
Abstract
Autophagy, a lysosomal degradative pathway, is potently stimulated in the myocardium by fasting and is essential for maintaining cardiac function during prolonged starvation. We tested the hypothesis that intermittent fasting protects against myocardial ischemia-reperfusion injury via transcriptional stimulation of the autophagy-lysosome machinery. Adult C57BL/6 mice subjected to 24-h periods of fasting, every other day, for 6 wk were protected from in-vivo ischemia-reperfusion injury on a fed day, with marked reduction in infarct size in both sexes as compared with nonfasted controls. This protection was lost in mice heterozygous null for Lamp2 (coding for lysosomal-associated membrane protein 2), which demonstrate impaired autophagy in response to fasting with accumulation of autophagosomes and SQSTM1, an autophagy substrate, in the heart. In lamp2 null mice, intermittent fasting provoked progressive left ventricular dilation, systolic dysfunction and hypertrophy; worsening cardiomyocyte autophagosome accumulation and lack of protection to ischemia-reperfusion injury, suggesting that intact autophagy-lysosome machinery is essential for myocardial homeostasis during intermittent fasting and consequent ischemic cardioprotection. Fasting and refeeding cycles resulted in transcriptional induction followed by downregulation of autophagy-lysosome genes in the myocardium. This was coupled with fasting-induced nuclear translocation of TFEB (transcription factor EB), a master regulator of autophagy-lysosome machinery; followed by rapid decline in nuclear TFEB levels with refeeding. Endogenous TFEB was essential for attenuation of hypoxia-reoxygenation-induced cell death by repetitive starvation, in neonatal rat cardiomyocytes, in-vitro. Taken together, these data suggest that TFEB-mediated transcriptional priming of the autophagy-lysosome machinery mediates the beneficial effects of fasting-induced autophagy in myocardial ischemia-reperfusion injury.
Abbreviations
| CMA | = | chaperone-mediated autophagy |
| CQ | = | chloroquine |
| GFP | = | green fluorescent protein |
| LAD | = | left anterior descending |
| LAMP2 | = | lysosomal-associated membrane protein 2 |
| MOI | = | multiplicity of infection |
| NRCMs | = | neonatal rat ventricular cardiac myocytes |
| q4D | = | quaque qutra die/every fourth day |
| qOD | = | quaque otra die/every other day |
| TFEB | = | transcription factor EB |
| MAP1LC3B (also abbreviated as LC3) | = | microtubule-associated protein 1 light chain 3, isoform B |
| TTC | = | triphenyl tetrazolium chloride |
| LV | = | left ventricle |
| AAR | = | area at risk |
| WT | = | wild type. |
Introduction
Therapeutic interventions to promote myocardial salvage in ischemia-reperfusion injury remain impractical, as they need to be applied before or simultaneously with onset of injury to achieve clinically significant reduction in cell death. Therefore, ongoing research efforts are focused upon preconditioning the myocardium to prevent cardiomyocyte death and/or dysfunction with myocardial infarction.Citation1 Contemporary approaches to ischemic preconditioning have limited translational application given the unpredictability of spontaneous atherosclerotic coronary plaque rupture and myocardial infarction. It is with this perspective that calorie restriction needs to be explored as a combinatorial strategy for prevention of cardiovascular events and preconditioning the myocardium to attenuate ischemia-reperfusion injury, if and when it were to occur.
Sustained calorie restriction attenuates atherosclerosis in humans;Citation2 markedly reduces the incidence of cardiovascular disease, diabetes and cancer; slows aging in primates;Citation3,4 and prolongs life span in multiple species.Citation5 Autophagy, an intracellular lysosomal degradative pathway, plays a central mechanistic role in the life-span prolongation benefits of calorie restriction;Citation6 and pharmacologic induction of autophagy with rapamycin, resveratrol, and spermidine has similar effects in model organisms as Drosophila melanogaster and Caenorhabditis elegans.Citation7-10 Pertinent to this discussion is the observation that starvation (i.e. total calorie restriction) is a potent stimulus for induction of myocardial macroautophagy (henceforth referred to as ‘autophagy’).Citation11-13 Indeed, autophagy is essential for cardiac homeostasis in the perinatal starvation period at birth, prior to establishment of maternal milk supply; as mice with genetic ablation of autophagy proteins ATG5 and ATG7 cannot form autophagosomes and develop fatal myocardial ischemia.Citation14,15 Autophagy is also essential for maintenance of cardiac structure and function during prolonged starvation in mice, as concomitant impairment of autophagy with genetic ablation of Foxo1, haploinsufficiency of Becn1Citation11 or pharmacological inhibition with bafilomycin A1,Citation16 an inhibitor of lysosome acidification and function, results in rapid development of cardiomyopathy with starvation. Activation of autophagy is also essential for cytoprotection during myocardial ischemia,Citation17 and pharmacologic stimulation of autophagy with rapamycin protects against postinfarction remodeling.Citation18 Myocardial autophagy is also induced with regimens of ischemic preconditioning;Citation19,20 and appears essential for the observed ischemic cytoprotection in ex-vivo ischemia-reperfusion injury.Citation19 Whether calorie restriction-induced autophagy confers ischemic cardioprotection is not known.
To overcome the behavioral challenges of maintaining persistent calorie restriction, periodic calorie restriction has been explored as a strategy to treat obesity;Citation21 and experimental studies have demonstrated life-span prolongation with intermittent fasting to an extent comparable to sustained calorie restriction in rodents.Citation22 Interestingly, intermittent fasting on an alternate day schedule preconditions the myocardium to cardiac ischemic injury with total coronary occlusion; and attenuates postinfarction remodeling.Citation23 In this study, we evaluated whether intermittent fasting stimulates cardiomyocyte autophagy-lysosome machinery as an essential mechanism for the observed cytoprotection against ischemia; and whether similar benefits are observed with reperfusion injury that often coexists with ischemic insult as a result of revascularization and is paradoxically suspected to be worsened by induction of autophagy.Citation17 Our data demonstrate a cyclical induction of autophagy with fasting accompanied by transcriptional induction of autophagy-lysosome machinery; and implicate a critical functional role for the autophagy-lysosome machinery in transducing the beneficial effects of intermittent fasting on ischemia-reperfusion injury.
Results
Intermittent fasting confers cardioprotection to ischemia-reperfusion injury
Previous studies have demonstrated a reduction in infarct size with attenuated post myocardial infarction remodeling and improved survival in rats subjected to every other day intermittent fasting for 3 mo, as compared to ad libitum fed rats followed by chronic total left anterior descending (LAD) coronary artery occlusion to create permanent ischemia.Citation23,24 To investigate whether reduced frequency of fasting or shorter duration of intermittent fasting regimens confer similar cardioprotection; and to determine whether the observed benefits extend to ischemia-reperfusion injury, we subjected adult male wild-type mice to 24 h of fasting on an every other day schedule (qOD; quaque otra die) for 2 and 6 wk; or on an every 4th d schedule (q4D; quaque qutra die) for 6 wk; and provided ad libitum access to standard chow to nonfasted controls; followed by ischemia-reperfusion modeling with reversible LAD ligation performed on a fed day to induce ischemia (30 min) followed by reperfusion (for 23.5 h; see Fig. S1A for experimental scheme). Intermittent fasting did not affect body weight (), as mice commensurately increased their food intake on fed days (), such that average food intake over a fasting-refeeding cycle was not different between any of the groups (), as previously described.Citation25 Mice subjected to 6 wk of qOD intermittent fasting demonstrated a ∼50% reduction in infarct size after ischemia-reperfusion injury, as compared with ad libitum fed controls (); whereas shorter duration qOD regimen (for 2 wk) or a less frequent fasting schedule for the 6 wk duration did not confer similar cardioprotection. We confirmed that the observed reduction in infarct size with 6 wk of intermittent fasting was not related to differences in area at risk, with concomitant Evans Blue staining of nonischemic myocardium, in a separate cohort of mice ().
Figure 1 (See previous page). Intermittent fasting confers protection to ischemia-reperfusion injury in wild-type mice, with upregulation of autophagic flux on fasting days. (A–C) Mean body weight (A), average daily food intake (B) and daily food intake averaged over each fasting-refeeding cycle (C) in adult male C57BL/6 mice subjected to every other day fasting (qOD, n = 21; black triangles and blue dotted line); every 4th day fasting (q4D, n = 5 ; black squares and red dashed line); and nonfasted (ad libitum fed) controls (n = 20 ; open circles and bold black line). (D) Representative images of TTC-stained sections of hearts (at 24 h after surgical modeling) from intermittently fasted (for 6 wk) and nonfasted male mice (as controls) subjected to reversible LAD ligation for 30 min followed by reperfusion on a fed day. (E) Infarct size (at 24 h), quantified as unstained area (white)/total left ventricular myocardial area in male mice subjected to LAD territory ischemia (30 min) followed by reperfusion on a fed day, after intermittent fasting on a q4D schedule (for 6 wk) or qOD schedule for 2 or 6 wk as compared to nonfasted (ad libitum) fed controls (as in A to C). P value depicted is by post-hoc test after one-way ANOVA. (F and G) Representative images of TTC and Evans blue-stained sections of hearts subjected to ischemia-reperfusion (IR) injury, as in (D), on a fed day after 6 wk of intermittent fasting (or ad libitum feeding as control; F), with quantification of area-at-risk (AAR) and infarct size as a percent of AAR (G). P value is by t test. (H, I) Representative gray-scale images (at 630X magnification; H) and quantification of punctate GFP-LC3 (autophagosomes; I) in mice with cardiomyocyte-specific expression of GFP-LC3 after various durations of fasting, refeeding and intermittent fasting (for 6 wk, assessed on a fed day). N = 3 or 4/group. (J) Assessment of autophagic flux by ratio of punctate GFP-tagged LC3 in mice subjected to 24 h of fasting, 6 weeks of intermittent fasting (IF) on a fed-day or provided ad libitum access to food (nonfasted) and treated with chloroquine (CQ) or diluent, 4 h prior to sacrifice (n = 3 or 4/group). (K) Representative immunoblot depicting endogenous LC3 and SQSTM1 in mice treated as in J. (L and M) Assessment of autophagic flux by ratio of LC3-II (L) or SQSTM1 (M) in CQ versus diluent-treated mice, modeled as in (J). N=3 /group. P values depicted are by post-hoc test after one-way ANOVA in I to M.
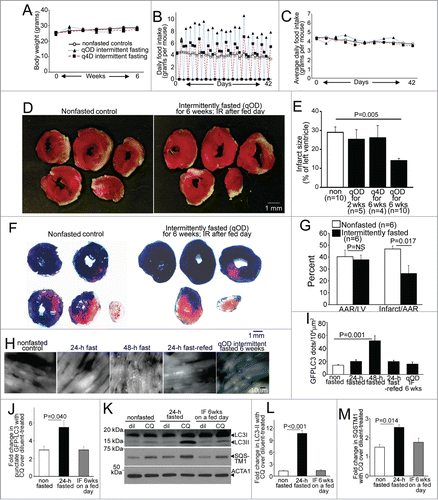
Fasting-refeeding cycles modulate myocardial autophagy
Since fasting stimulates myocardial autophagy,Citation11-13,16 we evaluated cardiomyocyte autophagosome abundance in male mice carrying cardiomyocyte autophagy reporter GFP-tagged MAP1LC3B (green fluorescent protein-tagged microtubule-associated protein 1 light chain 3) transgeneCitation13 subjected to intermittent (qOD) fasting for 6 wk (on a fed day). Mice subjected to 24 and 48 h of fasting, or to 24 h of fasting followed by 24 h of refeeding to mimic a fasting-refeeding cycle; and nonfasted age-matched controls were studied in parallel. Autophagosome abundance was not increased after a 24-h fast; but markedly increased after 48 h of fasting (), which is consistent with previous observationsCitation12,13,17 and suggests that 24 h of fasting induces autophagy with efficient flux (with prompt processing of autophagosomes). Indeed, assessment of autophagic flux by concomitant treatment with CQ (chloroquine) to inhibit lysosome acidification and prevent autophagosome processing;Citation26 and evaluate the ratio of autophagosomes (assessed with punctate GFP-LC3 imaging, ; and autophagosome-bound LC3-II by immunoblotting, ) and abundance of an autophagy substrate SQSTM1/p62 () in CQ-treated over diluent-treated animals, revealed marked enhancement of autophagic flux with 24 h of fasting as compared with basal state in nonfasted mice. Accumulation of autophagosomes after 48 h () likely reflects an imbalance between the rates of autophagosome generation and processing by the lysosomal machinery. Importantly, autophagosome accumulation was not observed after a fed day in the mice subjected to 6 wk of every other fasting, mimicking the observations in mice refed for 24 h after a 24 h fast (); and autophagic flux assessed with concomitant CQ treatment was not increased above the basal state observed in fed mice (). These data indicate that intermittent fasting stimulates cardiomyocyte autophagy in a cyclical fashion with each episode of fasting, with return in autophagic flux to the basal levels on the fed days.
Intermittent fasting-induced protection against ischemia-reperfusion injury is lost in Lamp2 heterozygous null mice, with impaired fasting-induced autophagy
To determine whether the lysosomal degradative machinery is essential for the observed benefits of intermittent fasting, we first studied mice with heterozygous ablation of Lamp2, an X-linked gene which encodes LAMP2 (lysosomal-associated membrane protein 2); loss of which results in impaired autophagosome-lysosome fusion and Danon disease.Citation27 We examined the effects of partial loss of LAMP2 on autophagy in the myocardium, which is a critical lysosomal pathway stimulated in response to starvation,Citation11,14,15 and is impaired in mice with complete loss of LAMP2.Citation28 Female Lamp2 heterozygous null mice (bearing the X/null genotype at the Lamp2 locus), demonstrate no difference in body weight or heart weight () as compared with littermate wild-type females. Interestingly, as compared with littermate wild-type mice, we observed a modest (but statistically nonsignificant) increase in punctate GFP-LC3 () and a significant increase in autophagosome-bound LC3-II (fold change: 2.4± vs.1.0± in wild-type, P < 0.001, N = 5 /group; see ) and SQSTM1 (fold change: 1.3 ± 0 .1 vs. 1.0 ± 0 .1 in wild type, P = 0.038, N = 5 /group, see ), accompanied by a 53% reduction in myocardial LAMP2 protein (fold change: 0.47 ± 0 .09 vs. 1.0 ± 0 .03 in wild-type mice, P = 0 .008, N = 5 /group; ), in the myocardium from 8-wk-old Lamp2 heterozygous null mice. To determine if this was as a result of impaired autophagic flux with Lamp2 deficiency, we treated a cohort of 8-wk-old Lamp2 heterozygous null and littermate wild-type mice with CQ (or diluent) in the fed state and assessed the ratio of autophagosome-bound LC3-II and SQSTM1 in CQ-treated over diluent-treated mice as an index of autophagic flux. As observed in the male mice (), CQ treatment resulted in accumulation of LC3-II and SQSTM1 in wild-type female myocardium indicating intact basal autophagic flux (). Interestingly, autophagic flux was not significantly different in hearts from nonfasted Lamp2 heterozygous null mouse hearts as compared with nonfasted wild-type littermates, suggesting that the reduced levels of LAMP2 in Lamp2 heterozygous null mice were sufficient to maintain basal autophagic flux in the myocardium (). It is conceivable that the observed accumulation of autophagosomes in the fed state in Lamp2 heterozygous null females is due to mosaic loss of Lamp2 allele due to random X chromosome inactivation in cells and tissues.Citation27,29 Importantly, the observed autophagosome accumulation in Lamp2 heterozygous null mouse myocardium did not impair resting cardiac structure and function in these mice in the unstressed state as compared with littermate wild types () confirming previous observations.Citation28,30
Figure 2 (See previous page). Intermittent fasting is associated with impaired autophagic flux in Lamp2 heterozygous null mice. (A) Representative gray-scale images (at 630X magnification) of female Lamp2 heterozygous null and littermate wild-type female mice bearing the GFP-LC3 transgene, after 48 h of fasting or in a fed (nonfasted) state. (B) Quantification of cardiomyocyte GFP-LC3 in Lamp2 heterozygous null mice or littermate control females after fasting and/or refeeding for the indicated duration; n = 3 or 4/group. P values depicted are by post-hoc test after one-way ANOVA. (C) Immunoblots depicting LC3 processing and SQSTM1 accumulation in Lamp2 heterozygous null mice with progressively longer periods of fasting, as compared with nonfasted littermate wild-type female controls. ACTA1 was employed as loading control. (D and E) Representative immunoblots (D) and quantification of ratio of LC3-II (E, top) and SQSTM1 (E, bottom) in cardiac extracts from Lamp2 heterozygous null mice and female littermate wild-type controls subjected to 24 h fasting or provide ad libitum access to diet and treated for CQ or diluent (for 4 h prior to sacrifice). N = 3 /group. P values are by post-hoc test after one-way ANOVA. (F) Representative immunoblots depicting LC3 and SQSTM1 in Lamp2 heterozygous null mice and female littermate wild-type mice subjected to intermittent fasting for 6 wk, and treated for CQ or diluent (for 4 h prior to sacrifice) on a fed day. (G) Representative transmission electron microscopy images of cardiac tissues from Lamp2 heterozygous null mice and female littermate wild-type mice subjected to intermittent fasting, or provided ad libitum access to food for 6 wk, on a fed day. N = 3 /group. Arrows indicate mitochondria with loss of cristal architecture, and arrowheads point to autophagic structures.
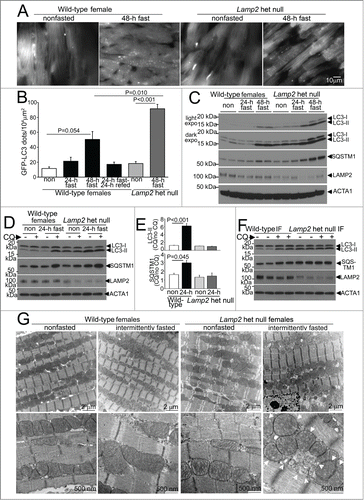
Table 1. Morphometric and echocardiographic characteristics of Lamp2 heterozygous null female mice at 8–10 wk of age, prior to starting intermittent fasting
We next examined whether heterozygous ablation of Lamp2 affects autophagic flux under fasting stress. Indeed, as compared with littermate wild-type females, the accumulation of GFP-LC3-labeled autophagosomes was markedly worsened when Lamp2 heterozygous null females were subjected to 48 h of fasting (); with progressive accumulation of autophagosome-bound LC3-II, SQSTM1 (a receptor protein which is also a substrate for autophagy;Citation31 ) and polyubiquitinated proteins (which accumulate with autophagy impairment;Citation32 Fig. S2) in the Lamp2 heterozygous null myocardium, provoked by increasing duration of fasting. Indeed, while 24 h of fasting significantly upregulated autophagic flux in wild-type female mice as compared to the nonfasted controls (mimicking the observations in male mice, ), fasting did not further stimulate autophagic flux above the basal state in the Lamp2 heterozygous null mice (). These data suggest that the consequences of LAMP2 deficiency (in Lamp2 heterozygous null mice) on global autophagic flux are only unmasked in the presence of a stressor such as fasting, which rapidly stimulates formation of autophagosomes, which are thereafter not processed efficiently, due to impaired autophagosome-lysosome fusion. Therefore, Lamp2 heterozygous null mice are well suited as a model system to evaluate the effects of impaired myocardial lysosome function and consequently impaired autophagy specifically under stress; as they do not display the disadvantages observed in other model systems employed to study the effects of impaired autophagy-lysosomal machinery, namely presence of cardiomyopathy in the unstressed state (as in mice with cardiac myocyte specific or germline ablation of proteins essential for autophagosome formation, ATG5, ATG7, and PIK3C3;Citation14,15,33,34 and in models of lysosomal dysfunction [reviewed in ref. Citation35]); or the paradoxical effects on autophagy observed with haploinsufficiency of BECN1.Citation13,17,36
To determine if LAMP2 deficiency impairs autophagic flux with repetitive fasting, we subjected Lamp2 heterozygous null mice and littermate wild types for 6 wk of intermittent fasting followed by assessment of autophagic flux with injection of CQ (or diluent) on a fed day. Lamp2 heterozygous null mice demonstrated impaired autophagic flux on a fed day after being subjected to the intermittent fasting protocol, assessed as CQ-induced accumulation of LC3-II (fold change in CQ/dil over nonfasted mice: 0.97 ± 0.06 vs.1.87 ± 0.32 in wild types, P = 0.036, N = 4 /group, ) and SQSTM1 (fold change in CQ/dil over nonfasted mice: 0.95 ± 0.16 vs.1.46 ± 0.01 in wild types, P < 0.001, N = 4 /group, ). This was associated with accumulation of autophagic structures (arrowheads, ) and abnormal appearing mitochondria with rarified cristae architechture (arrows) on transmission electron microscopy analysis in Lamp2 heterozygous null mice (and not wild-type controls) subjected to intermittent fasting for 6 wk, on a fed day. At this time point, we also observed impaired left ventricular ejection performance on echocardiographic assessment in intermittently fasted Lamp2 heterozygous null mice (with increased left ventricular end-systolic diameter and reduced % fractional shortening as compared with similarly treated wild type, see ) which is not observed in ad libitum fed Lamp2 heterozygous null females at this age (data not shown). Taken together, these data indicate that intermittent fasting-induced impairment of autophagic flux results in accumulation of abnormal mitochondria, which require intact autophagy-lysosomal machinery for their removal,Citation37 with consequent deterioration of myocardial function.
Table 2. Morphometric and echocardiographic characteristics of Lamp2 heterozygous null female mice subjected to 6 wk of intermittent fasting, on a fed day
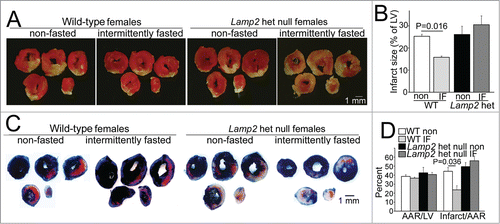
Importantly, intermittent fasting conferred cardioprotection in wild-type female mice, with a ∼50% reduction in infarct size as compared with nonfasted matched controls (), which was not due to differences in area at risk (), paralleling the observations in male mice (); and this cardioprotection was lost in Lamp2 heterozygous null mice (). Taken together, these data indicate that the impairment in autophagic flux with each fasting episode in Lamp2 heterozygous null mice results in accumulation of damaged mitochondria, with impaired basal autophagic flux even on a fed day after 6 wk, likely resulting in the loss of cardioprotection observed with this regimen in wild-type mice.
Figure 3. Intermittent fasting does not confer protection to cardiac ischemia-reperfusion injury in Lamp2 heterozygous null mice. (A and B) Representative images of TTC-stained sections of hearts (A) and infarct size (at 24 h, B) after in vivo LAD territory ischemia (30 min) followed by reperfusion in Lamp2 heterozygous null mice and littermate wild-type (WT) females subjected to intermittent fasting for 6 wk (with surgical modeling on a fed day) or provided ad libitum access to standard chow; n = 4 to 7/group. P values depicted are by post-hoc test after one-way ANOVA. (C and D) Representative images (C) of hearts stained with Evans blue to determine area at risk (AAR, quantified in D) simultaneously with TTC staining to assess infarct size (quantified as fraction of AAR, D) in mice modeled as in A. N = 3 to 6/group. P values depicted are by post-hoc test after one-way ANOVA. LV, left ventricle.
Intermittent fasting induces adverse ventricular remodeling and cardiomyocyte death in lamp2 null mice, associated with impaired autophagic flux
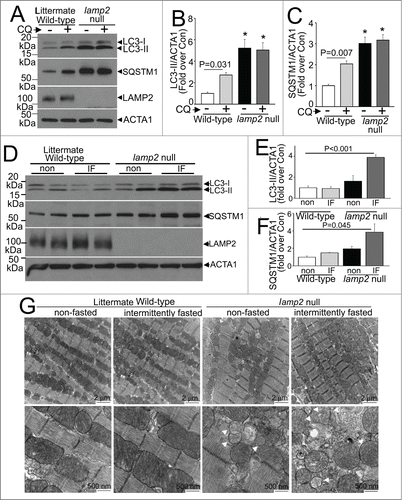
We next examined the effects of intermittent fasting on the myocardium with complete loss of LAMP2. lamp2 null mice demonstrate increased mortality in the neonatal period with autophagosome accumulation in cardiomyocytes and multiple other tissues with multisystem abnormalities;Citation28 and surviving lamp2 null mice demonstrate cardiomyopathy with impaired cardiac and myofiber contractility at ∼6 mo of age,Citation30 mimicking the observations of cardiomyopathy in humans with Danon disease and hypertrophic cardiomyopathy resulting from mutations in the LAMP2 gene and loss of the LAMP2 protein.Citation27 To determine if repetitive stimulation of autophagosome formation with fasting affects the development of cardiomyopathy in the absence of LAMP2, we subjected 8- to 10-wk-old (young adult) male lamp2 null mice and littermates to intermittent fasting, with serial weekly echocardiographic determination of cardiac structure and function, before and during, and immediately after the period of intermittent fasting. At this age, surviving lamp2 null mice demonstrated cardiomyocyte autophagosome accumulation (; Fig. S3), as previously described,Citation28 with accumulation of SQSTM1, an autophagy substrate in the myocardium (). Importantly, assessment of autophagic flux with CQ (or diluent) treatment revealed that basal autophagic flux was impaired in lamp2 null mice in the fed state (in contrast to intact flux in littermate wild types, ), prior to initiation of the intermittent fasting protocol. Intermittent fasting provoked further accumulation of autophagosome-bound LC3-II () and SQSTM1 () in intermittently fasted lamp2 null mice on a fed day, as compared with ad libitum fed lamp2 null mice as well as intermittently fasted wild-type controls (), indicating impaired autophagy resulting from impaired autophagosome-lysosome fusion in the absence of LAMP2. Myocardial ultrastructure examination at this point in time revealed accumulation of autophagosomes in ad libitum fed lamp2 null mice (as compared with wild-type controls in either group; arrowheads, ), which was markedly worsened with appearance of damaged mitochondria (appearing swollen with loss of cristal architecture, arrows) in intermittently fasted lamp2 null mice.
Figure 4 (See previous page). Intermittent fasting (IF) worsens the autophagic impairment observed in lamp2 null mice. (A) Representative immunoblot depicting LC3 processing and SQSTM1 in cardiac extracts from young adult lamp2 null males (and littermate wild-type controls) treated with CQ or diluent to assess autophagic flux. (B and C) Quantification of LC3-II (B) and SQSTM1 (C) in mice treated as in A. N = 4 /group. * indicated P < 0.05 versus diluent treated wild-type control. P values are by one-way ANOVA. (D–F) Representative immunoblots (D) with quantitative analysis of LC3-II (E) and SQSTM1 (F) in lamp2 null mice subjected to intermittent fasting (for 5 wk) as compared with similarly treated littermate controls, and age-matched nonfasted lamp2 null and littermate wild-type mice. N = 4 to 6/group. P values depicted are by post-hoc test after one-way ANOVA. (G) Representative transmission electron microscopy images of cardiac tissues from lamp2 null mice and littermate wild-type mice subjected to intermittent fasting, or provided ad libitum access to food for 5 wk, on a fed day. N = 3 /group. Arrows indicate mitochondria with loss of cristal architecture, and arrowheads point to autophagic structures.
Interestingly, surviving young adult lamp2 null mice did not demonstrate significant echocardiographic changes in cardiac structure and function as compared with controls (), before initiation of the intermittent fasting regimen. Importantly, while intermittent fasting did not affect cardiac structure or function or organ weights in wild-type mice, as compared with ad libitum fed wild-type mice (; ), the repetitive fasting protocol provoked progressive left ventricular dilation (), decline in ejection performance () and progressive increase in left ventricular mass () with increased heart weight; and increased liver and lung weights (suggesting presence of heart failure with congestion, ) in lamp2 null mice after 5 wk, with clinical signs of heart failure (such as hunched posture and labored breathing) observed in some animals. Importantly, these findings were not accompanied by a change in body weight (, ), or spontaneous development of cardiomyopathic changes in ad libitum fed lamp2 null mice followed for the same period (Fig. S4), as compared with similarly treated age-matched wild-type controls. We also observed increased cardiomyocyte cell death, with increased evidence for programmed apoptosis (TUNEL positivity, ) and necrosis (c5b-9 positivity, ) in intermittently fasted lamp2 null mice, as compared with ad libitum fed lamp2 null or wild-type mice from both ad libitum and intermittently fasted groups; as the likely mechanism for the observed adverse ventricular remodeling in these mice. Taken together, these data indicate an essential role for the lysosome machinery in maintaining autophagic flux, cardiomyocyte viability, and cardiac structure and function during intermittent fasting. Not surprisingly, therefore, intermittent fasting did not confer cardioprotection to ischemia-reperfusion injury in the lamp2 null mice (); which was confirmed with markedly increased left ventricular infarct size despite comparable area-at-risk in intermittently fasted lamp2 null mice as compared with similarly treated littermate controls ().
Figure 5 (See previous page). Intermittent fasting provokes worsening cardiomyopathy in lamp2 null mice. (A) Representative 2D-directed M mode echocardiographic images ± from lamp2 null mice and littermate wild-type (WT) mice subjected to intermittent fasting, or provided ad libitum access to food for 5 wk, on a fed day. (B–E) Serial echocardiographic evaluation of LVEDD (left ventricular end-diastolic diameter; B), % fractional shortening (C), left ventricular mass (LVM) (D) and body weight (E) in male lamp2 null (black squares and dotted line) and littermate wild-type males (open circles and solid line) subjected to intermittent fasting (IF); N = 5 to 7/group. ‘*’ indicates P < 0.05 for lamp2 null vs. wild type by post-hoc test after 2-way ANOVA.
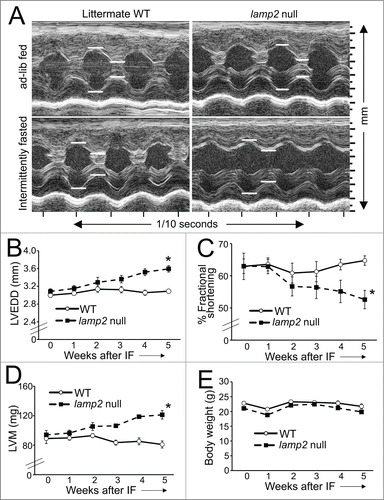
Figure 6. Intermittent fasting provokes cardiomyocyte death in lamp2 null mice and loss of cardioprotection to ischemia-reperfusion injury. (A and B) Representative images of TUNEL-labeled cardiomyocyte nuclei (green, arrowheads; A) with quantification of TUNEL-positive nuclei (blue, DAPI) as percent of total (B; N = 3 to 5 mice/group) in lamp2 null mice or littermate controls subjected to 5 wk of intermittent fasting (IF) or provided ad libitum access to standard chow. ACTA1 staining (red) was employed to delineate cardiomyocytes. P value is by post-hoc analysis after one-way ANOVA. (C and D) Representative images of c5b-9-labeled cardiomyocyte nuclei (green, arrowheads; C) with quantification of C5-C9-positive nuclei (blue, DAPI) as percent of total (D; N = 3 or 4 mice/group) in mice treated as in (A). ACTA1 staining (red) was employed to delineate cardiomyocytes. P value is by post-hoc analysis after one-way ANOVA. (E and F) Representative images of TTC-stained sections of hearts (E) and infarct size (at 24 h; F) after in-vivo LAD territory ischemia (30 min) followed by reperfusion, in lamp2 null mice subjected to intermittent fasting for 5 wk (with surgical modeling on a fed day), nonfasted lamp2 null mice and littermate controls. (G and H) Representative images of TTC and Evans blue-stained sections of hearts subjected to ischemia-reperfusion injury (G) on a fed day after 5 wk of intermittent fasting in lamp2 null mice or wild-type controls, with quantification of area-at-risk and infarct size (H). P value is by t test. LV, left ventricle.
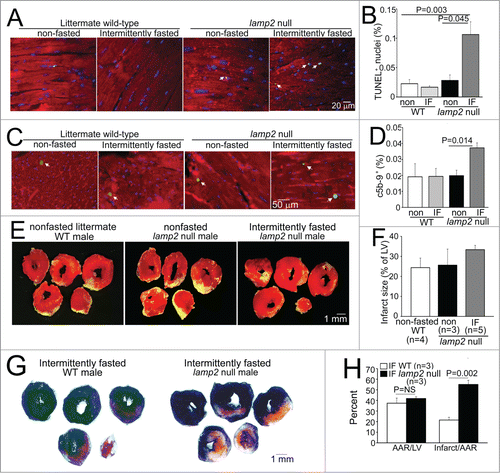
Table 3. Morphometric data on lamp2 null male mice subjected to intermittent fasting for 5 wk
Intermittent fasting modulates oxidative stress in the myocardium
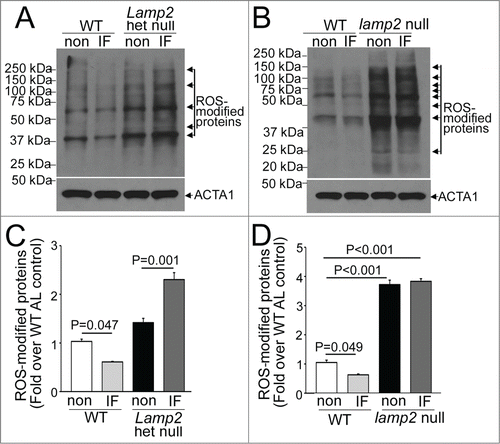
Our observations indicate that whereas autophagic flux was not upregulated on a fed day after a 6-wk period of intermittent fasting (above the basal levels), a remarkable (∼50%) infarct size reduction was observed in wild-type mice subjected to ischemia-reperfusion injury at this time ( and ), indicating the intermittent fasting preconditions the myocardium to the upcoming injury. Since ablation of LAMP2 prevented this preconditioning effect with accumulation of abnormal mitochondria (), and damaged mitochondria provoke increased generation of reactive oxygen species (ROS) in the myocardium,Citation38 we hypothesized that intermittent fasting regulates myocardial ROS generation via effects on mitochondria, which is lost in the setting of Lamp2 ablation due to impairment in mitochondrial autophagy. Indeed, we observed a significant reduction in myocardial levels of oxidatively modified proteins in intermittently fasted wild-type mice of both sexes as compared with ad libitum fed controls (), suggesting that intermittent fasting lowers myocardial ROS levels. Importantly, the abundance of oxidatively modified proteins was markedly increased in heterozygous Lamp2 null as well as lamp2 null myocardium (), with further worsening upon intermittent fasting in the Lamp2 heterozygous null mice (), paralleling the observed impairment in autophagic flux with accumulation of abnormal appearing mitochondria in these models. Taken together, these observations suggest that intermittent fasting preconditions the myocardium via enhanced autophagy of damaged mitochondria,Citation39 with consequent reduction in oxidative stress.
Figure 7. Intermittent fasting (IF) modulates myocardial ROS levels. (A and B) Representative immunoblot demonstrating abundance of ROS-modified (carbonylated) proteins in cardiac extracts from Lamp2 heterozygous null (A) and lamp2 null mice (B), together with respective wild-type controls, on a fed day after intermittent fasting or in nonfasted ad libitum fed controls (non). (C and D) Quantification of prominent bands (marked with arrows) from immunoblots in (A and B), respectively. N = 3 /group. P is by post-hoc analysis after one-way ANOVA.
Chaperone-mediated autophagy (CMA) is another well described lysosomal degradative pathway that is postulated to be impaired in mice with deficiency of LAMP2A isoform.Citation40 This pathway has been shown to be activated in the myocardium by fasting, in vivo, and by oxidative stress.Citation41 To examine its role in intermittent-fasting induced cardioprotection, we assessed the abundance of candidate proteins that are known chaperone-mediated autophagy substratesCitation40 in the myocardium, in these models. Interestingly, our data do not demonstrate any significant alteration in abundance of these proteins with intermittent fasting or Lamp2 ablation (which also results in loss of the LAMP2A isoform;Citation28 see Fig. S5), which suggests that alterations in CMA are unlikely to play a central role in transducing the cardioprotective benefits of intermittent fasting.
Given that autophagy plays an essential role for removal of damaged mitochondria during starvation,Citation42,43 we hypothesized that repetitive enhancement of autophagy would reduce prevalence of depolarized mitochondria; whereby simultaneous inhibition of autophagy would provoke accumulation of depolarized mitochondria. To test this hypothesis, we subjected neonatal rat ventricular cardiac myocytes (NRCMs) to 2 12-h periods of starvation, interspersed with 12 h periods of exposure to a nutrient-rich environment (see schematic in Fig. S1B), in the presence of shRNA targeting Becn1 (to inhibit autophagosome formation), Lamp2 (to inhibit autophagosome-lysosome fusion) and shLacZ as control; and evaluated mitochondrial quality, mass, and ultrastructure. As shown, near-complete knockdown of BECN1 prevented LC3-II generation with accumulation of SQSTM1 (), whereas knockdown of LAMP2 resulted in accumulation of autophagosome-bound LC3-II and SQSTM1 () confirming the inhibition of macroautophagy. Repetitive starvation resulted in significant reduction in prevalence of depolarized mitochondria assessed with JC-1 fluorescence (), without a change in mitochondrial mass () and with preserved mitochondrial ultrastructure (). In contrast, inhibition of autophagy with either BECN1 or LAMP2 knockdown resulted in increased prevalence of depolarized mitochondria; which was significantly worsened in cells subjected to repetitive starvation () consistent with lack of removal of starvation-damaged mitochondria when the autophagy-lysosome machinery was inhibited in this setting. Indeed, mitochondrial mass was also increased with autophagy inhibition (). Also, ultrastructural analysis demonstrated structurally damaged mitochondria, which appeared fragmented with BECN1 knockdown; and swollen with loss of cristal architecture and within autophagosomes with LAMP2 knockdown (). These findings are consistent with a preconditioning effect of repetitive starvation on the cells with improved mitochondrial quality (as observed in vitro, ) and reduced ROS (as observed in vivo, ), in a manner dependent upon presence of an intact autophagy-lysosome machinery.
Figure 8. Repetitive starvation-induced autophagy controls mitochondrial quality. (A) Representative immunoblot (left) and quantification of LC3-II (middle) and SQSTM1 (right) 12 h in the presence of shBecn1 or shLacZ as control (all at multiplicity of infection [MOI] = 10). N = 4/group. P values shown are by t test. (B) Representative immunoblot (left) and quantification of LC3-II (middle) and SQSTM1 (right) 12 h in the presence of shLamp2 or shLacZ as control (all at MOI = 10 ). N = 4 /group. P values shown are by t test. (C) Representative flow cytometric analyses of JC-1 expression in NRCMs subjected to 2 12-h cycles of starvation interspersed with 12 h periods of culture in a nutrient replete medium; or provided continual access to nutrients (as depicted in Fig. S1B), in the presence of shBecn1 (as in A), shLamp2 (as in B) and shLacZ as control. (D) Quantification of cells expressing predominantly depolarized mitochondria (right lower quadrant; N = 4 /group. P values shown are by post-hoc test after one-way ANOVA. (E) Mitochondrial DNA content in cells treated as in A. N = 6 /group. P values shown are by post-hoc test after one-way ANOVA. For (D and E) * indicates P < 0.05 vs. shLacZ, nurtrient replete group; NR, nutrient replete; RS, repetitive starvation. (F) Representative transmission electron micrographs demonstrating mitochondrial ultrastructure in NRCMs treated with shLamp2, shBecn1 or shLacZ as control and subjected to repetitive starvation as in (A), or cultured in nutrient-replete medium in the presence of shLacZ as control. Mitochondrial ultrastructure appears normal in shLacZ cells subjected to nutrient replete and repetitive starvation conditions, whereas mitochondria appear fragmented with shBecn1 (arrowheads) and within autophagosomes with shLamp2 (arrows). Scale bar: 500 nm.
![Figure 8. Repetitive starvation-induced autophagy controls mitochondrial quality. (A) Representative immunoblot (left) and quantification of LC3-II (middle) and SQSTM1 (right) 12 h in the presence of shBecn1 or shLacZ as control (all at multiplicity of infection [MOI] = 10). N = 4/group. P values shown are by t test. (B) Representative immunoblot (left) and quantification of LC3-II (middle) and SQSTM1 (right) 12 h in the presence of shLamp2 or shLacZ as control (all at MOI = 10 ). N = 4 /group. P values shown are by t test. (C) Representative flow cytometric analyses of JC-1 expression in NRCMs subjected to 2 12-h cycles of starvation interspersed with 12 h periods of culture in a nutrient replete medium; or provided continual access to nutrients (as depicted in Fig. S1B), in the presence of shBecn1 (as in A), shLamp2 (as in B) and shLacZ as control. (D) Quantification of cells expressing predominantly depolarized mitochondria (right lower quadrant; N = 4 /group. P values shown are by post-hoc test after one-way ANOVA. (E) Mitochondrial DNA content in cells treated as in A. N = 6 /group. P values shown are by post-hoc test after one-way ANOVA. For (D and E) * indicates P < 0.05 vs. shLacZ, nurtrient replete group; NR, nutrient replete; RS, repetitive starvation. (F) Representative transmission electron micrographs demonstrating mitochondrial ultrastructure in NRCMs treated with shLamp2, shBecn1 or shLacZ as control and subjected to repetitive starvation as in (A), or cultured in nutrient-replete medium in the presence of shLacZ as control. Mitochondrial ultrastructure appears normal in shLacZ cells subjected to nutrient replete and repetitive starvation conditions, whereas mitochondria appear fragmented with shBecn1 (arrowheads) and within autophagosomes with shLamp2 (arrows). Scale bar: 500 nm.](/cms/asset/40953c60-5904-4c07-b6c9-0c5de25ab009/kaup_a_1063768_f0008_c.gif)
Fasting and refeeding transcriptionally modulate the autophagy-lysosome machinery
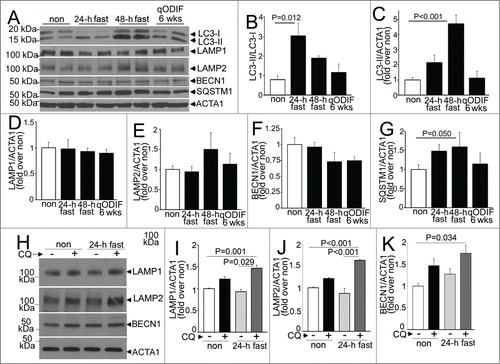
Consumption of lysosomes and components of the autophagy machinery occurs with starvation-induced autophagy; with prompt replenishment observed with continued starvation stress.Citation44 To determine the transcriptional regulation of autophagy-lysosome proteins in response to fasting, in vivo, we performed quantitative PCR analysis for candidate autophagy-lysosome genes. Our data demonstrate a robust transcriptional induction of Map1lc3b, Sqstm1, Lamp2a, and Lamp2b isoforms, with a strong trend toward upregulation of Becn1 and Lamp1 after 24 h of fasting (as compared with nonfasted controls; ). Although, we did not observe transcriptional regulation of Gapdh in the fasted myocardium; we also confirmed the observations with Rpl32 coding for a ribosomal protein, employed as a housekeeping control; and found significant transcriptional induction of Becn1 and Lamp1 too, in myocardium from mice subjected to a 24-h fast as compared with nonfasted controls (Fig. S6A and B). Refeeding fasted mice (for 24 h) restored the expression of these candidate gene transcripts to levels comparable to or lower than those observed in nonfasted mice. Importantly, longer durations of fasting (48 h) or intermittent fasting for 3 or 6 wk did not upregulate the expression of these candidate genes on a fed day; as compared with matched ad libitum fed controls. These data attest to a tight transcriptional regulation of autophagy-lysosome machinery in the myocardium that closely tracks activation of autophagy with fasting and decline to basal levels (or below) with refeeding. We next examined the abundance of proteins coded for by these transcripts. Fasting for 24 h increased the ratio of LC3-II to LC3-I implying induction of autophagy (), without a change in the abundance of any of the proteins evaluated, as compared with the nonfasted state (). Concomitant treatment with CQ revealed significantly increased abundance of total LC3 (1.91 ± 0.26 fold, N = 3 /group, P = 0.028, see ) and SQSTM1 (3.01 ± 0 .fold13-, N = 3 /group, P < 0.001, see ); and that of LAMP1, LAMP2 and BECN1 with 24 h of fasting, as compared with nonfasted diluent-treated controls (), indicating accumulation of these proteins with inhibition of lysosome acidification during fasting. Taken together with the transcriptional upregulation of their cognate genes with 24 h of fasting, the lack of a commensurate increase in the abundance of these autophagy-lysosome proteins in the myocardium (as compared with nonfasted controls), implies ongoing consumption of the autophagy-lysosome machinery components with fasting-induced upregulation of autophagic flux (see ).
Figure 9. Fasting and refeeding result in transcriptional regulation of autophagy-lysosome machinery genes. (A–F) Quantitative PCR analysis of relative abundance of representative autophagy-lysosome machinery gene transcripts, namely Map1lc3b (coding for LC3B; A), Sqstm1 (B), Becn1 (C), Lamp1 (D), Lamp2a (E) and Lamp2b (F) in adult male C57BL/6 mice subjected to 24 h of fasting, fasting for 24 h followed by refeeding for 24 h (fasted-refed), 48 h of fasting; and intermittent fasting for 3 and 6 wk (with samples procured on a fed day). Age-matched nonfasted controls were employed for each group. Control RNA for comparing the 24- and 48-h fasted RNA was prepared from the same nonfasted mouse hearts, and ran separately with each experimental group. N = 4 to 8/group. P value is by t test versus respective controls.
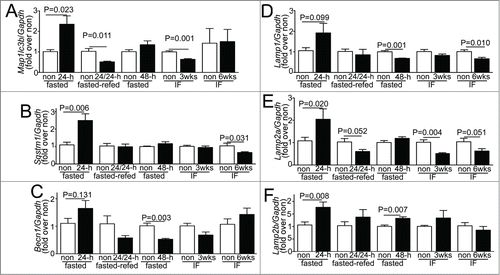
Figure 10. Intermittent fasting induces autophagy without depletion of myocardial autophagy-lysosome machinery proteins. (A–G) Representative immunoblots (A) and quantification of LC3 processing (ratio of LC3-II to LC3-I; B); and abundance of total LC3 (C), LAMP1 (D), LAMP2 (E), BECN1 (F) and SQSTM1 (G) in C57BL/6 male mice subjected to fasting for 24 h, 48 h and intermittently fasted for 6 wk (with samples procured on a nonfasted day) as compared with nonfasted controls. N = 4 /group. P value is by post-hoc test after one-way ANOVA. ACTA1 was employed as loading control. (H–K) Representative immunoblot (H) and quantification of LAMP1 (I), LAMP2 (J) and BECN1 (K) in myocardial extracts from wild-type mice treated with CQ or diluent, concomitantly with 24 h of fasting or with ad libitum feeding (also see ). N = 3 /group. P values are by post-hoc test after one-way ANOVA.
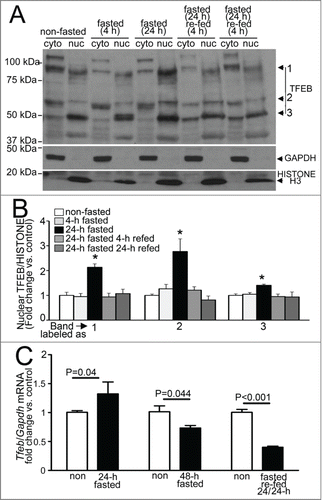
Fasting stimulates activation of TFEB in the myocardium
Recent studies have uncovered a potential mechanism for transcriptional replenishment of the autophagy-lysosome machinery with starvation; and ascribed a central role to the rapid starvation-induced dephosphorylation and cytoplasm to nuclear translocation of TFEB (transcription factor EB),Citation45-48 a basic helix-loop-helix transcription factor of the MiTF family, with resultant upregulation of its target genes.Citation47,49,50 Of note, TFEB is unique in coordinately upregulating the autophagy-lysosome machinery,Citation47 and is expressed in the adult mammalian myocardium.Citation51 We examined whether fasting, in vivo, is a stimulus for activation of TFEB in the myocardium. Given a lack of consensus on the suitability of the available antibodies in identifying the endogenous TFEB protein,Citation46,48,52 we first evaluated the specificity of a polyclonal anti-TFEB antibody employed in recent publications to detect endogenous human TFEB;Citation46 which is predicted to detect murine TFEB based on sequence similarity within the epitope employed in its generation; in a murine atrial myocyte cell line (HL-1 myocytes) and mouse hearts (Fig. S7). The antibody detected a band migrating between molecular mass markers of 50 kDa and 75 kDa in the nuclear fraction (labeled as 2; ), which likely corresponds to murine TFEB protein encoded by transcript variants 2 and 3 (NP_001155194.1, see Fig. S7 for controls); along with 2 other prominent bands close to these molecular mass standards (labeled as 1 and 3, ), which could represent the alternative isoform (NP_035679.3) or posttranslationally modified forms of the protein. To determine whether fasting activates TFEB with cytoplasm to nuclear translocation, in vivo (mirroring the observations in vitro),Citation45,46,48 we examined the relative abundance of endogenous TFEB in nuclear and cytoplasmic fractions of myocardial extracts from fasted and control mice. Fasting for 24 h resulted in marked accumulation of TFEB protein in the nuclear subfraction, with a rapid decline observed after refeeding (). This closely tracks the observed transcriptional changes of the candidate autophagy-lysosome genes (), which are all putative TFEB targets.Citation47,49,50 Commensurate with the observation that TFEB autoregulates its expression,Citation53 transcript levels for Tfeb gene also increased and declined significantly with a fasting-refeeding cycle, as compared with respective controls (). These data suggest a prominent role for TFEB in coordinating the transcriptional response to replenish the lysosomal degradative machinery, with fasting, in the myocardium, in vivo. Interestingly, Tfeb transcripts were significantly upregulated after 3 wk of intermittent fasting, on a fed day (), suggesting a possible role for enhanced TFEB activity in transducing the cardioprotective effects of intermittent fasting. Taken together, these data indicate that TFEB activation and transcriptional upregulation may mechanistically drive the cyclical transcriptional induction of the lysosomal degradative machinery with each fasting-refeeding cycle, facilitating beneficial autophagy.
Figure 11. Fasting induces nuclear translocation of TFEB in the myocardium. (A and B) Representative immunoblot (A) with quantification of nuclear TFEB (B, bands numbers from A) in nuclear and cytoplasmic subfractions from wild-type adult male C57BL/6 mouse hearts subjected to indicated durations of fasting and refeeding. Immunoblotting for GAPDH and Histone H3 was employed to detect enrichment of cytoplasmic and nuclear subfractions, respectively. N = 3 /group. P value is by post-hoc analysis after one-way ANOVA. (C) Quantitative PCR analysis of relative abundance of Tfeb transcript in mice subjected to 24 or 48 h of fasting (with age matched nonfasted controls); fasting for 24 h followed by refeeding for 24 h (fasted-refed). N = 4 to 8/group. P value is by t test vs. control. RNA from a common group of nonfasted mice was employed as control to compare transcript abundance in 24- and 48-h fasted samples.
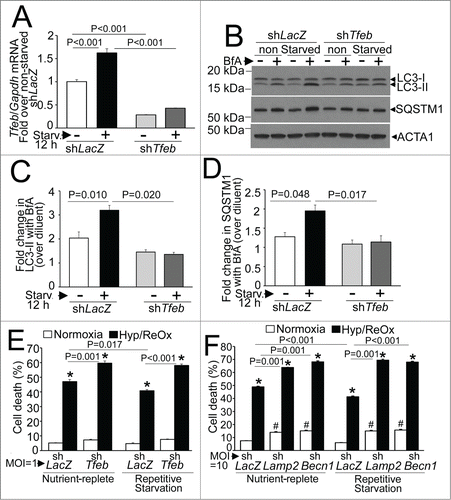
Endogenous TFEB-mediated stimulation of autophagic flux is essential for cytoprotective effects of repetitive starvation on hypoxia-reoxygenation injury
Given that our in vivo observations suggest that fasting induced activation of TFEB and up regulation of autophagic flux may transduce the beneficial effects of intermittent fasting, we examined whether repetitive starvation preconditions NRCMs to protect against hypoxia-reoxygenation (HR) injury, via endogenous TFEB (see schematic in Fig. S1B). Starvation in NRCMs transcriptionally upregulated TFEB expression, indicating its activation, as observed in multiple other cell types,Citation45,46,48,53 in a manner dependent upon endogenous TFEB (). Importantly, knockdown of endogenous TFEB prevented starvation-induced upregulation of autophagic flux, assessed as the ratio of LC3-II () and SQSTM1 () in the presence of bafilomycin A1 (to inhibit lysosomal processing) versus diluent. We next determined whether repetitive starvation would offer cytoprotection to HR-induced cell death.Citation36 Interestingly, 2 cycles of repetitive starvation were associated with a modest (and statistically significant) ∼13% to 15% reduction in HR-induced cell death (). Knockdown of endogenous TFEB also increased HR-induced cell death and completely prevented the cytoprotective effects of preceding repetitive starvation stimuli (). Importantly, the autophagy-lysosome machinery played a critical role in this observation, as the cytoprotection against HR-induced cell death was also lost with concomitant knockdown of LAMP2 to prevent autophagosome processing (as in ) or BECN1 to prevent autophagosome formation (as in ; see ). These data indicate that endogenous TFEB is essential for enhancement of autophagic flux with fasting, which transduces the preconditioning effect of repetitive starvation in cardiac myocytes.
Figure 12. Endogenous TFEB is essential for starvation-induced upregulation of autophagic flux and its cytoprotective preconditioning effect on hypoxia-reoxygenation injury in NRCMs. (A) Tfeb transcripts in NRCMs adenovirally transduced with shRNA targeting rat Tfeb (or shLacZ as control, both at MOI = 1 for 60 h) and subjected to starvation (or cultured in nutrient-rich medium) for 12 h. N = 3 /group. P value is by post-hoc test after one-way ANOVA. (B–D) Representative immunoblot (B) with quantification of LC3-II (C) and SQSTM1 (D) in NRCMs adenovirally transduced with shRNA targeting rat Tfeb (or shLacZ as control, both at MOI = 1 for 60 h) and subjected to starvation (or cultured in nutrient rich medium) for 12 h, in the presence of bafilomycin A1 (or diluent) to inhibit lysosmal acidification. N = 3 /group. P value is by post-hoc test after one-way ANOVA. (E) Cell death in NRCMs adenovirally transduced with shRNA targeting rat Tfeb (or shLacZ as control, all at MOI = 1 ) and subjected to 2 12-h periods of starvation interspersed to 12 h periods of feeding, followed by exposure to hypoxia (6 h) and reoxygenation (18 h) after the final 12 h of feeding, to simulate repetitive fasting conditions, in vitro (schematic in Figure S1B). (F) Cell death in NRCMs adenovirally transduced with shRNA targeting rat Becn1 or Lamp2 (or shLacZ as control, all at MOI = 10 ) and treated as in (E). N = 14 to 16/group; and P values are by post-hoc test after 2-way ANOVA for E, F. ‘*’ indicates P < 0.05 vs. respective normoxia group; and ‘#’ indicates P < 0.05 vs. respective shLacZ-treated group within normoxic nutrient replete and repetitive starvation conditions.
Intermittent fasting stimulates cellular degradative pathways
The benefits of intermittent fasting (or repetitive starvation) on cytoprotection against ischemia-reperfusion (or hypoxia-reoxygenation) are observed in a fed state, while autophagic flux is not upregulated above the basal state. These benefits are not accrued if lysosome function is impaired with heterozygous knockout of Lamp2 or its ablation, in vivo (or knockdown of a master regulator of lysosome function, TFEB, in vitro), resulting in impairment of autophagic flux and accumulation of damaged mitochondria () and upregulation of myocardial oxidative stress () on a fed day. It is conceivable that other lysosomal pathways are mechanistically involved in concert with, or independently of autophagy, in transducing the observed preconditioning effects. To explore potential alternative mechanisms, we examined the myocardial gene expression changes on a fed day in wild-type mice after intermittent fasting, and compared them with similarly modeled lamp2 null mice (as in , , and ; Fig. S4); at a time point immediately predating the observed benefits on cardioprotection at 6 wk after the intermittent fasting regimen (as in ; ). Nonfasted age-matched Lamp2 null and wild type were studied in parallel, in a 2-by-2 design, as controls.
Changes in gene expression with intermittent fasting (vs. nonfasted controls) were uniform within each genotype (wild-type and lamp2 null mice) but markedly divergent between the 2 (), providing an opportunity to dissect the intermittent fasting-induced changes observed only in the wild type mice, but not with concomitant lysosomal dysfunction in lamp2 null mice ( and Table S1; see the nonoverlapping set of 1584 genes). Analysis of functional pathways revealed major changes in genes governing transcription and metabolism (; also see list of genes separated by genotype in supplementary data), with upregulation of intracellular degradative and metabolic processes; and downregulation of cell death pathways () as likely mechanisms for the observed cardioprotective effects of intermittent fasting. Taken together with the effects of intermittent fasting on autophagy, these data raise the intriguing hypothesis that global regulation of lysosome function with intermittent fasting is mechanistically involved in preconditioning the myocardium via effects on organelle quality, metabolism, and cell death.
Figure 13. Intermittent fasting (IF) transcriptionally regulates cellular degradative, metabolic pathways, and cell death pathways. (A) Principal component analysis demonstrating markedly divergent regulation of gene transcription in hearts from lamp2 null and littermate male wild-type mice subjected to intermittent fasting or provided ad libitum access to standard chow (as in ); n = 4 to 6/group. IF stands for intermittent fasting. (B) Venn diagram depicting distribution of differentially regulated genes in wild-type and lamp2 null male mice, as compared with respective age-matched nonfasted controls, as in (A). (C) Pie chart depicting relative distribution of genes by function in intermittently fasted mice (as compared with nonfasted mice) specifically in the subset that does not overlap with genes differentially regulated in similarly treated lamp2 null mice (as in B).
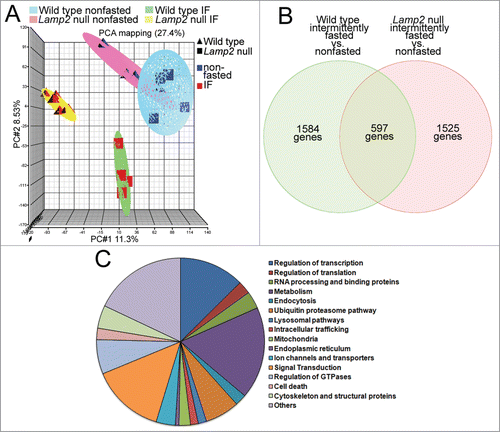
Table 4. List of top 15 GO (Gene Ontology) pathways selectively up- and downregulated only in wild-type mice subjected to intermittent fasting (vs. nonfasted controls) on a fed day
Discussion
Calorie restriction and stimulation of autophagy have salutary effects on life-span extension and cardioprotection. Our study indicates a central role for transcriptional stimulation of the autophagy-lysosome machinery in intermittent fasting-induced protection against ischemia-reperfusion injury based on the following observations. First, intermittent fasting preconditions the myocardium with a ∼50% reduction in infarct size with experimental ischemia-reperfusion injury in both sexes; and this benefit is lost in Lamp2 heterozygous null mice, wherein lysosomal function and consequently autophagy are impaired, principally under stress. Second, intermittent fasting stimulates autophagosome accumulation in the setting of lysosomal dysfunction, which provokes adverse ventricular remodeling and heart failure in lamp2 null mice, wherein basal autophagic flux is impaired. Third, intermittent fasting transcriptionally stimulates the autophagy-lysosomal machinery in a cyclical fashion to keep the machinery primed with repetitive cycles of fasting-induced autophagy; associated with nuclear translocation of transcription factor EB, which is a master regulator that coordinates the biogenesis of autophagy-lysosome machinery. Fourth, endogenous TFEB plays a critical role in enhancement of autophagic flux and starvation-induced cytoprotection to hypoxia-reoxygenation injury, in cardiac myocytes, in vitro. Last, intermittent fasting attenuates myocardial oxidative stress and transcriptionally reprograms the myocardium, even on a fed day, to stimulate degradative processes and suppress cell death pathways, likely contributing to the observed beneficial effects.
Previous studies have described multiple salutary effects of intermittent fasting on life-span prolongation in C. elegansCitation54 and rodents;Citation22,55 reduction in infarct size with improved mortality and attenuation of postinfarction remodeling in the heart;Citation23,24 improved glucose regulation and attenuation of hypoxic insult in neuronsCitation25 in experimental models; and remarkable effects on weight loss and improved glucose-regulation in obese women.Citation21 Our study extends these observations by defining the need for an optimal frequency and duration of the fasting regimen to derive the cardioprotective effects; and suggests the hypothesis that frequency and duration of fasting episodes have additive benefits that accumulate with continuation of an intermittent fasting regimen. These observations are particularly relevant, as while sustained calorie restriction has well-established effects on preventing cardiovascular disease and aging,Citation3,4 and confers cardioprotection to ischemia-reperfusion injury.Citation56 Clinical studies have repeatedly demonstrated an inability to maintain sustained calorie restriction for prolonged periods in obese subjects,Citation57,58 who are at markedly elevated risk for development of cardiovascular disease. Conversely, intermittent fasting is routinely practiced by diverse human social groups for religious and/or cultural reasons. Early clinical studies suggest excellent short-term efficacy in treating obesity and its attendant metabolic risks,Citation21 improving glucose regulation and favorably altering the cardiovascular risk profile (reviewed in ref.),Citation59 thus making it an attractive alternative that needs to be evaluated for deriving benefits of calorie restriction.
Our study implicates an essential role for normal lysosomal function, particularly with relevance to efficient execution of macroautophagy; and hints at a prominent role of TFEB-mediated transcriptional replenishment of the autophagy-lysosome machinery in transducing the beneficial effects of intermittent fasting. It is important to note that other mechanisms may contribute to the observed benefits, too. In this regard, we have examined the role of CMA. Our data do not suggest an accumulation of CMA substrates,Citation40 as would be expected with ablation of the LAMP2A isoform in lamp2 null mice),Citation28 which could conceivably be due to upregulation of alternate protein degradation mechanisms. Importantly, the levels of these substrates were not altered by the intermittent fasting regimen, suggesting that CMA is unlikely to be a significant contributor to the observed cardioprotective effects. In contrast, we observed accumulation of abnormal mitochondria, which are substrates for macroautophagy and increased oxidative stress in LAMP2 deficient mice on a fed day, paralleling impairment in macroautophagy, which is the major pathway for removal of damaged mitochondria.Citation60 LAMP2, together with LAMP1, plays an important role in cholesterol efflux,Citation61 which also may be mechanistically important in the observed benefits of intermittent fasting, and needs to be explored further. In addition, we did not observe any obvious effect of intermittent fasting or LAMP2 ablation on microautophagyCitation62 in the ultrastructural studies, but definitive studies on its role will require development of tools to modulate this pathway.
We observe that autophagic flux is not upregulated above basal levels on a fed day after intermittent fasting (or in culture in nutrient replete medium after starvation, in vitro), at a time when the benefits of cytoprotection are observed. This suggests that the preconditioning effect of the lysosomal degradative pathways on maintenance of mitochondrial quality (as demonstrated by our data in ),Citation19 and perhaps on maintenance of ER homeostasisCitation63 plays a central role in shoring up cellular defenses to withstand the stress.Citation64 Indeed, we observe a reduction in prevalence of depolarized mitochondria in repetitively starved cardiomyocytes, in vitro () and reduced abundance of carbonylated proteins in the myocardium of intermittently fasted mice on a fed day (), suggesting that intermittent fasting stimulates lysosome function to remove damaged organelles that are the source for deleterious ROS generation,Citation38,64 as a mechanism for preconditioning the myocardium to ischemia-reperfusion injury.Citation39 Additional upstream signaling events that link fasting to stimulation of lysosomal pathways, including autophagy, may also participate in preconditioning responses. Fasting-induced activation of AMPK and suppression of MTOR signaling, are 2 candidate pathways that have been previously demonstrated to have no effect (as with AMPKCitation65) or facilitate ischemic preconditioining (as with rapamycin-mediated inhibiton of MTORCitation66) in the myocardium. Fasting-induced upregulation of SIRT/sirtuin signaling and protein deacetylation are also implicated in provoking myocardial autophagy;Citation11 and future studies are required to determine the role of these signaling pathways in intermittent fasting-induced cardioprotection.
Interestingly, we observe nuclear translocation of TFEB with fasting in the myocardium; paralleling the observations with starvation in various noncardiomyocyte cell types, in vitro.Citation45-47,50 It is therefore conceivable that the mechanism for TFEB activation with fasting, in vivo, is similar to that observed in vitro, with starvation-induced MTOR inhibition resulting in dephosphorylation and nuclear translocation of TFEB. However, while both fasting and starvation induce autophagy, it is likely that fasting is sensed as a metabolic shift in the myocardium, unlike total nutrient deprivation employed in the in vitro modeling of starvation; which points to the need for elucidating unique signaling pathways that drive TFEB activation with fasting, in vivo. Our findings suggest a critical role for TFEB in transducing starvation-induced upregulation of autophagic flux, in vitro. In this context, given that germline ablation of TFEB results in embryonic lethality,Citation67 generation of conditional cardiomyocyte TFEB knockout mice will be required to confirm its critical role in fasting-induced cardioprotection, in vivo.
Our data suggest the hypothesis that the transcriptional replenishment of the autophagy-lysosome machinery by fasting (and starvation as previously described) may be a critical determinant of beneficial autophagy; which permits living organisms to survive what is likely to have been one of the earliest evolutionary stresses accompanying the origin of life. Indeed, we have previously found that transcriptional suppression of the autophagy machinery with reperfusion-induced accumulation of BECN1 explains the lack of a protective role of the observed extent of autophagy induction in cardiac ischemia-reperfusion injury.Citation36,68 Further insights into this process could be gleaned via careful examination of the effects of intermittent fasting on the transcriptional program of the injured myocardium, in future studies.
In summary, our data implicate a central role for lysosomal degradative pathways in facilitating the previously described cardioprotective effects of intermittent fasting. Observations made herein also point to the need for exploring the transcriptional regulation of the cytoprotective lysosomal pathways to foster development of translational strategies to derive the benefits of calorie restriction in promoting cardiovascular health.
Materials and Methods
Studies with mice
Wild-type mice of the C57BL/6 strain were obtained from Jackson Laboratories. Mice with cardiomyocyte-specific expression of GFP tagged LC3 transgeneCitation13 and those with Lamp2 ablationCitation28 have been described before. Echocardiographic studies were performed as previously described in conscious mice.Citation69 All animal studies were approved by the Animal Studies Committee at the Washington University School of Medicine and the Institutional Animal Care and Use Committee at the John Cochran VA Medical Center.
Intermittent fasting
Adult mice were housed in groups of up to n = 5 mice/cage and fed standard chow (Lab Diet, 5053; providing 3.4 Kcal/g with 62.1% Kcal derived from carbohydrates, 13.2% from fats, and 24.6% from protein) on a 6:00 PM to 6:00 AM night-day cycle, while housed on cedar pine chip bedding. Intermittent fasting was performed with total food deprivation and ad libitum access to water from 12:00 PM to 12:00 PM of the following day to implement alternate periods of 24 h fasting and feeding, with change in bedding. Nonfasted control mice were simultaneously provided fresh food with change in bedding. Daily food intake per cage was weighed to calculate average daily food intake per mouse. Mice were weighed at weekly intervals on fed days. Terminal studies on mice were initiated between 8:00 to 10:00 AM after an overnight period of feeding (i.e., on a fed day).
Assessment of autophagic flux, in vivo
Mice subjected to various fastng regimens were injected with CQ (40 mg/kg intraperitoneally) 4 h prior to sacrifice to assess autophagic flux, in vivo, as previously described.Citation26
Ischemia-reperfusion modeling
Mice were subjected to reversible left anterior descending artery ligation for 30 min followed by reperfusion, in an open-chest procedure, as described.Citation36 Overall surgical mortality was very low (<3 %). At 24 h post induction of ischemia, cardioplegic solution was injected retrogradely through the aorta in situ, followed by sectioning of the left ventricle in slices; which were incubated in triphenyl tetrazolium chloride for 30 min at 37°C. All surgeries were performed by one surgeon (C.J.W.) and uniform LAD ligation technique was confirmed by assessment of area at risk with injection of Evans Blue dye in separate cohort of animals. For infarct size calculation, triphenyl tetrazolium chloride (TTC)-stained slices were imaged and infarct area quantified as non-TTC-stained (pale) area as a ratio of total myocardial area, as previously described.Citation70
TUNEL and c5b-9 staining: Evaluation of cardiomyocyte TUNEL positivity and c5b-9 staining was performed, as previously described.Citation70,71
Quantitative PCR analysis
Total RNA was extracted from mouse hearts using RNAeasy Fibrous Tissue Minikit (Qiagen). Quantitative PCR analysis was performed as described.Citation36 Primers employed were as follows: Map1lc3b: forward 5′- CGTCCTGGACAAGACCAAGT-3′, reverse 5′- ATTGCTGTCCCGAATGTCTC-3′Citation72 ; Sqstm1: forward 5′- GCTGCCCTATACCCACATCT-3′, reverse 5′-CGCCTTCATCCGAGAAAC-3′ Citation73; Becn1: forward 5′- AATCTAAGGAGTTGCCGTTATAC-3′ , reverse 5′-CCAGTGTC-TTCAATCTTGCC-3′Citation74; Lamp1: forward 5′- ACATCAGCCCAAATGACACA-3′, reverse 5′- GGCTAGAGCTGGCATTCATC-3; Lamp2a: forward 5′- CCAAATTGGGATCCTAACCT-AA-3′, reverse 5′-TGGTCAAGCAGTGTTTATTAATTCC-3; Lamp2b: forward 5′- GGTGCTGGTCTTTCAGGCTTGATT-3′, reverse 5′- ACCACCCAATCTAAGAGCAGGACT-3Citation75; Tfeb: forward 5′- GTCTAGCAGCCACCTGAACGT-3, reverse 5′-CAGGTCACAGCCTCCATGGT-3Citation76; Gapdh: forward 5′- ACTCCCACTCTTCCACCTTC-3′, reverse 5′-TCTTGCTCA-GTGTCCTTGC-3′Citation75; and Rpl32: forward 5′- CCTCTGGTG AAGCCCAAGATC-3′, reverse 5′- TCTGGGTTTCCGCCA-GTTT-3′.
Studies with NRCMs
Neonatal rat cardiac myocytes were isolated and cultured, as previously described.Citation77 Starvation was induced by culturing in serum-free HBSS (Invitrogen, 24020117). For assessment of mitochondrial polarization, NRCMs were incubated with JC-1 (10 mg/mL for 10 min) at 37°C in 5% CO2, the cells were trypsinized and subjected to flow cytometry on FACScan instrument (Becton-Dickinson, NJ), as previously described.Citation68,78 Cyflogic software (CyFlo) was employed to analyze 20,000 events per run. Mitochondrial DNA content was assessed with quantitative PCR analysis as previously described.Citation68,78 Transmission electron microscopy was performed on NRCMs fixed in a modified Karnovsky's fixative, as previously described.Citation68,78 Hypoxia was induced by culturing cells in an oxygen control cabinet (Coy Laboratories, Grass Lake, MI) mounted within an incubator and equipped with oxygen sensor for continuous oxygen level monitoring. A mixture of 95% nitrogen and 5% CO2 was infused and oxygen levels in the chamber were monitored and maintained at <1%, as described.Citation36
Generation of adenoviral constructs
Adenoviral particles for expression of shRNA targeting murine Tfeb, and a scrambled shRNA control were generated with BLOCKiT™ adenoviral system (Invitrogen, K4941–00). Specific oligo sequences employed were as follows: 1) shRNA targeting mouse Tfeb (with targeted coding sequence underlined) – top strand oligo: 5′-CACCGCGGCAGTACTATGACTATGATCGAAATCATAGTCATAGTACTGCCG-3′; bottom strand oligo: 5′- AAAACGGCAGTACTATGACTATGATT-TCGATCATAGTCATAGTACTGCCGC-3′.
Two) Scrambled shRNA as control to Tfeb shRNA- top strand oligo: 5′- CACCGGCGCTCATCGAATTAATAGTCGAAACTATTAATTCGATGAGCGCC-3′; bottom strand oligo: 5′- AAAAAGGCGCTCATCGAATTAATAGTTTCGA-CTATTAATTCGATGAGCGCC-3′.
Adenoviral particles for shRNA targeting rat Lamp2 were generated with the following oligos: top strand oligo: 5′-CACCGCCTTTAACCTGAAGGTGCACGAATGCACCTTCAGGTTAAAGG-3′; bottom strand oligo: 5′-AAAAAGGCGCTCATCGAATTAATAGTTTCGACTATTAATTCGATGA-GCGCC-3′. Adenoviral particles for shRNA targeting Becn1 (shBecn1),Citation68 shLacZ and exogenous expression of human TFEB have been previously described.Citation77
Generation of conditional cardiac Tfeb transgenic mice
The coding sequence of mouse Tfeb (translated from transcript variant 2, NP_001155194.1, that encodes a protein similar in size to human TFEB) with a N-terminal FLAG tag was cloned into the α-Myosin Heavy Chain promoter –TetOMin construct generously provided by Jeffery Robbins, Cincinnati Children's Hospital. Resulting founders were mated with mice carrying the MYH6 promoter driven tTA transgene (also generously provided by Dr. Robbins); and pregnant and lactating mothers with pups were maintained on doxycycline-mixed chow (El-Mel diets, St. Charles, MO) until 8 wk of age. Myocardial extracts were prepared after 2 wk of being switched over to a standard chow diet in mice from founder line 4239, and employed as positive control for immunodetection of endogenous myocardial TFEB in mice.
Immunoblotting
Hearts were fractionated into nuclear enriched and cytoplasmic samples using CelLytic NuCLEAR Extraction kit (NXTRACT, Sigma); or crude extracts prepared followed by immunoblotting as described.Citation36 Antibodies employed were as follows: LAMP2, mouse monoclonal (Developmental Studies Hybridoma Bank, ABL-93), LAMP1 (Santa Cruz Biotechnology, sc-19992) and (Abcam, ab24170); anti-LC3 (encoding for MAP1LC3B subunit; Novus Biologicals, NB100–2220); SQSTM1 (Abcam, ab5416); BECN1/BECLIN-1 (Abcam, ab16998); TFEB (Bethyl Labs, A303–673A), LRRK2 (Cell Signaling Technology, 5559), NFKBIA/IκBα (Cell Signaling Technology, 4814), ALDOA/aldolase (Abcam, ab169544), GAPDH (Abcam, ab22555), and ACTA1/α-sarcomeric actin (Abcam, ab7799). ImageJ software was employed for quantitative analysis. Protein abundance was normalized to ACTA1 protein expression and reported as fold change vs. control.
Microarray analysis
Total RNA was extracted from mouse ventricles as above and hybridized to a Mouse Ref-6 Illumina BeadChip by the Genome Technology Access Center (GTAC) at Washington University School of Medicine and scanned with the BeadStation system from Illumina, Inc. Each chip was subjected to verification for quality standards for hybridization, labeling, staining, background signal, and basal level of housekeeping gene expression. After scanning the probe array, the resulting image was analyzed using the GenomeStudio software (Illumina), background was subtracted; and log transformation and quantile normalization were performed. Changes in gene expression were determined using Partek Genomics Suite on quantile-normalized and background-subtracted microarray data; and principal components analysis was performed. Significant gene lists were generated using an unadjusted P value <0.05 and fold change greater than 1.2 or less than −1.2. Functional analysis of pathways was performed using Database for Annotation Visualization and Integrated Discovery (DAVID).Citation79,80 List of genes were also annotated for functional significance based on Genecards (Weizmann Institute Database) and grouped under functional categories.
Assessment of myocardial ROS
ROS was monitored by detection of carbonylated proteins with Oxyblot™ Protein Oxidation Detection Kit (Chemicon/Millipore, S7150) following the manufacturer's instructions, as described.Citation81 Briefly, 20 µg protein of cardiac extract was derivatized from carbonyl groups to a 2,4-dinitrophenylhydrazone (DNP) moiety with 1X 2,4-dinitrophenylhydrazine and denatured with SDS at the same time. After the protein was separated on a 12% precast gel (Bio-Rad, 456–1043) using standard SDS polyacrylamide electrophoresis under reducing conditions, the gels were transferred to a PVDF membrane and probed with primary rabbit anti-DNP and secondary goat anti-rabbit IgG (HRP-conjugated) antibodies provided in the kit and developed using enhanced chemiluminescence (Perkin Elmer, NEL103001EA). Then the DNP moiety is detected to measure the oxidative damage to a protein. Image J software was employed to quantify the relative abundance of carbonylated proteins.
Statistical analysis
Results are expressed as mean±SEM (unless otherwise specified). Statistical differences were assessed with the unpaired Student t test for 2 independent groups, one-way or 2-way ANOVA for comparing one or 2 variables, respectively, across multiple groups with SPSS software. The Bonferroni post-hoc test was employed after ANOVA for testing all pair-wise comparisons. A 2-tailed value of P < 0.05 was considered statistically significant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1063768_supplemental_files.zip
Download Zip (1.2 MB)Acknowledgements
We thank Joseph A. Hill, University of Texas Southwestern Medical Center, Dallas, TX, for providing the GFP-LC3 mice and insightful comments; Jeffrey Robbins, Cincinnati Children's Hospital for providing the MYH6 promoter based conditional transgene construct; Junichi Sadoshima, Rutgers New Jersey Medical School, Newark, NJ, for providing adenoviral particles coding for shBecn1; Sarah Evans, Washington University, St. Louis, MO, for assistance with microarray analysis; Peter A. Crawford, Washington University, St. Louis, MO, for helpful discussions; Andrea Ballabio, Telethon Institute of Genetics and Medicine, Napoli, Italy for critical reading of the manuscript; Wandy Beatty, for technical and scientific assistance; and Douglas L. Mann, Washington University, for his support. We also thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis.
Funding
The center is partially supported by NCI Cancer Center Support Grant #P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA Grant #UL1RR024992 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research. This study was supported by grants from National Institutes of Health (HL107594) and Department of Veterans Affairs (I01BX000448, 1I01BX001969) to A.D; and the Deutsche Forschungsgemeinschaft (DFG) to P.S.
Related Research Data
References
- Schwartz LL, Kloner RA, Arai AE, Baines CP, Bolli R, Braunwald E, Downey J, Gibbons RJ, Gottlieb RA, Heusch G, et al. New horizons in cardioprotection: recommendations from the 2010 National Heart, Lung, and Blood Institute Workshop. Circulation 2011; 124:1172-9; PMID:21900096; http://dx.doi.org/10.1161/CIRCULATIONAHA.111.032698
- Fontana L, Meyer TE, Klein S, Holloszy JO. Long-term calorie restriction is highly effective in reducing the risk for atherosclerosis in humans. Proc Natl Acad Sci USA 2004; 101:6659-63; PMID:15096581; http://dx.doi.org/10.1073/pnas.0308291101
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, et al. Caloric restriction delays disease onset and mortality in rhesus monkeys. Science 2009; 325:201-4; PMID:19590001; http://dx.doi.org/10.1126/science.1173635
- Mattison JA, Roth GS, Beasley TM, Tilmont EM, Handy AM, Herbert RL, Longo DL, Allison DB, Young JE, Bryant M, et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 2012; 489:318-21; PMID:22932268; http://dx.doi.org/10.1038/nature11432
- Fontana L, Partridge L, Longo VD. Extending healthy life span–from yeast to humans. Science 2010; 328:321-6; PMID:20395504; http://dx.doi.org/10.1126/science.1172539
- Hansen M, Chandra A, Mitic LL, Onken B, Driscoll M, Kenyon C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet 2008; 4:e24.
- Eisenberg T, Knauer H, Schauer A, Buttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, et al. Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 2009; 11:1305-14.
- Madeo F, Eisenberg T, Buttner S, Ruckenstuhl C, Kroemer G. Spermidine: a novel autophagy inducer and longevity elixir. Autophagy 2010; 6:160-2; PMID:20110777; http://dx.doi.org/10.4161/auto.6.1.10600
- Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de CR, Fernandez E, Flurkey K, Javors MA, Nelson JF, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 2011; 66:191-201; PMID:20974732; http://dx.doi.org/10.1093/gerona/glq178
- Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, Criollo A, Galluzzi L, Malik SA, Vitale I, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis 2010; 1:e10; PMID:21364612; http://dx.doi.org/10.1038/cddis.2009.8
- Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res 2010; 107:1470-82.
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11.
- Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA, Hill JA. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest 2007; 117:1782-93.
- Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J Cell Biol 2005; 169:425-34; http://dx.doi.org/10.1083/jcb.200412022
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/10.1038/nature03029
- Kanamori H, Takemura G, Maruyama R, Goto K, Tsujimoto A, Ogino A, Li L, Kawamura I, Takeyama T, Kawaguchi T, et al. Functional significance and morphological characterization of starvation-induced autophagy in the adult heart. Am J Pathol 2009; 174:1705-14; PMID:19342365; http://dx.doi.org/10.2353/ajpath.2009.080875
- Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 2007; 100:914-22.
- Buss SJ, Muenz S, Riffel JH, Malekar P, Hagenmueller M, Weiss CS, Bea F, Bekeredjian R, Schinke-Braun M, Izumo S, et al. Beneficial effects of Mammalian target of rapamycin inhibition on left ventricular remodeling after myocardial infarction. J Am Coll Cardiol 2009; 54:2435-46; PMID:20082935; http://dx.doi.org/10.1016/j.jacc.2009.08.031
- Huang C, Yitzhaki S, Perry CN, Liu W, Giricz Z, Mentzer RM, Jr., Gottlieb RA. Autophagy induced by ischemic preconditioning is essential for cardioprotection. J Cardiovasc Transl Res 2010; 3:365-73; PMID:20559777; http://dx.doi.org/10.1007/s12265-010-9189-3
- Yan L, Vatner DE, Kim SJ, Ge H, Masurekar M, Massover WH, Yang G, Matsui Y, Sadoshima J, Vatner SF. Autophagy in chronically ischemic myocardium. Proc Natl Acad Sci USA 2005; 102:13807-12; PMID:16174725; http://dx.doi.org/10.1073/pnas.0506843102
- Harvie MN, Pegington M, Mattson MP, Frystyk J, Dillon B, Evans G, Cuzick J, Jebb SA, Martin B, Cutler RG, et al. The effects of intermittent or continuous energy restriction on weight loss and metabolic disease risk markers: a randomized trial in young overweight women. Int J Obes(Lond) 2011; 35:714-27; PMID:20921964; http://dx.doi.org/10.1038/ijo.2010.171
- Goodrick CL, Ingram DK, Reynolds MA, Freeman JR, Cider N. Effects of intermittent feeding upon body weight and lifespan in inbred mice: interaction of genotype and age. Mech Ageing Dev 1990; 55:69-87.
- Ahmet I, Wan R, Mattson MP, Lakatta EG, Talan M. Cardioprotection by intermittent fasting in rats. Circulation 2005; 112:3115-21; PMID:16275865; http://dx.doi.org/10.1161/CIRCULATIONAHA.105.563817
- Katare RG, Kakinuma Y, Arikawa M, Yamasaki F, Sato T. Chronic intermittent fasting improves the survival following large myocardial ischemia by activation of BDNF/VEGF/PI3K signaling pathway. J Mol Cell Cardiol 2009; 46:405-12.
- Anson RM, Guo Z, de CR, Iyun T, Rios M, Hagepanos A, Ingram DK, Lane MA, Mattson MP. Intermittent fasting dissociates beneficial effects of dietary restriction on glucose metabolism and neuronal resistance to injury from calorie intake. Proc Natl Acad Sci USA 2003; 100:6216-20; http://dx.doi.org/10.1073/pnas.1035720100
- Iwai-Kanai E, Yuan H, Huang C, Sayen MR, Perry-Garza CN, Kim L, Gottlieb RA. A method to measure cardiac autophagic flux in vivo. Autophagy 2008; 4:322-9; PMID:18216495; http://dx.doi.org/10.4161/auto.5603
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000; 406:906-10; PMID:10972294; http://dx.doi.org/10.1038/35022604
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature 2000; 406:902-6; PMID:10972293; http://dx.doi.org/10.1038/35022595
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature 1961; 190:372-3; PMID:13764598; http://dx.doi.org/10.1038/190372a0
- Stypmann J, Janssen PM, Prestle J, Engelen MA, Kogler H, Lullmann-Rauch R, Eckardt L, von FK, Landgrebe J, Mleczko A, et al. LAMP-2 deficient mice show depressed cardiac contractile function without significant changes in calcium handling. Basic Res Cardiol 2006; 101:281-91; http://dx.doi.org/10.1007/s00395-006-0591-6
- Bjorkoy G, Lamark T, Pankiv S, Overvatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol 2009; 452:181-97; PMID:19200883
- Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell 2009; 33:517-27.
- Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci USA 2012; 109:2003-8; http://dx.doi.org/10.1073/pnas.1112848109
- Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 2007; 13:619-24.
- Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy 2012; 8:719-30; http://dx.doi.org/10.4161/auto.19469
- Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation 2012; 125:3170-81; PMID:22592897; http://dx.doi.org/10.1161/CIRCULATIONAHA.111.041814
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 2013; 20:31-42; PMID:22743996; http://dx.doi.org/10.1038/cdd.2012.81
- Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res 2014; 114:524-37; PMID:24481843; http://dx.doi.org/10.1161/CIRCRESAHA.114.300559
- Huang C, Andres AM, Ratliff EP, Hernandez G, Lee P, Gottlieb RA. Preconditioning involves selective mitophagy mediated by Parkin and p62/SQSTM1. PLoS One 2011; 6:e20975; http://dx.doi.org/10.1371/journal.pone.0020975
- Schneider JL, Suh Y, Cuervo AM. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 2014; 20:417-32; PMID:25043815; http://dx.doi.org/10.1016/j.cmet.2014.06.009
- Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell 2004; 15:4829-40.
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Carreira RS, Lee Y, Ghochani M, Gustafsson AB, Gottlieb RA. Cyclophilin D is required for mitochondrial removal by autophagy in cardiac cells. Autophagy 2010; 6:462-72; PMID:20364102; http://dx.doi.org/10.4161/auto.6.4.11553
- Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010; 465:942-6; PMID:20526321; http://dx.doi.org/10.1038/nature09076
- Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012; 8:903-14; PMID:22576015; http://dx.doi.org/10.4161/auto.19653
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The Transcription Factor TFEB Links mTORC1 Signaling to Transcriptional Control of Lysosome Homeostasis. Sci Signal 2012; 5:ra42.
- Settembre C, Di MC, Polito VA, Garcia AM, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/10.1126/science.1204592
- Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 2012; 31(5):1095-108; PMID:22343943
- Palmieri M, Impey S, Kang H, di RA, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011; 20:3852-66.
- Sardiello M, Palmieri M, di RA, Medina DL, Valenza M, Gennarino VA, Di MC, Donaudy F, Embrione V, Polishchuk RS, et al. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473-7; PMID:19556463
- Kuiper RP, Schepens M, Thijssen J, Schoenmakers EF, van Kessel AG. Regulation of the MiTF/TFE bHLH-LZ transcription factors through restricted spatial expression and alternative splicing of functional domains. Nucleic Acids Res 2004; 32:2315-22; PMID:15118077; http://dx.doi.org/10.1093/nar/gkh571
- Pena-Llopis S, Vega-Rubin-de-Celis S, Schwartz JC, Wolff NC, Tran TA, Zou L, Xie XJ, Corey DR, Brugarolas J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J 2011; 30:3242-58; PMID:21804531; http://dx.doi.org/10.1038/emboj.2011.257
- Settembre C, De CR, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Jurgen KT, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. NatCell Biol 2013.
- Honjoh S, Yamamoto T, Uno M, Nishida E. Signalling through RHEB-1 mediates intermittent fasting-induced longevity in C. elegans. Nature 2009; 457:726-30; PMID:19079239; http://dx.doi.org/10.1038/nature07583
- Arum O, Bonkowski MS, Rocha JS, Bartke A. The growth hormone receptor gene-disrupted mouse fails to respond to an intermittent fasting diet. Aging Cell 2009; 8:756-60; PMID:19747233; http://dx.doi.org/10.1111/j.1474-9726.2009.00520.x
- Ahmet I, Tae HJ, de CR, Lakatta EG, Talan MI. Effects of calorie restriction on cardioprotection and cardiovascular health. J Mol Cell Cardiol 2011; 51:263-71.
- Sumithran P, Prendergast LA, Delbridge E, Purcell K, Shulkes A, Kriketos A, Proietto J. Long-term persistence of hormonal adaptations to weight loss. N Engl J Med 2011; 365:1597-604; http://dx.doi.org/10.1056/NEJMoa1105816
- Wing RR. Long-term effects of a lifestyle intervention on weight and cardiovascular risk factors in individuals with type 2 diabetes mellitus: four-year results of the Look AHEAD trial. Arch Intern Med 2010; 170:1566-75.
- Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab 2014; 19:181-92; PMID:24440038; http://dx.doi.org/10.1016/j.cmet.2013.12.008
- Lemasters JJ. Variants of mitochondrial autophagy: Types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol 2014; 2:749-54; PMID:25009776; http://dx.doi.org/10.1016/j.redox.2014.06.004
- Eskelinen EL, Schmidt CK, Neu S, Willenborg M, Fuertes G, Salvador N, Tanaka Y, Lullmann-Rauch R, Hartmann D, Heeren J, et al. Disturbed cholesterol traffic but normal proteolytic function in LAMP-1/LAMP-2 double-deficient fibroblasts. Mol Biol Cell 2004; 15:3132-45.
- Mijaljica D, Prescott M, Devenish RJ. Microautophagy in mammalian cells: revisiting a 40-year-old conundrum. Autophagy 2011; 7:673-82; PMID:21646866; http://dx.doi.org/10.4161/auto.7.7.14733
- Nishida K, Kyoi S, Yamaguchi O, Sadoshima J, Otsu K. The role of autophagy in the heart. Cell Death Differ 2009; 16:31-8; PMID:19008922; http://dx.doi.org/10.1038/cdd.2008.163
- Gottlieb RA, Finley KD, Mentzer RM, Jr. Cardioprotection requires taking out the trash. Basic Res Cardiol 2009; 104:169-80; http://dx.doi.org/10.1007/s00395-009-0011-9
- Kim AS, Miller EJ, Wright TM, Li J, Qi D, Atsina K, Zaha V, Sakamoto K, Young LH. A small molecule AMPK activator protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol 2011; 51:24-32; http://dx.doi.org/10.1016/j.amjcard.2011.02.053
- Khan S, Salloum F, Das A, Xi L, Vetrovec GW, Kukreja RC. Rapamycin confers preconditioning-like protection against ischemia-reperfusion injury in isolated mouse heart and cardiomyocytes. J Mol Cell Cardiol 2006; 41:256-64.
- Steingrimsson E, Tessarollo L, Reid SW, Jenkins NA, Copeland NG. The bHLH-Zip transcription factor Tfeb is essential for placental vascularization. Development 1998; 125:4607-16; PMID:9806910
- Ma X, Liu H, Murphy JT, Foyil SR, Godar RJ, Abuirqeba H, Weinheimer CJ, Barger PM, Diwan A. Regulation of TFEB-PGC1alpha axis by beclin-1 controls mitochondrial quality and cardiomyocyte death under stress. Mol Cell Biol 2015; 35(6):956-76.
- Syed F, Diwan A, Hahn HS. Murine echocardiography: a practical approach for phenotyping genetically manipulated and surgically modeled mice. J Am Soc Echocardiogr 2005; 18:982-90.
- Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, et al. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 2007; 117:2825-33.
- Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW. Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci USA 2010; 107:9035-42; http://dx.doi.org/10.1073/pnas.0914013107
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472-83; PMID:18054316; http://dx.doi.org/10.1016/j.cmet.2007.11.004
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/10.1016/j.cell.2007.10.035
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283:10892-903.
- Yanagawa M, Tsukuba T, Nishioku T, Okamoto Y, Okamoto K, Takii R, Terada Y, Nakayama KI, Kadowaki T, Yamamoto K. Cathepsin E deficiency induces a novel form of lysosomal storage disorder showing the accumulation of lysosomal membrane sialoglycoproteins and the elevation of lysosomal pH in macrophages. J Biol Chem 2007; 282:1851-62.
- Sferruzzi-Perri AN, Macpherson AM, Roberts CT, Robertson SA. Csf2 null mutation alters placental gene expression and trophoblast glycogen cell and giant cell abundance in mice. Biol Reprod 2009; 81:207-21.
- Ma X, Godar RJ, Liu H, Diwan A. Enhancing lysosome biogenesis attenuates BNIP3-induced cardiomyocyte death. Autophagy 2012; 8:297-309; PMID:22302006; http://dx.doi.org/10.4161/auto.18658
- Yang KC, Ma X, Liu H, Murphy J, Barger PM, Mann DL, Diwan A. TNF-receptor associated factor-2 mediates mitochondrial autophagy. Circ Heart Fail 2014; 8(1):175-87.
- Huang dW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4:44-57.
- Huang dW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37:1-13; PMID:19033363; http://dx.doi.org/10.1093/nar/gkn923
- Goonasekera SA, Hammer K, Auger-Messier M, Bodi I, Chen X, Zhang H, Reiken S, Elrod JW, Correll RN, York AJ, et al. Decreased cardiac L-type Ca(2)(+) channel activity induces hypertrophy and heart failure in mice. J Clin Invest 2012; 122:280-90.