINK1G109L-expressing cells showed more than 3 autophagosomes in approximately 50% of the cells, whereas PINK1I111S only showed a modest mitophagy activation in the range of the IMS-targeted LETM1NTD-PINK1CTD chimera or the PINK1 WT construct ( and Fig. S6B). This striking difference between cleavage-deficient PINK1 alleles suggests that the I111S mutation specifically prevents retrograde translocation of unprocessed PINK1–66 to the outer membrane. In accordance with an altered functionality of PINK1I111S, using the Thr257 phospho-specific PINK1 antibody,Citation14 no CCCP-induced activation was observed, whereas PINK1R98W and PINK1G109L reacted to uncoupling like the WT protein (). Likewise, also TOMM20-PINK1CTD and LETM1NTD-PINK1CTD were only marginally detected by the phospho-specific PINK1 antibody (). Of note, PINK1 autophosphorylation has previously been shown not to be strictly required for PARK2 activation,Citation14 and consistent with a previous study outer membrane-targeted TOMM20-PINK1CTD chimera efficiently triggered LC3B-dependent mitophagy ( and Fig. S6B).Citation12 Altogether, our analysis of PINK1 TM domain mutants confirms that PARL-catalyzed processing plays a central role in the control of mitophagy. Moreover, our results on the PD-associated heterozygous I111S mutant suggest that specific TM features are responsible for the previously unrecognized PINK1 retrograde translocation mechanism. The exact molecular mechanism of how the PINK1 signal anchor is rerouted from the inner mitochondrial membrane to the outer membrane, however, is an important question that remains to be addressed in future.
Figure 6. PD-associated PINK1 TM domain mutants show altered activity. (A) TM domain (TMD) sequences of PINK1 WT, PINK1R98W, PINK1I111S, and PINK1G109L are shown. Mutated residues are highlighted in red. (B) PARL cleavage deficient mutants PINK1R98W and PINK1G109L trigger mitochondrial PARK2 recruitment without dissipation of the membrane potential, whereas PINK1 WT and PINK1I111S require uncoupling of the membrane potential (+CCCP). HA-PARK2 was transfected in inducible stable cell lines expressing the indicated PINK1 constructs. As control, cells were cotransfected with human PARLS277A-GFP or treated with CCCP in order to trap the PINK1 precursor in the inner or outer membrane, respectively. m, mitochondrial fraction; c, cytosol. (C) Expression of PINK1R98W led to formation of autophagosomes, whereas expression of PINK1 WT and PINK1I111S did not. Autophagy induction was determined in the GFP-LC3B expressing stable cell line transiently expressing mCherry-tagged PINK1 constructs as indicated. As control, cells expressing PINK1 WT were treated with CCCP for 3 h. Scale bars: 10 μm. (D) Quantification of experiments shown in (C) and Figure S6B reveals number of cells with more than 3 autophagosomes formed. As control, CCCP was applied for cells harboring PINK1 WT. Per sample 100 cells were analyzed (means ±SEM, n = 3 ). Significant changes versus PINK1 WT are indicated (*P < 0.05; **P < 0.01; ***P < 0.001; Student t test); ns, nonsignificant. (E) PINK1 TM domain mutations impair PINK1 autophosphorylation at Thr257 as assessed by immunoprecipitation (IP) and western blot (WB) analysis.
Discussion
Here we show that knockdown of PARL stabilizes PINK1–66, leading to mitochondrial recruitment of the E3 ubiquitin-protein ligase PARK2 and activation of mitophagy. Since it has been suggested previously that PARL activity has the opposite effect in triggering mitophagy,Citation31 and alternative mitochondrial proteases have been implicated in PINK1–66 processing,Citation39 this provides an important validation of the pivotal impact of PARL on the PINK1 abundance.Citation28,29 Here, we confirm that upon import into healthy mitochondria, the main fraction of PINK1–66 is cleaved by PARL. Consistent with previous reports,Citation12,28,29,34 this intramembrane cut triggers rapid turnover of the cleavage fragment PINK1–55 by the proteasome, thereby suppressing PARK2 translocation. However, we now show that when PARL activity becomes rate limiting, PINK1–66 initially integrated into the inner membrane stays mobile and can reach the outer membrane. As it has been observed for CCCP-induced autophagy, this leads to recruitment of cytosolic PARK2 to mitochondria and mitophagy. Importantly, we observe PINK1 and PARK2-triggered activation of canonical LC3B-dependent autophagy irrespective of mitochondrial uncoupling or any other drastic treatment. Since reduced protease activity is a hallmark of dysregulated and aging cells, this provides additional strong evidence for a physiological relevance of PINK1-PARK2 as a checkpoint in mitochondrial quality control. Consistent with this, reduced mitochondrial mass has been reported upon muscle knockdown of PARL in miceCitation49 and expression of a cleavage-deficient PINK1 mutant in tissue culture cells.Citation30
Although signal sequences commonly target proteins only to one distinct cellular destination, dual targeting expands functionality of certain proteins. Likewise, PINK1–66 is subject to competing targeting events to the inner and outer mitochondrial membrane.Citation12,26,27,50 The N-terminal 77 amino acids of PINK1 are essential and sufficient for targeting to the inner membrane via the canonical TOMM-TIMM machinery ()Citation26 with the PINK1 TM domain acting as stop transfer signal.Citation24 On the other hand, interaction of the PINK1 C terminus with the cytosolic chaperone HSP90 counteracts full import of the kinase domain into the IMS.Citation25 Despite that cytosolic retention, it is still a matter of debate how PINK1–66 associates with the outer membrane.Citation35–37 Here we show that PINK1–66 inserted into the inner membrane, where it interacts with PARL, still bridges the IMS by exposing parts of its C-terminal domain toward the cytosol. As a consequence of this unusual 2-membrane-spanning topology, upon PARL-catalyzed removal of the signal anchor PINK1–55 slides back into the cytosol where it gets degraded by the proteasome ().Citation28,29 In this study we extend this model by demonstrating that upon knockdown of PARL also the unprocessed PINK1–66 is able to retrotranslocate (). Since the unprocessed PINK1–66 precursor still has its TM domain, this leads to anchorage into the outer membrane and activation of PARK2-dependent mitophagy. Similarly, retrograde translocation has been observed for import intermediates of soluble IMS proteins,Citation51,52 but to our knowledge PINK1–66 is the first protein with a signal anchor sequence that gets extracted from the inner membrane. This previously unrecognized PINK1 activating mechanism in mitochondria with intact mitochondrial membrane potential mimics the CCCP-induced effect in depolarized organelles ().Citation12 The mechanism of how the PINK1 signal anchor is integrated into the lipid bilayer of the outer membrane is not known yet. Cell-free mitochondrial import assays and BN-PAGE suggest that PINK1–66 does not accumulate in the protein-conducting channel formed by TOMM40, but creates a stable complex with the substrate receptor TOMM20.Citation35 However, more recent studies propose TOMM70Citation36 and TOMM7Citation37 to govern this noncanonical outer membrane insertion route. Despite that controversy, a putative role of a substrate receptor in the outer membrane insertion pathway is consistent with our observation that PINK1–66 trapped by the PARLS275A mutant at the inner membrane blocks CCCP-mediated PARK2 translocation by newly synthesized PINK1 ().
Figure 7. Model for the PARL-PINK1-PARK2 relay in mitochondrial quality control. (A) Due to prolonged interaction with the cytosolic chaperone HSP90, PINK1–66 forms an import intermediate spanning the IMS and the TOMM complex. PARL-catalyzed cleavage of the PINK1 TM domain in the inner mitochondrial membrane generates soluble PINK1–55, which due to lack of its membrane anchor slides back into the cytosol, where it becomes rapidly degraded by the proteasome. (B) Unprocessed PINK1–66, observed upon PARL ablation, stays competent for sliding back to the outer membrane where it is subsequently integrated into the lipid bilayer by an unknown mechanism. Outer membrane targeting is accompanied by PINK1 autophosphorylation, PARK2 recruitment and its PINK1-dependent phosphorylation. (C) Upon loss of the inner mitochondrial membrane potential (Δψ Upon lo–66 accumulates in the outer membrane similar to the situation in PARL-ablated cells. Although integration of the TM domain into the inner membrane is blocked, we postulate that the PINK1CTD transiently inserts into the TOMM channel leading to stripping of cytosolic HSP90 and subsequent efficient maturation of outer membrane targeted PINK1–66. (D) Direct targeting of PINK1CTD to the outer membrane by fusion to TOMM20 (black) is sufficient to trigger PARK2 recruitment, but lack of autophosphorylation suggests that due prolonged interaction with cytosolic factors such as HSP90 folding of the kinase domain is hampered probably leading to reduced activation.
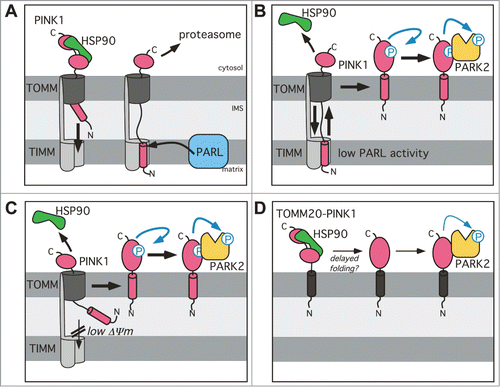
Why would cells constantly import PINK1–66 to the inner membrane in order to trigger its degradation? Although it has initially been suggested that this futile PINK1–66 import senses the integrity of the inner membrane potential,Citation12 treatment of cells with uncouplers such as CCCP is a rather harsh condition and the physiological relevance of this mechanism in mitochondrial quality control remains to be shown. Instead of sensing efficiency of protein import, we hypothesize that the PARL-PINK1-PARK2 relay allows integration of information from the matrix and the inner membrane. This may explain how accumulation of unfolded proteins in the matrix is capable of triggering PINK1 activation independent of changes in the mitochondrial membrane potential.Citation38 Another potential advantage of sequential targeting to the inner and outer membrane may be to prevent premature phosphorylation of PINK1 substrates. The molecular mechanism of how the PINK1 kinase activity is regulated is not known. Upon translation of its precursor at cytosolic ribosomes, however, PINK1 does not show any significant activity.Citation14,53 Likewise, our data suggest that direct outer membrane targeting of PINK1, without preceding ‘diving’ into the TOMM channel and retrograde translocation, leads to a reduced kinase activity as assessed by the lack of autophosphorylation (). This is consistent with previous reports demonstrating that despite strong accumulation at the mitochondrial surface, ectopically expressed PINK1 has less activity than low levels of endogenous PINK1 in chemically uncoupled mitochondria.Citation20,53 Although the precise molecular mechanism remains to be determined, it is attractive to speculate that cytosolic factors such as HSP90 have to be stripped off by TOMM, in order to allow efficient folding of PINK1 to the fully active kinase ().
Although rhomboid proteases are commonly believed to be constitutively active enzymes,Citation54 PARL has been suggested to be subject to direct post-translational control,Citation40 thereby potentially serving as a molecular switch in the PINK1-PARK2 pathway. Such a putative central role of PARL in controlling mitochondrial integrity is supported by several reports linking mutations in the PARL gene to mitochondrial dysfunction in human diseases ranging from type-2 diabetes to PD.Citation55,31 Here we demonstrate that 2 PD-associated mutations in the PINK1 TM domain affect PARK2 translocation and mitophagy, although with opposing effects. To determine the precise role these PINK1 alleles play in the pathophysiology of PD, it will be important to study the impact of the TM domain mutations in neuronal cells. However, our study in immortalized HEK293 cells provides the proof of concept that altered PARL-catalyzed PINK1–66 cleavage has the potential to trigger PARK2-dependent mitophagy. Since defects in the mitophagy pathway have been linked to neurodegenerative disorders,Citation56 inhibition of PARL and the subsequent boost of PINK1-PARK2-mediated mitophagy emerge as a promising new therapeutic intervention strategy to suppress deleterious effects causing dysfunctional mitochondria such as accumulation of toxic protein aggregates or increased production of reactive oxygen species.
Materials and Methods
Plasmids and RNA interference
Construction of pcDNA3.1/PINK1 (WT and TM domain mutants), pCDH/PINK1-IRES-GFP, pEGFP/PINK1-GFP, pcDNA3.1/mPARL, pcDNA3.1/hPARL, pcDNA3.1/mPARLS275A, pcDNA3.1/hPARLS277A and pCDH/HA-PARK2/Parkin-IRES-GFP expression plasmids has been described previously.Citation29,57 For attachment of a C-terminal FLAG tag, PINK1 was subcloned into pFLAG-N1.Citation29 Human TOMM20 and LETM1 were amplified by PCR from IMAGE clones 925859 and 4126510, respectively, and subcloned into pcDNA3.1 and pFLAG-N1, respectively. For TOMM20-PINK1CTD, the N-terminal 111 amino acids of PINK1 comprising its mitochondrial targeting signal and TM domain were replaced by TOMM20. The construct LETM1NTD-PINK1CTD was generated by overlap extension PCR, fusing residues 1 to 229 of LETM1 containing its matrix-targeting sequence and TM domain N-terminally to the PINK1CTD. To generate HA-PARK2 we ligated oligo DNA encoding the N-terminal HA epitope tag into pcDNA3.1 and subsequently introduced the PARK2 open reading frame downstream. C-terminally-fluorescently-labeled PINK1, PINK1R98W, PINK1I111S, PINK1G109L, TOMM20-PINK1CTD, LETM1, LETM1NTD-PINK1CTD, mPARLS275A and hPARLS277A were generated by subcloning into monomeric variants of pmCherry-N1 and pEGFP-N1, respectively. N-terminally labeled PARK2 was generated by subcloning into pmCherry-C1 and pEGFP-C1. Expression vectors for mito-mCherry and CD3D-mCherry have been described previously.Citation42 For stable expression, PINK1, PINK1R98W, PINK1I111S, PINK1G109L, TOMM20-PINK1CTD, LETM1NTD-PINK1CTD, the respective mEGFP-tagged variants and mPARLS275A-GFP were subcloned into pcDNA5/FRT/TO. Likewise, GFP-LC3B described previouslyCitation58 was subcloned into pcDNA5/FRT/TO for stable expression.
Small interfering RNA (siRNA)-oligonucleotides PARL-1 (ID s30889), PARL-2 (ID s30890) and a nontargeting control siRNA (ID 4390843) were purchased from Ambion.
Cell lines and transfection
HEK293T cells were grown in DMEM supplemented with 10% (v/v) fetal bovine serum at 37°C in 5% (v/v) CO2. Inducible HEK293 T-REx-shRNA cells expressing a small hairpin RNA (shRNA) with the PARL-1 targeting sequence (5′-GGCAUGAAAUAAGGACUAATT-3′),Citation29 were grown in DMEM supplemented with 10% (v/v) fetal bovine serum, blasticidin (10 µg/ml; Gibco, A1113903) and G418 (0.5 µg/µl; Gibco, 11811031). Knockdown was performed with 1 µg/ml doxycycline (Applichem, A2951). To prepare inducible stable cell lines, Flp-In HEK293 T-REx cells were cotransfected with the respective pcDNA5/FRT/TO constructs (described above) and pOG44 (Invitrogen, K65000), followed by selection with blasticidin (10 µg/ml) and hygromycin B (125 µg/ml; Invitrogen, 10687). Positive clones were selected by western blot analysis. To test for functional GFP-LC3B clones, doxycycline-induced cells were starved in phosphate-buffered saline (50 mM potassium phosphate, 137 NaCl, 2.7 mM KCl, pH 7.4) for 20 h and analyzed for formation of autophagosomes by fluorescence microscopy. Disruption of the mitochondrial membrane potential was achieved by incubating the cells with 10 µM CCCP (Sigma, C2759) for 3 h. For inhibition of the proteasome, 20 h postinduction HEK293 T-REx-PINK1 cells were treated for 16 h with 2 µM MG132 (Calbiochem, 474790) added from a 5 mM stock dissolved in DMSO. Transient transfections were performed using 25-kDa linear polyethylenimine (Polysciences, 9002–98–6).Citation59 If not otherwise indicated, 1 µg plasmid encoding PINK1, 200 ng plasmid encoding PARL and 250 ng plasmid encoding PARK2 were used. Total transfected DNA (2 µg/well) was held constant by addition of empty plasmid. If not otherwise stated, cells were harvested 36 h after transfection. For transfection of siRNA, 2 × 105 HEK293 T-REx-GFP-LC3B cells were seeded per 6-well. After 24 h, cells were transfected with 10 nM siRNA-oligonucleotide using Oligofectamine (Invitrogen, 12252011).
Flow cytometry
Detection of mitochondrial membrane potential was performed using a JC-1 based mitochondrial staining kit (Sigma-Aldrich, MAK160) according to the manufacturer's instructions. For FACS analysis, 100,000 cells/sample were analyzed using a BD FACSCanto II (Becton Dickinson, Franklin Lakes, New Jersey, USA); excitation with 488 nm, filter: 502-LP/530–30 and 556-LP/585–42.
Subcellular fractionation
Subcellular fractions were prepared by cell disruption followed by differential centrifugation. In brief, cells were detached by incubation in phosphate-buffered saline-EDTA and resuspended in isolation buffer (250 mM sucrose, 10 mM HEPES pH 7.4, 0.1 mM EGTA, EDTA-free complete protease inhibitor cocktail [PI; Roche; 05892953001]). After 10 min incubation at 4°C, cells were lyzed by passing 6 times through a 27-gauge needle. Cellular debris and nuclei were discarded after centrifugation at 200 g for 5 min at 4°C. The supernatant fraction was spun at 10,000 g for 10 min at 4°C. For detection of endogenous PARK2 in the mitochondrial fraction, the postnuclear supernatant fraction was supplemented with 100 mM KOAc prior to centrifugation. The mitochondrial membrane pellet was resuspended in isolation buffer, whereas the supernatant fraction, referred to as the nonmitochondrial fraction, was precipitated with 10% (w/v) trichloracetic acid, washed with acetone and resuspended in SDS sample buffer.
For detection of endogenous PINK1–66 and PARL, mitochondrial membranes were extracted in 100 µl freshly prepared 100 mM Na2CO3 (pH 11.3) solution. After 30 min incubation on ice, extracted membranes were overlaid on a sucrose cushion (100 mM Na2CO3, 250 mM sucrose) and centrifuged at 130,000 g for 15 min at 4°C. Membrane pellets were resuspended in SDS sample buffer.
Immunoprecipitation
If not indicated differently, all steps were performed at 4°C. For substrate trapping, the mitochondrial fraction from hPARLS277A-GFP expressing HEK293T cells was solubilized with 2% Triton X-100 in IP buffer (50 mM HEPES-KOH, pH 7.4, 150 mM NaCl, 2 mM MgOAc2, 10% glycerol, 1 mM EGTA), containing 1xPI and 10 µg/ml PMSF. Detergent-solubilized membrane proteins were cleared by centrifugation at 20,000 g for 10 min and subsequently preincubated for 1 h on BSA (Roth, 8076)-coupled sepharose beads (GE Healthcare, 17–0906–01). Anti-GFP immunoprecipitation was performed using a recombinant and purified GFP-specific single chain antibody fragmentCitation60 coupled to NHS-activated sepharose beads (GE Healthcare, 17–0906–01) as described.Citation44 Immunoprecipitates were washed 3 times in IP buffer, containing 0.1% Triton X-100 (Applichem, A1287). The immunocomplexes were eluted in SDS sample buffer.
For immunoprecipitation of phosphorylated FLAG-tagged PINK1, HEK293T cells were lysed in fractionation buffer (250 mM sucrose, 30 mM HEPES-KOH, pH 7.4, 3 mM EDTA, 10 mM sodium orthovanadate, 10 mM sodium-β-glycerophosphate, 50 mM NaF, 5 mM sodium pyrophosphate) supplemented with 1xPI. Cellular debris and nuclei were removed by centrifugation at 500 g for 10 min. The supernatant fraction was spun at 10,000 g for 10 min. The mitochondrial membrane pellet was washed and resuspended in 500 µl fractionation buffer containing 1% Triton-X 100. After 30 min incubation insoluble material was removed by centrifugation at 10,000 g for 10 min. The supernatant fraction was supplemented with 500 µl fresh fractionation buffer, the FLAG specific antibody and protein A beads (GE Healthcare, 17–0469–01) for overnight incubation. Immunoprecipitates were washed 3 times in fractionation buffer containing 0.1% Triton-X 100 and then resuspended in SDS sample buffer followed by SDS-PAGE and western blotting.
Western blotting
Proteins were resolved on Tris-glycine acrylamide gels followed by western blot analysis. The following antibodies were used at dilutions recommended by the manufacturer: polyclonal rabbit PINK1 (Novus Biologicals, BC100–494), polyclonal rabbit VDAC (Pierce, PA1–954A), monoclonal mouse AIFM/AIF (Santa Cruz Biotechnology, SC-13116), monoclonal mouse FLAG (M2; Sigma, F3165), monoclonal mouse HA (Covance, MMS-101P), polyclonal rabbit PARL (Abcam, ab45231) and ACTA/α-actin (Abcam, ab1801). Antibody against GFP was a gift from D. Görlich. The phospho-PINK1 (Thr257) antibody was already described in Kondapalli et al.Citation14 Bound antibodies were detected by enhanced chemiluminescence. For detection, films or the LAS 4000 system (Fuji, Tokyo, Japan) were used. Data shown are representative of 3 independent experiments. For quantification, we used the Multi Gauge software (Fuji) and data acquired from the LAS 4000.
Microscopy and fluorescence protease protection assay
For wide-field microscopy, images were acquired on an xcellence IX81 inverted microscope system (Olympus, Tokyo, Japan) equipped with an UPLSAPO 60x/1.35 numerical aperture (NA) oil objective (Olympus) and a ORCA-R2 camera (Hamamatsu, Hamamatsu City, Japan). Different fluorochromes with overlapping spectra in multilabeled samples were spectrally unmixed by using the xcellence software (Olympus). Experiments were performed at 37°C and 5% CO2/95% air using an incubation chamber.
For FPP assays, confocal microscopy was performed on an LSM 780 system (Carl Zeiss). Images were taken with a 40x/1.2 NA water C-Apochromat objective lens (Carl Zeiss, Oberkochen, Germany) and pinhole settings between 1.5 and 2 airy units. Z-stacks of images were recorded over time and displayed as maximum intensity projections. The assay was performed as described previously.Citation42 In brief, cells were first permeabilized with 20 µM digitonin (Calbiochem, 300410) in KHM buffer (110 mM potassium acetate, 20 mM HEPES, 2 mM MgCl2), then washed with KHM buffer to remove digitonin and finally treated with 4 mM trypsin (Gibco, 25300) in KHM buffer for the indicated time. For quantitative FPP assays, Z-stacks of 10 images over 20 µm along the Z-axis were recorded with open pinhole settings. All acquisition parameters, time frames and digitonin and trypsin applications were kept identical for all measurements. Maximum intensity projections of the Z-stacks were background subtracted and corrected for acquisition bleaching over the course of the image series. The mean intensities of 40+ cells from 3 or more independent experiments per condition were quantified by using ImageJ (Rasband, W.S., ImageJ, U. S. National Institutes of Health, Bethesda, Maryland, USA, http://imagej.nih.gov/ij/, 1997–2014), and normalized and plotted with Microsoft Excel. All images for FPP assays were acquired by a microscopist without knowledge of the actual sample ID, in order to guarantee unbiased data collection.
Image processing including background subtraction, cropping, and montage creation was performed using ImageJ.
Protease protection assays
Isolated mitochondria were resuspended in isolation buffer (supplemented with 10 µM E64 [Sigma, E3132], 1 µM pepstatin A [Sigma, P5318] and 2 µg/ml o-phenanthroline [Sigma, P9375]) and treated with the indicated amount of proteinase K (30 units/mg; Applichem, A3826) for 30 min on ice. Subsequently 10 µg/ml phenylmethylsulfonyl fluoride (Applichem, A0999) and SDS sample buffer were added.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1063763_supplemental_files.zip
Download Zip (1.9 MB)Acknowledgments
We thank Bernhard Dobberstein, Andreas Reichert, Sebastian Schuck, and Dönem Avci for advice and critical reading of the manuscript.
Funding
The work was supported by a PhD student fellowship by the Hartmut Hoffman-Berling International Graduate School to CM, funds from the Baden-Württemberg Stiftung within the Network of Aging Research (NAR, University of Heidelberg) and the Deutsche Forschungsgemeinschaft (SFB 1036, TP 12) to MKL.
Related Research Data
References
- Gasser T. Genomic and proteomic biomarkers for Parkinson disease. Neurology 2009; 72:S27-31; PMID:19221311; http://dx.doi.org/10.1212/WNL.0b013e318198e054
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392:605-8; PMID:9560156; http://dx.doi.org/10.1038/33416
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. Hereditary early-onset Parkinson disease caused by mutations in PINK1. Science 2004; 304:1158-60; PMID:15087508; http://dx.doi.org/10.1126/science.1096284
- Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 2011; 12:9-14; PMID:21179058; http://dx.doi.org/10.1038/nrm3028
- Schapira AH. Mitochondrial diseases. Lancet 2012; 379:1825-34; PMID:22482939; http://dx.doi.org/10.1016/S0140-6736(11)61305-6
- Exner N, Lutz AK, Haass C, Winklhofer KF. Mitochondrial dysfunction in Parkinson disease: molecular mechanisms and pathophysiological consequences. EMBO J 2012; 31:3038-62; PMID:22735187; http://dx.doi.org/10.1038/emboj.2012.170
- Karbowski M, Neutzner A. Neurodegeneration as a consequence of failed mitochondrial maintenance. Acta Neuropathol 2012; 123:157-71; PMID:22143516; http://dx.doi.org/10.1007/s00401-011-0921-0
- Kitada T, Pisani A, Porter DR, Yamaguchi H, Tscherter A, Martella G, Bonsi P, Zhang C, Pothos EN, Shen J. Impaired dopamine release and synaptic plasticity in the striatum of PINK1-deficient mice. Proc Natl Acad Sci U S A 2007; 104:11441-6; PMID:17563363; http://dx.doi.org/10.1073/pnas.0702717104
- Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A 2008; 105:11364-9; PMID:18687901; http://dx.doi.org/10.1073/pnas.0802076105
- Haque ME, Thomas KJ, D'Souza C, Callaghan S, Kitada T, Slack RS, Fraser P, Cookson MR, Tandon A, Park DS. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci U S A 2008; 105:1716-21; PMID:18218782; http://dx.doi.org/10.1073/pnas.0705363105
- Gispert S, Ricciardi F, Kurz A, Azizov M, Hoepken HH, Becker D, Voos W, Leuner K, Müller WE, Kudin AP, et al. Parkinson phenotype in aged PINK1-deficient mice is accompanied by progressive mitochondrial dysfunction in absence of neurodegeneration. PloS One 2009; 4:e5777; PMID:19492057; http://dx.doi.org/10.1371/journal.pone.0005777
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 2010; 8:e1000298; PMID:20126261; http://dx.doi.org/10.1371/journal.pbio.1000298
- Sha D, Chin LS, Li L. Phosphorylation of parkin by Parkinson disease-linked kinase PINK1 activates parkin E3 ligase function and NF-kappaB signaling. Hum Mol Genet 2010; 19:352-63; PMID:19880420; http://dx.doi.org/10.1093/hmg/ddp501
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol 2012; 2:120080; PMID:22724072; http://dx.doi.org/10.1098/rsob.120080
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 2014; 205:143-53; PMID:24751536; http://dx.doi.org/10.1083/jcb.201402104
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 2014; 510:162-6; PMID:24784582
- Shiba-Fukushima K, Imai Y, Yoshida S, Ishihama Y, Kanao T, Sato S, Hattori N. PINK1-mediated phosphorylation of the Parkin ubiquitin-like domain primes mitochondrial translocation of Parkin and regulates mitophagy. Sci Rep 2012; 2:1002; PMID:23256036; http://dx.doi.org/10.1038/srep01002
- Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem 2013; 288:22019-32; PMID:23754282; http://dx.doi.org/10.1074/jbc.M113.467530
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 2010; 191:1367-80; PMID:21173115; http://dx.doi.org/10.1083/jcb.201007013
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 2010; 189:211-21; PMID:20404107; http://dx.doi.org/10.1083/jcb.200910140
- Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 2010; 107:378-83; PMID:19966284; http://dx.doi.org/10.1073/pnas.0911187107
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 2010; 12:119-31; PMID:20098416; http://dx.doi.org/10.1038/ncb2012
- Unoki M, Nakamura Y. Growth-suppressive effects of BPOZ and EGR2, two genes involved in the PTEN signaling pathway. Oncogene 2001; 20:4457-65; PMID:11494141; http://dx.doi.org/10.1038/sj.onc.1204608
- Mills RD, Sim CH, Mok SS, Mulhern TD, Culvenor JG, Cheng HC. Biochemical aspects of the neuroprotective mechanism of PTEN-induced kinase-1 (PINK1). J Neurochem 2008; 105:18-33; PMID:18221368; http://dx.doi.org/10.1111/j.1471-4159.2008.05249.x
- Lin W, Kang UJ. Structural determinants of PINK1 topology and dual subcellular distribution. BMC Cell Biol 2010; 11:90; PMID:21092208; http://dx.doi.org/10.1186/1471-2121-11-90
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet 2005; 14:3477-92; PMID:16207731; http://dx.doi.org/10.1093/hmg/ddi377
- Zhou C, Huang Y, Shao Y, May J, Prou D, Perier C, Dauer W, Schon EA, Przedborski S. The kinase domain of mitochondrial PINK1 faces the cytoplasm. Proc Natl Acad Sci U S A 2008; 105:12022-7; PMID:18687899; http://dx.doi.org/10.1073/pnas.0802814105
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 2010; 191:933-42; PMID:21115803; http://dx.doi.org/10.1083/jcb.201008084
- Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem 2011; 117:856-67; PMID:21426348; http://dx.doi.org/10.1111/j.1471-4159.2011.07253.x
- Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 2011; 20:867-79; PMID:21138942; http://dx.doi.org/10.1093/hmg/ddq526
- Shi G, Lee JR, Grimes DA, Racacho L, Ye D, Yang H, Ross OA, Farrer M, McQuibban GA, Bulman DE, et al. Functional alteration of PARL contributes to mitochondrial dysregulation in Parkinson disease. Hum Mol Genet 2011; 20:1966-74; PMID:21355049; http://dx.doi.org/10.1093/hmg/ddr077
- Lin W, Kang UJ. Characterization of PINK1 processing, stability, and subcellular localization. J Neurochem 2008; 106:464-74; PMID:18397367; http://dx.doi.org/10.1111/j.1471-4159.2008.05398.x
- Takatori S, Ito G, Iwatsubo T. Cytoplasmic localization and proteasomal degradation of N-terminally cleaved form of PINK1. Neurosci Lett 2008; 430:13-7; PMID:18031932; http://dx.doi.org/10.1016/j.neulet.2007.10.019
- Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy 2013; 9:1758-69; PMID:24121706; http://dx.doi.org/10.4161/auto.24633
- Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 2012; 22:320-33; PMID:22280891; http://dx.doi.org/10.1016/j.devcel.2011.12.014
- Kato H, Lu Q, Rapaport D, Kozjak-Pavlovic V. Tom70 is essential for PINK1 import into mitochondria. PloS one 2013; 8:e58435; PMID:23472196; http://dx.doi.org/10.1371/journal.pone.0058435
- Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman-Ackah SM, Hessa T, Guha R, et al. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 2013; 504:291-5; PMID:24270810; http://dx.doi.org/10.1038/nature12748
- Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 2013; 9:1750-7; PMID:24149988; http://dx.doi.org/10.4161/auto.26122
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 2012; 13:378-85; PMID:22354088; http://dx.doi.org/10.1038/embor.2012.14
- Chan EY, McQuibban GA. The mitochondrial rhomboid protease: its rise from obscurity to the pinnacle of disease-relevant genes. Biochim Biophys Acta 2013; 1828:2916-25; PMID:24099009; http://dx.doi.org/10.1016/j.bbamem.2013.05.012
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3B, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805-12; PMID:15169837; http://dx.doi.org/10.1242/jcs.01131
- Lorenz H, Hailey DW, Wunder C, Lippincott-Schwartz J. The fluorescence protease protection (FPP) assay to determine protein localization and membrane topology. Nat Protoc 2006; 1:276-9; PMID:17406244; http://dx.doi.org/10.1038/nprot.2006.42
- Santos JM, Ferguson DJ, Blackman MJ, Soldati-Favre D. Intramembrane cleavage of AMA1 triggers Toxoplasma to switch from an invasive to a replicative mode. Science 2011; 331:473-7; PMID:21205639; http://dx.doi.org/10.1126/science.1199284
- Fleig L, Bergbold N, Sahasrabudhe P, Geiger B, Kaltak L, Lemberg MK. Ubiquitin-Dependent Intramembrane Rhomboid Protease Promotes ERAD of Membrane Proteins. Mol Cell 2012; 47:558-69; PMID:22795130; http://dx.doi.org/10.1016/j.molcel.2012.06.008
- Lee JR, Urban S, Garvey CF, Freeman M. Regulated intracellular ligand transport and proteolysis control EGF signal activation in Drosophila. Cell 2001; 107:161-71; PMID:11672524; http://dx.doi.org/10.1016/S0092-8674(01)00526-8
- Urban S, Wolfe MS. Reconstitution of intramembrane proteolysis in vitro reveals that pure rhomboid is sufficient for catalysis and specificity. Proc Natl Acad Sci USA 2005; 102:1883-8; PMID:15684070; http://dx.doi.org/10.1073/pnas.0408306102
- Ye Y, Shibata Y, Yun C, Ron D, Rapoport TA. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004; 429:841-7; PMID:15215856; http://dx.doi.org/10.1038/nature02656
- Lemberg MK. Sampling the Membrane: Function of Rhomboid-Family Proteins. Trends Cell Biol 2013; 23:210-7; PMID:23369641; http://dx.doi.org/10.1016/j.tcb.2013.01.002
- Civitarese AE, MacLean PS, Carling S, Kerr-Bayles L, McMillan RP, Pierce A, Becker TC, Moro C, Finlayson J, Lefort N, et al. Regulation of skeletal muscle oxidative capacity and insulin signaling by the mitochondrial rhomboid protease PARL. Cell Metabol 2010; 11:412-26; PMID:20444421; http://dx.doi.org/10.1016/j.cmet.2010.04.004
- Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 2009; 48:2045-52; PMID:19152501; http://dx.doi.org/10.1021/bi8019178
- Sass E, Karniely S, Pines O. Folding of fumarase during mitochondrial import determines its dual targeting in yeast. J Biol Chem 2003; 278:45109-16; PMID:12960177; http://dx.doi.org/10.1074/jbc.M302344200
- Ben-Menachem R, Regev-Rudzki N, Pines O. The aconitase C-terminal domain is an independent dual targeting element. J Mol Biol 2011; 409:113-23; PMID:21440554; http://dx.doi.org/10.1016/j.jmb.2011.03.045
- Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 2012; 3:1016; PMID:22910362; http://dx.doi.org/10.1038/ncomms2016
- Lemberg MK. Intramembrane Proteolysis in Regulated Protein Trafficking. Traffic 2011; 12:1109-18; PMID:21585636; http://dx.doi.org/10.1111/j.1600-0854.2011.01219.x
- Walder K, Kerr-Bayles L, Civitarese A, Jowett J, Curran J, Elliott K, Trevaskis J, Bishara N, Zimmet P, Mandarino L, et al. The mitochondrial rhomboid protease PSARL is a new candidate gene for type 2 diabetes. Diabetologia 2005; 48:459-68; PMID:15729572; http://dx.doi.org/10.1007/s00125-005-1675-9
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; http://dx.doi.org/10.1038/nm.3232
- Weihofen A, Ostaszewski B, Minami Y, Selkoe DJ. Pink1 Parkinson mutations, the Cdc37/Hsp90 chaperones and Parkin all influence the maturation or subcellular distribution of Pink1. Hum Mol Genet 2008; 17:602-16; PMID:18003639; http://dx.doi.org/10.1093/hmg/ddm334
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3B, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/10.1093/emboj/19.21.5720
- Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res 2002; 30:E9; PMID:11788735; http://dx.doi.org/10.1093/nar/30.2.e9
- Rothbauer U, Zolghadr K, Muyldermans S, Schepers A, Cardoso MC, Leonhardt H. A versatile nanotrap for biochemical and functional studies with fluorescent fusion proteins. Mol Cell Proteomics 2008; 7:282-9; http://dx.doi.org/10.1074/mcp.M700342-MCP200