
Abstract
RNA viruses modify intracellular membranes to produce replication scaffolds. In pancreatic cells, coxsackievirus B3 (CVB3) hijacks membranes from the autophagy pathway, and in vivo disruption of acinar cell autophagy dramatically delays CVB3 replication. This is reversed by expression of GFP-LC3, indicating that CVB3 may acquire membranes from an alternative, autophagy-independent, source(s). Herein, using 3 recombinant CVB3s (rCVB3s) encoding different proteins (proLC3, proLC3G120A, or ATG4BC74A), we show that CVB3 is, indeed, flexible in its utilization of cellular membranes. When compared with a control rCVB3, all 3 viruses replicated to high titers in vivo, and caused severe pancreatitis. Most importantly, each virus appeared to subvert membranes in a unique manner. The proLC3 virus produced a large quantity of LC3-I which binds to phosphatidylethanolamine (PE), affording access to the autophagy pathway. The proLC3G120A protein cannot attach to PE, and instead binds to the ER-resident protein SEL1L, potentially providing an autophagy-independent source of membranes. Finally, the ATG4BC74A protein sequestered host cell LC3-I, causing accumulation of immature phagophores, and massive membrane rearrangement. Taken together, our data indicate that some RNA viruses can exploit a variety of different intracellular membranes, potentially maximizing their replication in each of the diverse cell types that they infect in vivo.
Abbreviations
| ALV | = | autophagosome-like vesicle |
| CANX | = | calnexin |
| Ct | = | C-terminal |
| Cre | = | recombinase that causes recombination |
| CVB3 | = | coxsackievirus B3 |
| DMV | = | double-membrane vesicle |
| DPBS | = | Dulbecco's PBS |
| EL | = | elastase |
| ERAD | = | endoplasmic reticulum-associated degradation |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MOI | = | multiplicity of infection |
| PE | = | phosphatidylethanolamine |
| Pfu | = | plaque-forming unit(s) |
| p.i. | = | postinfection |
| rCVB | = | recombinant coxsackievirus B3 |
| RT | = | room temperature |
| SEL1L | = | sel-1 suppressor of lin-12-like (C. elegans) |
| siRNA | = | small interfering RNA |
| tg | = | transgenic |
| WT | = | wild type. |
Introduction
Viruses depend on the host cell for growth and replication. During infection, the virus initiates spatio-temporally ordered sequences of signaling events that alter cellular architecture and physiology. The host cell not only provides the energy and materials for viral multiplication, but also supplies structural scaffolds for replication and viral assembly.Citation1 Positive-strand RNA viruses replicate their RNA genomes on the cytosolic faces of invaginated cytoplasmic host membranesCitation2,3 or on abundant small (70 to 400 nm diameter) double-membrane vesicles that have been variously termed compound membrane vesicles, double-membrane vesicles (DMVs) or autophagosome-like vesicles (ALVs), first identified by electron microscopy studies of cells infected by poliovirus or coxsackievirus, both enteroviruses in the family Picornaviridae.Citation4,5 As the initialism ALV implies, the virus-induced vesicles have morphological similarities to components of the autophagy pathway, a highly conserved recycling process in which double-membrane vesicles, termed autophagosomes, shuttle cellular debris to lysosomes for degradation. It is now clear that the autophagy pathway can be the source of at least some of the DMVs that are present in RNA virus-infected cells.Citation6,7 Further work with poliovirus has confirmed that the membrane reorganizations supply topological platforms for oligomeric 3D structures that promote replication,Citation6,8 and elegant studies using 3D electron tomography to analyze coxsackievirus B3 (CVB3)-infected tissue culture cells have revealed multiple closely-apposed DMVs, whose appearance is preceded by transitory single-membrane structures.Citation9 Our laboratory has evaluated the relationship between enteroviruses and autophagy in vivo, demonstrating that, in C57BL/6 mice, CVB3 infection of pancreatic acinar cells causes upregulation of early steps in the autophagy pathway, as well as downstream blockadeCitation10 which, we have proposed,Citation11 together cause the observed accumulation of DMVs that is exploited by the virus. Consistent with this notion, our subsequent work using ElCre-Atg5f/f mice shows that the acinar cell-specific deletion of the autophagy-related protein 5 (Atg5) gene (which is vital for the development of autophagy-related DMVs) reduces not only the abundance of the vesicles but also CVB3 replication; the viral titer is ∼2000-fold lower compared with normal acinar cells.Citation12 At least some of these enterovirus-driven changes in host cell membranes are quite specific, relying on the effects of individual viral proteins. For example, formation of autophagy-like vesicles can be induced by cotransfection of 2 nonstructural polioviral proteins 2BC and 3A,Citation13 and plasmids encoding the CVB3 2A, 2B, or 2BC proteins can cause radical intracellular membrane reorganization, including wholescale disruption of the Golgi apparatus.Citation14-19
Thus, the autophagy pathway can contribute positively to CVB3 replication, and—at least in part—this appears to involve the virus's hijacking of the pathway's vesicular assembly process. However, autophagy is not absolutely required. In ATG5-deficient pancreata—in which the pathway is disrupted at a very early stage (see below)—CVB3 replication, although ∼2000-fold lower at d 1 postinfection (p.i.), nevertheless recovers relatively well over the ensuing days and reaches peak titers of ∼5 × 108 plaque-forming units (pfu)/gram of pancreas.Citation12 Thus, either (i) enteroviruses must be able to replicate without membrane support or (ii) pathways other than autophagy must be able to contribute to the generation of such scaffolds. In regard to the latter possibility, as noted above, CVB3 proteins dramatically modulate the secretory machinery,Citation18,19 and proteins from the secretory pathway have been identified in enteroviral replication loci,Citation6,20,21 suggesting that this pathway may contribute to viral replication. Viruses from other families (e.g., flaviviruses and nidoviruses) certainly take advantage of the secretory pathway, and recent data indicate that one facet of the secretory apparatus—the endoplasmic reticulum-associated degradation (ERAD) pathway—may be actively exploited by coronaviruses,Citation22-24 which seem to subvert one particular protein, LC3, which conventionally has been considered as being limited to the autophagy pathway. We found these observations particularly interesting because, while using a recombinant CVB3 expressing the fluorophore dsRed (dsRed-CVB3) in in vivo studies, we have reported a positive effect of LC3 expression on CVB3 replication and pathogenicity.Citation12 Like other rCVB3s, dsRed-CVB3 is highly attenuated in wild-type mice, but this virus replicated to higher titers, and causes substantial disease in GFP-LC3 mice, in which the LC3 protein is abundantly expressed in all cell types.Citation10 In the present report we have followed up on this observation, by generating a number of recombinant viruses that encode proteins that modulate the abundance or functionality of LC3, and evaluating their effects on viral replication in wild-type (WT) and in ATG5-deficient mice. We have drawn 4 conclusions from the resulting data. First, that the effect of LC3 on CVB3 replication varies depending on the precise form of the protein that is present in the infected cell. Second, that some of the “proviral” effects of LC3 are dependent upon autophagy, while others are not. Third, that CVB3 appears to use LC3 as a common entry point by which to gain access to 2 discrete cellular pathways that involve membrane reorganization. Fourth, that the requirement for LC3 may not be absolute, as described below. Our findings indicate that CVB3—and, perhaps other RNA viruses—may be more flexible in their use of membranes than is currently thought. This has substantial implications not only for our understanding of viral pathogenesis, but also for the design and use of future antiviral therapies.
Results
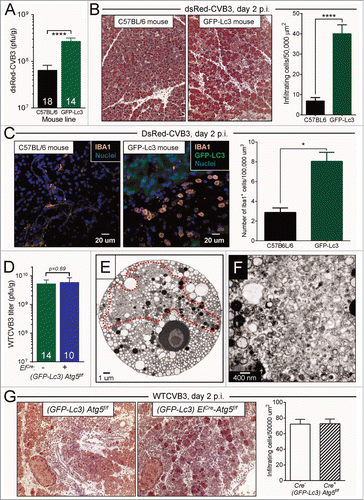
GFP-LC3 facilitates CVB3 replication, and reverses the antiviral effect of ATG5 deficiency
We began by confirming and expanding our observations that GFP-LC3 can support enteroviral replication in vivo. First, we evaluated replication and pathogenesis of dsRed-rCVB3 in WT (C57BL/6) mice, and in congenic mice carrying a single allele of GFP-LC3. Both mouse strains were infected with dsRed-CVB3 and, 2 d later, were sacrificed. Pancreatic viral titers were ∼6-fold higher in GFP-LC3 mice (P < 0 .0001, ), and pancreatitis was correspondingly increased; Masson trichrome staining showed that pancreatic acinar cells in these mice were hypochromic, and that there was a greater degree of inflammation, revealed by edema and an ∼8-fold increase in cellular infiltration (). Confocal microscopy of vibratome sections () identified many of the infiltrating cells as monocytes or macrophages, shown by their expression of IBA1, a protein that is expressed predominantly on cells of this lineage.Citation25,26 In contrast, in infected C57BL/6 mice, AIF1/IBA1-positive cells were less frequent and had the morphology of resting macrophages. Thus, the presence of a single allele of GFP-LC3 is sufficient to increase rCVB3 replication and disease in the pancreas. Next, we used WTCVB3, the early in vivo replication of which is severely disrupted by the ablation of the autophagy pathway; we have shown that, at d 1 p.i., pancreatic titers of WTCVB3 are ∼2000-fold lower in ElCre-Atg5f/f mice when compared to their Cre− littermates.Citation12 We wished to determine if the availability of GFP-LC3 would alter this antiviral effect of ATG5 deletion and, to do so, we generated triple-transgenic mice ([GFP-LC3]-ElCre-Atg5f/f), along with their Cre− littermates. These mice were infected with WTCVB3 and, one d later, were sacrificed and viral titers were measured in their pancreata. As shown in , and as expected, WTCVB3 replicated to a high titer in the autophagy-intact (GFP-LC3)-Atg5f/f mice (green bar). Of greater interest, there was no detectable deficit in WTCVB3 replication in their Cre+ (i.e., ATG5-deficient) counterparts; thus, the ∼2000-fold difference that we observed in ATG5-deficient WT mice is completely abolished by the provision of GFP-LC3. This normal level of replication was reflected by the ultrastructural morphology of (GFP-LC3)-ElCre-Atg5f/f acinar cells following WTCVB3 infection. The cells contained large tracts of DMVs (, enclosed in red-dashed line; and at higher magnification in ). Finally, we evaluated early pancreatitis, which had been substantially mitigated in ElCre-Atg5f/f mice following infection with WTCVB3;Citation12 as shown in , infiltration of inflammatory cells was similar in the double-transgenic (GFP-LC3)-Atg5f/f and triple-transgenic (GFP-LC3-ElCre-Atg5f/f) animals, confirming that provision of GFP-LC3 could functionally complement the deficiency of ATG5. The data in show clearly that both rCVB3 and WTCVB3 can benefit in vivo from GFPLC3, contrasting with studies involving coronaviruses, arteriviruses and flaviviruses, in which the authors found no effect of GFP-LC3.Citation22,27,28 Moreover, the results obtained in the triple-transgenic mice () strongly support the hypothesis that pathways other than autophagy must be able to provide membrane scaffolds that allow efficient enteroviral replication. The studies below were carried out to better define how CVB3 exploits the various possible forms of LC3 in vivo, and to better understand the roles of autophagy-dependent and -independent replication in the CVB3 life cycle.
Figure 1. For figure legend, see page 1392.
Design and characterization of 3 rCVB3s to dissect the impact of various forms of LC3
We began by generating 3 new rCVB3s, each of which had a unique effect of the availability of autophagy-related proteins in virus-infected cells. shows the 3 forms of LC3, and their interrelationships. The precursor form, proLC3, is encoded by mammalian orthologs of the yeast Atg8 gene and, in mice, is a 125 amino-acid precursor protein, with the indicated C-terminal residues FGTAMAV. ProLC3 is cleaved by a cysteine protease, ATG4B, after the Gly120 residue (shown in blue), producing LC3-I. The newly exposed Gly120 residue on LC3-I becomes covalently attached to ATG7, then is transferred to ATG3. Assisted by the ATG12-ATG5 complex, LC3-I-ATG3 is recruited to the ER membrane (shown diagrammatically as a lipid bilayer; the black dot indicates phosphatidyl ethanolamine (PE)). There, the LC3-I moiety undergoes a final transfer, forming an amide bond between its Gly120 residue and PE. This lipidated, membrane-resident, form of LC3 is termed LC3-II and, as explained in more detail below, its formation drives the expansion of the phagophore, a transient compartment that engulfs cytosolic materials and, ultimately, closes to form a double-membrane vesicle, the autophagosome. As indicated in the diagram (dashed arrow), LC3-II can be “recycled” by ATG4B, replenishing LC3-I levels. This occurs after formation of the mature autophagosome, at which time LC3-II becomes functionally redundant; the amide bond is cleaved by ATG4B, releasing LC3-I that can be reincorporated into developing autophagosomes.Citation29 In order to determine the importance of LC3 in CVB3 replication, we generated 3 different recombinant coxsackieviruses, each of which should have a different effect on LC3 content within an infected cell. First, proLC3-CVB3; this virus should produce abundant proLC3, which should feed into the normal cellular processing pathway, potentially increasing the intracellular quantities of proLC3, LC3-I and LC3-II. Second, proLC3G120A-CVB3, which encodes a mutated form of proLC3 in which the Gly120 residue is replaced by an alanine (G120A); this mutation prevents cleavage by ATG4B.Citation30 The resulting virus should produce a large quantity of the mutated protein, but this should not be processed to LC3-I and LC3-II; hence, only endogenous (host) LC3-I and LC3-II should be present. Finally, third, a virus (Atg4BC74A-CVB3) that encodes an enzymatically-inactive form of ATG4B. This protein, carrying a C74A mutation, binds to and sequesters any available LC3-I, limiting its conversion to LC3-II.Citation31 In cells infected by this virus, the endogenous (host) ATG4B should cleave the endogenous proLC3, generating LC3-I; the virus-produced ATG4BC74A protein should sequester this molecule, causing it to accumulate at the expense of LC3-II.
Figure 2. Design and characterization of 3 rCVB3s to dissect the impact of various forms of LC3. (A) A schematic summary of proLC3 processing into LC3-I and LC3-II is shown, and includes the relevant amino acid residue at the Ct of the protein (see text). The Ct residues of the mutated form, proLC3G120A, is shown at the top-right. (B) One-step growth curves of rCVB3s and WTCVB3 are shown (HeLa cells, MOI = 10). (C) Protein was harvested from HeLa cells (8 h p.i.), and the expression of the viral protein VP1, and of GAPDH control, were assessed by western blot. (D) The samples were applied on a separate gel, this time using differing loading amounts, for reasons explained in the text. The relative loading amounts can be determined from the GAPDH data (lower blot). For all viruses, the quantities of autophagy-related products were determined (upper blot) and, for the Atg4BC74A virus, the amount of ATG4BC74A expression was evaluated (middle blot). Control samples were included from uninfected mouse embryonic fibroblasts (MEF), some of which had been incubated with the late autophagy inhibitor bafilomycin A1 (Baf). (E) Equal quantities of protein from cells infected either with proLC3-CVB3, or proLC3G120A-CVB3, were applied to a polyacrylamide gel, and for each, the gel region containing proteins of 12 to 18 KDa was excised. Proteins were extracted, and analyzed using nano LC-MS/MS. The predicted protein sequences from the N termini of the recombinant polyproteins are shown. Native viral amino-acids are in lower case, and recombinant (proLC3 or proLC3G120A) residues are in upper case. Red font indicates peptides that were identified by nano LC-MS/MS; the uncleavable FATAMAV sequence in the proLC3G120A protein (see text) is underlined. (F) Summary of the observed autophagy-related changes for each of the 3 new rCVB3s.
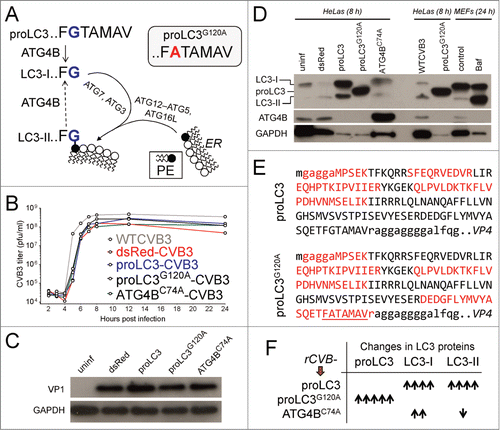
HeLa cells were infected with these 3 rCVB3s, as well as with a control recombinant expressing dsRed, or with WTCVB3, and one-step growth curves were carried out. As shown in , the 3 new recombinants grew ∼3-fold better than the existing dsRed rCVB3 but, compared with WT virus, titers of all 3 new rCVB3s were slightly reduced (by ∼3-fold). Next, we evaluated protein synthesis by western blot. HeLa cells were infected at high multiplicity of infection (MOI; 10) and cell lysates were prepared 8 h later. As shown in , when approximately equal amounts of protein extract were applied to the gel (see GAPDH loading control), all of the viruses appeared to make similar quantities of viral protein (as reflected by VP1), consistent with the similar titers of infectious virus that were produced. Next, we evaluated the abundance of the various virus-encoded recombinant proteins, and of the host-derived autophagy-related and ATG4B proteins. When this was done as in , by applying equal quantities of protein to a gel, the vast differences in abundance of virus-encoded proteins compared to cell-encoded proteins made it impossible to visualize them all on the same blot. For this reason, using the same cell extracts as shown in , we carried out a western blot using a gel () in which the lanes were intentionally loaded with different amounts of total protein, as demonstrated by the GAPDH data. Using this approach, we conclude that the proLC3 virus is making protein which is being processed (by endogenous ATG4B) to generate LC3-I which, in turn, undergoes normal processing to generate LC3-II. proLC3G120A-CVB3 as expected, shows a single autophagy-related band (). It is notable that the proLC3G120A protein—which is longer than the LC3-I protein—migrates somewhat faster. This is consistent with reports from other laboratories, which have shown that proLC3 from mouse, rat, and human all migrate more rapidly than their processed (LC3-I) forms;Citation32,33 a recent study attributes this aberrant migration to the C-terminal (Ct) amino acids (distal to Gly120).Citation33 Although those published data suggested that the protein made by the proLC3G120A virus was likely to be intact proLC3G120A, we chose to further examine this virus-encoded recombinant protein. Proteins were isolated from HeLa cells infected by either the proLC3 or proLC3G120A viruses, separated by gel electrophoresis, and subjected to nanoscale liquid chromatography coupled to tandem mass spectrometry. The relevant sequence from proLC3-CVB3 is shown (), and the protein isolated from the infected cells contained several of the expected peptides (red lettering; upper case = LC3 amino acids, lower case = viral amino acids). The protein isolated from cells infected with the proLC3G120A virus (bottom sequence), contained a peptide (DEDGF… … …FATAMAVr) that spanned the FATAMAV sequence (underlined); the equivalent peptide encoded by proLC3-CVB3 (DEDGF… … …FGTAMAVr) was not detected by mass spectrometry because the proLC3 protein had been cleaved by host cell ATG4B. Thus, we conclude that the anomalously migrating band is, indeed, proLC3G120A. The absence of a band at the equivalent position in cells infected with the proLC3 virus (lane 3) suggests that the virally-produced proLC3 is being very rapidly processed into the slower-migrating LC3-I. Finally, the Atg4BC74A virus produced abundant protein of the correct size (). In these cells, all LC3 proteins are host-encoded and, as predicted, the presence of the mutated ATG4B led to an increase in LC3-I (compare lane 5 to lane 1 [uninfected], lane 2 [dsRed-CVB3] or lane 7 [WTCVB]); the increase in LC3-I content in cells infected by the Atg4BC74A virus is much more marked than it may at first appear, because one must factor in the relative quantities of total protein that had been applied to each of these lanes (see GAPDH, bottom row). Taken together, these results demonstrate that the new rCVB3s replicate efficiently, and that the recombinant proteins that they encode function as intended in tissue culture. The effects of the 3 new rCVB3s on the abundance of autophagy-related proteins in infected cells are summarized in .
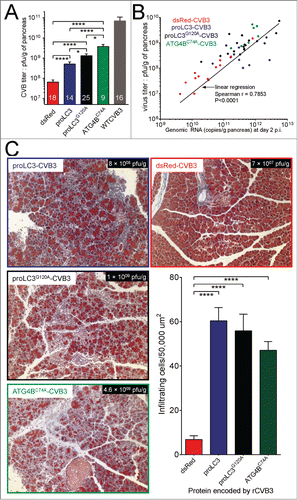
Virus-encoded autophagy-related proteins increase viral replication and pancreatitis, in WT mice
Next, we initiated the in vivo evaluation of the viruses. C57BL/6 mice were infected intraperitoneally either with 107 pfu of the rCVB3s or with 104 pfu of WT CVB3, and pancreata were harvested 2 d later for analysis. Pancreatic titers of proLC3-CVB3, proLC3G120A-CVB3 and Atg4BC74A-CVB3 were substantially higher (by ∼9-, 17- and 55- fold, respectively) than the titer of the control rCVB3 encoding dsRed (), suggesting that the “proviral” in vivo effect of an additional host allele encoding GFP-LC3 () can be recapitulated in WT mice by viral delivery of autophagy-related proteins. Although none of the rCVB3s encoding autophagy-related proteins achieved WT levels of replication, when compared with dsRed-CVB3 their increases in replication were highly statistically significant (P < 0 .0001; WT CVB3 titers were significantly different from all rCVB3s, P values not shown). Quantitative PCR was carried out, and showed that, for all 4 rCVB3s, there was a near-linear relationship between genomic RNA abundance and infectious virus titer (). Finally, the presence of excess autophagy-related proteins had a dramatic effect on the extent of pancreatitis (; Masson trichrome stain). dsRed-CVB3, like most rCVB3s, is attenuated in vivo, and did not cause severe pancreatitis in WT mice. In contrast, the pancreata of mice infected with any of the remaining 3 rCVB3s showed substantial inflammatory infiltration, and acinar cell hypochromicity, reminiscent of the florid disease that is induced by WT CVB3.
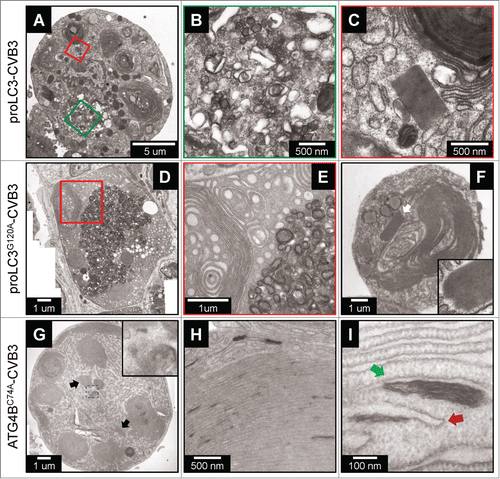
Ultrastructural changes in cells infected with rCVB3s expressing autophagy-related proteins
Picornaviral infection induces a number of characteristic ultrastructural changes that can be identified using transmission electron microscopy. We have previously reported that WTCVB3 triggers dramatic increases in the abundance of autophagy-like vesicles and single membrane vesicles, and causes extensive ER membrane rearrangements; moreover, paracrystalline lattices are visible in WTCVB3-infected cells.Citation10,12 The above changes are much less prevalent in cells infected with dsRed-CVB3, suggesting that they may develop in parallel with the extent of virus replication. Therefore, we next evaluated the ultrastructural changes that occur in pancreatic acinar cells infected with the 3 rCVB3s. Acini were isolated from WT mice, and were infected at MOI=100. Eighteen h later, cells were processed for, then analyzed by, transmission electron microscopy. Representative results are shown (). All 3 of these rCVB3s caused dramatic structural changes that, for the proLC3-CVB3 and proLC3G120A-CVB3 viruses, were very similar to those previously reported for WTCVB3.Citation10,12 In acinar cells infected with proLC3-CVB3, DMVs were abundant (), and we observed many paracrystalline lattices, one of which is shown in . DMVs also were numerous in cells infected with the proLC3G120A virus, and there was extensive ER reorganization and “swirling” (); these cells, too, contained many large lattices (, indicated by a white arrow, and enlarged in inset). Pancreatic acinar cells infected with the Atg4BC74A virus also contained many lattices (black arrows in ), and this virus caused some characteristic changes, distinct from cells infected with the WT virus, or with the other rCVB3s. DMVs, although not entirely absent, were substantially less abundant; and, in these cells, much of the cytosol was taken up by swirled membranous material, with electron-dense “flecks” of ∼300 to 400 nm in length (inset in shows 2 such structures, adjacent to a lattice). These flecks are quite frequent, and are intimately associated with the ER membranous structures (). In a healthy cell, autophagosome biogenesis begins with the emergence, from the ER, of lipid structures named omegasomes, which develop into phagophores and these, in turn, close to form double-membrane autophagosomes. When visualized at still-greater magnification (), the morphology of some of these flecks is consistent with their being omegasomes (red arrow) or phagophores that have failed to properly mature (green arrow). Under normal circumstances, omegasomes and phagophores are transitory, rapidly developing into mature autophagosomes. However, accumulation of omegasomes and phagophores has been previously reported in cells transfected with a plasmid encoding the C74A mutant of ATG4B, and is thought to be caused by the sequestration of endogenous LC3-I and paralogs; the consequent reduction in LC3-II formation retards the growth and ultimate closure of phagophores, thereby inhibiting autophagy.Citation31 Thus, the Atg4BC74A-CVB3 virus mirrors these data, causing the accumulation of omegasomes and immature phagophores, together with the massive reorganization of intracellular membranes (). Compared to the other 2 rCVB3s (and to WTCVB3), there is a relative lack of DMVs but, despite this, the replication of the Atg4BC74A virus is very substantially enhanced compared to the dsRed-CVB3 virus control ().
Figure 4. Ultrastructural changes in cells infected with rCVB3s expressing autophagy-related proteins. Acinar cells isolated from pancreata of WT mice were infected (MOI 100) with each of 3 indicated rCVB3s: proLC3-CVB3 (A-C), proLC3G120A-CVB3 (D-F) or Atg4BC74-CVB3 (G-I). Eighteen h later, the cells were evaluated by transmission electron microscopy. n = 80 acinar cells infected with proLC3-CVB3; n = 85 acinar cells infected with proLC3G120A-CVB3; and n = 92 acinar cells infected with Atg4BC74A-CVB3 from 3 independent experiments. For each of the 3 different virus-infected cell types, the diameters of numerous DMVs (38 to 90) were measured; there was no significant difference in the sizes of the DMVs. Scale bars are included for all panels. Colored box(es) (if present) in the left-hand panel are shown at higher magnification in the adjacent panel(s). The white arrow in (F) indicates a paracrystalline lattice, shown at higher magnification in the inset. In (G), the 2 black arrows show paracrystalline lattices, and the area enclosed by the small dashed box is shown at higher magnification in the inset. In panel I, the red arrow points to an omegasome, and the green arrow to a developing phagophore.
Viruses differ in their ability to exploit GFP-LC3, and there appears to be a threshold effect
The foregoing data show that excess autophagy-related proteins provided either by the host () or by the virus ( and ) can upregulate CVB3 replication in vivo. Next, we determined if the effects of host-derived and virus-derived LC3 proteins were additive, by inoculating the 3 autophagy-related rCVB3s into mice carrying either one or 2 alleles of GFP-LC3. First, WT mice, or mice carrying one allele of GFP-LC3 were inoculated intraperitoneally with 104 pfu of WT virus, or with 107 pfu of the rCVBs, and pancreatic virus titers were determined at 2 d p.i. (). As was described in , dsRed-CVB3 showed a highly significant increase in the GFP-LC3 animals. The proLC3-CVB3 virus, too, showed a statistically-significant increase in titer. Thus, like WTCVB3,Citation12 these viruses appear to exploit the early stage of the autophagy pathway. However, neither proLC3G120A-CVB3 nor Atg4BC74A-CVB3 showed a statistically significant change in titer, suggesting that neither of these viruses are as capable of exploiting the autophagy pathway. Similar to WT mice (), the ratio of genomic RNA:infectious virus remained relatively linear in GFP-LC3 mice for all rCVB3s (). Finally, even for the 2 viruses (dsRed-CVB3 and proLC3-CVB3) that had benefited from the presence of one allele of GFP-LC3, the addition of a second host allele had no statistically significant impact on titers (), indicating that there is an upper threshold of LC3 content, above which virus can accrue no further benefit. The increased amount of GFP-LC3 protein in the tg/tg mice, compared to their hemizygous (WT/tg) littermates, was confirmed by western blot ().
Figure 5. Viruses differ in their ability to exploit GFP-LC3, and there appears to be a threshold effect. Two approaches were used to determine if the beneficial effects of host-derived and virus-delivered LC3 were additive. First, WT mice (black bars) or GFP-Lc3 mice (green hatched bars) carrying a single allele of GFP-LC3 were infected with the 4 indicated rCVB3s, or with WT CVB3, and were sacrificed at d 2 p.i. (A) Pancreatic viral titers were determined, and (B) for the rCVB3s, RNA genome quantities were measured using RT-PCR. Second (C), the replication of the 4 rCVB3s was evaluated in mice homozygous for GFP-Lc3 (i.e, carrying 2 alleles of the transgene, solid green bars) and was compared to the titers in mice with a single gene copy. (D) Upper panel: Mice hemizygous (WT/tg) and homozygous (tg/tg) for GFP-Lc3 were genotyped as described.Citation62 To determine if the GFP-Lc3 gene copy number was reflected in the amount of GFP-LC3 protein that was present, western blots were carried out on pancreatic protein extracts, and probed using an antibody to LC3, or to GAPDH (loading control). After normalizing for the amounts of GAPDH, the amount of GFP-LC3 in pancreata of tg/tg mice was found to be ∼2.8x higher than that of their WT/tg counterparts.
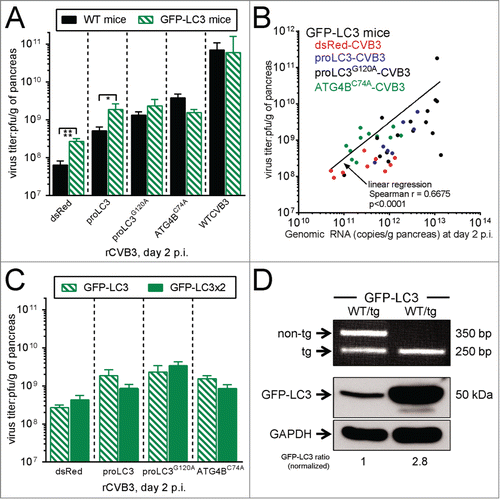
rCVB3-encoded ATG5 restores the functional deficiency in ElCre-Atg5f/f acinar cells, and enhances viral replication and pathogenesis
The above data provided a hint that WTCVB3 and the various rCVB3s might differ in their abilities to exploit the autophagy pathway. Therefore, we have asked if the in vivo effects of the various autophagy-related proteins were reliant upon an intact autophagy pathway, or conversely, if some of the autophagy-related proteins' effects might be independent of autophagy. We wished to compare the autophagy-dependence of our rCVB3s using our ElCre-Atg5f/f mice, and we considered it important to first determine if these mice were a valid model. We took a straightforward approach (), generating a new rCVB3 that encoded ATG5; this virus should rectify the genetic defect in the pancreatic acinar cells. We first confirmed the extent, and the cell-specificity, of Cre recombinase activity in uninfected ElCre-Atg5f/f mice, by backcrossing the mice against a Cre reporter line (JAX 7914,Citation34) in which Cre activity activates the transcription and translation of the Tomato fluorescent protein. Vibratome sections of liver (negative control) and pancreas were taken from the resulting ElCre-Atg5f/WT-7914 mice, and from their 7914+ littermates lacking ElCre. The sections were incubated with fluor-tagged phalloidin (which binds to F-actin, revealing the cytoskeleton, green) and Hoechst 33342 (staining nuclei blue), and images were captured by confocal microscopy. As shown in , in reporter mice lacking ElCre, the pancreas was macroscopically normal (inset), and Tomato signal was undetectable by fluorescence microscopy. In contrast, the pancreata of mice expressing ELCre (2 center panels) were visibly red (inset), and cytosolic Tomato signal (red) was present throughout the acinar pancreas, but not in the islets of Langerhans. This high degree of cell-specificity is reflected by the absence of Tomato signal in the liver (right panel). Thus, Cre activity is ubiquitous in, and specific to, pancreatic acinar cells. Next, we generated the new rCVB3 encoding the murine ATG5 protein, and determined if this could overcome the genetic defect, restore autophagy, and enhance viral replication and pathogenesis in vivo. Atg5-CVB3 infection of HeLa cells showed that this virus makes abundant ATG5, far in excess of the quantity of endogenous protein in uninfected cells, or in cells infected by dsRed-CVB3 (). Both ATG5, and cleaved ATG5 (cATG5) were present, and the ATG5 appeared to be functional, as there was an increase in the abundance of ATG12-ATG5 conjugate. To confirm that this virus-encoded ATG5 retained biological function, ElCre-Atg5f/f mice were infected with Atg5-CVB3 or with dsRed-CVB3 and, 2 d later, the pancreata were harvested. Western blot analysis () showed that, as expected, the lack of host ATG5 in the pancreata of dsRed-CVB3 infected ElCre-Atg5f/f mice prevented ATG12-ATG5 complex formation but, in contrast, this complex was readily detected in the pancreata of animals infected with Atg5-CVB3. Thus, not only is the ATG5 protein made in abundance by the recombinant virus; it also retains the key functional attribute of covalent association with ATG12. When compared to dsRed-CVB3, this recombinant virus replicated to higher titers in ElCre-Atg5f/f mice () and, unlike dsRed-CVB3,Citation12 Atg5-CVB3 also caused marked pancreatitis (; the number of infiltrating cells per 50,000 μm2 is shown). Thus, ElCre-Atg5f/f mice are a valid model in which to evaluate the autophagy-dependence of virus-delivered autophagy-related proteins.
Figure 6. rCVB3-encoded ATG5 restores the functional deficiency in ElCre-Atg5f/f acinar cells, and enhances viral replication and pathogenesis. (A) The extent, and cell-specificity, of ELCre activity was determined by confocal microscopy of vibratome sections from the F1 cross of ElCre-Atg5f/f mice against JAX 7914 mice (which carry a reporter cassette that, when cleaved by Cre recombinase, causes expression of the Tomato fluorescent protein). Genotyping showed that, as expected, all mice in the resulting litter were Atg5f/WT-7914+; and ∼50% also carried ElCre. Pancreatic samples were taken from mice expressing ELCre (2 central images, the rightmost of which shows a higher magnification including an islet of Langerhans). Negative controls were (i) pancreatic tissue from a mouse lacking ElCre (left image) and (ii) liver from an ELCre-expressing mouse. Red = Tomato; Blue = nuclei (Hoechst 33342 dye); Green = F-actin (phalloidin staining; phalloidin binds to F-actin, thereby revealing the cytoskeleton). Images are representative, and are from 2 independent experiments (n = 4 mice per group). Twenty-six pancreatic fields were evaluated for ElCre mice, and 2 fields for the Cre-negative control mice. (B) HeLa cells were infected with Atg5-CVB3 or with dsRed-CVB3, and a western blot was probed for ATG5, or for GAPDH. (C) ElCre-Atg5f/f mice were infected with Atg5-CVB3 or with dsRed-CVB3 and, 2 d later, their pancreata were harvested, and the amount of ATG12-ATG5 complex was determined by western blot. The complex is absent from the pancreata of mice infected with dsRed-CVB3, but is abundant in the pancreata of Atg5-CVB3-infected animals, demonstrating that the virus-encoded ATG5 is biologically active. (D). Virus titers show that the provision of ATG5 by rCVB3 increases viral replication in the pancreata of ElCre-Atg5f/f mice. (E). This increased replication is reflected by marked pancreatitis (Masson trichrome stain; n = 7 mice, in 5 independent experiments; quantitation shown in bar graph).
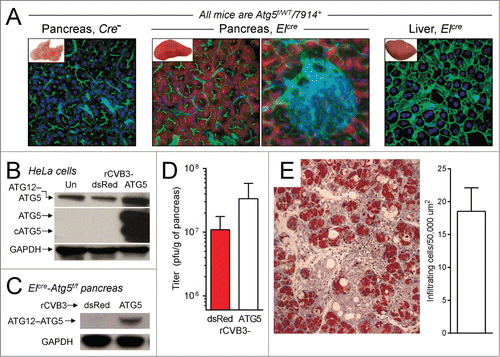
rCVB3s encoding different autophagy-related proteins show differing dependence on an intact autophagy pathway: Evaluating the involvement of SEL1L
Next, we evaluated the autophagy requirements of the 3 rCVB3s that encode autophagy-related proteins. ElCre-Atg5f/f mice were infected with dsRed-CVB3 (control), or with each of the 3 autophagy-related rCVB3s, and pancreata were harvested 24 or 48 h later, for titration (). At d 1 p.i., when compared to the dsRed virus, the proLC3G120A and Atg4BC74A viruses had replicated to significantly higher titers (0.01 ≥ P > 0 .001), indicating that the increased abundance of proLC3G120A, or of sequestered LC3-I, was beneficial even in the absence of ATG5. These data, combined with the observation that neither of these 2 viruses could exploit GFP-LC3 (), strengthen the conclusion that the enhanced replication of these 2 viruses may rely on something other than the autophagy pathway. In contrast, at d 1 p.i. the proLC3 virus titer was almost identical to that of the dsRed control virus; and by d 2 p.i., the titer of proLC3-CVB3 was marginally higher than that of dsRed-CVB3. This suggests that the positive impact of virus-delivered proLC3 is substantially—but not wholly—reliant on ATG5, indicating that the autophagy pathway plays a significant part in the life cycle of this agent.
Figure 7. rCVB3s encoding different autophagy-related proteins show differing dependence on an intact autophagy pathway: evaluating the involvement of SEL1L. (A) To determine if the beneficial impact of autophagy-related proteins was dependent on an intact autophagy pathway, ElCre-Atg5f/f mice were infected with each of the 3 new rCVB3s, or with dsRed-CVB3 as a control. Pancreata were harvested at d 1 or 2 p.i., and virus titers were determined. Next, for reasons explained in the text, we investigated the binding of LC3-related proteins to the ERAD pathway protein SEL1L. HeLa cell lysates were prepared from uninfected cells (Un) and from cells infected by each of the 4 indicated rCVB3. (B) Aliquots of these lysates were subjected to gel electrophoresis, and western blots were carried out using antibody to LC3, or to GAPDH (loading control). As was explained for , the great abundance of LC3-related proteins produced by the proLC3 and proLC3G120A viruses required that less total protein be applied to those lanes. Identification of each band is facilitated by the inclusion of a colored dot; the data are nearly identical to those shown in . (C) Equal amounts of each protein lysate were immunoprecipitated with antibody to SEL1L. The precipitates were subjected to gel electrophoresis, and western blots were carried out using antibody to LC3, or to SEL1L. The observed coimmunoprecipitation of LC3 and SEL1L was confirmed in 5 independent experiments. (D) As an additional control for the specificity of immunoprecipitation (see text), the lysates were immunoprecipitated using an antibody to the ER resident protein CANX then, after electrophoresis and blotting, were probed for the presence of LC3-related proteins (top panel) or CANX (bottom panel).
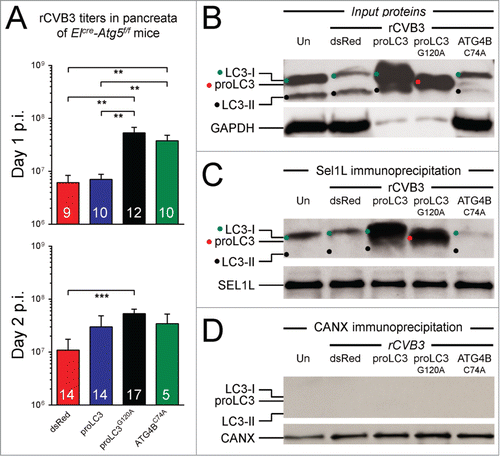
The data in , together with the EM data from and the GFP-LC3 data from , show that the rCVB3s encoding autophagy-related proteins differ in at least 2 ways: first, in the intracellular membrane changes that they induce (both of the LC3-encoding viruses induce many DMVs, while the Atg4BC74A virus induces fewer, and instead increases the frequency of immature lipid structures, along with massive membrane reorganizations); and, second, in their ability to exploit the autophagy pathway via ATG5 (strong for the proLC3 virus; weak for the proLC3G120A and Atg4BC74A viruses). The ability of the proLC3 virus to exploit the early stage of the autophagy pathway is probably mediated by the large quantities of LC3-I (and LC3-II) in cells infected by this virus (), which leads to the formation of DMVs. However, it is difficult to understand how the proLC3G120A virus could induce DMVs via the autophagy pathway, because cells infected with this virus do not contain large amounts of LC3-I or LC3-II (). Therefore, we sought an explanation for how this virus could induce DMV formation in the absence of abundant LC3-I, and in a manner that is relatively independent of ATG5. Furthermore, we wondered how the sequestration of host LC3-I by the ATG4BC74A protein could lead to an ATG5-independent improvement in replication.
Others have shown that DMVs can be induced by some viruses in an autophagy-independent manner. As noted in the Introduction, like enteroviruses, coronaviruses use LC3-coated DMVs as replication scaffolds. However, these DMVs appear to be derived not from the autophagy pathway, but instead from the ERAD pathway. These DMVs develop from small ER-derived vesicles termed EDEMosomes,Citation22 so named because they contain abundant quantities of the protein EDEM1 which forms a macromolecular complex with additional proteins, including the ER-resident transmembrane protein SEL1L. Using a plasmid encoding proLC3 carrying a G120R mutation (which, like the G120A mutation employed herein, prevents cleavage by ATG4B), it was shown that proLC3G120A can bind to EDEMosomes.Citation22,23,25 SEL1L plays a key role in ER homeostasis in vivo, particularly in pancreatic acinar cells,Citation36 which are primary targets of CVB3. Therefore, we hypothesized that SEL1L might represent a binding target for nonlipidated forms of LC3.
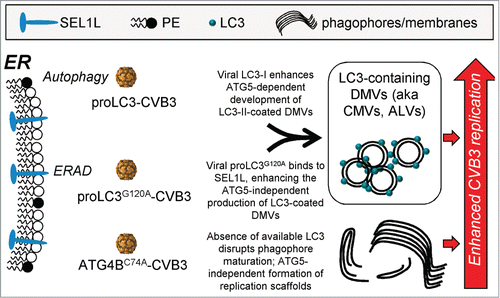
To test the above hypothesis, HeLa cells were infected for 8 h with each of the 3 new rCVB3s, or with dsRed virus, and protein lysates were prepared. Aliquots were analyzed by western blot for their content of LC3-related proteins () and, as expected, the results paralleled those shown in . Next, aliquots of the same lysates were precipitated using antibody against SEL1L. The precipitated proteins were subjected to gel electrophoresis, and the presence of LC3 proteins, and of SEL1L protein, was determined by western blot. As shown in , the quantity of SEL1L was near-identical in all 5 samples, demonstrating the efficiency and fidelity of the immunoprecipitation. Probing for LC3 () showed that some of this protein coimmunoprecipitated in pancreatic cells from uninfected mice; thus, the SEL1L-LC3 interaction is physiological, occurring without viral intervention. There was no significant change in the amount of coimmunoprecipitated LC3-I from cells infected with dsRed-CVB3. In contrast, a large quantity of LC3-I was captured from cells infected with proLC3-CVB3, indicating that some of the overabundant LC3-I that is produced by ATG4B cleavage is diverted to SEL1L. The proLC3G120A protein, too, was readily detected in SEL1L immunoprecipitates, thus potentially providing an alternative route by which LC3 can stably attach to the ER membrane. Thus, the ATG5-independent production of DMVs in cells infected with proLC3G120A-CVB3 presumably proceeds by this alternate route, i.e., the ERAD pathway. However, in cells infected with Atg4BC74A-CVB3, the sequestered host LC3-I—although present at an elevated level ()—did not bind, at an increased level, to SEL1L. These data suggest that the enhanced replication of this virus can be attributed to neither the autophagy nor the ERAD pathways, and may be completely independent of LC3. Note that the LC3-II that was present in the original lysates (panel B) is absent from the SEL1L precipitates (panel C). This has 2 implications. First, that this protein—which, by definition, should be covalently attached to the membrane of developing autophagosomes—does not associate with SEL1L; and, second, that the observed coprecipitation of the LC3-I proteins with SEL1L represents a specific protein-protein interaction, rather than the nonspecific precipitation of ER-associated proteins. However, to confirm the specificity of the LC3 precipitation, we also subjected the protein lysates to a different immunoprecipitation, this time using antibody to the ER integral membrane protein CANX (calnexin). CANX was efficiently precipitated, but the LC3-related proteins did not coprecipitate () confirming that the coimmunoprecipitation of SEL1L and LC3 represents a specific interaction. These data, combined with others in this paper, have led us to develop an overall hypothesis that is represented diagrammatically in , and is discussed below.
Figure 8. Diagrammatic summary: CVB3 can exploit cellular membranes from a variety of sources. We propose that CVB3 can exploit 3 distinct sources of membrane scaffolds, all arising from the ER. Two of them are constitutive cellular pathways (autophagy and ERAD), and both of them (i) involve the attachment, covalently or otherwise, of LC3 to the ER membrane; and (ii) culminate in the production of abundant DMVs. The third pathway is extant only when LC3-I is sequestered by ATG4BC74A, giving rise to immature phagophores and reorganized membranes. Only the first of these 3 pathways is ATG5-dependent.
Discussion
RNA viruses frequently co-opt cellular proteins and lipid membranes to support their replication, and several cellular pathways have been implicated, serving either as prefabricated scaffolds, or as sources to be remodeled to suit the virus's needs (reviewed in ref. 37). Viruses can alter cellular membranes not only physically, but also chemically, by changing their lipid composition. Phosphatidylinositol-4-kinase type III β plays a key role in modifying host cell membranes, catalyzing the production of the lipid phosphatidylinositol-4-phosphate, which facilitates signaling and vesicle formation, and the enzyme also is recruited, by the CVB3 3A protein, to the sites of RNA replication, creating a microenvironment rich in phosphatidylinositol-4-phosphate lipids.Citation21 Virus-driven modification of lipid composition also can occur indirectly; Dengue virus regulates cellular lipid metabolism by modulating the autophagy pathway.Citation38
Our laboratory has previously shown that the enterovirus CVB3 can modify the autophagy pathway in vivo and that, in the absence of ATG5, replication of WT CVB3, and dsRed-CVB3, are markedly diminished.Citation10,12 In carrying out those studies, we observed that mice expressing a single copy of the GFP-LC3 protein were better able to support the replication of normally attenuated rCVB3, suggesting that an excess of that protein might directly benefit the virus. Here, we have confirmed this observation (), and extended it by showing that provision of the GFP-LC3 protein allows WTCVB3 to efficiently circumvent the potentially disruptive impact of ATG5 depletion (). These findings indicate that GFP-LC3 mice, which have been used extensively to characterize the effect of deletions of autophagy-related genes, should be used with caution when evaluating viral pathogenesis; the same argument applies to immortalized tissue culture cells in which GFP-LC3 quite commonly has been expressed to facilitate evaluation of viral effects on autophagy. From those data, and guided also by recent publications that have shown that the nonlipidated form of LC3 may be exploited by several different RNA viruses, we have evaluated the outcomes of in vivo infections with rCVB3s encoding different autophagy-related proteins (proLC3, proLC3G120A, or ATG4BC74A). All 3 of these viruses are at a substantial replicative advantage in autophagy-intact cells when compared to a control recombinant virus dsRed-CVB3, multiplying to higher titers, and causing more severe disease (). Furthermore, paracrystalline arrays were abundant in acinar cells infected with each of the 3 viruses ( and not shown). However, the viruses displayed unique features in vivo. proLC3-CVB3 produced abundant LC3-I and LC3-II (), induced DMVs (), could exploit an excess of GFP-LC3 (), was relatively dependent on ATG5 (), and the processed protein (LC3-I) bound to SEL1L (). proLC3G120A-CVB3 produced abundant uncleaved protein (), induced DMVs (), was unable to benefit from GFP-LC3 (), was independent of ATG5 (), and the encoded protein bound to SEL1L (). Finally, ATG4BC74A-CVB3 caused sequestration of host LC3-I (), induced immature lipid structures and extensive membrane reorganization (), did not utilize GFP-LC3 (), was independent of ATG5 (), and the sequestered host LC3-I did not show increased binding to SEL1L (). Based on these findings, we have developed the following hypothesis, which is represented diagrammatically in .
In broad terms, we propose that CVB3 is biologically flexible in vivo, being able to co-opt the cell membranes that will provide it with the greatest advantage within the particular cell type in which it is present. This explains why depletion of ATG5 reduces, but by no means prevents, the replication of WTCVB3;Citation12 the virus can utilize a number of different membrane sources, so the loss of one of them (in that case, early autophagy) is not a terminal event. We suggest that CVB3 can employ LC3 as the entry point to 2 different cellular pathways, autophagy and ERAD, and that both pathways are exploited in acinar cells. Recent in vivo work has shown that the SEL1L protein is especially abundant, and functionally important, in pancreatic acinar cells.Citation36 An inducible knockout approach was used to delete SEL1L from most somatic cells in adult mice, after which the animals developed pancreatic pathology leading to profound, and ultimately lethal, malabsorption. These data suggest some intriguing possibilities. Perhaps the abundance of SEL1L in acinar cells contributes to the tropism of coxsackievirus for these cells. Conversely, perhaps the interactions between SEL1L and the recombinant LC3 proteins in proLC3-CVB3 and proLC3G120A-CVB3 viruses disrupts SEL1L function, contributing to ER cell death.
We propose that both the autophagy and the ERAD pathways culminate in the generation of abundant LC3-containing double-membrane vesicles that can support viral replication complexes. By this hypothesis, the proLC3-CVB3 virus exploits the autophagy pathway by providing abundant substrate for host cell ATG4B, producing a large amount of LC3-I that, by binding to PE, accelerates the ATG5-dependent development of DMVs. At day 1 p.i. of ElCre-Atg5f/f mice, the titer of proLC3-CVB3 lagged behind those of the other 2 rCVB3s, and was almost identical to the titer of the dsRed control (). At first blush, the reason for this delay is obvious: in the absence of ATG5, the virus-derived LC3-I cannot be processed into LC-II. However, the data in show that the virus-derived LC3-I can bind to SEL1L, so one must ask: why did this interaction not allow the virus to replicate to a higher titer on d 1? During normal autophagy, LC3-I is only transiently available; after being generated by ATG4B, it is covalently attached to ATG7, and then is passed on (again, covalently) to ATG3, before being linked to PE. In ATG5-deficient cells, this chain is broken; in the absence of ATG5, the LC3-I-ATG3 fusion protein will accumulate, because transfer to PE cannot occur. Hence, we speculate that, in the ElCre-Atg5f/f mice, the ATG7-ATG3 pathway serves as a sink that, until saturated, consumes the virus-derived LC3-I, thereby preventing its binding to SEL1L. Only when the sink is full does “free” LC3-I become available for SEL1L binding, allowing the proLC3 virus to slightly outstrip the dsRed control by d 2 p.i. (). This argument also may explain our published observations in which we compared WTCVB3 titers in the pancreata of WT mice to those in ElCre-Atg5f/f mice, and found a large (>2000-fold) difference at d 1 p.i., which declined over the ensuing days, reaching only ∼10-foldby d 4.Citation12 Those findings also reinforce the general conclusion we reach herein: removal of a favored replication substrate may delay viral replication, but it does not abort it altogether. The enhanced replication of proLC3G120A-CVB3 (compared to the dsRed-CVB3 control) can be ascribed to the increased protein binding to SEL1L (), driving the generation of EDEMosome-like DMVs. Moreover, our hypothesis explains why replication of this virus—unlike proLC3-CVB3—is not delayed in ATG5-deficient mice: the proLC3G120A protein cannot be drawn into the ATG7-ATG3 sink described above, and so it can immediately attach to SEL1L.
We argue that, in normal cells, the proLC3 and proLC3G120A viruses both cause a rapid increase in the quantity of membrane-bound LC3 (either as LC3-II, or complexed to SEL1L). What happens next? A very recent studyCitation39 has shown that, in uninfected cells, autophagy begins at the ER membrane, with the loose attachment of the ATG12-ATG5 complex. This recruits commonly termed “free” LC3-I (although in reality, it is covalently attached, via its Gly120 residue, to ATG3), and the enzymatic activity of the ATG12-ATG5 complex transfers the LC3 moiety to PE, generating LC3-II. This covalently-attached version of LC3 then attracts more ATG12-ATG5 to the membrane, leading to a positive feedback loop that causes the very rapid accumulation of ATG12-ATG5 and LC3-II on the membrane. This, in turn, triggers the attachment of ATG16, forming a “mesh”, and subsequent deformation of the membrane leads to the expansion of the phagophore and, ultimately, its closure to form an autophagosome. Applying these observations to cells infected with our rCVB3s, we consider it probable that, for both proLC3-CVB3 and proLC3G120A-CVB3, the increased abundance of membrane-bound LC3 (either as LC3-II, or as proLC3G120A-SEL1L) acts as a “nucleation site” that recruits the ATG12-ATG5 complex; this triggers the positive feedback loop involving LC3I, ultimately leading to DMV formation. What of the Atg4BC74A virus, which appears incapable of exploiting either route? We think it likely that, for this virus, the electron-dense lipid structures (omegasomes or immature phagophores, ) and/or the extensive whorled membrane serve as the supports for replication in an LC3-independent manner.
In all of the above cases, the nature of the interaction between CVB3 and the various membranes remains to be determined. One obvious candidate for this function is the viral 3A protein, which—in the case of poliovirus—is membrane-tethered and was proposed, some time ago, as the lipophilic anchor of the viral replication complex.Citation40 More recent work has proven that the CVB3 3A protein is, indeed, located at viral replication sitesCitation21 and, as noted above, our laboratory has shown that this protein can have dramatic effects on intracellular membranes.Citation18,19 Interestingly, others have shown that a single mutation in the CVB3 3A protein allows the virus to bypass its use of PI4KIIIβ, yet still replicate quite well;Citation41 this, too, strongly suggests that coxsackievirus can use a number of different strategies to maximally exploit the host cell. Finally, the golgi brefeldin A resistant guanine nucleotide exchange factor 1 protein is known to interact with the 3A protein, and prepares ER membranes for CVB3 replication, yet siRNA inhibition of this protein has been reported to cause only an ∼2- to ∼4-fold reduction in replication in tissue culture cells.Citation42 These findings are consistent with growing evidence that is blurring the lines of demarcation that were thought to limit a given RNA virus to only one particular source of cellular membrane. Such plasticity was first noted for tombusviruses, positive-sense RNA viruses that infect plants. These viruses normally utilize peroxisome membranesCitation43 but, in the absence of that source, they can switch to ER membranesCitation44 and, perhaps, even to mitochondrially derived lipids.Citation45 The concept may apply, too, to coronaviruses. These viruses are thought to replicate independently of the autophagy pathway, and the role of EDEMosomes and LC3 in coronavirus replication has been reported by the Molinari group.Citation22,23 Nevertheless, careful evaluation of published data suggests that a role for autophagy should not be completely excluded. An initial report concluded that viral replication was defective in embryonic stem cells deficient of ATG5, and was restored by transfection of an ATG5-encoding plasmid.Citation46 However, a subsequent tissue culture analysis, using a variety of different cell types, came to the opposite conclusion,Citation47 and a more recent study using ATG7-deficient cells showed that replication of the coronavirus mouse hepatitis virus was not inhibited.Citation22 These more recent data suggest that autophagy is not generally required for coronavirus replication, but it remains possible that the virus can exploit the autophagy pathway if forced to do so (e.g., in specific cell types, or in circumstances where the normal source of membranes is less available). LC3 also is utilized in an autophagy-independent manner by other RNA viruses, including an arterivirusCitation27 and a flavivirus.Citation28 In the latter case, siRNA against SEL1L had no demonstrable effect on JEV yield, while siRNA targeted to LC3 had a significant effect. The factor common to all of the observations, in the present study and in the cited papers, is that autophagy-related proteins can be targeted by a variety of very different RNA viruses. Perhaps the most remarkable evidence that LC3 can play a profound role in RNA virus biology comes from studies of pestiviruses, some of which actually incorporate host LC3-coding sequences into their RNA genome, thereby increasing viral pathogenicity.Citation48,49 Herein, we propose that enteroviruses, coronaviruses, flaviviruses, and pestiviruses (and, perhaps, other RNA viruses) may use LC3 as a skeleton key to access 2 different pathways, each of which can provide DMVs. However, our observations with the Atg4BC74A virus indicate that CVB3 may be even more eclectic in its ability to exploit abundant intracellular membranes, and may be entirely independent of LC3. Taken together, these data suggest that CVB3 may exploit any available membranes, regardless of their source. This, in turn, has an important therapeutic implication: it complicates any attempt to mitigate viral diseases by targeting individual pathways, such as autophagy or ERAD, because the virus may simply use other membranes to support its replication. In this light, it is interesting to consider a recent study in which the authors screened a library of small-molecule inhibitors in an attempt to identify candidate drugs that would target host proteins involved in enteroviral replication, thereby preventing the related diseases.Citation50 The authors identified several candidates that quite effectively blocked the replication of CVB3 and poliovirus in vitro but, when one of the drugs was evaluated in vivo using the mouse model of paralytic poliomyelitis, in which control mice developed the disease on d3, the authors observed only a delay in paralysis; 100% of mice developed paralysis on d4 p.i. This brief delay in disease did not result from the emergence of drug-resistant viral mutants, indicating that the virus had evaded the drug's effect by other means, perhaps by exploiting an alternative, noncanonical, replication pathway. The “proviral” effect of LC3 and related proteins, too, has clinical implications. LC3 is upregulated in a number of pathological conditions and, in theory, this may render the host more vulnerable to positive strand RNA virus infection. Evidence exists to support this contention. LC3 accumulates in Paneth cells from patients with Crohn disease,Citation51 and this may be explained by a reduction in ATG4B, which has been reported to occur in affected areas of the colon in inflammatory bowel disease.Citation52 Intriguingly, a recent investigation of enteroviral infection during Crohn disease identified CVB or echovirus in the crypt epithelial cells all 24 patients; no virus was detected in the intestine of any of the 6 controls who had undergone emergency surgery for acute volvulus.Citation53 Additional indirect evidence supports the notion that chronic inflammation may upregulate LC3, rendering individual host cells more susceptible to productive RNA virus infection. Increased autophagy, and higher levels of proLC3 expression, have been observed in lungs of patients with chronic obstructive pulmonary disease,Citation54 and respiratory viral infections, especially with rhinoviruses, are a major cause of disease exacerbations.Citation55 Finally, ATG4B is an emerging target for treatment of various cancers, and inhibitors are being actively developed.Citation56 If the above speculations prove correct, they counsel caution in the use of such compounds, as the resulting increase in intracellular proLC3 may increase susceptibility to RNA virus infections.
Materials and Methods
Generation of pancreatic acinar ATG5-deficient mice
C57BL/6 mice were purchased from the Scripps Research Institute (TSRI) breeding facility or from Jackson Laboratories. Mice with pancreatic acinar cell-specific deletion of Atg5 (ElCre-Atg5f/f mice) were generated as we have previously described.Citation12 Homozygous GFPLc3 transgenic mice (GFP-LC3Tg/Tg)Citation57 were obtained from Riken Bio Resource Center (Koyadai, Japan). For some experiments, these mice were bred with wild-type C57BL/6 mice to generate heterozygous transgenic (GFP-LC3Tg/WT) mice. Additional breeding, with appropriate screening, allowed us to generate (GFPLc3)-ElCre-Atg5f/f mice and (GFPLc3)-Atg5f/f mice. To evaluate the extent and cell-specificity of Cre expression within the pancreas, ElCre-Atg5f/f mice were crossed with a Crereporter strain (A114, JAX 007914)Citation34 to obtain ElCre-Atg5f/WT-7914 animals. Mice were genotyped by PCR as previously described (Alirezaei et al., 2012; Alirezaei et al., 2010; Hara et al., 2006).
Viruses and infections
The wild-type CVB3 used in these studies is a plaque-purified isolate (designated H3) of the myocarditic Woodruff variant of CVB3.Citation58 Plasmid pH3, encoding a full-length infectious clone of this virus,Citation59 was provided by Dr. Kirk Knowlton (University of California, San Diego). Four rCVB3 (proLC3-CVB3, proLC3G120A-CVB3, Atg4BC74A-CVB3 and Atg5-CVB3), encoding respectively mouse proLC3B, mouse proLC3B carrying a G120A mutation, mouse ATG4B carrying a C74A mutation, and mouse ATG5 are used in the experiments described herein. All plasmids have been purchased form OriGene Technologies Inc. (Rockville, MD, USA) and the point mutations G120A (for proLC3G120A) and C74A (for ATG4BC74A) were introduced using the QuikChange Site-Directed mutagenesis system (Agilent Technologies, 200523). In all cases, the intended autophagy-related genes were inserted immediately downstream of the N terminus of the CVB3 polyprotein, using the cloning approaches developed by our laboratory. rCVB3s were grown in HeLa cells, and virus stocks were generated as described previously (Kemball et al., 2010; Slifka et al., 2001). The recombinant CVB3 encoding dsRed was previously reported.Citation60 Naïve adult mice (8- to 12-wk old) were inoculated intraperitoneally with 104 pfu of WT CVB3 or with 107 pfu of the various rCVB3s.
One-step growth curve
HeLa cell monolayers (∼75% confluent in 6-well plates) were infected with rCVB3 or WTCVB3 at a multiplicity of infection of 10. After 1 h at 37°C, monolayers were washed with saline, and then 3 ml of complete DMEM was added to each well. Cells and supernatant fractions were harvested at the indicated time points, the culture supernatant fractions and freeze-thaw-clarified media were pooled, and titers (pfu/ml) were determined by plaque assay.
Plaque assays
Mice were perfused with Dulbecco's phosphate-buffered saline (DPBS, Life Technologies, 14190-144) to clear blood from the tissues and to prevent overestimation of organ virus titers due to CVB3 in the blood. Samples were weighed, homogenized in 1 ml DMEM (Life Technologies, 100313-021), and the lysates were titered for infectious virus. Plaque assays were performed on subconfluent HeLa cell monolayers as described,Citation61 and the virus titers (pfu/g) were calculated for each sample.
Isolation of dispersed pancreatic acini
Dispersed pancreatic acini were prepared using a modified collagenase digestion protocol. In brief, pancreas was isolated from 10- to 12-wk-old (20 to 22 g) mice, and cut into small pieces which were incubated 3 times (15 min each) with freshly prepared 2 mg/ml collagenase D (Roche/Genentech, 11088866001) at 37°C in a shaking incubator. Before and after the third digestion, the cells were shaken vigorously by hand. Cells were washed 3 times with different concentrations of bovine serum albumin (4% BSA, 4% BSA+0.2% BSA and 4% BSA; Sigma, A7030), then resuspended in 199 Medium containing soybean trypsin inhibitor (Gibco/LifeTechnologies, 59A11016). The cells were maintained in culture for no more than 24 hrs, during which time they were used for CVB3 infection and electron microscopy analysis.
Analysis of protein expression by western blot
Mice were perfused with DPBS and the pancreata were collected and snap-frozen in liquid nitrogen. Samples were thawed, weighed, and homogenized in freshly prepared RIPA buffer (EMD Millipore, 20-188) containing the following inhibitors: 30 mM p-nitrophenyl phosphate (MP Biomedicals, 980701), 1X protease inhibitor cocktail (Roche/Genentech, 11697498001), and 1x Halt Phosphatase Inhibitor Cocktail (Pierce/Thermo Scientific, 1862495). Homogenates were spun at max speed in a cold microcentrifuge for 25 min, and the clarified supernatant fraction was collected. The total protein concentration of each lysate was determined by the Pierce BCA Protein Assay (Thermo Scientific, PI-23227), using bovine serum albumin (BSA, Pierce, 23209) as a standard. Lysates were diluted in denaturing sample buffer, boiled for 5 min, and 40 μg of each sample was separated by SDS-PAGE on 4-15% Criterion precast Tris-HCl polyacrylamide gels (Bio-Rad, 345-0028); 18% gels (Bio-Rad, 345-0024) were used to separate LC3 proteins. Proteins were then transferred onto a 0.45 μm PVDF membrane (Bio-Rad, 1704085), following the manufacturer's instructions. Membranes were blocked with 5% nonfat milk in TBS (137 mM sodium chloride, 20 mM Tris. at pH 7.6) + 0.1% Tween-20 (Sigma-Aldrich, P1379) (TBST) for 1 h at room temperature (RT). Next, membranes were washed in TBST and incubated with primary antibody in 1% milk with TBST overnight at 4°C. The following primary antibodies were used: polyclonal rabbit anti-human LC3B (MBL International Corporation, PM036); polyclonal rabbit anti anti-Enterovirus VP1 antibody (provided by Dr. Klingel, University Hospital Tübingen, Germany); polyclonal rabbit anti SEL1L and anti CANX (calnexin) antibodies (Sigma-Aldrich, S3699 and C4731); rabbit anti-human ATG5 (Cosmo Bio, TMD-PH-AT5). Membranes were washed again and then incubated with HRP-conjugated secondary antibody (Amersham donkey anti-rabbit IgG or sheep anti-mouse IgG; GE Healthcare Life Sciences, NA934, NA931) in 1% milk in TBST for 1 h at RT. After additional washing, membranes were incubated with Amersham™ ECL™ Prime (GE Healthcare Life Sciences, RPN2232) for 5 min, and then exposed to BioMax MR Film (Kodak). The membranes were then washed in TBST, stripped with Restore PLUS Western Blot Stripping Buffer (Pierce/Thermo Scientific, 46430) for 10 min at RT, then washed and blocked with 5% milk in TBST for 1 h at RT. Finally, to provide a control for protein loading of each sample, membranes were washed in TBST and reprobed with mouse GAPDH antibody (clone 6C5, EMD Millipore, MAB374) in 1% milk in TBST overnight at 4°C, and then processed as described above.
Coimmunoprecipitation
Cells were washed twice with DPBS and harvested in IP lysis buffer (Pierce/ThermoFisher, 87787) containing 1x protease inhibitors and 1x Halt Phosphatase Inhibitor Cocktail. Supernatant fractions were obtained by centrifuging the lysates for 25 min at max speed in a cold microcentrifuge, and were subsequently incubated with SEL1L or CANX antibodies for 4 h at 4°C on a platform rocker. Protein-A Sepharose beads were added and the lysates incubated overnight at 4°C. The beads were then washed 5 times in IP lysis buffer and eluted with 40 µl 2X Laemmli buffer at 100°C for 5 min. The immunoprecipitates were analyzed by SDS-PAGE and western blotting using the Criterion Tris-HCl polyacrylamide gel system. The membranes were probed with antibody against LC3, and bands were visualized as described above. The membranes were then stripped and reprobed with polyclonal rabbit SEL1L or CANX antibodies.
RT-PCR
Pancreatic tissue was collected, and immediately stored in RNAlater stabilization reagent (Qiagen, 1017980). The tissue was weighed, and RNA was isolated using an RNeasy Mini Kit (Qiagen, 74104), following the manufacturer's instructions. Alternatively, RNA was obtained from acinar cells using the same kit. 0.25 μg total RNA from the samples was reverse transcribed using SuperScript III Reverse Transcriptase (Life Technologies, 18080-051) according to the manufacturer's protocol; a CVB3-specific genome complementary oligonucleotide 5′GAACGCTTTCTCCTTCAACC3′ was used as primer for the RT reaction. The RT reactions were carried out in a thermocycler as follows: 65°C for 5 min, 50°C for 45 min, 70°C for 15 min. Samples were then treated with 1μl Ribonuclease H (Life Technologies, 18021-071) to remove RNA complementary to the cDNA. Next, Taqman quantitative real time PCR was performed using CVB3-specific primers (forward primer 5′CACACTCCGATCAACAGTCA3′; reverse primer 5′GAACGCTTTCTCCTTCAACC3′) and a FAM/TAMRA-labeled probe (5′CGTGGCACACCAGCCATGTTT3′) as previously described (Tabor-Godwin et al., 2010). PCR amplification was done using Platinum Quantitative PCR SuperMix-UDG ready to use cocktail (Life Technologies, P/N55294) as described by the manufacturer. Quantitative analysis of viral RNA was carried out using a Bio-Rad iQ5 Real-Time PCR System (Hercules, CA) in 96-well optical reaction plates heated to 50°C for 2 min to digest dUTP-containing contaminants, 95°C for 2 min to deactivate UNG and activate Platinum Taq DNA polymerase, followed by 40 cycles of: denaturation at 95°C for 15s and annealing and extension at 60°C for 30s. All samples were evaluated in triplicate amplification reactions. In order to assign a genome copy number to the cycle threshold value, a standard curve was generated: a known quantity of in vitro transcribed CVB genomic RNA was serially diluted, and all dilutions were subjected to the above reverse transcriptase and qPCR reactions. Additional control reactions were set up that omitted RT, and were invariably negative. Values are expressed as the average number of CVB genome copies per gram of tissue.
Vibratome sectioning and immunostaining
Mice were perfused with DPBS, and the pancreas was harvested and fixed in buffered zinc formalin (Anatech, 174) at RT overnight. The fixative was replaced with DPBS on the following day, and tissues were preserved at 4°C. Sections of pancreas (70 μm) were cut with a VT1000 S vibratome (Leica Microsystems Inc., Germany). Free-floating sections were washed 3 times in PBS and permeabilized in PBS with 0.5% Triton X-100 (Sigma-Aldrich, T8787) for 30 min at RT. After incubation, the sections were washed once in PBS with 0.3% Triton X-100 and then blocked in PBS with 5% normal goat serum (Life Technologies, PCN5000) and 0.3% Triton X-100 for 2 h at RT. Next, the sections were washed 3 times in PBS with 0.3% Triton X-100 and then incubated either with polyclonal rabbit anti-mouse IBA1 (Wako, 019-19741) in PBS with 1% normal goat serum and 0.3% Triton X-100 or with with Alexa Fluor 647 phalloidin overnight at 4°C. The sections were then washed 3 times in PBS with 0.3% Triton X-100 and the ones exposed to IBA1 antibody were incubated for 1 h at RT with goat anti-rabbit IgG Alexa Fluor 647 dye (Life Technologies, A21245). After incubation, the sections were washed once in PBS with 0.3% Triton X-100 and 3 times in PBS, then were counterstained with Hoechst 33342 in PBS for 5 min at RT (to label nuclei). After 3 additional PBS washes, the sections were mounted on glass coverslips with ProLong Gold (Life Technologies, P36930) mounting medium.
Confocal laser scanning microscopy
Three or 4 color images were captured using an LSM 710 laser scanning confocal microscope equipped with Zen 2009 software (Carl Zeiss, Inc., Germany). Representative regions within each vibratome section of pancreas or liver were scanned at 40X or 63X magnification as serial 8-bit optical sections with 0.3- to 0.5-μm-interval step slices (1,024 by 1,024 image sizes). Representative images shown are flattened maximum intensity projections of 3D deconvolved z-stacks using Imaris (Bitplane, Switzerland).
Histology
Mice were perfused with DPBS and the pancreas was harvested and fixed in buffered zinc formalin at RT overnight. Tissues were paraffin-embedded and 3 μm sections were cut and stained with Masson trichrome. The sections were observed at 20X magnification with an Axiovert 200 inverted microscope and images were acquired using AxioVision 4.7.1 software (Carl Zeiss, Inc., Germany).
Regular transmission electron microscopy from mouse tissue
Anesthetized mice were perfused with 0.9% saline, followed by 4% paraformaldehyde, 1.5% glutaraldehyde in 0.1 M cacodylate buffer with 1 mM CaCl2. Following removal of the pancreas, the tissue pieces were immersed in fixative on ice for 6 h and then were transferred to 2.5% glutaraldehyde in 0.1 M cacodylate buffer + 1 mM CaCl2 for overnight fixation. After a buffer wash, the tissue was further fixed in 1% OsO4 with 1.5% potassium ferricyanide in 0.1 M Na cacodylate and again washed in cacodylate buffer, dehydrated in graded ethanols and transitioned in propylene oxide. Pancreatic tissues were embedded in Embed 812/Araldite (Electron Microscopy Sciences, 13940). Thick sections (1 to 2 µm) were cut, mounted on glass slides and stained in toluidine blue for general assessment in the light microscope. Subsequently, 70-nm thin sections were cut, mounted on copper slot grids (Ted Pella, 1QC12H) coated with parlodion (Ladd, 10800) and stained with uranyl acetate and lead citrate for examination on a Philips CM100 electron microscope (FEI, Hillsbrough OR) at 80kv and images collected using a Megaview III ccd camera (Olympus Soft Imaging Solutions, Lakewood, CO).
Regular transmission electron m icroscopy from acini primary cultures
Uninfected or virus-infected acinar cells were gently spun down, the culture medium supernatant fraction removed and the cells fixed in 2.5% glutaraldehyde in 0.1 M cacodylate buffer pH 7.3 with the addition of 1 mM CaCl2. The cells were then buffer washed, fixed in 1% osmium tetroxide in cacodylate buffer for 2 h. Following dehydration in a graded ethanol series, the pellet fractions were transitioned in propylene oxide (Electron Microscopy Sciences, 20401) and embedded in EMbed 812/Araldite (Electron Microscopy Sciences, 13940). Thin sections (70 nm) were cut with a diamond knife (Diatome, Hatfield PA) on a Reichert Ultracut E microtome (Leica Microsystems, Buffalo Grove, IL), mounted on parlodion coated copper slot grids and stained with uranyl acetate and lead citrate for examination on a Philips CM100 electron microscope (FEI, Hillsbrough, OR). Images were documented using a Megaview III ccd camera (Olympus Soft Imaging Solutions, Lakewood, CO).
Statistics
A 2-tailed unpaired nonparametric Mann-Whitney test was used to determine statistical significance between 2 groups of data (GraphPad Prism, CA, USA). P values less than 0.05 were considered significant and, when represented by asterisks, are shown as follows: *0.05 ≥ P > 0 .01; **0.01 ≥ P > 0 .001;*** 0.001 ≥ P > 0 .0001; ****P≤0 .0001.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Drs. Noburu Mizushima (Tokyo Medical and Dental University) and Herbert W. “Skip” Virgin (Washington University) for providing Atg5f/f mice; Dr. Eric P. Sandgren (University of Wisconsin-Madison) for providing EL-Cre transgenic mice; and Dr. Karin Klingel (University Hospital Tübingen) for providing antibodies against VP1. We thank Dr. G Suizdak and LT Hoang of the Scripps Center for Metabolomics and Mass Spectrometry.
Funding
This work was supported by NIH R01 awards AI103218, AI114615 and AI110621 (to J.L.W.). This is manuscript number 28047 from the Scripps Research Institute.
References
- Uetz P, Dong YA, Zeretzke C, Atzler C, Baiker A, Berger B, Rajagopala SV, Roupelieva M, Rose D, Fossum E, et al. Herpesviral protein networks and their interaction with the human proteome. Science 2006; 311:239-42; PMID:16339411; http://dx.doi.org/10.1126/science.1116804
- Miller S, Krijnse-Locker J. Modification of intracellular membrane structures for virus replication. Nat Rev Microbiol 2008; 6:363-74; PMID:18414501; http://dx.doi.org/10.1038/nrmicro1890
- Kirkegaard K. Subversion of the cellular autophagy pathway by viruses. Curr Top Microbiol Immunol 2009; 335:323-33; PMID:19802573
- Dales S, Eggers HJ, Tamm I, Palade GE. Electron microscopic study of the formation of poliovirus. Virology 1965; 26:379-89; PMID:14319710; http://dx.doi.org/10.1016/0042-6822(65)90001-2
- Jezequel AM, Steiner JW. Some ultrastructural and histochemical aspects of Coxsackie virus-cell interactions. Lab Invest 1966; 15:1055-83; PMID:5911943
- Jackson WT, Giddings TH Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol 2005; 3:e156; PMID:15884975; http://dx.doi.org/10.1371/journal.pbio.0030156
- Jackson WT. Poliovirus-induced changes in cellular membranes throughout infection. Curr Opin Virol 2014; 9:67-73; PMID:25310497; http://dx.doi.org/10.1016/j.coviro.2014.09.007
- Lyle JM, Bullitt E, Bienz K, Kirkegaard K. Visualization and functional analysis of RNA-dependent RNA polymerase lattices. Science 2002; 296:2218-22; PMID:12077417; http://dx.doi.org/10.1126/science.1070585
- Limpens RW, van der Schaar HM, Kumar D, Koster AJ, Snijder EJ, van Kuppeveld FJ, Bárcena M. The transformation of enterovirus replication structures: a three-dimensional study of single- and double-membrane compartments. MBio 2011; 2:e00166-11; PMID:21972238
- Kemball CC, Alirezaei M, Flynn CT, Wood MR, Harkins S, Kiosses WB, Whitton JL. Coxsackievirus infection induces autophagy-like vesicles and megaphagosomes in pancreatic acinar cells in vivo. J Virol 2010; 84:12110-24; PMID:20861268; http://dx.doi.org/10.1128/JVI.01417-10
- Alirezaei M, Whitton JL. Studying RNA viruses and autophagy in vivo. In: Jackson WT, Swanson MS, eds. Autophagy, Infection, and the Immune Response. John Wiley & Sons, 2014:49-65
- Alirezaei M, Flynn CT, Wood MR, Whitton JL. Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo. Cell Host & Microbe 2012; 11:298-305; PMID:22423969; http://dx.doi.org/10.1016/j.chom.2012.01.014
- Suhy DA, Giddings TH, Jr., Kirkegaard K. Remodeling the endoplasmic reticulum by poliovirus infection and by individual viral proteins: an autophagy-like origin for virus-induced vesicles. J Virol 2000; 74:8953-65; PMID:10982339; http://dx.doi.org/10.1128/JVI.74.19.8953-8965.2000
- Wessels E, Notebaart RA, Duijsings D, Lanke K, Vergeer B, Melchers WJ, van Kuppeveld FJ. Structure-function analysis of the coxsackievirus protein 3A: identification of residues important for dimerization, viral RNA replication, and transport inhibition. J Biol Chem 2006; 281:28232-43; PMID:16867984; http://dx.doi.org/10.1074/jbc.M601122200
- Wessels E, Duijsings D, Notebaart RA, Melchers WJ, van Kuppeveld FJ. A proline-rich region in the coxsackievirus 3A protein is required for the protein to inhibit endoplasmic reticulum-to-golgi transport. J Virol 2005; 79:5163-73; PMID:15795300; http://dx.doi.org/10.1128/JVI.79.8.5163-5173.2005
- de Jong AS, Melchers WJ, Glaudemans DH, Willems PH, van Kuppeveld FJ. Mutational analysis of different regions in the coxsackievirus 2B protein: requirements for homo-multimerization, membrane permeabilization, subcellular localization, and virus replication. J Biol Chem 2004; 279:19924-35; PMID:14976211; http://dx.doi.org/10.1074/jbc.M314094200
- van Kuppeveld FJ, Melchers WJ, Kirkegaard K, Doedens JR. Structure-function analysis of coxsackie B3 virus protein 2B. Virology 1997; 227:111-8; PMID:9007064; http://dx.doi.org/10.1006/viro.1996.8320
- Cornell CT, Kiosses WB, Harkins S, Whitton JL. Inhibition of protein trafficking by coxsackievirus B3: multiple viral proteins target a single organelle. J Virol 2006; 80:6637-47; PMID:16775351; http://dx.doi.org/10.1128/JVI.02572-05
- Cornell CT, Kiosses WB, Harkins S, Whitton JL. Coxsackievirus B3 proteins directionally complement each other to downregulate surface major histocompatibility complex class I. J Virol 2007; 81:6785-97; PMID:17442717; http://dx.doi.org/10.1128/JVI.00198-07
- Schlegel A, Giddings TH, Jr., Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol 1996; 70:6576-88; PMID:8794292
- Hsu NY, Ilnytska O, Belov G, Santiana M, Chen YH, Takvorian PM, Pau C, van der Schaar H, Kaushik-Basu N, Balla T, et al. Viral reorganization of the secretory pathway generates distinct organelles for RNA replication. Cell 2010; 141:799-811; PMID:20510927; http://dx.doi.org/10.1016/j.cell.2010.03.050
- Reggiori F, Monastyrska I, Verheije MH, Calì T, Ulasli M, Bianchi S, Bernasconi R, de Haan CA, Molinari M. Coronaviruses Hijack the LC3-I-positive EDEMosomes, ER-derived vesicles exporting short-lived ERAD regulators, for replication. Cell Host & Microbe 2010; 7:500-8; PMID:20542253; http://dx.doi.org/10.1016/j.chom.2010.05.013
- Bernasconi R, Noack J, Molinari M. Unconventional roles of nonlipidated LC3 in ERAD tuning and coronavirus infection. Autophagy 2012; 8:1534-6; PMID:22895348; http://dx.doi.org/10.4161/auto.21229
- Noack J, Bernasconi R, Molinari M. How Viruses Hijack the ERAD Tuning Machinery. J Virol 2014; 88:10272-5; PMID:24990995; http://dx.doi.org/10.1128/JVI.00801-14
- Utans U, Arceci RJ, Yamashita Y, Russell ME. Cloning and characterization of allograft inflammatory factor-1: a novel macrophage factor identified in rat cardiac allografts with chronic rejection. J Clin Invest 1995; 95:2954-62; PMID:7769138; http://dx.doi.org/10.1172/JCI118003
- Kohler C. Allograft inflammatory factor-1/Ionized calcium-binding adapter molecule 1 is specifically expressed by most subpopulations of macrophages and spermatids in testis. Cell Tissue Res 2007; 330:291-302; PMID:17874251; http://dx.doi.org/10.1007/s00441-007-0474-7
- Monastyrska I, Ulasli M, Rottier PJ, Guan JL, Reggiori F, de Haan CA. An autophagy-independent role for LC3 in equine arteritis virus replication. Autophagy 2013; 9:164-74; PMID:23182945; http://dx.doi.org/10.4161/auto.22743
- Sharma M, Bhattacharyya S, Nain M, Kaur M, Sood V, Gupta V, Khasa R, Abdin MZ, Vrati S, Kalia M. Japanese encephalitis virus replication is negatively regulated by autophagy and occurs on LC3-I- and EDEM1-containing membranes. Autophagy 2014; 10:1637-51; PMID:25046112
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/10.4161/auto.4451
- Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem 2004; 279:47704-10; PMID:15355958; http://dx.doi.org/10.1074/jbc.M407016200
- Fujita N, Hayashi-Nishino M, Fukumoto H, Omori H, Yamamoto A, Noda T, Yoshimori T. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell 2008; 19:4651-9; PMID:18768752; http://dx.doi.org/10.1091/mbc.E08-03-0312
- Marino G, Fernández AF, Cabrera S, Lundberg YW, Cabanillas R, Rodríguez F, Salvador-Montoliu N, Vega JA, Germanà A, Fueyo A, et al. Autophagy is essential for mouse sense of balance. J Clin Invest 2010; 120:2331-44; PMID:20577052; http://dx.doi.org/10.1172/JCI42601
- Wang W, Chen Z, Billiar TR, Stang MT, Gao W. The carboxyl-terminal amino acids render pro-human LC3B migration similar to lipidated LC3B in SDS-PAGE. PLoS One 2013; 8:e74222; PMID:24040206; http://dx.doi.org/10.1371/journal.pone.0074222
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci 2010; 13:133-40; PMID:20023653; http://dx.doi.org/10.1038/nn.2467
- Cali T, Galli C, Olivari S, Molinari M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem Biophys Res Commun 2008; 371:405-10; PMID:18452703; http://dx.doi.org/10.1016/j.bbrc.2008.04.098
- Sun S, Shi G, Han X, Francisco AB, Ji Y, Mendonça N, Liu X, Locasale JW, Simpson KW, Duhamel GE, et al. Sel1L is indispensable for mammalian endoplasmic reticulum-associated degradation, endoplasmic reticulum homeostasis, and survival. Proc Natl Acad Sci U S A 2014; 111:E582-E591; PMID:24453213; http://dx.doi.org/10.1073/pnas.1318114111
- Nagy PD, Pogany J. The dependence of viral RNA replication on co-opted host factors. Nat Rev Microbiol 2012; 10:137-49; PMID:22183253
- Heaton NS, Randall G. Dengue virus-induced autophagy regulates lipid metabolism. Cell Host & Microbe 2010; 8:422-32; PMID:21075353; http://dx.doi.org/10.1016/j.chom.2010.10.006
- Kaufmann A, Beier V, Franquelim HG, Wollert T. Molecular mechanism of autophagic membrane-scaffold assembly and disassembly. Cell 2014; 156:469-81; PMID:24485455; http://dx.doi.org/10.1016/j.cell.2013.12.022
- Towner JS, Ho TV, Semler BL. Determinants of membrane association for poliovirus protein 3AB. J Biol Chem 1996; 271:26810-8; PMID:8900162; http://dx.doi.org/10.1074/jbc.271.43.26810
- van der Schaar HM, van der Linden L, Lanke KH, Strating JR, Pürstinger G, de Vries E, de Haan CA, Neyts J, van Kuppeveld FJ. Coxsackievirus mutants that can bypass host factor PI4KIIIbeta and the need for high levels of PI4P lipids for replication. Cell Res 2012; 22:1576-92; PMID:22945356; http://dx.doi.org/10.1038/cr.2012.129
- Lanke KH, van der Schaar HM, Belov GA, Feng Q, Duijsings D, Jackson CL, Ehrenfeld E, van Kuppeveld FJ. GBF1, a guanine nucleotide exchange factor for Arf, is crucial for coxsackievirus B3 RNA replication. J Virol 2009; 83:11940-9; PMID:19740986; http://dx.doi.org/10.1128/JVI.01244-09
- McCartney AW, Greenwood JS, Fabian MR, White KA, Mullen RT. Localization of the tomato bushy stunt virus replication protein p33 reveals a peroxisome-to-endoplasmic reticulum sorting pathway. Plant Cell 2005; 17:3513-31; PMID:16284309; http://dx.doi.org/10.1105/tpc.105.036350
- Jonczyk M, Pathak KB, Sharma M, Nagy PD. Exploiting alternative subcellular location for replication: tombusvirus replication switches to the endoplasmic reticulum in the absence of peroxisomes. Virology 2007; 362:320-30; PMID:17292435; http://dx.doi.org/10.1016/j.virol.2007.01.004
- Xu K, Huang TS, Nagy PD. Authentic in vitro replication of two tombusviruses in isolated mitochondrial and endoplasmic reticulum membranes. J Virol 2012; 86:12779-94; PMID:22973028; http://dx.doi.org/10.1128/JVI.00973-12
- Prentice E, Jerome WG, Yoshimori T, Mizushima N, Denison MR. Coronavirus replication complex formation utilizes components of cellular autophagy. J Biol Chem 2004; 279:10136-41; PMID:14699140; http://dx.doi.org/10.1074/jbc.M306124200
- Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E, Mizushima NN, Denison MR, Virgin HW 4th. Coronavirus replication does not require the autophagy gene ATG5. Autophagy 2007; 3:581-5; PMID:17700057; http://dx.doi.org/10.4161/auto.4782
- Meyers G, Stoll D, Gunn M. Insertion of a sequence encoding light chain 3 of microtubule-associated proteins 1A and 1B in a pestivirus genome: connection with virus cytopathogenicity and induction of lethal disease in cattle. J Virol 1998; 72:4139-48; PMID:9557703
- Fricke J, Voss C, Thumm M, Meyers G. Processing of a pestivirus protein by a cellular protease specific for light chain 3 of microtubule-associated proteins. J Virol 2004; 78:5900-12; PMID:15140988; http://dx.doi.org/10.1128/JVI.78.11.5900-5912.2004
- Ford Siltz LA, Viktorova EG, Zhang B, Kouiavskaia D, Dragunsky E, Chumakov K, Isaacs L, Belov GA. New small-molecule inhibitors effectively blocking picornavirus replication. J Virol 2014; 88:11091-107; PMID:25008939; http://dx.doi.org/10.1128/JVI.01877-14
- Thachil E, Hugot JP, Arbeille B, Paris R, Grodet A, Peuchmaur M, Codogno P, Barreau F, Ogier-Denis E, Berrebi D, et al. Abnormal activation of autophagy-induced crinophagy in Paneth cells from patients with Crohn's disease. Gastroenterology 2012; 142:1097-9; PMID:22285936; http://dx.doi.org/10.1053/j.gastro.2012.01.031
- Cabrera S, Fernández AF, Mariño G, Aguirre A, Suárez MF, Español Y, Vega JA, Laurà R, Fueyo A, Fernández-García MS, et al. ATG4B/autophagin-1 regulates intestinal homeostasis and protects mice from experimental colitis. Autophagy 2013; 9:1188-200; PMID:23782979; http://dx.doi.org/10.4161/auto.24797
- Nystrom N, Berg T, Lundin E, Skog O, Hansson I, Frisk G, Juko-Pecirep I, Nilsson M, Gyllensten U, Finkel Y, et al. Human enterovirus species B in ileocecal Crohn's disease. Clin Transl Gastroenterol 2013; 4:e38; PMID:23804031; http://dx.doi.org/10.1038/ctg.2013.7
- Chen ZH, Kim HP, Sciurba FC, Lee SJ, Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ, Yousem SA, et al. Egr-1 regulates autophagy in cigarette smoke-induced chronic obstructive pulmonary disease. PLoS One 2008; 3:e3316; PMID:18830406; http://dx.doi.org/10.1371/journal.pone.0003316
- Wedzicha JA. Role of viruses in exacerbations of chronic obstructive pulmonary disease. Proc Am Thorac Soc 2004; 1:115-20; PMID:16113423; http://dx.doi.org/10.1513/pats.2306030
- Tran E, Chow A, Goda T, Wong A, Blakely K, Rocha M, Taeb S, Hoang VC, Liu SK, Emmenegger U. Context-dependent role of ATG4B as target for autophagy inhibition in prostate cancer therapy. Biochem Biophys Res Commun 2013; 441:726-31; PMID:24184480; http://dx.doi.org/10.1016/j.bbrc.2013.10.117
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/10.1091/mbc.E03-09-0704
- van Houten N, Bouchard PE, Moraska A, Huber SA. Selection of an attenuated Coxsackievirus B3 variant, using a monoclonal antibody reactive to myocyte antigen. J Virol 1991; 65:1286-90; PMID:1847455
- Knowlton KU, Jeon ES, Berkley N, Wessely R, Huber SA. A mutation in the puff region of VP2 attenuates the myocarditic phenotype of an infectious cDNA of the Woodruff variant of coxsackievirus B3. J Virol 1996; 70:7811-8; PMID:8892902
- Tabor-Godwin JM, Ruller CM, Bagalso N, An N, Pagarigan RR, Harkins S, Gilbert PE, Kiosses WB, Gude NA, Cornell CT, et al. A novel population of myeloid cells responding to coxsackievirus infection assists in the dissemination of virus within the neonatal CNS. J Neurosci 2010; 30:8676-91; PMID:20573913; http://dx.doi.org/10.1523/JNEUROSCI.1860-10.2010
- Hunziker IP, Cornell CT, Whitton JL. Deletions within the 5′UTR of coxsackievirus B3: consequences for virus translation and replication. Virology 2007; 360:120-8; PMID:17084431; http://dx.doi.org/10.1016/j.virol.2006.09.041
- Kuma A, Mizushima N. Chromosomal mapping of the GFP-LC3 transgene in GFP-LC3 mice. Autophagy 2008; 4:61-2; PMID:17786029; http://dx.doi.org/10.4161/auto.4846