
Abstract
Autophagy is an essential process for eliminating ubiquitinated protein aggregates and dysfunctional organelles. Defective autophagy is associated with various degenerative diseases such as Parkinson disease. Through a genetic screening in Drosophila, we identified CG11148, whose product is orthologous to GIGYF1 (GRB10-interacting GYF protein 1) and GIGYF2 in mammals, as a new autophagy regulator; we hereafter refer to this gene as Gyf. Silencing of Gyf completely suppressed the effect of Atg1-Atg13 activation in stimulating autophagic flux and inducing autophagic eye degeneration. Although Gyf silencing did not affect Atg1-induced Atg13 phosphorylation or Atg6-Pi3K59F (class III PtdIns3K)-dependent Fyve puncta formation, it inhibited formation of Atg13 puncta, suggesting that Gyf controls autophagy through regulating subcellular localization of the Atg1-Atg13 complex. Gyf silencing also inhibited Atg1-Atg13-induced formation of Atg9 puncta, which is accumulated upon active membrane trafficking into autophagosomes. Gyf-null mutants also exhibited substantial defects in developmental or starvation-induced accumulation of autophagosomes and autolysosomes in the larval fat body. Furthermore, heads and thoraxes from Gyf-null adults exhibited strongly reduced expression of autophagosome-associated Atg8a-II compared to wild-type (WT) tissues. The decrease in Atg8a-II was directly correlated with an increased accumulation of ubiquitinated proteins and dysfunctional mitochondria in neuron and muscle, which together led to severe locomotor defects and early mortality. These results suggest that Gyf-mediated autophagy regulation is important for maintaining neuromuscular homeostasis and preventing degenerative pathologies of the tissues. Since human mutations in the GIGYF2 locus were reported to be associated with a type of familial Parkinson disease, the homeostatic role of Gyf-family proteins is likely to be evolutionarily conserved.
Abbreviations
| Atg | = | autophagy-related |
| dsRNA | = | double-stranded RNA |
| 4EHP | = | eukaryotic translation initiation factor 4E-homologus protein |
| GMR | = | glass multiple reporter |
| GRB10 | = | growth factor receptor-bound protein 10 |
| MiMIC | = | Minos-mediated integration cassette |
| PD | = | Parkinson disease |
| PtdIns3P | = | phosphatidylinositol 3-phosphate |
| ROS | = | reactive oxygen species |
| s.p.m. | = | single pair mating |
| TORC1 | = | TOR complex 1 |
| TUNEL | = | terminal deoxynucleotidyl transferase dUTP nick end labeling |
| UAS | = | upstream activator sequence. |
Introduction
Autophagy is a highly conserved homeostatic process essential for bulk degradation of cytoplasmic contents. It is critical for elimination of toxic cytoplasmic inclusions, dysfunctional organelles such as damaged mitochondria,Citation1 excessive nutrient deposits such as lipid droplets,Citation2 and invading microorganisms.Citation3 Defective regulation of autophagy in animal models can result in diverse pathological phenomena including neurodegenerationCitation4 and muscular dystrophyCitation5 that are associated with accumulation of protein aggregates and reactive oxygen species (ROS)-producing dysfunctional mitochondria. In humans, genetic mutations leading to autophagy defects have been reported to be associated with hereditary neurodegenerative diseases such as several types of familial Parkinson disease (PD).Citation6
The molecular basis for autophagy has been explored most thoroughly in yeast, which offers a simple and straightforward genetic model. Genetic screening in yeast has identified many components constituting the core autophagy-controlling machinery named autophagy-related (ATG) genes. Particularly, it has been well established that ATG genes play a central role in autophagy processes as they conjugate ATG12 and ATG8 or MAP1LC3/LC3 onto target proteins and lipids in a mechanism analogous to the ubiquitin conjugating system.Citation7 The conjugation of ATG8 or LC3 into target lipid membranes induces the formation of an autophagosome that will fuse with the lysosome, in which the cargo will become degraded.Citation8 Subsequent biochemical and genetic studies in mammalian cells and animal models revealed that this core machinery is surprisingly well conserved in higher animals as well, including both vertebrates and invertebrates.Citation7
However, unlike unicellular yeast, multicellular animals have developed more sophisticated molecular mechanisms for controlling autophagy, as autophagy needs to respond to a greater variety of physiological inputs such as hormonal regulation and developmental signaling. Furthermore, tissues that are quiescent and have a low cell turnover rate such as neurons and muscle cells will need to maintain their intracellular homeostasis by removing unnecessary and toxic cell constituents through autophagy. Indeed, a recent genetic screening in C. elegans and Drosophila identified several autophagy components that are not present in yeast cells, demonstrating the existence of metazoan-specific autophagy components.Citation9,10 Still, the metazoan-specific autophagy pathway awaits more rigorous investigation.
To discover additional autophagy-controlling genetic components, we conducted our own genetic screening in Drosophila, from which we isolated Gyf/CG11148, whose human ortholog is mutated in a type of familial PD, as a new autophagy-regulating gene. We showed that Gyf is essential for starvation-induced and developmental autophagy, as well as for physiological autophagy which is critical for eliminating protein aggregates and dysfunctional organelles. Correspondingly, null mutants of Gyf exhibited a dramatically reduced life span and early impairment of mobility, which is associated with neurodegeneration, muscle degeneration, and accumulation of ubiquitinated proteins and dysfunctional mitochondria in the tissues. Taken together, our results establish Gyf as a new mediator of autophagy that protects tissue homeostasis and further prevents development of diverse degenerative pathologies in Drosophila.
Results
Identification of Gyf/CG11148 as a new autophagy-regulating gene
In order to discover new autophagy regulators, we constructed a Drosophila line that stably expresses both Atg1 and Atg13 specifically in the eye using the upstream activator sequence (UAS)-Atg1 and UAS-Atg13 transgenes and a glass multiple reporter (GMR)-Gal4 driver. Simultaneous transgenic expression of Atg1 and Atg13 provoked excessive autophagy in the developing eye, resulting in easily noticeable eye degeneration (), as formerly reported.Citation11 We conducted a genetic screening by crossing the stable GMR>Atg1+Atg13 line (the fly line with UAS-Atg1 and UAS-Atg13 transgenes whose expression is driven by GMR-Gal4) with double-stranded RNA (dsRNA) transgenic Drosophila lines obtained from the established libraries of dsRNA transgenic fliesCitation12,13 and searching for dsRNA lines that can suppress the phenotype of the GMR>Atg1+Atg13 transgene. A dsRNA line that targets an unnamed gene temporarily annotated as CG11148 was identified to completely suppress the eye-degeneration phenotype of the GMR>Atg1+Atg13 flies (). The level of suppression by CG11148 silencing was even comparable to the level conferred by silencing of Atg1 itself (). Silencing of CG11148 alone did not affect eye development and morphology, similar to other autophagy regulators such as Atg1 ().
Figure 1. Isolation of Gyf/CG11148 as a genetic modifier of the Atg1-Atg13-gain-of-function eye phenotype. (A and B) Eyes from the flies expressing indicated transgenes were imaged using light dissection microscopy. (C) Schematic representation of the comparison among Drosophila and human Gyf orthologs. Amino acid sequence similarity is displayed as a percentage. (D) Phylogenic tree analysis of Gyf family proteins from Drosophila melanogaster (Dme), Caenorhabditis elegans (Cel), Anopheles gambiae (Aga), Apis mellifera (Ame), Danio rerio (Dre), Mus musculus (Mmu), and Homo sapiens (Hsa), constructed by the neighbor-joining algorithm. (E) Expression of Gyf at different developmental stages of Drosophila. Em, embryo; 1st, first instar larva; 2nd, second instar larva; 3rd, wandering-stage third instar larva; P, pupa; F, adult female; M, adult male. Quantitative reverse transcriptase (RT)-PCR was performed, and Gyf mRNA expression was normalized to ribosomal protein 49 (rp49) expression (upper panel, n = 3). Quantification data are represented as means ± standard error. Immunoblot analysis was performed to monitor Gyf (180 kDa) and Actin (40 kDa) expression. (F-I) Expression of Gyf in indicated 1-wk-old adult flies was examined through quantitative RT-PCR (F) and immunoblotting (G). Expression of 4EHP and pico in indicated 1-wk-old adult flies (H) or wandering-stage third instar larvae (I) was examined through quantitative RT-PCR. Tub>picodsRNA flies cannot develop into adulthood. Quantification data are represented as means ± standard error (n = 3). P values were calculated using the Student t test. *, P < 0.05; **, P < 0.01; NS, not significant. Immunoblot analysis was performed to monitor Gyf (180 kDa) and Actin (40 kDa) expression. (J) Eyes from the flies expressing the indicated transgenes were imaged using light dissection microscopy.
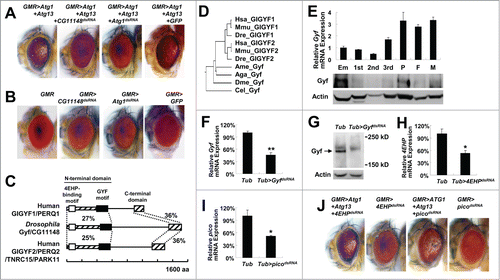
A sequence database search identified the protein product of CG11148 as an ortholog of mammalian proteins named GIGYF1 (GRB10 interacting GYF protein 1) and GIGYF2. GIGYF2 is also known as KIAA0642, PERQ2, PERQ3, TNRC15, and PARK11.Citation14 Considering that the protein product of CG11148 is orthologous to both GIGYF1 and GIGYF2, we named this newly discovered gene Drosophila Gigyf (hereafter Gyf). Strong primary sequence homology (approximately 30% similarity) was detected at both terminal regions of the protein, while the central region did not show any significant similarity between the mammalian and Drosophila orthologs (). We subsequently designated the 2 homologous terminal regions as the N-terminal and C-terminal domain, respectively. The N-terminal domain of mammalian GIGYF2 contains 2 important protein-protein interaction motifs, namely EIF4E2/4EHP (eukaryotic translation initiation factor 4E-homologous protein)-binding motifCitation15 and GRB10 (growth factor receptor-bound protein 10)-binding GYF motif.Citation16 GIGYF2 can regulate protein translation and insulin signal transduction through these 2 motifs.Citation15,16 Such motifs are conserved in Drosophila Gyf as well (). Gyf-family proteins are found in most metazoan organisms (), but no specific orthologs exist in fungi and plants. Expression of Drosophila Gyf mRNA and Gyf protein is detected throughout all developmental stages, although the expression was relatively low during larval development (). Unlike Gyf whose silencing () can strongly suppress the eye phenotype of Atg1-Atg13 overexpression (), silencing of 4EHP () or pico/Grb10 () did not suppress the Atg1-Atg13 effect (), suggesting that Gyf may control autophagy independently of these formerly known interacting partners. In addition, although mammalian GIGYF2 was shown to modulate insulin signaling,Citation16,17 the Gyf silencing construct did not produce any genetic interaction with insulin-target of rapamycin (TOR) signaling components in the eye system (Fig. S1).
Gyf silencing does not inhibit Atg1-Atg13 expression and activity
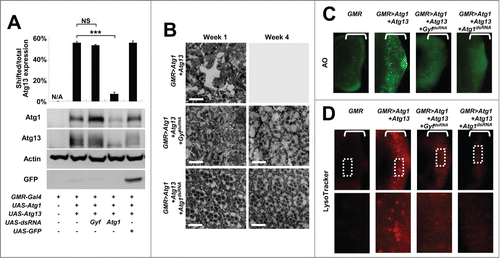
We further characterized the phenotypic interaction between Atg1-Atg13 activation and Gyf silencing through biochemical and histological methods. First, we checked the molecular status of overexpressed Atg1 and Atg13 in eye protein lysates. Gyf silencing did not reduce transgenic Atg1 expression; rather it strongly increased the Atg1 level by more than 2-fold in comparison to the control (). It is formerly reported that, under the conditions of autophagy inhibition, there is compensatory upregulation of Atg1 protein expression.Citation11,18 Thus, it is possible that Gyf silencing inhibited autophagy. In contrast, Atg1 silencing (positive control) strongly reduced the Atg1 expression level as expected (). The amount of Atg13 expression was not altered by silencing of either Atg1 or Gyf. However, the electromobility retardation (gel shift) of Atg13 was diminished by Atg1 silencing () since the gel shift reflects the Atg1-mediated phosphorylation of Atg13.Citation11 The Atg13 gel shift is maintained after Gyf silencing (), suggesting that Gyf silencing does not inhibit the catalytic activity of the Atg1 kinase.
Figure 2. Gyf silencing suppresses Atg1-Atg13-induced eye degeneration and ectopic cell death. (A) Fly heads expressing indicated transgenic elements were subjected to immunoblotting to monitor the level of myc-Atg1 (Atg1, 120 kDa), Atg13-GFP (Atg13, 80 kDa), Actin (40 kDa), and GFP (25 kDa) expression. The level of Atg13 shift was quantified by densitometry of total and shifted Atg13 expression. Quantification data are represented as means ± standard error of the shifted/total Atg13 protein ratios (n=10). P values were calculated using the Student t test. *** P < 0.001; NS, not significant. (B) Eyes from the flies expressing indicated transgenes were analyzed by electron microscopy to visualize photoreceptor structures. Scale bars: 10 μm. (C and D) Developing eye discs from wandering-stage third instar larvae expressing indicated transgenes were analyzed by acridine orange (AO) staining (C) and LysoTracker Red staining (D) to visualize dying cells and autolysosomes, respectively. Brackets indicate the areas of differentiated ommatidia that express GMR-Gal4. Boxed regions in the top panel of (D) are magnified in the bottom panel to visualize the size of LysoTracker Red-positive vesicles.
Gyf silencing does not completely suppress morphogenetic abnormalities of ommatidia caused by Atg1-Atg13 activation
We then examined the histological morphology of Atg1-Atg13-overexpressing and Gyf-silenced adult eyes. Overexpression of Atg1-Atg13 induced pronounced degeneration of retinal cells such as photoreceptors and cone cells, in addition to a complete disarray of the ommatidia structure and accumulation of membrane-dense vacuoles (). After 4 wk, the retinal structure was no longer detectable inside the eye; the ommatidial structure was completely degenerated except for the corneal lens exocuticle (). The silencing of Atg1 completely prevented eye degeneration and restored ommatidial morphology at either one or 4 wk (). Gyf silencing also dramatically suppressed an Atg1-Atg13-induced progressive eye degeneration phenotype; however, it failed to fully restore the photoreceptor morphology (). It has been formerly shown that Atg1 regulates neuronal development independent of its autophagy-regulating activities.Citation18-21 Therefore, it is possible that, although Gyf is required for Atg1-Atg13-induced autophagic eye degeneration, it does not play a critical role in regulating Atg1's neuronal morphogenesis-regulating activities.
Atg1-Atg13-induced ectopic cell death is suppressed by Gyf silencing
To decipher the underlying basis of Atg1-Atg13-induced eye degeneration and Gyf-silencing-mediated suppression of the degenerative phenotype, we examined the cell biological effects of Atg1-Atg13 and Gyf in developing eye discs. Because the GMR-Gal4 driver is only expressed in differentiated ommatidia, Atg1-Atg13 overexpression and Gyf silencing occurred only posterior to the morphogenetic furrow (brackets in ). As formerly reported, Atg1-Atg13 overexpression induced prominent cell death in differentiated ommatidia (), monitored by acridine orange (AO) staining. However, the ectopic cell death was completely suppressed by silencing of Gyf (), demonstrating that Gyf is necessary for the Atg1–Atg13 signaling module to induce ectopic cell death. Atg1–Atg13-induced expansion of acidic compartments, which was visualized by the LysoTracker Red reagent to represent active autophagy, was also substantially reduced in size by silencing of Gyf (), supporting the idea that Gyf is an autophagy controller downstream of, or in parallel with, Atg1-Atg13.
Atg1–Atg13-induced autophagic flux is inhibited by Gyf silencing
We directly measured the autophagic activity of developing eye disc using an autophagic flux marker GFP-mCherry-Atg8a ().Citation22 Due to the intrinsic nature of fluorescence proteins, mCherry matures, fluoresces, and fades slower than GFP ().Citation23,24 Because mCherry-GFP is conjugated with Atg8a which becomes integrated into autophagosomes, autophagy can induce specific loss of GFP fluorescence; upon fusion with lysosomes, exposure to an acidic environment denatures GFP and quickly diminishes its fluorescence, while mCherry fluorescence can be maintained even in the acidic condition. Therefore, in conditions of active autophagy, GFP fluorescence is dramatically suppressed while mCherry fluorescence persists ().
Figure 3. Gyf silencing interferes with Atg1-Atg13-induced autophagic flux. (A) Schematic representation of the GFP-mCherry-Atg8a autophagic flux reporter. (B) Due to the intrinsic nature of fluorescence proteins, GFP matures, fluoresces, and fades faster than mCherry. (C) Incorporation of GFP-mCherry-Atg8a into autolysosomes diminishes the fluorescence of GFP, but not that of mCherry. (D) Developing eye discs from wandering-stage third instar larvae expressing indicated transgenes and the GFP-mCherry-Atg8a reporter were observed using laser confocal microscopy. Brackets indicate the areas of differentiated ommatidia. (E–H) GFP and mCherry fluorescence intensities were measured from individual ommatidia expressing indicated transgenes and plotted into a graph against estimated time (h) after expression (n ≥ 15 in each time period) (E–G). GFP/mCherry fluorescence ratio at each time period was calculated (H). Data are represented as means ± standard error.
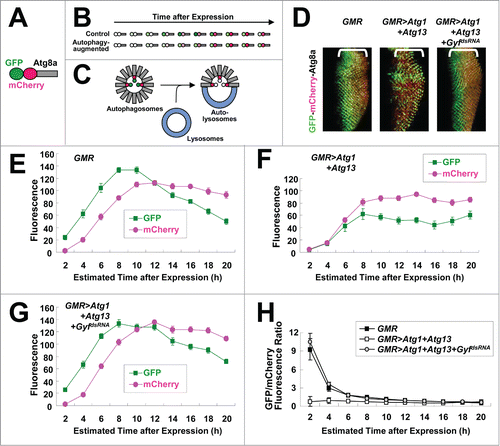
We expressed GFP-mCherry-Atg8a marker in differentiated ommatidia of the developing eye disc using a GMR-Gal4 driver. In the disc, ommatidia differentiation is initiated at the morphogenetic furrow advancing in the anterior direction at the rate of approximately 2 h per column.Citation25 Therefore, by measuring the distance between ommatidia and the morphogenetic furrow, we are able to estimate how long GMR-Gal4 has been expressed in the ommatidia.Citation25 GFP signal was strong in the control eye disc as expected because there is minimal autophagic activity in ommatidia during eye development.Citation26 (). When overexpressed, expression of Atg1-Atg13 in the eye disc strongly interfered with GFP fluorescence emission while it did not affect mCherry fluorescence substantially (), implying that Atg1-Atg13-induced autophagic flux reduced GFP fluorescence by degrading it in autolysosomes. However, silencing of Gyf completely restored GFP fluorescence (), demonstrating that Gyf is required for Atg1-Atg13-induced stimulation of autophagic flux.
Gyf silencing does not interfere with PtdIns3P accumulation
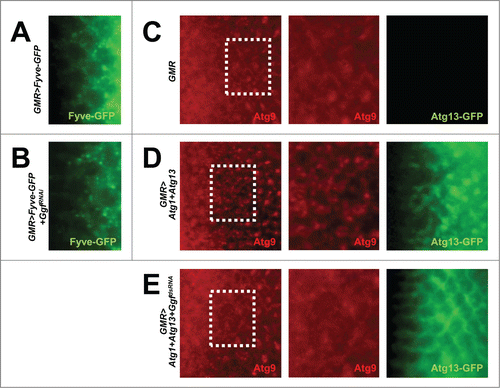
It has been formerly shown that GIGYF2, the mammalian ortholog of Gyf, is present in endosomal compartments.Citation27 Because the activity of Atg6-Pi3K59F (class III PtdIns3K) is important for both endosomal trafficking and autophagosome formation, we tested if Gyf is necessary for the proper functionality of Atg6-Pi3K59F, which produces phosphatidylinositol 3-phosphate (PtdIns3P). The PtdIns3P level inside the tissue was monitored by Fyve-GFP, which has a PtdIns3P-binding domain conjugated with a GFP fluorophore.Citation28 Gyf silencing in the eye disc does not reduce the level of Fyve-GFP puncta ( and Fig. S2A), suggesting that Gyf is not essential for the gross activity of Atg6-Pi3K59F in Drosophila eyes.
Figure 4. Gyf silencing does not interfere with the Atg6-Pi3K59F activity but inhibits Atg1-Atg13-induced Atg9 trafficking. (A and B) Developing eye discs from wandering-stage third instar larvae expressing indicated transgenes were observed under a fluorescence microscope to visualize Fyve-conjugated GFP (green) that monitors PtdIns3P. (C–E) Developing eye discs from wandering-stage third instar larvae expressing indicated transgenes were analyzed by anti-Atg9 (red) staining, which monitors trafficking of endosomes to autophagosomes. Boxed areas in the left-most panels are magnified in the middle panels. The right-most panels display fluorescence of Atg13-conjugated GFP (green) in the region corresponding to the left-most panel.
Gyf silencing inhibits Atg1-Atg13-induced Atg9 trafficking
During active autophagy in mammalian cells, endosomally localized Atg1-Atg13 induces trafficking of Atg9-labeled endosomal membranes to autophagosomes.Citation29,30 Indeed, overexpression of Atg1-Atg13 in the developing eye disc induced formation of Atg9-positive puncta (), which are known to be accumulated during active autophagy.Citation31,32 We tested the requirement of Gyf in this process. Silencing of Gyf strongly abrogated the Atg9 puncta formation ( and Fig. S2B), suggesting that Gyf is required for Atg1-Atg13-induced trafficking of the endosomal membrane and Atg9. Interestingly, Atg13 puncta formation, which is observed in Atg1-Atg13-overexpressing eye disc, was also completely prevented by silencing of Gyf ( and Fig. S2C). This result suggests that Gyf is required for proper subcellular localization of the Atg1-Atg13 complex, which is required for instigation of active Atg9 trafficking and autophagosome formation.
Gyf silencing does not interfere with classical apoptosis
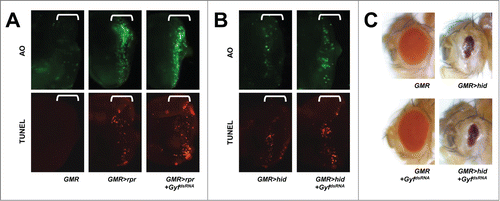
Because Gyf silencing inhibited Atg1-Atg13-induced cell death and eye degeneration, we speculated on whether Gyf can also serve as a general inhibitor of cell death such as apoptosis. Rpr/Reaper and Hid proteins in Drosophila antagonize the inhibitor of apoptosis (Thread/dIAP1) proteins, thereby inducing activation of caspase cascades that lead to apoptotic cell death.Citation33 Eye-specific overexpression of either of these proteins induced significant cell death, as monitored by acridine orange (AO) or terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining, in the developing eye disc (), and reduced eye size in adults (). These apoptotic phenotypes were not suppressed at all by Gyf silencing (), suggesting that Gyf specifically regulates the autophagic signaling pathway.
Figure 5. Gyf silencing does not interfere with apoptotic cell death. (A and B) Developing eye discs from wandering-stage third instar larvae expressing indicated transgenes were analyzed by acridine orange (AO, upper panels) and TUNEL (lower panels) staining to visualize apoptotic cells. Brackets indicate the areas of differentiated ommatidia. (C) Eyes from the flies expressing indicated transgenes were imaged using light dissection microscopy. GMR>rpr flies cannot develop into adulthood.
Identification and characterization of Gyf-null mutant flies
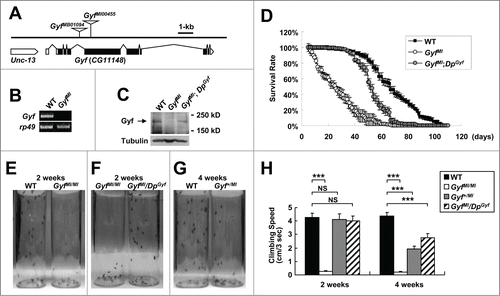
To further study the physiological function of the Gyf gene, we isolated Gyf-null mutant flies. A FlyBase search identified 2 Minos transposon insertions, GyfMB01094 and GyfMI00455 (designated as GyfMB and GyfMI), in the fourth intron and fifth exon of the Gyf gene respectively (). These mutants were formerly generated by the large-scale Drosophila gene disruption projects.Citation34,35 Although the GyfMB mutation did not affect the expression of Gyf mRNA (data not shown), the GyfMI mutation, which disrupts the coding sequence of the Gyf gene, completely prevented Gyf mRNA () and Gyf protein () expression. Thus, we characterized GyfMI as the null mutant of Gyf gene.
Figure 6. Gyf mutant flies exhibit shortened life span and mobility defects. (A) Schematic genomic organization of the Gyf (CG11148) locus and Gyf mutants. Triangles, transposon insertions; open boxes, untranslated exons; closed boxes, protein-coding exons. Scale bar, relative length of 1-kb genomic span. (B and C) Absence of Gyf expression in Gyf-null flies (GyfMI). RT-PCR of Gyf and rp49 (B) and immunoblotting of Gyf (180 kDa) and Tubulin (50 kDa) (C) were conducted from the flies of indicated genotypes. (D) Survivorship of WT (w1118 control), Gyf-null mutant (GyfMI) and Gyf-null mutant with Gyf genomic rescue (GyfMI; DpGyf) male flies (n ≥ 180). Survival of 4 independent cohorts is presented as mean ± standard error. (E–G) Photographs of the vials containing 2- or 4-wk-old adult male flies of indicated genotypes taken at 3 sec after negative geotaxis induction. (H) Quantification of the climbing speeds of indicated 2- or 4-wk-old adult male flies (n ≥ 80). Climbing speed is presented as mean ± standard error. P values were calculated using the Student t test. *** P < 0.001; NS, not significant.
Gyf-null mutants are short lived
GyfMI mutants developed into adulthood well without any noticeable developmental problems. Because many Drosophila mutants with autophagy defects are short-lived,Citation18,36 we measured longevity of WT control and GyfMI mutants under the standard laboratory condition. Interestingly, GyfMI mutants exhibited a high mortality rate in the early ages of their life, making their life span drastically shorter than WT counterparts (). We then confirmed whether the early onset of mortality was due to the mutation of the Gyf gene. The genomic sequence including and surrounding Gyf gene on the fourth chromosome was cloned into a bacterial artificial chromosome (BAC) vector and transgenically inserted into an independent genomic locus on the third chromosome by phiC31 integrase-mediated recombination. This allele, which was formerly generated by Duplication Consortium,Citation37 was designated as the DpGyf duplication. Using the DpGyf strain, we tested if transgenic expression of Gyf can suppress the longevity defects of GyfMI mutants. DpGyf was able to partially restore Gyf protein expression in GyfMI mutant (). This expression was enough to substantially prevent an early rise in the mortality rate of GyfMI mutant (), suggesting that the absence of Gyf protein is indeed responsible for the observed life-span phenotype.
Gyf-null mutants exhibit mobility defects
We observed that the early mortality rate of GyfMI mutants is associated with a dramatic decline in locomotor ability, as most of the flies were found at the bottom of vials with a largely impaired ability to fly or climb the walls. A negative geotaxis assay confirmed the loss of mobility in GyfMI mutants, which was already prominent at 2 wk of age (). The mobility defect was again substantially restored by DpGyf genomic rescue (). Interestingly, the heterozygotic GyfMI/+ flies and rescued flies showed a slight decrease in climbing ability at 4 wk of age (), suggesting that the reduced expression of Gyf in these flies may have caused a motor deficit in later ages. These results indicate that Gyf is important for preservation of mobility in adult flies.
Gyf-null mutants exhibit autophagy defects
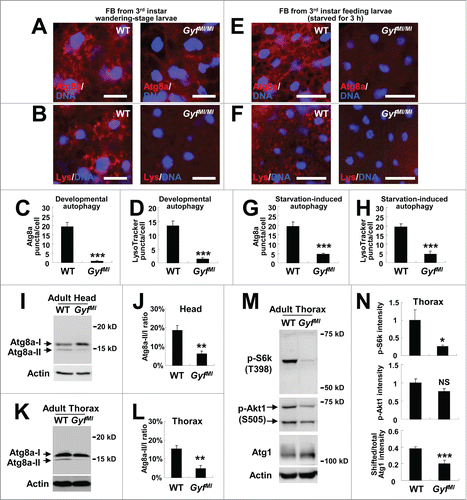
We then returned to the question of whether Gyf is a genuine regulator of autophagy. Because Atg1-Atg13, which showed strong genetic interaction with the Gyf-silencing construct, is essential for developmental and starvation-induced autophagy, we questioned the contribution of Gyf to these processes. As previously reported,Citation36,38 late wandering-stage third instar WT larvae exhibited prominent autophagic activities, such as accumulation of autophagosomes (monitored by Atg8a staining), and acidic vesicles such as autolysosomes (monitored by LysoTracker Red) in their fat bodies (). However, these autophagic activities were strongly abrogated by the Gyf-null mutation (). Gyf-null mutant larvae were also defective in starvation-induced autophagy (). Because the Gyf-null mutant showed degenerative phenotypes at the adult stage, we were also curious if Gyf controls autophagy at the adult stage. To biochemically monitor the autophagic activity in adult tissues, we have utilized an anti-Atg8 antibody, which can detect an endogenous level of Atg8a protein expression in Drosophila (Fig. S3). Head and thorax tissues of WT adults exhibited considerable Atg8a-II expression () which correlates with the basal level of physiological autophagy that is necessary for maintaining tissue homeostasis. However, Gyf-null mutants showed substantial decreases in Atg8a-II expression in both head and thorax tissues (). These results collectively indicate that Gyf is a gene that is critical for developmental, starvation-induced and physiological autophagy processes.
Figure 7. Gyf is essential for developmental, starvation-induced and physiological autophagy. (A–H) Fat bodies (FB) of wandering-stage third instar larvae (A–D) or feeding-stage third instar larvae that were placed on 20% sucrose solution for 3 h (E–H) of WT and GyfMI/MI flies were subjected to anti-Atg8a immunostaining (A, C, E and G) or LysoTracker Red (Lys) staining (B, D, F and H). Hoechst 33258 (DNA) was used to visualize nuclei. Scale bars: 50 μm. Number of Atg8a or LysoTracker Red puncta per cell in fat bodies are presented as means ±standard error (n ≥ 40) (C, D, G and H). (I–L) Heads (I and J) and thoraxes (K and L) of 1-wk-old WT and GyfMI/MI flies were analyzed through immunoblotting of Atg8a-I (16 kDa), Atg8a-II (14 kDa) and Actin (40 kDa). The ratios of Atg8a-II to Atg8a-I are presented as means ± standard error (n = 7) (J and L). (M and N) Thoraxes of 1-wk-old WT and GyfMI/MI flies were analyzed through immunoblotting of phospho-Thr398 S6k (70 kDa), phospho-Ser505 Akt1 (60 and 70 kDa), Atg1 (120 kDa) and Actin (40 kDa). The immunoblot results were quantified and presented as means ± standard error (n = 4). P values were calculated using the Student t-test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant.
Gyf loss downregulates TOR signaling in adult tissues
As briefly mentioned above, GIGYF2, the mammalian ortholog of Gyf, was formerly shown to regulate the insulin-TOR signaling pathway.Citation16,17 Because TOR complex 1 (TORC1) signaling antagonizes autophagy and inhibits Atg8-II processing through downregulation of Atg1/ULK1,Citation39 we were curious whether the organism-level Gyf deficiency affects autophagy through activation of TORC1. Phosphorylation of the TORC1 substrate S6k (RPS6-p70-protein kinase) was, however, dramatically downregulated in Gyf-null mutant tissues (), suggesting that TORC1 signaling was inhibited, not activated, by the Gyf loss. Phosphorylation of a TORC2 substrate Akt1 was also downregulated, but to a less extent when compared to the S6k phosphorylation (). TORC1-induced inhibitory phosphorylation of Atg1, which induces a gel shift of Atg1 in Drosophila,Citation11 was also substantially reduced by Gyf loss (). This TORC1 downregulation could be as an unsuccessful compensatory response to restore autophagy in Gyf-null mutant tissues.
Gyf-null mutants accumulate large amounts of ubiquitinated proteins and dysfunctional mitochondria
Autophagy is important for clearance of ubiquitinated proteins and maintenance of protein homeostasis. Mice and flies with autophagy defects accumulate a large amount of ubiquitinated proteins inside their body, provoking neurodegeneration and muscular dysfunctionCitation4,5 which can together interfere with mobility. As the data indicate that Gyf is important for physiological autophagy in neuron and skeletal muscle tissues (), we inferred that Gyf-null mutants may have defects in eliminating ubiquitinated proteins in these tissues. Indeed, 2-wk-old Gyf-null mutants accumulated a highly elevated amount of ubiquitinated proteins inside the body, which was suppressed by DpGyf genomic rescue ().
Figure 8. Accumulation of ubiquitinated proteins and damaged mitochondria in Gyf mutant flies. (A and B) 2-wk-old adult male flies of indicated genotypes were subjected to immunoblot analyses with ubiquitin and tubulin (50 kDa) antibodies (A). Relative protein level was quantified and presented as mean ±standard error (B). (C and E) Electron micrographs reveal the presence of dysfunctional mitochondria (green arrows) in the brain (C, optic lobe medulla) and the skeletal muscle (E) of 4-wk-old Gyf mutant flies. Boxed areas with normal or damaged mitochondria are magnified in lower (C) or right (E) panels. In the skeletal muscle of Gyf mutant flies, broadened Z-bands (black arrows) were frequently observed. Scale bars: 500 nm. (D, F and G) Quantification data are represented as means ± standard error (n ≥ 3). P values were calculated using the Student t test. **, P < 0.01; ***, P < 0.001; NS, not significant.
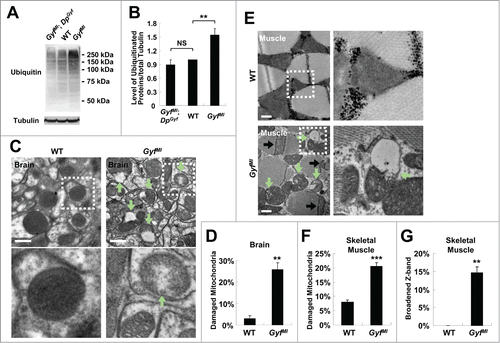
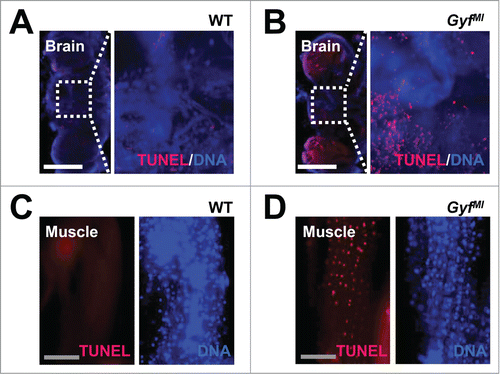
In addition to ubiquitinated proteins, dysfunctional organelles such as damaged mitochondria are physiologically important substrates of autophagy.Citation40 Defective clearance of ROS-producing mitochondria can also facilitate neurodegeneration and muscle degeneration in autophagy-defective flies and mice.Citation5,41-44 Brain (, optic lobe medulla) and skeletal muscle tissues () of Gyf-null mutants exhibited a number of damaged mitochondria, characterized by vacuolization and disorganized cristae structure which are rarely observed in WT control flies. The mitochondrial dysfunctions were also associated with extensive apoptotic cell death in both tissues (). In muscle, the Z band in the sarcomere structure was frequently broadened (), indicative of muscle dysfunction and degeneration.Citation45-47 These results highlight the critical role of Gyf in maintenance of tissue homeostasis that prevents degeneration of neuron and muscle tissues.
Figure 9. Degenerative phenotypes of brain and muscle tissues of Gyf mutant flies. (A–D) TUNEL (red) and DAPI (blue, DNA) staining reveals apoptotic cells in the brain and skeletal muscle of 4-wk-old Gyf mutant flies. Right panel images are corresponding to the boxed areas in the left panel images (A and B). Scale bars: 200 μm (white), 50 μm (gray).
Discussion
Through screening for mediators of Atg1-Atg13-induced autophagy, we have isolated Gyf as a new autophagy-regulating gene. Gyf is required for the Atg1-Atg13 complex to stimulate autophagic flux, autolysosome formation and cell death in the case of excessive autophagy. It is also physiologically essential for developmental and starvation-induced autophagy as well as autophagic elimination of ubiquitinated proteins and dysfunctional mitochondria from neuron and skeletal muscle tissues. This physiological role of Gyf is critical for preventing neurodegeneration and myopathy as well as for preserving life span and mobility.
Although Gyf is required for Atg1-Atg13-induced autophagy, Gyf-null mutant phenotypes are milder than those of Atg1-, Atg13-, or Atg17/Fip200-null mutants, which all fail to develop into viable adult flies.Citation11,18,48,49 Thus, it is likely that Gyf does not mediate all biological functions of the Atg1-Atg13 complex. Consistently, we observed that Gyf silencing did not restore some phenotypic outputs of the Atg1-Atg13 overexpression, such as interference with photoreceptor morphogenesis. Nevertheless, accumulation of autophagy substrates in Gyf-null mutants, such as ubiquitinated proteins and dysfunctional mitochondria, shows that Gyf is indeed required for the physiological autophagy that is dependent on Atg1-Atg13. Accumulation of ubiquitinated proteins was also formerly observed in flies deficient in several autophagy-regulating gene products, such as Atg7,Citation36 Atg8a,Citation50 and Bchs/ALFY.Citation51 However, mitochondrial dysfunction or muscle degeneration was not observed in some of these flies,Citation36 and it has been determined that these components are not necessarily essential for mitochondrial autophagy (mitophagy) that functions to eliminate damaged mitochondria.Citation52-55 On the contrary, the Atg1-Atg13-Atg17 system is essential for mitophagy,Citation52-56 and hypomorphic mutations of Atg1 or Atg17 provoke accumulation of damaged mitochondria,Citation18,44 similar to the currently described Gyf-null mutant phenotypes. Thus, Gyf is a genetic component that is essential for Atg1-Atg13-dependent autophagy of both ubiquitinated proteins and dysfunctional mitochondria in the physiological context.
Although our current study uncovered the genetic function of Gyf in regulating autophagy and neuromuscular homeostasis, the exact biochemical role of Gyf in autophagy process still awaits further investigation. As there is no known catalytic activity or motif in Gyf or its related mammalian orthologs, it is likely that Gyf works with other proteins to regulate autophagy. For example, formerly known interacting partners of Gyf, such as 4EHP and Pico/GRB10,Citation15,16 may play some roles in Gyf-mediated autophagy. However, as these molecules are largely dispensable for Atg1-Atg13-induced autophagy, additional molecules should be also involved in Gyf-regulated autophagy process. One report shows that GIGYF2 in human cells is localized in endosomal compartments.Citation27 As Atg1/ULK1 protein kinase is known to localize into endosomes and endosomes contribute to the growth of the autophagosomal membrane,Citation30 Gyf/GIGYF2 may promote autophagosome formation by regulating Atg1-Atg13-mediated trafficking of endosomal membrane constituents into autophagosomes. Supporting this idea, we have found that Gyf is required for Atg1-Atg13-induced Atg9 puncta formation, which reflects active membrane trafficking to autophagosomes.Citation31,32 In addition, although Gyf does not control Atg1-induced Atg13 phosphorylation, it was necessary for Atg1-induced Atg13 puncta formation in developing eye discs, suggesting that Gyf may control autophagy by regulating subcellular localization of the Atg1-Atg13 protein complex.
It should be noted that mutations in GIGYF2 gene, one of the 2 human orthologs of Gyf, are associated with several cases of familial PD.Citation14 Although GIGYF2/PARK11 mutations are actually excluded from the frequent causes of PD in the general human population,Citation57-60 GIGYF2 may still play important roles in maintaining neuronal homeostasis and preventing neurodegeneration. Gigyf2−/− mice are reported to exhibit perinatal mortality and Gigyf2+/− mice display early onset of age-associated neurodegenerative phenotypes that include neuronal protein inclusion and a significant decline in mobility.Citation17 Our results of Gyf+/− flies exhibiting a slight motor deficit and Gyf−/− flies displaying an early mortality rate and neurodegeneration-associated mobility defects are highly reminiscent of the mouse Gigyf2 and human PD phenotypes.Citation14,17,61 The prominent cell death observed in neuron and muscle tissues of Gyf−/− flies () is most likely due to excessive accumulation of dysfunctional mitochondria and protein aggregates () as a consequence of chronic autophagy downregulation (). Thus, it should be investigated in the future whether mammalian GIGYF1 and GIGYF2 proteins play roles in neuronal autophagy as Drosophila Gyf does. In addition, since many autophagy-defective mouse mutants are lethal at early neonatal stage due to their defects in responding to neonatal starvation,Citation62,63 it would be interesting to investigate later whether perinatal lethality of Gigyf2−/− miceCitation17 is associated with autophagic defects. Based on the current results, it is highly likely that Gyf-family proteins are evolutionarily conserved regulators of neuronal autophagy that is critical for prevention of neurodegenerative phenotypes.
Human mutations in several genes have been implicated as playing a causal role in familial PD. These genes include SNCA/PARK1, PARK2/Parkin, UCHL1/PARK5, PINK1/PARK6, PARK7/DJ-1, LRRK2/PARK8 and ATP13A2/PARK9. It is interesting to note that all of these genes play some roles in autophagic elimination of ubiquitinated proteins or damaged mitochondria.Citation6,53,64,65 Because our current report suggests GIGYF2 as another potential regulator of the autophagy process, autophagy defects that impede cellular protein and oxidative metabolism homeostasis seem to be a general underlying pathogenetic cause of the familial PD syndrome. The genetic role of PARK genes also seems to be evolutionarily conserved because Drosophila strains bearing mutations of SNCA,Citation66 park/Parkin,Citation67,68 Pink1,Citation41,42 Dj-1Citation69-71 and LrrkCitation72-74 genes show neurodegenerative phenotypes similar to those of PD patients, and Gyf-null mutants also exhibited such phenotypes. Therefore, Gyf is among the family of neuroprotective proteins that are important for preventing PD-like degenerative pathologies.
Materials and Methods
Fly strains and culture
Atg8ad4, UAS-myc-Atg1 (UAS-Atg1) and UAS-Atg13-GFP (UAS-Atg13) were gifts from Dr. Thomas Neufeld (University of Minnesota). GMR-Gal4, UAS-Atg1dsRNA, UAS-GyfdsRNA, UAS-4EHPdsRNA, GyfMB01094 (GyfMB), GyfMI00455 (GyfMI) and PBac[y+ w+ Dp(4;3)RC049]VK00033 (DpGyf), UAS-GFP, as well as balancer and WT strains, were obtained from Bloomington Drosophila Stock Center (BDSC). UAS-picodsRNA was obtained from Vienna Drosophila RNAi Center (VDRC, 16369). The flies were reared on standard cornmeal-agar medium with humidity (70%), temperature (25°C or as indicated) and light (12/12 h light/dark cycle) control. For negative geotaxis assays, pictures were taken 3 sec after negative geotaxis induction, and climbing speed was calculated from the pictures.Citation18
Construction of the GMR>Atg1+Atg13 line
UAS-Atg1 and UAS-Atg13 were interbred to generate UAS-Atg1/UAS-Atg13 females, which were then crossed with TM3Sb/TM6B balancer male flies. The male progenies from this cross were mated with GMR-Gal4; TM2/TM6B virgins (˜50 single pair mating (s.p.m.)). GMR-Gal4/+; UAS-Atg1 UAS-Atg13/TM6B flies with noticeable eye degeneration were selected from the s.p.m crosses and interbred. Finally, a stable GMR-Gal4; UAS-Atg1 UAS-Atg13/TM6B line (GMR>Atg1+Atg13) was constructed, and expression of Atg1 and Atg13 was confirmed through immunoblotting. However, we noticed that GMR>Atg1+Atg13 flies exhibited reduced viability and fecundity. Raising the flies at 18°C restored fertility while maintaining the eye phenotypes that allowed us to distinguish the flies; therefore, all genetic experiments using the GMR>Atg1+Atg13 transgene were conducted at 18°C.
Gyf mutants
The Gyf gene locus is located on the fourth chromosome. GyfMB and GyfMI mutant alleles were generated by an insertion of genetically engineered Minos transposons called MiET1Citation34 and MiMIC,Citation35 respectively. The original GyfMB stock was maintained as homozygotes while the GyfMI stock was balanced with In(4)ciD inversion. Because the fourth chromosome does not undergo recombination, this genetic combination is enough to maintain GyfMI allele whose homozygosity produces early mortality and extreme sickness. Due to these defects, we were unable to establish a GyfMI homozygotic fly line. Therefore, all GyfMI homozygotes were collected from the progenies of GyfMI/In(4)ciD intercrosses by absence of ciD marker.
DpGyf allele was generated by the BDSC Duplication Consortium using a formerly described method.Citation37,75 A duplication of 165,448 bp-long sequence from the fourth chromosome (coordinates 4:798,603..964,051, Drosophila melanogaster genome release 5; encompassing Gyf (4:874,342..887,651) and several neighboring genes) derived from the BACR11A04 BAC clone was inserted at the PBac[y+-attP-3B]VK00033 docking site on the third chromosome using phiC31-mediated recombination.
DpGyf was incorporated into GyfMI mutants by means of the following genetic scheme. DpGyf contains y+ and w+ genetic markers. GyfMI contains y+ genetic marker only. w−/Y; DpGyf/TM6Sb males were crossed with y− w−; GyfMI/In(4)ciD females. The resulting y− w−/Y; DpGyf/+; GyfMI/+ males were backcrossed with y− w−; GyfMI/In(4)ciD females. Males with y+ w+ ci+ phenotypes, whose genotype should be either y− w−/Y; DpGyf/+; GyfMI/GyfMI or y− w−/Y; DpGyf/+; +/GyfMI, were collected and bred again with y− w−; GyfMI/In(4)ciD females in s.p.m. crosses. The males that produce progenies with y− w− ciD phenotypes in s.p.m. crosses are judged to have genotype of y− w−/Y; DpGyf/+; GyfMI/+, therefore removed from the study. y+ w+ ci+ male progenies from all the other s.p.m. crosses will be y− w−/Y; DpGyf/+; GyfMI/GyfMI flies (designated as GyfMI; DpGyf) that were found to be fully viable and fertile.
Antibodies
Guinea pig anti-Gyf antibodies were made as follows: a cDNA fragment encoding a.a. 500 to 1000 of Gyf was cloned into pGEX and transformed into E. coli BL21. Insoluble GST-fusion proteins were purified from SDS-PAGE gel bands and injected into guinea pigs (Pocono farms, Inc.). Sera were subjected to affinity purification using PVDF-immobilized proteins. Anti-Atg1 and anti-Atg9 antibodies were formerly described.Citation18,32 Atg13-GFP was detected using anti-GFP antibodies (Santa Cruz Biotechnology, sc-9996; 1:50). Anti-TUBA/α-tubulin (Sigma, T5168; 1:1000), anti-ACTA1/actin (DSHB, JLA20; 1:100), anti-ubiquitin (Santa Cruz Biotechnology, sc-8017; 1:100), anti-phospho Thr398 Drosophila S6k (Cell Signaling Technology, 9209; 1:1000) and anti-phospho-Ser505 Drosophila Akt1 (Cell Signaling Technology, 4054; 1:1000) antibodies were used for immunoblot analyses. Anti-GABARAP (anti-Atg8) antibody (Abcam, ab109364) was used to detect endogenous levels of Drosophila Atg8a in both immunostaining (1:200) and immunoblotting (1:1000) experiments. According to Abcam, this rabbit monoclonal antibody was raised against the evolutionarily conserved region of GABARAP, and therefore can cross-react with Atg8-family proteins from a variety of species including Drosophila and silkworm. In Drosophila, this antibody detected endogenous Atg8a in immunoblotting and immunostaining experiments (Fig. S3).
Histology
Immunostaining,Citation44 LysoTracker Red staining,Citation76 acridine orange staining,Citation77 Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining,Citation78 transmission electron microscopyCitation44 and observation of the GFP-mCherry-Atg8a autophagy flux indicatorCitation22 were done as previously described. Fluorescence images and electron micrographs were quantified using NIH ImageJ software or manually in a blinded manner. To simultaneously conduct autophagy phenotyping and PCR genotyping of Gyf from a single larva, we thoroughly rinsed the larvae in phosphate-buffered saline (PBS; Life Technologies, 14190-144) and dissected them individually in a drop of PBS, using fresh disposable slides and an 18-gauge needle every time. Fat bodies were transferred to a well in a 96-well plate filled with Brower FixCitation44 (for immunostaining)Citation79 or to a well in a 9-well glass plate filled with PBS plus 100 nM LysoTracker Red DND-99 (Invitrogen, L7528) and 20 μg/ml Hoechst 33258 (Sigma, 23491-45-4) (for LysoTracker Red staining),Citation76 and the remaining carcass was transferred to a microcentrifuge tube. For PCR genotyping of larval carcasses, an individual carcass was digested in 100 μL mouse tail digestion buffer (100 mM Tris-HCl, pH 8.5, 200 mM NaCl, 5 mM EDTA, 0.2% SDS [IBI Scientific, IB07060], 0.2 mg/mL proteinase K [Roche, 3115879]) at 65°C overnight, and diluted 2 fold with water. After centrifugation at 15,000 g for 10 min, 1 μL of the supernatant fraction was used for a PCR reaction with relevant primers as described below.
Biochemical analyses
Total RNA was isolated, reverse transcribed, and analyzed using quantitative RT-PCR method using relevant primers as previously described.Citation44 Protein lysate preparation and immunoblotting were performed using a standard cell lysis buffer or the indicated buffers as previously described.Citation44,80 Predicted protein molecular weights were calculated using the ExPASy molecular weight computation tool. Observed protein size was estimated by comparison of the protein band to protein size marker bands (Bio-Rad, 161-0374).
Primer sequences
For genotyping of the Gyf gene, 5′-GACCGAAAAGTAAAGACCTGTC-3′ (forward) and 5′-AGTGCAGCTGTAAACCCGT-3′ (reverse) primers were used. Homozygosity of GyfMI insertion abolishes PCR amplification from the Gyf locus with these primers. Primers detecting rp49 locus, 5′-ACGTTGTGCACCAGGAACTT-3′ (forward) and 5′- CCAGTCGGATCGATATGCTAA-3′ (reverse), were used as a positive control reaction. Quantitative RT-PCR of Gyf and rp49 (normalization control) was also done using the same primers described above. Quantitative RT-PCR of 4EHP and pico was done using 5′- GCGAGTAGAGCGACCACC-3′ (forward) and 5′-GAACAGCGCCACAGGAAC-3′ (reverse), and 5′-AGTGCCACGTCATCAGGTT-3′ (forward) and 5′-CGTCGGC-TGTTTGTCAAGG-3′ (reverse), respectively.
Statistical analysis
A 2-tailed Student t test was used to calculate statistical significance of differences between 2 groups. P values equal to or above 0.05 were judged to be not statistically significant (NS).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
1063766_supplemental_files.zip
Download Zip (144.4 KB)Acknowledgments
We thank Drs. T. Neufeld (UMN), R.J. Wessells, S. Pletcher, R.A. Miller (UM) and G.C. Chen (National Taiwan University), and DSHB (Iowa), DGRC (Indiana), VDRC (Austria), Santa Cruz Biotech. Inc., TRiP (Harvard, GM084947) and Bloomington stock centers (Indiana) for cell lines, fly strains, reagents and access to lab equipment. We thank S. Meshinchi for imaging assistance.
Funding
Work was supported by grants from Ellison Medical Foundation (AG-NS-0932-12) and NIH (R21OD018265, P30AG024824 and P30AG013283).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Yen WL, Klionsky DJ. How to live long and prosper: autophagy, mitochondria, and aging. Physiology 2008; 23:248-62; PMID:18927201; http://dx.doi.org/10.1152/physiol.00013.2008
- Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab 2011; 13:495-504; PMID:21531332; http://dx.doi.org/10.1016/j.cmet.2011.04.004
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/10.1038/nature04723
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507-15; PMID:19945408; http://dx.doi.org/10.1016/j.cmet.2009.10.008
- Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson's disease. Cold Spring Harb Perspect Med 2012; 2:a009357; PMID:22474616; http://dx.doi.org/10.1101/cshperspect.a009357
- Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res 2014; 24:24-41; PMID:24366339; http://dx.doi.org/10.1038/cr.2013.168
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol 2007; 9:1102-9; PMID:17909521; http://dx.doi.org/10.1038/ncb1007-1102
- Tian Y, Li Z, Hu W, Ren H, Tian E, Zhao Y, Lu Q, Huang X, Yang P, Li X, et al. C. elegans screen identifies autophagy genes specific to multicellular organisms. Cell 2010; 141:1042-55; PMID:20550938; http://dx.doi.org/10.1016/j.cell.2010.04.034
- Arsham AM, Neufeld TP. A genetic screen in Drosophila reveals novel cytoprotective functions of the autophagy-lysosome pathway. PLoS One 2009; 4:e6068; PMID:19562034; http://dx.doi.org/10.1371/journal.pone.0006068
- Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell 2009; 20:2004-14; PMID:19225150; http://dx.doi.org/10.1091/mbc.E08-12-1250
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007; 448:151-6; PMID:17625558; http://dx.doi.org/10.1038/nature05954
- Cook KR, Parks AL, Jacobus LM, Kaufman TC, Matthews KA. New research resources at the Bloomington Drosophila Stock Center. Fly (Austin) 2010; 4:88-91; PMID:20160480; http://dx.doi.org/10.4161/fly.4.1.11230
- Lautier C, Goldwurm S, Durr A, Giovannone B, Tsiaras WG, Pezzoli G, Brice A, Smith RJ. Mutations in the GIGYF2 (TNRC15) gene at the PARK11 locus in familial Parkinson disease. Am J Hum Genet 2008; 82:822-33; PMID:18358451; http://dx.doi.org/10.1016/j.ajhg.2008.01.015
- Morita M, Ler LW, Fabian MR, Siddiqui N, Mullin M, Henderson VC, Alain T, Fonseca BD, Karashchuk G, Bennett CF, et al. A novel 4EHP-GIGYF2 translational repressor complex is essential for mammalian development. Mol Cell Biol 2012; 32:3585-93; PMID:22751931; http://dx.doi.org/10.1128/MCB.00455-12
- Giovannone B, Lee E, Laviola L, Giorgino F, Cleveland KA, Smith RJ. Two novel proteins that are linked to insulin-like growth factor (IGF-I) receptors by the Grb10 adapter and modulate IGF-I signaling. J Biol Chem 2003; 278:31564-73; PMID:12771153; http://dx.doi.org/10.1074/jbc.M211572200
- Giovannone B, Tsiaras WG, de la Monte S, Klysik J, Lautier C, Karashchuk G, Goldwurm S, Smith RJ. GIGYF2 gene disruption in mice results in neurodegeneration and altered insulin-like growth factor signaling. Hum Mol Genet 2009; 18:4629-39; PMID:19744960; http://dx.doi.org/10.1093/hmg/ddp430
- Kim M, Park HL, Park HW, Ro SH, Nam SG, Reed JM, Lee JH. Drosophila Fip200 is an essential regulator of autophagy that attenuates both growth and aging. Autophagy 2013; 9:1201-13; PMID:23819996; http://dx.doi.org/10.4161/auto.24811
- Mochizuki H, Toda H, Ando M, Kurusu M, Tomoda T, Furukubo-Tokunaga K. Unc-51/ATG1 controls axonal and dendritic development via kinesin-mediated vesicle transport in the Drosophila brain. PLoS One 2011; 6:e19632; PMID:21589871; http://dx.doi.org/10.1371/journal.pone.0019632
- Toda H, Mochizuki H, Flores R, 3rd, Josowitz R, Krasieva TB, Lamorte VJ, Suzuki E, Gindhart JG, Furukubo-Tokunaga K, Tomoda T. UNC-51/ATG1 kinase regulates axonal transport by mediating motor-cargo assembly. Genes Dev 2008; 22:3292-307; PMID:19056884; http://dx.doi.org/10.1101/gad.1734608
- Ahantarig A, Chadwell LV, Terrazas IB, Garcia CT, Nazarian JJ, Lee HK, Lundell MJ, Cassill JA. Molecular characterization of Pegarn: a Drosophila homolog of UNC-51 kinase. Mol Biol Rep 2009; 36:1311-21; PMID:18636236; http://dx.doi.org/10.1007/s11033-008-9314-4
- Nezis IP, Shravage BV, Sagona AP, Lamark T, Bjorkoy G, Johansen T, Rusten TE, Brech A, Baehrecke EH, Stenmark H. Autophagic degradation of dBruce controls DNA fragmentation in nurse cells during late Drosophila melanogaster oogenesis. J Cell Biol 2010; 190:523-31; PMID:20713604; http://dx.doi.org/10.1083/jcb.201002035
- Khmelinskii A, Keller PJ, Bartosik A, Meurer M, Barry JD, Mardin BR, Kaufmann A, Trautmann S, Wachsmuth M, Pereira G, et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat Biotechnol 2012; 30:708-14; PMID:22729030; http://dx.doi.org/10.1038/nbt.2281
- Hebisch E, Knebel J, Landsberg J, Frey E, Leisner M. High variation of fluorescence protein maturation times in closely related Escherichia coli strains. PLoS One 2013; 8:e75991; PMID:24155882; http://dx.doi.org/10.1371/journal.pone.0075991
- Pollock JA, Ellisman MH, Benzer S. Subcellular localization of transcripts in Drosophila photoreceptor neurons: chaoptic mutants have an aberrant distribution. Genes Dev 1990; 4:806-21; PMID:2143163; http://dx.doi.org/10.1101/gad.4.5.806
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Higashi S, Iseki E, Minegishi M, Togo T, Kabuta T, Wada K. GIGYF2 is present in endosomal compartments in the mammalian brains and enhances IGF-1-induced ERK1/2 activation. J Neurochem 2010; 115:423-37; PMID:20670374; http://dx.doi.org/10.1111/j.1471-4159.2010.06930.x
- Wucherpfennig T, Wilsch-Brauninger M, Gonzalez-Gaitan M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J Cell Biol 2003; 161:609-24; PMID:12743108; http://dx.doi.org/10.1083/jcb.200211087
- Young AR, Chan EY, Hu XW, Kochl R, Crawshaw SG, High S, Hailey DW, Lippincott-Schwartz J, Tooze SA. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci 2006; 119:3888-900; PMID:16940348; http://dx.doi.org/10.1242/jcs.03172
- Longatti A, Lamb CA, Razi M, Yoshimura S, Barr FA, Tooze SA. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol 2012; 197:659-75; PMID:22613832; http://dx.doi.org/10.1083/jcb.201111079
- Tang HW, Liao HM, Peng WH, Lin HR, Chen CH, Chen GC. Atg9 interacts with dTRAF2/TRAF6 to regulate oxidative stress-induced JNK activation and autophagy induction. Dev Cell 2013; 27:489-503; PMID:24268699; http://dx.doi.org/10.1016/j.devcel.2013.10.017
- Nagy P, Hegedus K, Pircs K, Varga A, Juhasz G. Different effects of Atg2 and Atg18 mutations on Atg8a and Atg9 trafficking during starvation in Drosophila. FEBS Lett 2014; 588:408-13; PMID:24374083; http://dx.doi.org/10.1016/j.febslet.2013.12.012
- Steller H. Regulation of apoptosis in Drosophila. Cell Death Differ 2008; 15:1132-8; PMID:18437164; http://dx.doi.org/10.1038/cdd.2008.50
- Metaxakis A, Oehler S, Klinakis A, Savakis C. Minos as a genetic and genomic tool in Drosophila melanogaster. Genetics 2005; 171:571-81; PMID:15972463; http://dx.doi.org/10.1534/genetics.105.041848
- Venken KJ, Schulze KL, Haelterman NA, Pan H, He Y, Evans-Holm M, Carlson JW, Levis RW, Spradling AC, Hoskins RA, et al. MiMIC: a highly versatile transposon insertion resource for engineering Drosophila melanogaster genes. Nat Methods 2011; 8:737-43; PMID:21985007; http://dx.doi.org/10.1038/nmeth.1662
- Juhasz G, Erdi B, Sass M, Neufeld TP. Atg7-dependent autophagy promotes neuronal health, stress tolerance, and longevity but is dispensable for metamorphosis in Drosophila. Genes Dev 2007; 21:3061-6; PMID:18056421; http://dx.doi.org/10.1101/gad.1600707
- Venken KJ, Popodi E, Holtzman SL, Schulze KL, Park S, Carlson JW, Hoskins RA, Bellen HJ, Kaufman TC. A molecularly defined duplication set for the X chromosome of Drosophila melanogaster. Genetics 2010; 186:1111-25; PMID:20876565; http://dx.doi.org/10.1534/genetics.110.121285
- Berry DL, Baehrecke EH. Growth arrest and autophagy are required for salivary gland cell degradation in Drosophila. Cell 2007; 131:1137-48; PMID:18083103; http://dx.doi.org/10.1016/j.cell.2007.10.048
- Cornu M, Albert V, Hall MN. mTOR in aging, metabolism, and cancer. Curr Opin Genet Dev 2013; 23:53-62; PMID:23317514; http://dx.doi.org/10.1016/j.gde.2012.12.005
- Kanki T, Klionsky DJ. The molecular mechanism of mitochondria autophagy in yeast. Mol Microbiol 2010; 75:795-800; PMID:20487284; http://dx.doi.org/10.1111/j.1365-2958.2009.07035.x
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006; 441:1162-6; PMID:16672981; http://dx.doi.org/10.1038/nature04779
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006; 441:1157-61; PMID:16672980; http://dx.doi.org/10.1038/nature04788
- Liang CC, Wang C, Peng X, Gan B, Guan JL. Neural-specific deletion of FIP200 leads to cerebellar degeneration caused by increased neuronal death and axon degeneration. J Biol Chem 2010; 285:3499-509; PMID:19940130; http://dx.doi.org/10.1074/jbc.M109.072389
- Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, et al. Sestrin as a feedback inhibitor of TOR that prevents age-related pathologies. Science 2010; 327:1223-8; PMID:20203043; http://dx.doi.org/10.1126/science.1182228
- Price HM, Gordon GB, Pearson CM, Munsat TL, Blumberg JM. New evidence for excessive accumulation of Z-band material in nemaline myopathy. Proc Natl Acad Sci U S A 1965; 54:1398-406; PMID:5218258; http://dx.doi.org/10.1073/pnas.54.5.1398
- Maron BJ, Ferrans VJ, Roberts WC. Ultrastructural features of degenerated cardiac muscle cells in patients with cardiac hypertrophy. Am J Pathol 1975; 79:387-434; PMID:124533
- Friden J, Sjostrom M, Ekblom B. A morphological study of delayed muscle soreness. Experientia 1981; 37:506-7; PMID:7250326; http://dx.doi.org/10.1007/BF01986165
- Lee SB, Kim S, Lee J, Park J, Lee G, Kim Y, Kim JM, Chung J. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep 2007; 8:360-5; PMID:17347671; http://dx.doi.org/10.1038/sj.embor.7400917
- Scott RC, Juhasz G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol 2007; 17:1-11; PMID:17208179; http://dx.doi.org/10.1016/j.cub.2006.10.053
- Cumming RC, Simonsen A, Finley KD. Quantitative analysis of autophagic activity in Drosophila neural tissues by measuring the turnover rates of pathway substrates. Methods Enzymol 2008; 451:639-51; PMID:19185743
- Simonsen A, Cumming RC, Lindmo K, Galaviz V, Cheng S, Rusten TE, Finley KD. Genetic modifiers of the Drosophila blue cheese gene link defects in lysosomal transport with decreased life span and altered ubiquitinated-protein profiles. Genetics 2007; 176:1283-97; PMID:17435236; http://dx.doi.org/10.1534/genetics.106.065011
- Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 2009; 461:654-8; PMID:19794493; http://dx.doi.org/10.1038/nature08455
- Winklhofer KF. Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol 2014; 24:332-41; PMID:24485851; http://dx.doi.org/10.1016/j.tcb.2014.01.001
- Itakura E, Kishi-Itakura C, Koyama-Honda I, Mizushima N. Structures containing Atg9A and the ULK1 complex independently target depolarized mitochondria at initial stages of Parkin-mediated mitophagy. J Cell Sci 2012; 125:1488-99; PMID:22275429; http://dx.doi.org/10.1242/jcs.094110
- Chang TK, Shravage BV, Hayes SD, Powers CM, Simin RT, Wade Harper J, Baehrecke EH. Uba1 functions in Atg7- and Atg3-independent autophagy. Nat Cell Biol 2013; 15:1067-78; PMID:23873149; http://dx.doi.org/10.1038/ncb2804
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Di Fonzo A, Fabrizio E, Thomas A, Fincati E, Marconi R, Tinazzi M, Breedveld GJ, Simons EJ, Chien HF, Ferreira JJ, et al. GIGYF2 mutations are not a frequent cause of familial Parkinson's disease. Parkinsonism Relat Disord 2009; 15:703-5; PMID:19482505; http://dx.doi.org/10.1016/j.parkreldis.2009.05.001
- Zhang Y, Zheng L, Zhang T, Wang Y, Xiao Q, Fei QZ, Cui PJ, Cao L, Chen SD. GIGYF2 Asn56Ser mutation is rare in Chinese Parkinson's disease patients. Neurosci Lett 2009; 463:172-5; PMID:19638301; http://dx.doi.org/10.1016/j.neulet.2009.07.067
- Sutherland GT, Siebert GA, Newman JR, Silburn PA, Boyle RS, O'Sullivan JD, Mellick GD. Haplotype analysis of the PARK 11 gene, GIGYF2, in sporadic Parkinson's disease. Mov Disord 2009; 24:449-52; PMID:19117363
- Prestel J, Sharma M, Leitner P, Zimprich A, Vaughan JR, Durr A, Bonifati V, De Michele G, Hanagasi HA, Farrer M, et al. PARK11 is not linked with Parkinson's disease in European families. Eur J Hum Genet 2005; 13:193-7; PMID:15523496; http://dx.doi.org/10.1038/sj.ejhg.5201317
- Maraganore DM, de Andrade M, Lesnick TG, Strain KJ, Farrer MJ, Rocca WA, Pant PV, Frazer KA, Cox DR, Ballinger DG. High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet 2005; 77:685-93; PMID:16252231; http://dx.doi.org/10.1086/496902
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; PMID:15866887; http://dx.doi.org/10.1083/jcb.200412022
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/10.1038/nature03029
- Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, Bezard E. Lysosomal impairment in Parkinson's disease. Mov Disord 2013; 28:725-32; PMID:23580333; http://dx.doi.org/10.1002/mds.25462
- Kabuta T, Wada K. Insights into links between familial and sporadic Parkinson's disease: physical relationship between UCH-L1 variants and chaperone-mediated autophagy. Autophagy 2008; 4:827-9; PMID:18635949; http://dx.doi.org/10.4161/auto.6560
- Feany MB, Bender WW. A Drosophila model of Parkinson's disease. Nature 2000; 404:394-8; PMID:10746727; http://dx.doi.org/10.1038/35006074
- Greene JC, Whitworth AJ, Andrews LA, Parker TJ, Pallanck LJ. Genetic and genomic studies of Drosophila parkin mutants implicate oxidative stress and innate immune responses in pathogenesis. Hum Mol Genet 2005; 14:799-811; PMID:15689351; http://dx.doi.org/10.1093/hmg/ddi074
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A 2003; 100:4078-83; PMID:12642658; http://dx.doi.org/10.1073/pnas.0737556100
- Meulener M, Whitworth AJ, Armstrong-Gold CE, Rizzu P, Heutink P, Wes PD, Pallanck LJ, Bonini NM. Drosophila DJ-1 mutants are selectively sensitive to environmental toxins associated with Parkinson's disease. Curr Biol 2005; 15:1572-7; PMID:16139213; http://dx.doi.org/10.1016/j.cub.2005.07.064
- Park J, Kim SY, Cha GH, Lee SB, Kim S, Chung J. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene 2005; 361:133-9; PMID:16203113; http://dx.doi.org/10.1016/j.gene.2005.06.040
- Menzies FM, Yenisetti SC, Min KT. Roles of Drosophila DJ-1 in survival of dopaminergic neurons and oxidative stress. Curr Biol 2005; 15:1578-82; PMID:16139214; http://dx.doi.org/10.1016/j.cub.2005.07.036
- Lee SB, Kim W, Lee S, Chung J. Loss of LRRK2/PARK8 induces degeneration of dopaminergic neurons in Drosophila. Biochem Biophys Res Commun 2007; 358:534-9; PMID:17498648; http://dx.doi.org/10.1016/j.bbrc.2007.04.156
- Liu Z, Wang X, Yu Y, Li X, Wang T, Jiang H, Ren Q, Jiao Y, Sawa A, Moran T, et al. A Drosophila model for LRRK2-linked parkinsonism. Proc Natl Acad Sci U S A 2008; 105:2693-8; PMID:18258746; http://dx.doi.org/10.1073/pnas.0708452105
- Imai Y, Gehrke S, Wang HQ, Takahashi R, Hasegawa K, Oota E, Lu B. Phosphorylation of 4E-BP by LRRK2 affects the maintenance of dopaminergic neurons in Drosophila. EMBO J 2008; 27:2432-43; PMID:18701920; http://dx.doi.org/10.1038/emboj.2008.163
- Venken KJ, Carlson JW, Schulze KL, Pan H, He Y, Spokony R, Wan KH, Koriabine M, de Jong PJ, White KP, et al. Versatile P[acman] BAC libraries for transgenesis studies in Drosophila melanogaster. Nat Methods 2009; 6:431-4; PMID:19465919; http://dx.doi.org/10.1038/nmeth.1331
- Neufeld TP. Genetic manipulation and monitoring of autophagy in Drosophila. Methods Enzymol 2008; 451:653-67; PMID:19185744
- Lee JH, Lee E, Park J, Kim E, Kim J, Chung J. In vivo p53 function is indispensable for DNA damage-induced apoptotic signaling in Drosophila. FEBS Lett 2003; 550:5-10; PMID:12935877; http://dx.doi.org/10.1016/S0014-5793(03)00771-3
- Lee JH, Koh H, Kim M, Park J, Lee SY, Lee S, Chung J. JNK pathway mediates apoptotic cell death induced by tumor suppressor LKB1 in Drosophila. Cell Death Differ 2006; 13:1110-22; PMID:16273080; http://dx.doi.org/10.1038/sj.cdd.4401790
- Hunt LC, Demontis F. Whole-mount immunostaining of Drosophila skeletal muscle. Nat Protoc 2013; 8:2496-501; PMID:24232251; http://dx.doi.org/10.1038/nprot.2013.156
- Park HW, Park H, Semple IA, Jang I, Ro SH, Kim M, Cazares VA, Stuenkel EL, Kim JJ, Kim JS, et al. Pharmacological correction of obesity-induced autophagy arrest using calcium channel blockers. Nat Commun 2014; 5:4834; PMID:25189398; http://dx.doi.org/10.1038/ncomms5834