
Abstract
Autophagosome-lysosome fusion and autolysosome acidification constitute late steps in the autophagic process necessary to maintain functional autophagic flux and cellular homeostasis. Both of these steps are disrupted by the V-ATPase inhibitor bafilomycin A1, but the mechanisms potentially linking them are unclear. We recently revisited the role of lysosomal acidification in autophagosome-lysosome fusion, using an in vivo approach in Drosophila. By genetically depleting individual subunits of the V-ATPase, we confirmed its role in lysosomal acidification and autophagic cargo degradation. Surprisingly, vesicle fusion remained active in V-ATPase-depleted cells, indicating that autophagosome-lysosome fusion and autolysosome acidification are 2 separable processes. In contrast, bafilomycin A1 inhibited both acidification and fusion, consistent with its effects in mammalian cells. Together, these results imply that this drug inhibits fusion independently of its effect on V-ATPase-mediated acidification. We identified the ER-calcium ATPase Ca-P60A/dSERCA as a novel target of bafilomycin A1. Autophagosome-lysosome fusion was defective in Ca-P60A/dSERCA-depleted cells, and bafilomycin A1 induced a significant increase in cytosolic calcium concentration and disrupted Ca-P60A/SERCA-mediated fusion. Thus, bafilomycin A1 disrupts autophagic flux by independently inhibiting V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion.
Lysosomal proton pump V-ATPases are responsible for establishing the low lumenal pH indispensable for lysosomal enzyme activation and cargo degradation. The V-ATPase is a hetero-multimeric enzyme composed of a cytosolic complex V1 and a membrane-bound complex V0, with a total of 15 independent types of subunits assembled and docked on/in the lysosome membrane. We used a Drosophila in vivo model to genetically deplete each V-ATPase subunit and study the impact of their loss on autophagy. We found that depletion of any V-ATPase subunit led to 2 robust phenotypes in Drosophila fat body cells: an abnormal accumulation of autophagic structures in well-fed animals and an expansion of the autophagic vesicle compartment under starvation-induced autophagy. Using multiple established assays, we confirmed that the V-ATPase is involved, as expected, in lysosomal acidification, activation of lysosomal enzymes and autophagic-specific cargo degradation.
The role of the V-ATPase in promoting vesicle fusion is somewhat controversial and is based largely on studies using bafilomycin A1. We found that despite having a strong effect on lysosomal pH, genetic loss of V-ATPase subunits does not disrupt autophagosome-lysosome fusion. Using confocal, electron microscopy and quantitative image analysis, we established that in V-ATPase-depleted cells autolysosomes are indeed formed through a continuous and active fusion between autophagosomes and lysosomes. The resulting enlarged autolysosomes are nonfunctional, filled with intact cellular material, and they further expand under prolonged stress in a time-dependent manner. On the contrary, autolysosomes were not detectable in cells depleted for Syx17/Syntaxin17, a recently characterized Q-SNARE protein specifically involved in autophagosome-lysosome fusion. Our findings clearly indicate that autophagosome-lysosome fusion is not driven by V-ATPase-mediated lysosomal acidification, which contrasts with current views based on bafilomycin A1's known mode of action.
Bafilomycin A1 is widely used as an inhibitor of autophagosome-lysosome fusion in vitro to determine the activity of autophagic flux. Bafilomycin A1 is a macrolide that targets the V-ATPase ATP6V0C/V0 subunit c, thus inhibiting lysosomal acidification by preventing the passage of protons into the lysosomal lumen. Taking together these properties of bafilomycin A1, it has been generally assumed that bafilomycin A1 inhibits autophagosome-lysosome fusion by blocking V-ATPase pump activity. We found that bafilomycin A1 blocks autophagosome-lysosome fusion and lysosome acidification in Drosophila fat body cells, as it does in vitro in mammalian cell culture. However, our genetic results demonstrate that V-ATPase-dependent acidification is not a prerequisite for vesicle fusion. To explain why autophagosome-lysosome fusion is inhibited by bafilomycin A1 treatment but not by genetic depletion of V-ATPase, we speculated that bafilomcyin A1 might affect fusion through another target.
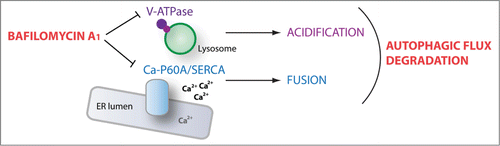
It was recently demonstrated that thapsigargin, an inhibitor of the Ca2+-transporting ATPase ATP2A/SERCA, blocks fusion between autophagosomes and late endosomes. Interestingly, in addition to its action on V-ATPases, bafilomycin A1 moderately inhibits other types of ATPases including ATP2A/SERCA. We found that depletion of the single Drosophila Ca-P60A/SERCA homolog phenocopies the effects of bafilomycin A1, inhibiting autophagosome-lysosome fusion in vivo. Conversely, activation of Ca-P60A/SERCA by overexpression of its positive regulator Seipin leads to the formation of elongated tubular autolysosome-like structures, suggesting that hyperactive Ca-P60A/SERCA promotes increased fusion and growth of autolysosomes. Interestingly, bafilomycin A1 treatment abolishes Seipin-induced elongation of autolysosomes, consistent with bafilomycin A1 acting as an inhibitor of Ca-P60A/SERCA activity. In further support of this idea, a significant increase in cytosolic calcium concentration is observed in cells treated with bafilomycin A1, similar to the effect of thapsigargin. In all, these data support a model whereby bafilomycin A1 targets the V-ATPase at the lysosome to prevent lumenal acidification, and independently inhibits Ca-P60A/SERCA to disrupt autophagosome-lysosome fusion, together resulting in a robust block of autophagic flux ().
Figure 1. Bafilomycin A1 disrupts autophagic flux through 2 independent targets. By inhibiting the lysosomal proton pump V-ATPase, bafilomycin A1 prevents lumenal acidification and lysosomal enzyme activation. Bafilomycin A1-dependent inhibition of the endoplasmic reticulum (ER) calcium pump Ca-P60A/SERCA induces a defect in autophagosome-lysosome fusion. Together, bafilomycin A1 prevents cargo degradation and functional autophagic flux by acting at 2 distinct steps of the autophagic process.
This study opens several new lines of investigation, including how bafilomycin A1 inhibits Ca-P60A/ATP2A/SERCA, how Ca-P60A/ATP2A/SERCA promotes vesicle fusion, and the nature of the intracellular pool(s) of Ca2+ relevant to autophagosome-lysosome fusion. Although high cytoplasmic Ca2+ concentration has long been associated with functional fusion events, it has recently been shown that abnormally high Ca2+ concentration can inhibit fusion. Lysosomes are also important stores of Ca2+, and recent data have linked lysosomal Ca2+ signaling to both PPP3/calcineurin regulation and autophagy induction. Precise analysis of Ca2+ microdomain regulation would be insightful, as Ca2+ gradients generated at the lysosome, the ER or the autophagosome might be part of the mechanisms driving autophagosome-lysosome fusion. Another recent publication highlighted the role of ATG14, an autophagic protein involved in autophagosome formation at the ER, in the coordination of the SNARE proteins essential for fusion between autophagosomes and lysosomes. Future studies focusing on the identification of the proteins regulating Ca2+-dependent vesicle fusion would be of interest.