
Abstract
Plasmodium parasites are transmitted by Anopheles mosquitoes to the mammalian host and actively infect hepatocytes after passive transport in the bloodstream to the liver. In their target host hepatocyte, parasites reside within a parasitophorous vacuole (PV). In the present study it was shown that the parasitophorous vacuole membrane (PVM) can be targeted by autophagy marker proteins LC3, ubiquitin, and SQSTM1/p62 as well as by lysosomes in a process resembling selective autophagy. The dynamics of autophagy marker proteins in individual Plasmodium berghei-infected hepatocytes were followed by live imaging throughout the entire development of the parasite in the liver. Although the host cell very efficiently recognized the invading parasite in its vacuole, the majority of parasites survived this initial attack. Successful parasite development correlated with the gradual loss of all analyzed autophagy marker proteins and associated lysosomes from the PVM. However, other autophagic events like nonselective canonical autophagy in the host cell continued. This was indicated as LC3, although not labeling the PVM anymore, still localized to autophagosomes in the infected host cell. It appears that growing parasites even benefit from this form of nonselective host cell autophagy as an additional source of nutrients, as in host cells deficient for autophagy, parasite growth was retarded and could partly be rescued by the supply of additional amino acid in the medium. Importantly, mouse infections with P. berghei sporozoites confirmed LC3 dynamics, the positive effect of autophagy activation on parasite growth, and negative effects upon autophagy inhibition.
Abbreviations
| ATG | = | autophagy related |
| atg5−/− | = | autophagy-related 5 knockout |
| Atg8 | = | (yeast) autophagy-related 8 |
| CLS | = | confocal line scanning |
| CSP | = | circumsporozoite protein |
| CTSD | = | cathepsin D |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| DMEM | = | Dulbecco's modified Eagle's medium |
| EBSS | = | Earle's balanced salt solution |
| ECACC | = | European Collection of Cell Cultures |
| EM | = | electron microscopy |
| e-schz | = | early schizont |
| Exp1 | = | exported protein 1 |
| FACS | = | fluorescent-activated cell sorting |
| FCS | = | fetal calf serum |
| GFP | = | green fluorescent protein |
| H&E, | = | hematoxylin and eosin |
| HepG2 | = | human hepatoma cells |
| hpi | = | hours postinfection |
| IFA | = | immunofluorescence assay |
| LAP | = | LC3-associated phagocytosis |
| LAMP1 | = | lysosomal-associated membrane protein 1 |
| LGALS, | = | galectins |
| l-schz | = | late schizont |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MEFs | = | mouse embryonic fibroblasts |
| MEM | = | minimum essential medium |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| Pb | = | Plasmodium berghei |
| PbmCherry | = | Plasmodium berghei expressing mCherry fluorescent protein |
| PBS | = | phosphate-buffered saline |
| PE | = | phosphatidylethanolamine |
| PM | = | parasite membrane |
| PtdIns3P | = | phosphatidylinositol-3-phosphate |
| PV | = | parasitophorous vacuole |
| PVM | = | parasitophorous vacuole membrane |
| RFP | = | red fluorescence protein |
| SD | = | standard deviation |
| spz | = | sporozoite |
| STED | = | stimulated emission depletion |
| SQSTM1 | = | sequestosome 1 |
| UIS4 | = | upregulated in infectious sporozoites gene 4 |
| WT | = | wild type. |
Introduction
Plasmodium parasites are the causative agents of malaria, a disease that still affects more than 300–500 million people and kills several hundred thousands yearly. Plasmodium parasites are transmitted by Anopheles mosquitoes and injected into the skin of the vertebrate host as motile sporozoites. For further development, these sporozoites first have to find their way to a blood vesselCitation1 before being transported passively by the blood stream to the liver. Within the liver, motile sporozoites migrate along the endothelia and pass through Kupffer cells or endothelial cells to reach their target hepatocyte.Citation1 Sporozoites transmigrate through a number of hepatocytes, until finally settling in one where they undergo growth and asexual replication. Sporozoites invade their host cell by invagination of the host cell plasma membrane and the formation of a parasitophorous vacuole membrane (PVM). Upon invasion, the parasite massively modulates the PVM by export of numerous proteins. Although it has been speculated that proteins are also secreted into the host cell, and that they can manipulate host cell signaling, so far this observation has been confirmed only for very few proteins.Citation2 Inside the parasitophorous vacuole (PV), rodent Plasmodium species transform within the first 16 h to a trophozoite, and later, a schizont stage. The liver schizont stage is characterized by an extraordinary growth and nuclear replication that lasts approximately 30 h. In the last few hours of liver stage development, the parasite plasma membrane starts to invaginate, forming thousands of merozoites that are released into the host cell cytoplasm upon PVM rupture. PVM rupture induces an ordered form of host cell death that is characterized by host nucleus condensation but not by fragmentation of the DNA.Citation3 Importantly, merozoites accumulate Ca2+ released from host cell stores during cell death and thus block exposure of phosphatidylserine residues to the outer leaflet of the membrane of the infected cell. Host cell death also allows the formation of merosomes, vesicles that bud from the host cell membrane and are filled with merozoites. By a so far unknown mechanism, merosomes are pushed into the blood vessel while merozoites continue to be transported in the budding vesicle. Merosomes are liberated and transported into the lung capillaries where they release merozoites to initiate infection of red blood cells, marking the beginning of the pathogenic phase of the infection.Citation4
In our present work, we studied 2 main types of autophagy that influence parasite establishment, growth and development: on the one hand, the host cell response selectively targeting Plasmodium parasites (selective autophagy), and on the other hand nonselective canonical autophagy potentially supporting parasite nourishment. Recently it has been shown that the autophagy marker protein LC3 is recruited to the PVM and to tubules budding from the PVMCitation5 but nothing is known about canonical host cell autophagy and its effect on parasite development.
Although intracellular pathogens like Plasmodium avoid direct contact with the host adaptive or innate immune responses, the invaded host cell is also equipped with a plethora of mechanisms to eliminate the invader.Citation6,7 These mechanisms can be considered to be cytosolic immune responses. One prominent cytosolic immune mechanism is selective autophagy, which also serves as a catabolic process to recycle damaged organelles or large protein complexes.Citation8 Selective autophagy differs from canonical autophagy in that it specifically targets intracellular pathogens or intracellular components by ubiquitination, and does not induce the general autophagy response of the cell that is elicited upon starvation. Similar to canonical autophagy, upon selective autophagy, double-membrane structures called phagophores form around invading pathogens or damaged organelles.
Another autophagy-related response of host cells to pathogens residing in a vacuole is to directly target the vacuolar membrane and label it for fusion with lysosomes in a process called LC3-associated phagocytosis (LAP).Citation9 LC3 targeting of membranes is a common feature of most autophagy-related processes.Citation10,11 LC3 is a ubiquitous cytosolic protein and, like other proteins of the autophagy machinery, it is constitutively expressedCitation8 and can be immediately recruited when autophagy is induced. LC3 is synthesized as a precursor and the C terminus is processed by the cysteine protease ATG4. This generates LC3-I, in which a C-terminal glycine becomes exposed, thus becoming available for the subsequent conjugation reactions. LC3-I is activated by ATG7, transferred to the E2-like enzyme ATG3, and finally, with the help of the ATG12–ATG5-ATG16L1 complex (which acts as an E3-like enzyme), conjugated with phosphatidylethanolamine (PE) to form LC3-II. In canonical autophagy, LC3-II localizes to target membranes such as autophagosomal membranes. In noncanonical autophagy processes such as LAP, LC3-II localizes to membranes of pathogen-containing compartments. Due to the different localization patterns of LC3-I and LC3-II, LC3-II is considered to be a key marker protein for all different forms of autophagy.Citation9-11
Several pathogens have developed strategies to circumvent autolysosomal degradation.Citation12-14 It has been shown that Toxoplasma gondii interferes with Akt signaling to block autophagy but it has also been suggested for the same parasite that it even can exploit autophagy as an additional source of nutrients.Citation13 An explanation for these results, which at first glance may seem contradictory, might be the difference of selective autophagy that needs to be controlled by the parasite to avoid elimination, and canonical autophagy that might be exploited by the pathogen.
In the present study, the autophagic response of hepatocytes to the invasion of Plasmodium sporozoites was examined. Since biochemical analysis of Plasmodium-infected hepatocytes is limited by the low infection rate, we developed long-term live imaging to follow individual infected cells. This approach allowed us to record for the first time the entire 64-h development period of individual parasites that successfully invaded a hepatocyte, developed into merozoites, and were released into the host cell cytoplasm, thereby inducing cell detachment in vitro. We found that the invading Plasmodium sporozoites failed to control the initial LC3 labeling of the PVM and some were even eliminated by autophagy. However, the surviving parasite majority appeared to be able to control the progression of the autophagic process by an as-yet unknown mechanism. Indeed, these surviving parasites appeared to benefit from nonselective canonical autophagy as an additional source of nutrients.
Results
The host cell recognizes and labels invading sporozoites
To analyze the selective autophagy-related mechanisms that arise while Plasmodium is infecting hepatocytes, we used the murine malaria model Plasmodium berghei. We first examined whether sporozoite infection of hepatocytes causes the cytosolic LC3 protein to localize to autophagosomes. Immunofluorescence analysis (IFA) of P. berghei-infected primary hepatocytes stained with an anti-LC3 antibody revealed that the localization of endogenous LC3 differed depending on the stage of parasite development (). Upon sporozoite invasion and early schizogony, LC3 accumulated in membranous structures around the parasite, whereas in late schizogony LC3-positive vesicles were more randomly distributed throughout the cytoplasm of the host cell.
Figure 1. LC3-II is recruited to the vicinity of Plasmodium parasites in primary mouse hepatocytes. (A) Primary mouse hepatocytes were freshly isolated, seeded on coverslips, and infected with P. berghei that expressed mCherry fluorescent protein (PbmCherry). The infected cells were fixed at different developmental stages and stained with an anti-LC3 antibody and the DNA dye DAPI. Labeled scale bars are included at the bottom-right of the images. (B) In vivo LC3 dynamics in P. berghei-infected cells. A GFP-LC3-expressing mouse (green) was infected with mCherry-expressing P. berghei parasites (red) and analyzed by intravital imaging at different developmental stages, as indicated. (C) Intravital imaging of the same mouse that shows a dying parasite at the late developmental stage. The dispersed parasite vesicles show a transient but strong association with GFP-LC3. Scale bars: 10 µm. See also Movie S1.
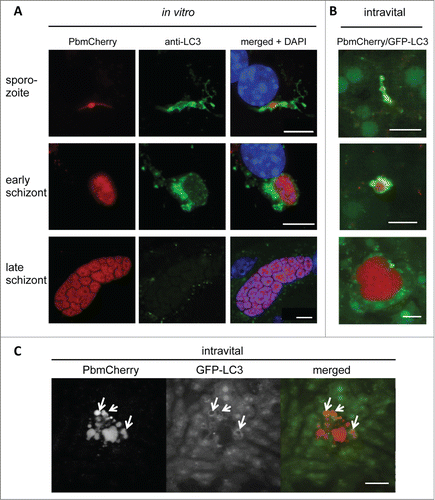
Although the original observation of LC3 accumulation around early parasite stages was made in primary mouse hepatocytes, it was essential to validate this observation in vivo. To follow the dynamic events of LC3 recruitment after sporozoite invasion, we used mCherry-expressing parasites to infect transgenic mice that express in all nucleated cells LC3 N-terminally tagged with green fluorescent protein (GFP). Intravital imaging of the liver of an infected mouse revealed a pattern of LC3 distribution around the parasite in the early developmental stages, which was not seen in the later stages () that resembled the pattern we also observed in vitro (). Importantly, intravital imaging indicated that even late-stage parasites are occasionally eliminated in an autophagy-typical manner (; Movie S1). This phenomenon was also observed in vitro (Movie S2). Since the same effect was observed in vitro and in vivo it could be concluded that LC3 accumulation around the parasite is indeed a physiological event and not restricted to in vitro cultivated cells. It further proves that both the in vitro analysis and the GFP-LC3 mouse are suitable and complementary models for studying autophagy in Plasmodium-infected hepatocytes.
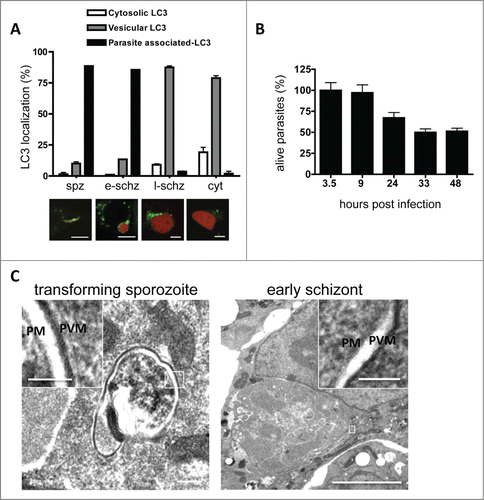
To study the dynamics of LC3 localization, we transfected the hepatoma cell line HepG2 with a plasmid encoding GFP-LC3 and infected these cells with mCherry-expressing P. berghei parasites. Live imaging approaches revealed that in the vast majority of infected cells, GFP-LC3 was immediately and dramatically recruited into a membrane around the parasite after invasion (; Movie S3). This confirms the IFA data () and suggests that the host cells recognize the invader and mount a strong cytosolic immune response against it. Thereafter, however, although a considerable number of parasites were indeed eliminated between 9 and 24 hpi (), the LC3 accumulation around the majority of parasites was progressively lost suggesting that the parasites escaped this host cell response (; Movies S4–S6).
Figure 2. In vitro LC3 dynamics in Plasmodium berghei-infected cells. (A) HepG2 cells were transfected with a plasmid encoding GFP-LC3, infected with P. berghei sporozoites expressing mCherry protein, and monitored at different phases of development. Spz: invaded sporozoites at 4 hpi; e-schz: early schizont at 24 hpi; l-schz: late schizont at 48 hpi; cyt: cytomere stage at 56 hpi. Representative images are shown below the graph. The infected host cells harboring LC3-associated parasites, vesicular LC3, or cytoplasmic LC3 were counted and expressed as percentages. Scale bars: 10 µm. (B) To study the dynamics of parasite elimination during liver-stage infection, HepG2 cells were seeded in 96-well plates and infected with mCherry-expressing parasites. To eliminate the extracellular parasites, the infected cells were treated with an anti-CSP antiserum. The parasites were then counted by using an IN Cell analyzer at the indicated time points. The average number of invaded parasites at the sporozoite stage (i.e. 3.5 hpi) was considered to be 100%. (C) Electron microscopy analysis of P. berghei-infected cells during sporozoite transformation and early schizogony. The overview and the magnifications clearly demonstrate that apart from the parasite membrane (PM) and the parasitophorous vacuole membrane (PVM), no other surrounding membranes are visible. The insets are magnifications of the areas of interest, including PM and PVM. Scale bars of the left image: 200 nm; inset: 100 nm. Scale bars of the right image: 2 µm; inset 100 nm.
Before wild-type (WT) LC3 can accumulate around the invaded sporozoite, it must be correctly processed, lipidated, and incorporated into a membrane. When HepG2 cells were transfected instead with a plasmid encoding the GFP-LC3G120A mutant, which cannot be processed and lipidated,Citation10 mutant LC3G120A was not mobilized around the invaded parasite (Fig. S1) suggesting that processing and lipidation are prerequisites for the observed LC3 accumulation around the parasite. It is important to note that the cells transfected with the GFP-LC3G120A mutant construct still express the endogenous and thus functional LC3. Therefore, it is unlikely that expression of the GFP-LC3G120A mutant would affect parasite development.
Next, we wanted to know whether the host cell isolates the parasite in an autophagosome. Electron microscopy (EM) analysis of P. berghei–infected HepG2 cells did not reveal the formation of a typical autophagosomal membrane around the invading parasite () confirming previous EM studies examining in great detail P. berghei development in vitro and in vivo.Citation15-17 This hinted that upon sporozoite invasion, LC3 must be incorporated into another membrane.
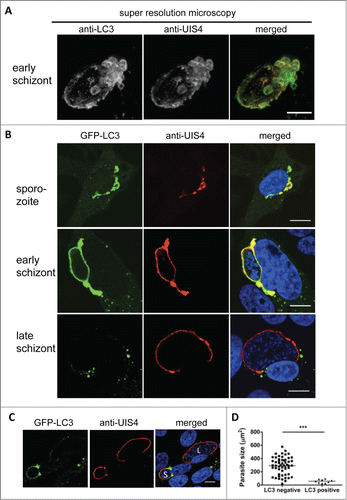
One such membrane may be the PVM, which is a membrane that surrounds parasites. Indeed, when P. berghei-infected HepG2 cells were stained with an antiserum against the PVM marker protein UIS4 and an anti-LC3 antibody, super resolution STED microscopy revealed a clear colocalization of both markers ().
LC3 labeling of the PVM during early parasite development was further confirmed by confocal microscopy of GFP-LC3-expressing HepG2 cells additionally stained with an anti UIS4 antiserum (, upper 2 panels). As observed before (), in the later stages, LC3 accumulation around the PVM diminished; instead, the protein formed LC3-positive aggregates around the PVM and throughout the cytoplasm (, lower panel). It is possible that the parasites actively clear the PVM of this autophagy marker protein, most likely to prevent progression of the destructive events in the PV that could result in parasite elimination. Supporting this view is the fact is that the parasites whose PVMs continued to bear ample amounts of LC3 in the later stages did not complete liver stage development (; Movie S7). Instead, they often underwent a kind of cell death that has been previously reported.Citation18 Indeed, the late liver-stage schizonts that continued to have an LC3-positive PVM were significantly smaller than the parasites whose PVM lost LC3 (). Thus, LC3 persistence in the PVM is associated with parasite growth arrest and elimination.
Figure 3. Host cell LC3 incorporation into the parasitophorous vacuole membrane (PVM). (A) Hepatocytes infected with a 24-h liver schizont were fixed and costained with anti-LC3 (green) and the PVM marker UIS4 (red). STED microscopy was perfomed and z-Stacks were obtained with increments of 0.22 µm and subsequently deconvolved. 3D-projection of the PVM shows a direct incorporation of the host cell autophagy marker LC3-II. Quantification of the colocalization applying the Pearson coefficient revealed a correlation value of ρ = 0.71. The inset shows a magnification of the PVM with strong LC3 incorporation. Scale bar: 2 µm. (B) GFP-LC3-transfected HepG2 cells were infected with P. berghei sporozoites, fixed at different time points after infection, and stained with antiserum against the PVM marker protein UIS4 (red). To detect GFP-LC3 localization, the cells were also stained with an anti-GFP antibody (green). For the merged picture, the DNA stain with DAPI (blue) was included. Scale bars: 10 µm. (C) LC3 persistence in the PVM during schizogony associates with impaired parasite growth. The immunofluorescence analysis images show neighboring infected cells harboring schizonts, one of which is GFP-LC3-positive and small (S). The second is GFP-LC3-negative and large (L). Staining was performed as described in (B). Scale bars: 10 µm. (D) The sizes of the LC3-positive and -negative parasites 48 hpi (late schizogony) were measured and compared statistically. P < 0.0001.
Lysosomes are attracted by early-stage and arrested parasites
Having confirmed that LC3 labeling of the PVM and subsequent autophagy-related events can limit parasite infection to a certain extent, we next investigated the events that occur up- and downstream of the labeling of the PVM in infected cells with LC3. A hallmark of autophagy is the fusion of LC3-positive autophagosomes with lysosomes, which generates lysogenic autolysosomes.Citation19 One study suggested that lysosomes fuse with the PVM in P. berghei-infected hepatocytes.Citation20 We therefore sought to determine i) the dynamics of lysosome fusion in relation to LC3 recruitment to the PVM in infected cells, ii) whether the pH drops in the PV, and iii) whether the lysosomal protease CTSD (cathepsin D) is recruited to the PV.
To follow the dynamics of lysosomes in infected cells and to quantify the corresponding observations, we used several live and indirect staining methods. LAMP1 is a protein of the lysosomal membraneCitation21 and can either be tagged by GFP or detected by IFA using antibodies. Other methods that we used for live imaging were to employ a RFP-tagged version of CTSD,Citation22 which is the main lysosomal protease and to stain cells with LysoTracker Red, which detects lysosomes because of the low pH in these compartments. Finally, to visualize protein degradation in lysosomes, DQ-BSA red was used. Unprocessed DQ-BSA red is colorless because its fluorogenic group is quenched. On processing, DQ-BSA red emits red fluorescence and thus labels degradation vesicles like lysosomes and autolysosomes.
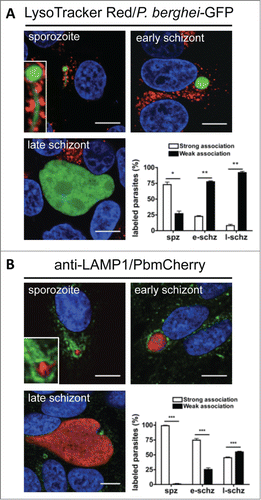
The main observation from the experiments using these different staining approaches was that while lysosomes accumulated around the early-stage parasites, this association was progressively lost during parasite development ( Figs. S2–S5). All lysosome markers, namely, LysoTracker Red, DQ-BSA red, CTSD, and LAMP1, appeared to be closely associated with the PVM and LC3 (Figs. S3–S5) at early parasite stages and show a more random distribution in the host cell at later developmental stages. Furthermore, in the early parasite stages LAMP1 was found to partially colocalize with UIS4 and thus the PVM (Fig. S3; Movie S8), which strongly suggests that the lysosomes fused with the PVM. However, this LAMP1 incorporation into the PVM was eventually lost, similar to what was observed for LC3.
Another important observation of these staining experiments was that the PV did not become positive for LysoTracker Red, CTSD-RFP, or DQ-BSA red (, Figs. S2 and S4). This suggests that even though the PV fused with lysosomes, this event did not enhance the degradation properties of the vacuole.
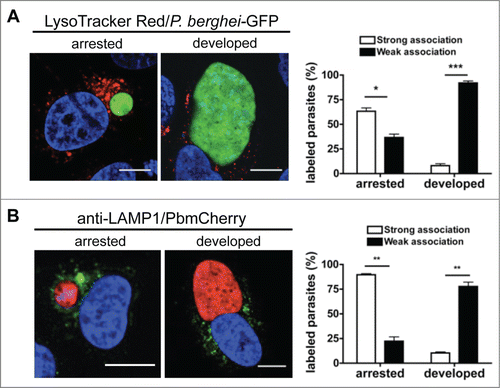
Figure 4. The PV is not a destructive vacuole. (A) HepG2 cells infected with P. berghei sporozoites expressing the GFP fluorescent protein were labeled with LysoTracker Red for 10 min. They were then monitored by live microscopy at different developmental stages. At the sporozoite stage, there was marked accumulation of LysoTracker Red-positive vesicles around the parasite, which met the criteria for strong association (the presence of >10 LysoTracker Red-positive vesicles around the parasites was deemed to indicate a strong association. A weak association was <10 vesicles). The parasites with strong and weak associations were counted and expressed as percentages. Standard deviations are depicted. DNA was stained with Hoechst 33342. Sporozoite (spz) P = 0.0148. Early schizont (e-schz) P = 0.0015. Late schizont (l-schz) P = 0.0011. (B) HepG2 cells infected with mCherry-expressing P. berghei parasites were fixed at different time points and stained with an antibody specific for LAMP1 (green). DNA was stained with DAPI. Scale bars: 10 µm. Stained cells were then monitored by live microscopy at different stages of parasite development. At the sporozoite stage, marked accumulation of LAMP1-positive vesicles around the parasite was observed. It was deemed to be a strong association if more than 30% of the parasite was covered by anti-LAMP1 antibody. The parasites with strong and weak associations were counted and expressed as percentages. Each bar indicates the average of 3 independent experiments. Standard deviations are depicted. P < 0.001 (spz), P < 0.001 (e-schz), P < 0.001 (l-schz).
Finally, we investigated the fate of the arrested parasites and compared them to normally developing parasites. At later stages, the arrested parasites remained LAMP1-, LysoTracker Red-, and LC3-positive, which indicated that these parasites were in the process of being eliminated (). Elimination of an arrested LC3-positive parasite can be observed in Movie S7.
Figure 5. Autophagic markers label arrested parasites. (A) HepG2 cells infected with fully virulent P. berghei-GFP sporozoites were labeled with LysoTracker Red for 10 min. At 48 hpi, the cells were monitored by live microscopy. LysoTracker Red-positive vesicles around the parasites were counted and scored as strong or weak associations as described in . The strongly and weakly associated parasites were then counted and expressed as percentages. Each bar indicates the average of 3 independent experiments. Standard deviations are depicted. DNA was stained with Hoechst 33342. Scale bars: 10 µm. Arrested P = 0.0298. Developed P = 0.0011. (B) HepG2 cells infected with mCherry-P. berghei sporozoites were fixed 48 hpi and stained with an antibody specific for LAMP1 (green). DNA was stained with DAPI. Scale bars: 10 µm. The association of LAMP1-positive vesicles with the parasites was scored as strong or weak as before (). The strongly and weakly associated parasites were then counted and expressed as percentages. Each bar indicates the average of 3 independent experiments. Standard deviations are depicted. P = 0.0023 (arrested), P = 0.002 (developed).
These observations together suggest that while the host cells try to isolate the invading parasites in LC3-positive compartments, they are not able to eliminate the majority of these parasites within these vacuoles by acidification through fusion with lysosomes. Thus, even though lysosomes associate with the PVM at an early stage, the majority of parasites avoid acidification, protease activation, and degradation.
LC3 recruitment to the PVM is the result of a novel form of autophagy
There are several pathways that could potentially trigger the observed LC3 labeling of the PVM in P. berghei-infected hepatocytes. One is LAP, which is characterized by a series of clearly defined molecular events.Citation9,23 A hallmark of LAP is the initial labeling of the target membrane by phosphatidylinositol 3-phosphate (PtdIns3P) and a transient labeling by BECN1/Beclin 1. Since we did not find PtdIns3P and BECN1 in the PVM (data not shown) and because we have identified ubiquitin and SQSTM1 localization to the PVM (see below), we excluded LAP as a possible mechanism.
Another process that targets LC3 to a membrane is selective autophagy, in which target membranes are labeled with ubiquitin and the receptor protein SQSTM1 that then recruits LC3.Citation24-27 However, in selective autophagy, a newly formed phagophore is generated to isolate the target compartment.Citation8 Since the presence of this additional membrane was excluded by EM analysis (), selective autophagy as it has been characterized so far was also excluded as a possible autophagic mechanism as well.
To our surprise, we then found that the PVM became positive for ubiquitin, SQSTM1, and LC3, all with very similar kinetics (; Movie S9). Still, so far it is not clear whether ubiquitination is connected with labeling of the PVM with LC3 and lysosomes. To exclude the possibility that ubiquitination of the PVM depends on LC3, we infected GFP-ubiquitin-expressing atg5−/− and control MEFs with P. berghei sporozoites. Although the atg5−/− MEFs are deficient in LC3 processing and lipidation,Citation28 both cell lines showed early ubiquitination of the PVM (). This further supports the hypothesis that a mechanism related to selective autophagy is activated on P. berghei invasion.
Figure 6. Ubiquitination and SQSTM1 labeling during early parasite development. (A) GFP-ubiquitin (green)-transfected HepG2 cells were infected with mCherry-P. berghei sporozoites (red) and monitored by live microscopy at different developmental stages. Each image depicts the main phenotype quantified at the indicated developmental stage. Scale bars: 10 µm. Quantification was performed visually by counting the parasites that associated strongly or weakly with ubiquitin. If more than 30% of the parasite's periphery was covered with ubiquitin-positive signals, the parasite was considered to be strongly associated. If less than 30% of the parasite's periphery was covered, the parasite was considered to be weakly associated. Each bar indicates the average of 3 independent experiments. Standard deviations are depicted. Sporozoite (spz) P = 0.002. Early schizont (e-schz) P = 0.0037. Late schizont (l-schz) P < 0.001. (B) HepG2 cells infected with mCherry-expressing P. berghei sporozoites were fixed at different developmental stages and stained with an antibody specific for SQSTM1 (green). DNA was stained with DAPI. Scale bars: 10 µm. Quantification of SQSTM1 signals was performed as described above for ubiquitin signals. Each bar indicates the average of 3 independent experiments. Standard deviations are depicted. Spz P < 0.001. E-schz P = 0.0021. L-schz P = 0.042. (C) WT and atg5−/− murine embryonic fibroblasts (MEFs) transfected with GFP-Ubiquitin plasmid were infected with mCherry-expressing Plasmodium sporozoites. Live microscopy was performed at 6 hpi. Scale bars: 10 µm.
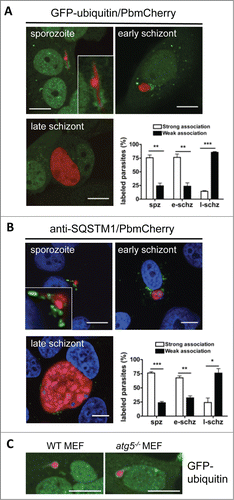
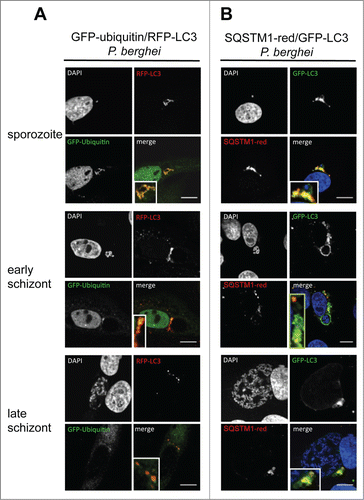
Figure 7. Colocalization of ubiquitin, SQSTM1, and LC3 in infected cells during parasite development. (A) GFP-Ubiquitin and RFP-LC3 double-transfected HepG2 cells were infected with wild-type P. berghei sporozoites and monitored by live microscopy at different developmental stages. The sporozoites are labeled by complete colocalization of ubiquitin and LC3, which is lost at later time points. DNA was stained with Hoechst 33342. (B) GFP-LC3 (green)-expressing HepG2 cells were infected, fixed at different developmental stages, and stained with an antibody specific for SQSTM1 (red). DNA was stained with DAPI (blue). Scale bars: 10 µm.
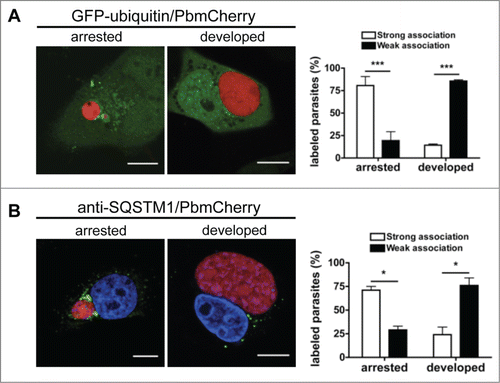
Importantly, in infected HepG2 cells the PVM of arrested parasites stayed positive for all markers, namely, ubiquitin, SQSTM1 (), and LC3 (). This suggests that this PVM labeling pattern associates with parasite elimination. However, the majority of parasites escaped this advanced stage of autophagy. The molecular basis of this escape mechanism is currently unknown.
Figure 8. Persistence of ubiquitination and SQSTM1 in arrested parasites. (A) GFP-ubiquitin-transfected HepG2 cells infected with mCherry-expressing P. berghei sporozoites were monitored by live microscopy 48 hpi. Scale bars: 10 µm. The strength of ubiquitin-positive staining around the parasite was scored as described for ubiquitin in the legend to . Each bar indicates the average of 2 independent experiments. Standard deviations are depicted. P < 0.0001. (B) HepG2 cells were infected with mCherry-expressing P. berghei sporozoites, fixed 48 hpi, and stained with antibodies specific for SQSTM1 (green). DNA was stained with DAPI. Scale bars: 10 µm. The strength of SQSTM1-positive staining around the parasite was scored as strong or weak as described for ubiquitin in the legend of . Each bar indicates the average of 2 independent experiments. Standard deviations are depicted. Arrested P = 0.0177. Developed P = 0.042.
Galectins do not mediate labeling of the PVM
Galectins have been shown to target damaged bacteria-containing vacuoles for autophagy, in order to defend cells against bacterial invasion.Citation29 To investigate whether galectins are also recruited to the PVM in P. berghei-infected cells, we transfected HepG2 cells with constructs that allow expression of fluorescently tagged galectins LGALS1, LGALS3, LGALS8 and LGALS9. When we infected these transfected cells with P. berghei sporozoites we did not observe a marked labeling of the PVM, suggesting that none of the galectins investigated play an important role the recruitment of LC3 to the PVM (Figs. S6–S8).
Starvation-induced autophagy promotes parasite growth and survival
To test whether enhanced autophagy has an effect on parasite development we wanted to analyze parasite development in vivo under starvation conditions. For that we generated a P. berghei ANKA reporter line (PbmCherryhsp70+Luceef1α), which expresses luciferase under the control of the eef1α (PBANKA_113330) promoter and, in addition, mCherry under the control of the hsp70 (PBANKA_071190) promoter (Fig. S9A–D). Parasites of this line express luciferase and mCherry throughout the life cycle. The life cycle progression of this line in the blood, mosquito, and liver stage development is comparable to that of wild-type P. berghei ANKA parasites (Fig. S9E).
Balb/c mice were starved and infected with the luciferase-expressing P. berghei line PbmCherryhsp70+Luceef1α, and parasite growth in the liver was monitored until transition to the blood stage (). We followed 2 different regimens: mice were either prestarved 12 h before infection or they were starved upon infection. Surprisingly, both starvation regimens resulted in an increased parasite load in the liver with a peak at 48 hpi (). To analyze whether starvation indeed induces canonical autophagy in hepatocytes, GFP-LC3 mice were starved and the liver was monitored by intravital imaging 30 h later (Fig. S10). Significantly higher numbers of GFP-LC3-positive autophagosomes upon starvation confirmed increased autophagy levels. Together, canonical nonselective autophagy appears to even support parasite development in hepatocytes.
Figure 9. Assessment of the effect of starvation on parasite liver-stage development by in vivo bioluminescence. Balb/c mice were infected by intravenous (i.v.) injection of 1 × 105 PbmCherryhsp70 + Luceef1α sporozoites. Mice were divided into 3 groups: a control group feeding ad libidum, a group of mice fasting 12 h prior to infection, and 18 hpi, and a group of mice fasting for 30 h following infection. (A) parasite development was assessed by whole body luminescence at the indicated time-points (hours post infection; hpi) in mice. Representative images of one mouse per group are shown. (B) ROI (region of interest) measurements for the liver area of all mice (n = 9) were recorded as photons per sec per area (photons/s/cm2) for firefly luciferase. (C) Balb/c and C57B/6 mice were infected with 2 × 105 sporozoites, and divided into a control group, or groups fasted for 24 or 36 h. For all mice, livers were removed at 40 hpi for histology analyses. Sections were stained with H&E, and parasite numbers and sizes quantified. Parasite sizes (C) and numbers (D) are expressed as percentage to the control. Mean number and parasite sizes of control mice were set as 100%. Experiments were repeated 3 to 5 times (error bars show SD; *P = 0.05; **, P = 0.01; ***, P < 0.001).
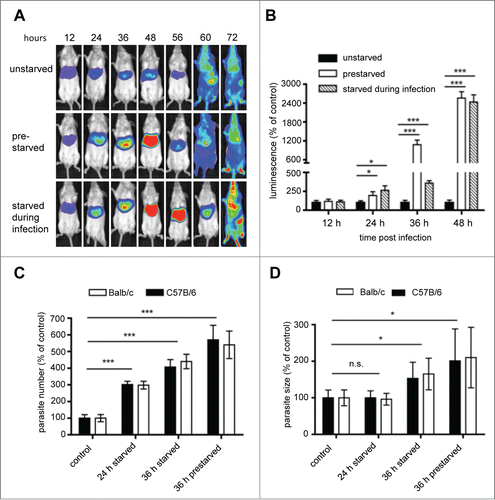
To examine whether the increased parasite load is due to more surviving parasites or increased parasite growth, Balb/c and C57B/6 mice were infected with P. berghei and the size and the numbers of intracellular parasites were analyzed on histological sections (). Liver sections of nonstarved mice served as control. At 24 hpi the number of parasites was significantly increased in the starved animals; this was even more pronounced at 36 and 44 hpi. The fact that in starved animals the relative number of parasites was higher at later stages indicates that the number of control parasites must have decreased over time. This confirms our original in vitro observations that a considerable number of parasites are eliminated during their development from sporozoite to liver schizont. Importantly, although the absolute number of infected hepatocytes differed substantially among Balb/c and C57B/6 mice, there was no significant difference in the relative number in both mouse strains when comparing starved and control mice. In conclusion starvation-induced autophagy had similar effects on parasite development independent of the mouse strain used suggesting very general autophagy mechanisms in different mouse stains. This confirms that both animal models are suited to study autophagy-related events upon Plasmodium infection.
Drug-mediated manipulation of autophagy has a profound effect on parasite growth and survival
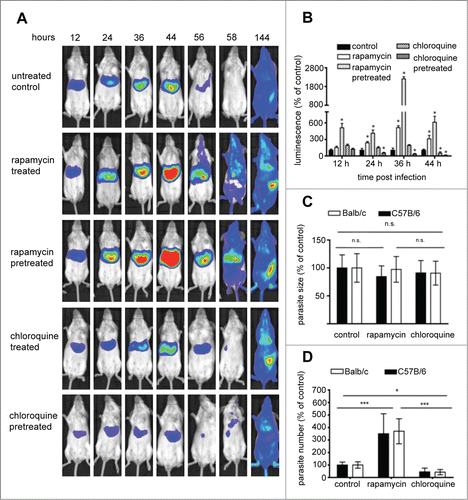
To confirm that inducing canonical autophagy has a positive effect on parasite development, an in vivo drug-based approach was employed. Rapamycin is a well-known inhibitor of the gatekeeper of autophagy, MTOR, and thus a strong inducer of autophagy. Chloroquine blocks autophagy by preventing the fusion of lysosomes with autophagosomes. Mice were treated with the either of the 2 drugs, before or after infection with luciferase-expressing P. berghei PbmCherryhsp70 + Luceef1α sporozoites. As observed in the starvation experiment, the autophagy-inducing drug rapamycin had a strong positive effect on parasite liver load whereas the autophagy inhibitor chloroquine had the opposite effect (, Fig. S11). The strongest effects were observed in mice pretreated with the drugs. In contrast to starvation-induced autophagy, increased parasite load at 40 hpi in rapamycin-treated mice were solely due to increased parasite numbers not to enhanced growth of the parasite in the liver (). Interestingly, chloroquine treatment also did not significantly affect parasite size at this time. Lower parasite load in the liver was due to fewer infected cells at the end of liver stage development. Intravital imaging of GFP-LC3 mice was again employed to confirm that rapamycin induces autophagy (Fig. S10). Actually, in comparison to starvation, rapamycin treatment resulted in a dramatically higher number of GFP-LC3-positive autophagosomes.
Figure 10. Assessment of the effect of autophagy induction and inhibition on parasite liver-stage development by in vivo bioluminescence. Balb/c mice were infected by intravenous (i.v.) injection of 1 × 105 PbmCherryhsp70+Luceef1α sporozoites. Mice were divided into: a control group untreated; a control group injected with solution media only; a group treated with rapamycin 12 and 24 hpi; a group pretreated with rapamycin 12 h prior to infection, and 12 hpi; a group treated with chloroquine 12 and 24 hpi; a group pretreated with chloroquine 12 h prior to infection and 12 hpi (A) parasite development was assessed by whole body luminescence at the indicated time-points (hpi) in mice. Representative images of one mouse per group are shown. (B) ROI measurements for the liver area of all mice (n = 15) were recorded as photons per sec per area (photons/s/cm2) for firefly luciferase. Balb/c and C57B/6 mice were infected with 5 × 104, 1 × 105, and 2 × 105 sporozoites. Histology sections were obtained from infected mice under 3 conditions, namely untreated controls, rapamycin-treated mice, and chloroquine-treated mice. Livers of all mice were removed 40 hpi, paraffin-fixed, H&E-stained and analyzed. Parasite sizes (C) and numbers (D) were quantified for the 3 groups. Average number and parasite sizes of control mice were set as 100%. Parasite numbers and sizes of rapamycin- and chloroquine-treated mice are expressed as percentage of controls. Experiments were repeated in triplicate (error bars show SD; *, P = 0.05; **, P = 0.01; ***, P < 0.001; for (B) for simplification * denotes P value is significant; details of significance are shown in Fig. S9).
Together, the in vivo experiments strongly suggest that manipulation of autophagy has profound effects on parasite development and growth.
Inhibition of LC3 recruitment to the PVM promotes parasite establishment in host cells
Since starvation and drug treatment can have many off-target effects in mice, we next aimed for a more specific in vitro approach to test whether blocking the autophagy machinery affects parasite growth and development. We took advantage of the fact that P. berghei sporozoites infect a wide variety of cells, including murine embryonic fibroblasts (MEFs). We used an atg5−/− MEF cell line in which the pathway that leads to LC3 lipidation is blocked.Citation28 Thus, in Atg5-deficient cells, all events that depend on correct LC3 lipidation and membrane association, including autophagy, are interrupted. First, we confirmed that atg5−/− MEFs are deficient in LC3 lipidation upon starvation of the cells by IFA with anti-LC3 antibodies. The knockout and WT MEFs were starved, fixed and stained to analyze LC3 localization (). In WT MEFs, the typical LC3 recruitment to autophagosomes was observed. By contrast, in atg5−/− MEFs, LC3 had a cytoplasmic localization (). Next, we analyzed the dynamics of LC3 in WT and atg5−/− MEFs after infection with mCherry-expressing sporozoites (). While LC3 accumulated in the classical pattern around the parasite in WT MEFs, in infected atg5−/− MEFs it stayed in the cytoplasm. This suggests that the LC3 recruitment observed in WT cells after sporozoite infection is dependent on the typical processing and lipidation of LC3 that is mediated by ATG5.
Figure 11. Parasite development after host cell autophagy is blocked. (A) Wild-type (WT) and atg5−/− MEFs were incubated in starvation media (EBSS) for 2 h or maintained in normal media (left panels) before they were fixed and stained then and cells were stained with a monoclonal anti-LC3 antibody, and analyzed by IFA. In subsequent experiments, WT and atg5−/− MEFs were infected with mCherry-expressing P. berghei (right panels). The infected cells were fixed 2 hpi. Then, cells were stained with a monoclonal anti-LC3 antibody, and analyzed by IFA. Scale bars: 10 µm. (B) WT and atg5−/− MEFs were infected with GFP-expressing P. berghei and fixed 48 hpi. The total parasites were counted and depicted in the figure. Each bar represents the average of 3 independent experiments. The standard deviation is indicated. P = 0.0091. (C) WT and atg5−/− MEFs were infected with P. berghei parasites. In the atg5−/− cells, the medium was either supplemented with additional amino acids (atg5−/− +aa) or left untreated (atg5−/−). Parasite sizes were measured 48 hpi. ***, P < 0.0001. ns, not significant.
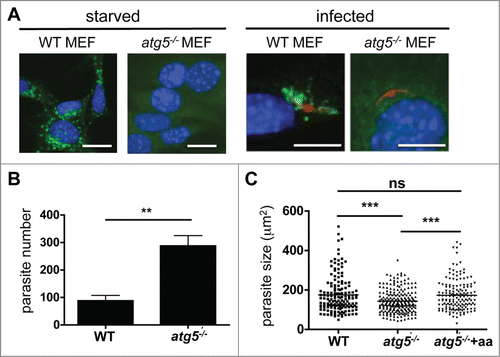
We next reasoned that if LC3-dependent autophagy is indeed an important mechanism by which the host restricts parasite establishment in its cells, infection of atg5−/− MEFs should result in higher parasite counts. Indeed, the atg5−/− MEF cultures had more than twice as many infected cells than the WT MEF cultures (). However, the parasites developing in the atg5−/− MEFs were significantly smaller than the parasites that had escaped elimination and continued to reside in the WT MEFs (). This suggests that host cell autophagy may also have beneficial effects for the parasites that manage to escape the first wave of the cytosolic immune response. Since we observed that infected cells not only recruited LC3 to the PVM but also constantly generated typical autophagosomes (Movie S10), we reasoned that successful Plasmodium parasites may induce and exploit host cell autophagy to obtain additional amino acids. To test this possibility, we supplemented the medium of sporozoite-infected atg5−/− cells with additional amino acids. Indeed, this rescued the parasite size in these cells ().
Discussion
Host cell response against Plasmodium infection
It has become increasingly clear that cells try to fight intracellular pathogens by many means.Citation12 Committing ‘suicide’ by inducing apoptosis is one possibility; another one is to isolate the pathogen in a compartment and then specifically eliminate it. The latter mechanism is particularly valuable for the host as the infected cell can then introduce pathogen peptides into the MHC presentation system.Citation30 Presentation via MHC has the additional advantage that it can help to induce a cell-mediated immune response against the infected cells.
Autophagy is an important and well-studied cytosolic immune response. In selective autophagy, newly formed membranes surround the pathogen that is to be eliminated.Citation12 These membranes are labeled with LC3-II and later fuse with lysosomes to form degradative autolysosomes. Aside from selective autophagy, recent studies have suggested that there are several other alternative pathways that have the capacity to eliminate pathogens.Citation31,32 Most of such pathways are closely related to autophagy and even share the same molecules to execute their tasks. In principle, their main role is to isolate the pathogen in a compartment, to label the membrane of the pathogen-containing compartment, to direct destructive organelles like lysosomes toward these compartments, and finally to fuse these organelles with the membrane of such compartments, thereby releasing their degradative content into the pathogen's hosting niche. Many pathogens, including Plasmodium sporozoites, enter the cell by invaginating the host cell membrane and forming a vacuolar membrane. Thus, they are already in an isolated compartment. It has been shown for several invasive bacteria that such already existing compartments can be recognized by the host cell and are labeled by LC3-II.Citation9,33,34 We have shown here that this also occurs with invading Plasmodium sporozoites. Although the entire mechanism for new membrane formation is not needed because the PVM is directly labeled, the pathways that result in LC3 processing and lipidationCitation35 still appear to be necessary. These pathways include 2 ubiquitin-like conjugation systems. If one of these conjugations systems is blocked or the C terminus of LC3 is altered, correct processing and lipidation will no longer be possible. Indeed, when we expressed the GFP-LC3G120A mutant in HepG2 cells and then infected them with mCherry-expressing parasites, we found that the mutant protein did not incorporate into the PVM. Thus, in Plasmodium infection of hepatocytes, like in other infections, LC3 must be properly processed and lipidated before it successfully incorporates into the PVM. We were also able to assess the functional significance of this event by using MEFs in which the atg5 gene has been deleted.Citation28 In these atg5−/− MEFs, we never observed incorporation of LC3 into the PVM and parasite elimination was significantly reduced, confirming that the autophagy-related pathway participated directly in parasite control. The fact that more parasites were detected in the autophagy-deficient MEFs than in the autophagy-competent MEFs clearly supports the notion that the host cell exerts an active autophagy-related process to eliminate viable parasites, rather than the dying parasites simply being removed by autophagy.
While LC3 incorporation into the PVM appears to be an autophagy-related process, it is not a typical selective autophagy since the latter is characterized by the formation of new phagophores. Instead, LC3 incorporation to the PVM resembles LAP more closely. However, LAP is clearly defined by a certain order of molecular events.Citation36 Since the invasion of bacteria and Plasmodium sporozoites are entirely different processes, we expected that the molecular events resulting in LC3 incorporation into the PVM would differ from the events in LAP. Indeed, results from our laboratory strongly suggest that LC3 incorporation into the PVM does not depend on PtdIns3P and BECN1 labeling (data not shown), which are hallmarks of LAP.Citation36
The exclusion of canonical autophagy and LAP suggests that P. berghei-infected hepatocytes must have an alternative pathway by which LC3 is incorporated into a membrane. Similar to selective autophagy, this new pathway appears to be initiated by ubiquitination, and subsequent association with the linker protein SQSTM1. However, unlike selective autophagy, no new membrane is built around the PVM. Instead, ubiquitin, SQSTM1 and LC3-II all appear to be directly incorporated into the PVM. The fact that atg5−/− MEFs allow ubiquitination but not LC3-II incorporation supports the idea that ubiquitination precedes LC3-II mobilization, perhaps via SQSTM1, which is incorporated into the PVM with similar kinetics as ubiquitin and LC3-II.
Parasite escape from the cytosolic immune response of the host cell
Although our data show clearly that host cells can actively eliminate a considerable number of parasites, more than half of them evade this response. We still do not know why some parasites are eliminated and others develop normally. We hypothesize that there may be a very fine balance between the parasite and the host cell that determines the outcome of the infection. During schizogony, successful parasites appear to actively remove LC3 from the PVM and avoid continued attack by lysosomes. There are several mechanisms by which the parasite could achieve this. First, it could activate a protease that cleaves LC3. One such protease may be the host-cell ATG4. This protease executes exactly this function during canonical autophagy.Citation37,38 Another option is that the labeled membrane is shed; the time-lapse movies provided in this study indeed suggest this is the case (in particular in the long-term; Movies S2–S4). However, this observation needs to be further validated experimentally.
Although it is clear that the parasite has developed strategies that allow it to survive within hepatocytes, many aspects remain to be analyzed. It is not known why the PV of successfully developing parasites does not acidify, despite fusion of the PVM with lysosomes, as shown in this and a previous study.Citation20 One possibility is that the PVM contains outward-directed proton pumps. However, this may not be necessary considering that the PVM contains pores for the easy uptake of nutrients of up to 855 Da,Citation39 and that proton gradients may not be able to form in such an environment because they need tight membranes without pores. It has also been suggested that the PVM fuses selectively with lysosomes with a mild pH. However, how the PVM can select between lysosomes with low and mild pH is not known. It is also not clear why we did not detect DQ-BSA red in the PV of normally developing parasites. If lysosomes fuse with the PVM, it can be assumed that their contents are released into the PV. Indeed, it has been shown that gold particles that are taken up by the endocytic pathway of infected host cells can be delivered via lysosomes to the PV and even to the parasite cytoplasm.Citation20 However, since we have shown in this study that parasites can be isolated and eliminated within autolysosomes, it remains to be determined whether the parasites containing gold particles are still viable.
A recent study shows that in Plasmodium yoelii-infected hepatocytes, the phosphorylation of AKT kinase and MTOR is elevated.Citation40 The authors speculate that this protects the parasites from host cell autophagy. Moreover, the expression levels of typical autophagy marker proteins like LC3 are not increased.Citation40 Transcriptome and proteome analysis in earlier reports also has failed to detect elevation of autophagy-related transcripts or proteins in Plasmodium-infected hepatocytes,Citation41,42 which strongly suggests that transcription of autophagy genes is at least not enhanced. These observations are not contradictory to the data provided in this study. Autophagy can be induced at different levels. On the one hand, selective autophagy can immediately be activated without transcriptional activation because all components are already present in the cell.Citation27 Induced autophagy, on the other hand, relies on transcriptional activation and the production of new proteins. Our results suggest that the new pathway observed in Plasmodium-infected hepatocytes does not depend on transcriptional activation of autophagy genes and is thus compatible with the data of the study on P. yoelii-infected hepatocytes described above.Citation40 It now remains to be determined how the parasite controls the induced autophagy of the host cell.
Canonical nonselective autophagy supports parasite survival and growth
Another interesting aspect of Plasmodium infection of hepatocytes was that the successful parasites appeared to benefit from host-cell autophagy, as it provided them with an additional source of nutrients. During schizont development, many autophagic events were observed in infected cells probably because the fast-growing parasite uses amino acids from the host cell. As such, amino acid starvation is a strong trigger of canonical nonselective autophagy. In fact our in vivo and in vitro data strongly suggest that activation of nonselective autophagy heavily supports parasite survival and growth. Starvation-mediated as well as drug-activated autophagy resulted in significantly enhanced parasite liver load whereas block of autophagy reduced parasite liver load. Nevertheless, we believe that the autophagy we observed is restricted to the already existing basal autophagy machinery in agreement with observations provided by earlier studies.Citation40-42
In summary, we suggest that hepatocytes counter Plasmodium parasite infection via a novel cytosolic immune reaction, and that the parasite responds by manipulating and modulating this host cell response. We believe that other intracellular parasites, bacteria, and viruses may employ similar mechanisms. How this knowledge can be translated into effective treatment strategies against these different pathogens remains to be explored.
Materials and Methods
Ethics statement
This study was carried out in strict accordance with the guidelines of the German Tierschutzgesetz (Animal Rights Laws). Mice were obtained from Charles River Laboratories and were between 6 and 10 wk of age. The protocol was approved by the Department of Veterinary Affairs of the Hamburg state authorities (Permit Number: FI 28/06). Blood feeding and intravital imaging was performed under ketavet/rompun anesthesia, and all efforts were made to minimize suffering. For the generation of the PbmCherryhsp70+Luceef1α parasite line Swiss mice (OF1 ico, Construct 242; 6 wk old; 25 to 26 g; Charles River) were used. All animal experiments to generate and characterize this line were performed at the LUMC and approved by the Animal Experiments Committee of the Leiden University Medical Center (DEC 12042). The Dutch Experiments on Animals Act was established under European guidelines (EU directive no. 86/609/EEC regarding the Protection of Animals used for Experimental and Other Scientific Purposes).
Parasites
P. berghei-ANKA lines were used to infect mice and cultured cells. P. berghei-mCherryCitation43 and P. berghei-GFPCitation44 constitutively express fluorescent proteins that localize to the cytosol of the parasite. P. berghei-Exp1-mCherryCitation45 express the PVM marker protein Exp1 fused to mCherry under the liver stage specific promoter (PBANKA_100300) at the parasite PVM.
The PbANKA-230p GIMO mother line (GIMOPbANKA; line 1596cl1) which contains the positive-negative selection marker (SM) cassette, a fusion gene of HsDHFR (Homo sapiens/human dihydrofolate reductase; positive SM), ScFCY1 (Saccharomyces cerevisiae/cytosine deaminase) and ScFUR1 (Saccharomyces cerevisiae/uracil phosphoribosyltransferase; negative SM) under control of the constitutive eef1a promoter, stably integrated into the neutral 230p locus (PBANKA_030600) through double crossover recombination.Citation46 GIMOPbANKA is a reference P. berghei ANKA line that is used to rapidly introduce transgenes without drug-resistance markers.
Generation and analysis of the reporter line PbmCherryhsp70+Luceef1α
Construct pL1720 was made for generation of reporter line mCherryhsp70+Luceef1α that expresses mCherry reporter under control of the hsp70 (PBANKA_071190) 5′- and 3′-regulatory sequences and luciferase under the control of the ee1fα (PBANKA_113330) 5′-promoter/regulatory sequences and the pbdhfr/ts (PBANKA_071930) 3′-regulatory sequences. This construct was used to target the GIMOANKA mother line using the ‘gene insertion/marker out’ (GIMO transfection) procedure.Citation46 Construct pL1720 was made as follows: first the selectable marker (SM) cassette from pL1661 (containing both 5′hsp70 and 3′hsp70 sequences, tgdhfr/ts SM, and 230p targeting regions) was removed by digesting the plasmid with SnaBI (Roche, 10997480001) and SbfI (New England Biolabs, R06425) followed by treatment of the DNA fragment with Klenow (Roche, 11008404001) and religation resulting in a plasmid that contains only the 5′hsp70–3′hsp70 sequences and the 230 p (PBANKA_030600) targeting regions; the mCherry coding sequences (excised from pL0047 which contains the 5′pbeef1α-mCherry-3′pbdhfr cassette) was then introduced into this plasmid using sites BamHI (Roche, 10220612001) and NotI (Roche, 11014706001). Next a luciferase expression cassette (5′pbeef1a-LucIAV-3′pbdhfr) excised from pL1098 using KpnI (Roche 11198939001) and AflII (Roche, 11198939001) was introduced into this vector using the same restriction sites. This final construct, pL1720, therefore contains the 5′hsp70-mCherry-3′hsp70 and 5′pbeef1a-LucIAV-3′pbdhfr expression cassettes and 230p targeting sequences but no SM. This construct was linearized by digesting with SacII (Roche, 11117807001) before transfection. The linear DNA construct was introduced into GIMOANKA parasites using standard methods of GIMO-transfection.Citation46 Transfected parasites were selected in mice by applying negative selection by providing 5-fluorocytosine (Sigma, F7129–5G) in the drinking water of mice.Citation47 Negative selection results in selection of parasites where the hdhfr-yfcu SM in the 230p locus is replaced by the mCherry reporter-cassette. Selected transgenic parasites (mCherryhsp70) were cloned by the method of limiting dilution.Citation48 Correct integration of the constructs into the genome of mCherryhsp70 parasites was analyzed by diagnostic PCR-analysis on gDNA and Southern analysis of pulsed field gel (PFG) separated chromosomes as described.Citation49 Growth of blood stages (asexual multiplication rate) of mCherryhsp70 parasites was determined during the cloning period as described previously.Citation50 Gametocyte production (gametocyte conversion rate) was determined as previously described.Citation51
Cell culture and infection of HepG2, MEF cells, and primary hepatocytes
Human hepatoma cells (HepG2) were provided by the European Collection of Cell Cultures (ECACC) and were maintained in minimum essential medium (MEM) with Earle's salts supplemented with 10% fetal calf serum (FCS), 100 U penicillin, 100 µg/ml streptomycin and 2 mM L-glutamine (all from PAA Laboratories, E15–024, A15–101, P11–010, M11–004). WT and atg5−/− murine embryonic fibroblasts (MEF, kindly provided by N. Mizushima, University of Tokyo) were maintained in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% FCS, 100 U penicillin, 100 µg/ml streptomycin, 2 mM L-glutamine and 1x non-essential amino acids (NEAA) (all from PAA Laboratories, E15–009, A15–101, P11–010, M11–004, M11–003). For the rescue experiments, MEFs were cultured in MEM supplemented as described above with or without extra amino acids (PAA Laboratories, M11–002). The cells were cultured at 37°C and 5% CO2 and were split using Accutase (PAA Laboratories, L11–007).
Primary mouse hepatocytes were isolated by using the protocol described by Wenshuo Zhang (http://www.mouselivercells.com) with modifications. Briefly, the liver of a freshly euthanized mouse was perfused via the portal vein with HEPES Buffer (137 mM NaCl [Sigma-Aldrich, S9888–1KG], 2.68 mM KCl [Fluka Chemie AG P9541–1KG], 10 mM HEPES [Sigma-Aldrich, H3375–500G], 0.7 mM Na2HPO4,[Fluka Chemie AG, 71633–250G], pH 7.6, 100 U penicillin, 100 µg/ml streptomycin), for 10 min followed by collagenase IV (100 U/ml; Worthington Biochemical Corporation, LS004188) in William medium E (Bioconcept, 1–48F01-l) containing 2 mM L-glutamine for 10 min. The perfusate was drained out of the body via the inferior vena cava, which was cut immediately after starting perfusion. During both perfusion periods, the inferior vena cava was clamped every 2 min for 10 sec to inflate the liver. The digested liver was then excised rapidly, the liver capsule was disrupted, and the hepatocytes were released by gently shaking the digested liver into William medium E with 2 mM L-glutamine. The cells were washed 3 times by 50 g centrifugation in William medium E with 2 mM L-glutamine for 2 min. Viability was checked by using the trypan blue exclusion method. Between 100,000 and 200,000 cells were seeded onto glass bottom dishes (MatTek corporation, P35G-1.5–20-C) in growth medium (William medium E, 2 mM L-glutamine, 10% FCS, 100 U penicillin, 100 µg/ml streptomycin). The growth medium was replaced 3 to 5 h after isolation. The cells were cultured at 37°C and 5% CO2.
When required, cells were treated with 10 µg/ml DQ™ Red BSA (DQ-BSA red; Molecular Probes, D12051) overnight or 75 nM LysoTracker Red DND-99 (Molecular Probes, L-7528) for 10 min. P. berghei-ANKA lines were used to infect mice and cell cultures. P. berghei WT does not express any fluorescent protein. P. berghei-mCherry and P. berghei-GFP are phenotypically not different from WT parasites apart from the fluorescence. Sporozoites were prepared from the salivary glands of infected female Anopheles stephensi mosquitoes and were incubated with the host cells for 2 to 4 h. To infect MEFs, the cell and sporozoite mixture underwent an additional centrifugation step (500 xg for 5 min) during the incubation. After washing, the infected cells were incubated in the medium described for each host cell type above, which was supplemented with 2.5 µg/ml amphotericin B (PAA Laboratories, P11–001).
Plasmids
The plasmid EGFP-LC3 was kindly supplied by Jonathan C. Howard, Cologne, Germany and the LAMP1-GFP construct was a kind gift from John Brumell (Hospital for Sick Children). pmRFP-LC3 (plasmid 21075, provided by Tamotsu Yoshimori)Citation52 and GFP-Ubiquitin (plasmid 11928 provided by Nico Dantuma)Citation53 were obtained from Addgene. The plasmid CTSD-mRFP was kindly provided by François Darchen (CNRS, Université Paris Descartes)Citation22 All LGALS/galectin constructs were kindly provided by Felix Randow (MRC Laboratory of Molecular Biology). To generate the plasmid EGFP-LC3G120A, the G120A mutation was introduced into the LC3 gene in the plasmid EGFP-LC3 by using the 5′-GGGCTCGAGATGCCG-TCCGAGAAGACC-3′ and 5′-GGGGTCGACTTACACAGCCATTGCTGTCGCGAATGTCTC-3′ primers and the restriction enzymes XhoI (New England Biolabs, R0146S) and SacI (New England Biolabs, R0156S).
Transfection of HepG2 and MEF cells
1 × 106 cells were pelleted by centrifugation. After resuspension in Nucleofector V solution (Lonza, VVCA-1003) for HepG2 cells or MEF1 solution for MEFs (Lonza, VPD-1004), the cells were transfected with 3 µg of either GFP-LC3 DNA, RFP-LC3 DNA, CTSD-mRFP DNA, or GFP-ubiquitin DNA by using a Nucleofector transfection device (program T-028 for HepG2 cells and T20 for MEFs) according to the manufacturer's instructions. The cells were then seeded onto glass bottom dishes for live cell imaging or onto 24-well plates on cover slips for IFA analysis.
Indirect immunofluorescence analysis
After the indicated time points, the infected cells were fixed in 4% paraformaldehyde in phosphate-buffered saline (PBS; 137 mM NaCl [Sigma-Aldrich, S9888–1KG], 2.7 mM KCl [Fluka Chemie AG P9541–1KG], 10 mM Na2HPO4 [Sigma-Aldrich, S5136–500G], 1.8 mM KH2PO4 [Sigma-Aldrich, P5655], pH 7.4) for 20 min at room temperature. Subsequently, they were either incubated in ice-cold methanol for 10 min at −20°C (for the anti-LAMP1 and -UIS4 antibodies) or permeabilized by using 10 µg/ml digitonin (for the anti-LC3 antibody; Sigma-Aldrich, D141–100MG) or 1% Triton X-100 (Fluka Chemie, T8787–250ML) in 10% FCS-PBS (for the anti-ubiquitin and anti-SQSTM1 antibodies). After washing with PBS, the nonspecific binding sites were blocked by incubation in 10% FCS-PBS for 1 h at room temperature. The primary antibodies were diluted in 10% FCS-PBS and then added to the culture. After 1 h at room temperature, the cells were washed with PBS. For detection, the cells were incubated with secondary antibodies for 30 to 60 min at ambient temperature. The nuclei were stained with 1 µg/ml DAPI (Invitrogen, D-1306). The cells were washed again with PBS and mounted on microscope slides with Dako Fluorescent Mounting Medium (Dako, S3023). For live imaging, the nuclei were stained with Hoechst 33342 (Sigma-Aldrich, B2261).
The primary antibodies used were mouse monoclonal anti-GFP (BD Biosciences, 565197), rat monoclonal anti-RFP (Chromotek, 5f8), mouse monoclonal anti-LC3 (MBL international, M152–3), rabbit polyclonal anti-UIS4 (kindly provided by P. Sinnis), chicken polyclonal anti-Exp1 (Heussler Lab, BNITM, Hamburg, Germany), mouse monoclonal anti-ubiquitin (Enzo life science, BML-PW8810, clone FK2), rabbit monoclonal anti-SQSTM1 (MBL international, PM045), and mouse monoclonal anti-LAMP1 (Developmental Studies Hybridoma Bank, H4A3). Secondary antibodies were anti-rabbit Alexa Fluor® 594 (Invitrogen, A21207), goat anti-rabbit Alexa Fluor® 488 (Invitrogen, A11034), anti-mouse Alexa Fluor® 488 (Life technologies, A21202), anti-mouse Alexa Fluor® 594 (Invitrogen, A11032), anti-rat Alexa Fluor® 594 (Invitrogen, A11007), anti-rat Alexa Fluor® 488 (Invitrogen, A21208), and anti-chicken Alexa Fluor® 594 (Invitrogen, A11042).
Live cell imaging and time lapse microscopy
Confocal laser line scanning (CLS) was performed with a Zeiss Observer Z1 inverted microscope integrated into a laser-scanning microscope (LSM 5 live, Zeiss, Germany). Images were acquired by using a Zeiss 63 × Plan-Apochromat 1.4 oil objective and the Zeiss Efficient Navigation 2008 and 2009 software. During imaging, the cells were maintained in a CO2 (5%) incubator at 37°C. Images of fixed cells were generated by a confocal laser point scanning microscope (FluoView 1000, Olympus, Germany) or by using the Leica DM12000B widefield epifluorescence microscope (Wetzlar, Germany). Contrast and brightness levels were optimized by using either the Zeiss Efficient Navigation 2009 (Jena, Germany), the ImageJ (NIH, USA) software, or Adobe Photoshop. Images were processed as a whole.
To monitor parasite development, the size of parasites was determined by using the density slice module of the OpenLab software version 5.0.2 or ImageJ.
Intravital microscopy was performed as previously describedCitation54 by using the LSM 510 Zeiss microscope (Zeiss, Germany) in the LSM 5 live mode. Images were acquired by CLS microscopy using a Zeiss Plan Apochromat 63x/1.40 oil DIC M27 objective and the Zeiss LSM 5 Duo Release Version 4.2. GFP-LC3 mice (kindly provided by K. MizushimaCitation11) were infected with mCherry-expressing parasites and intravital imaging of an infected liver lobe of an anesthetized mouse was performed at the indicated time points. Image processing was performed by using ImageJ.
Super-resolution microscopy
For dual-color stimulated emission depletion (STED) microscopy, hepatocytes infected with P. berghei sporozoites were fixed 24 hpi with 4% (w/v) paraformaldehyde in PBS followed by 15 min permeabilization in 0.1% Triton-X and 1 h incubation in 10% FCS-PBS blocking buffer. Cells were stained over night by 4°C with 10 µg/ml of anti-LC3 (MBL international, M152–3) and anti-UIS4 (kindly provided by Photini Sinnis) and labeled with 2 µg/ml Oregon Green-488 goat anti-mouse IgG (H+L) (Molecular Probes, Invitrogen, O-6380) and Abberior STAR 440SX goat anti-rabbit IgG (Abberior GmbH, 2–0012–003–4) at room temperature for 1 h. Samples were embedded in Mowiol® 4–88 (Roth, 0713.1) containing 2.5% DABCO® (Roth, 0718.1) antifade.
Microscopy was conducted on the Leica TCS SP8 inverted microscope equipped with gated STED system and a HC PL APO STED WHITE 100x/1.4 oil immersion objective (Leica Microsystems, Wetzlar, Germany). Depletion was performed with a 592-nm depletion laser and images acquired with a time delay of T g = 0.5 ns. Z-Stacks were obtained with increments of 0.22 µm and subsequently deconvolved with the Huygens STED Deconvolution software (Scientific Volume Imaging, Hilversum, Netherlands) and further processed with the image analysis software FIJI.Citation55
Electron microscopy
Infected HepG2 cells were FACS-sorted to enrich for infected cells and seeded on Thermanox coverslips. At 48 hpi, the cells were washed twice with PBS, fixed with 2% glutaraldehyde (Electron Microscopy Sciences, 16210) in sodium-cacodylate buffer, pH 7.2 and then fixed with 1% osmium tetroxide (Sigma-Aldrich, 201030–1G). The samples were then dehydrated by using a series of increasing ethanol concentrations and propylene oxide. The cells were embedded in an epoxy resin (Epon E812 resin; EMS, 14120). Ultrathin sections were made by using an Ultra Cut E microtome (Reichert-Leica, Germany) and stained with uranyl acetate and lead citrate. The sections were then examined with a Tecnai Spirit microscope (FEI, USA) at an acceleration voltage of 80 kV.
Quantification of viable P. berghei parasites
HepG2 cells seeded in 96-well μClear® plates (Greiner-Bio-one, 655097) were infected with P. berghei expressing mCherry. Two hpi, excess parasites were washed away twice with medium. Rabbit polyclonal anti-CSP antibody (diluted 1:1000) was added to the medium and incubated for 1 h to eliminate extracellular parasites. Cells were washed and the plate was imaged with an IN Cell Analyzer 2000 automated cell imaging system (GE Healthcare Life sciences, Glattbrugg, Switzerland) (10× objective, 9 fields/well) at 3.5, 9, 24, 33 and 48 hpi. The mCherry signal was detected using excitation and emission filters for TexasRed (emission: 579 nm; excitation: 605 nm). The exposure time was 15 to 30 ms (30 ms for the 3.5 h and 9 h time points and 15 ms for the later time points). IN Cell Developer Toolbox 1.7 was used to analyze the acquired images. Intensity segmentation was used to identify P. berghei mCherry. A post segmentation process (Sieve) was used to exclude 10 μm2 objects (3.5 to 24 h) and 20 μm2 objects (33 to 48 h). The output was the number of parasites per well in the 9 fields. The results of quadruplicate experiments are displayed graphically (the number of parasites per well at 3.5 h was set at 100%).
Quantification of autophagy and degradation markers
HepG2 cells were infected with P. berghei sporozoites and labeled with antibodies specific for different degradation markers, LysoTracker Red, or DQ-BSA red before being monitored by microscopy. Quantification was performed visually by counting the parasites that were stained strongly or weakly by the dye or antibody. For LysoTracker Red-, and DQ-BSA red-stained HepG2 cells, and HepG2 cells transfected with CTSD-RFP, a strong association was deemed to be the presence of 10 or more positive vesicles in the immediate proximity of the parasite. Fewer than 10 vesicles around the parasite was defined as a weak association. In infected HepG2 cells that were transfected with ubiquitin-GFP or LC3 or stained with anti-SQSTM1 antibody or anti-LAMP1 antibody, if the signal covered more than 30% of the proximity of the parasite, the association was considered to be strong. If less than 30% of the parasite was covered with these signals, it was deemed to be a weak association. Arrested parasites were those parasites with clear growth impairment, as indicated by their small size.
Preparation of P. berghei parasites and mice for bioluminescence imaging
For bioluminescence imaging of liver stage development, 1 × 105 P. berghei sporozoites were collected from salivary glands of infected Anopheles stephensi mosquitoes and injected intravenously into female Balb/c mice 8 to 10 wk of age. For starvation assays, mice were separated into a group feeding ad libidum, a 30h starvation group after infection, and a group starved for 12 h prior to infection followed by 18 hpi. For autophagy inhibition and induction assays, mice were separated into an untreated control group, a group treated with 50 mg/kg chloroquine (Sigma-Aldrich, C6628) at 12, 24 and 36 hpi; a group treated with 2.5 mg/kg of rapamycin (Enzo Life Sciences, BML-A275–0005) at 12, 24, and 36 hpi; a pretreated group, treated with 50 mg/kg chloroquine 2 h prior to sporozoite injection, and then 12, 24, and 36 hpi; and a pretreated group, treated with 2.5 mg/kg rapamycin 12 h prior to sporozoite injection followed by consecutive treatments at 12, 24 and 36 hpi.
Chloroquine was diluted in 0.9% NaCl to a final concentration of 20 mg/ml. Rapamycin was dissolved in 100% ethanol to a final concentration of 10 mg/ml. For luminescence assays, rapamycin was dissolved in a solution medium consisting of 5% PEG-400 (Sigma-Aldrich, 81170), 5% Tween 80 (Sigma-Aldrich, P4780–500 ML), and 4% ethanol (Merk Millipore, 100983). Dilutions of both drugs were prepared at the time of administration. Separate control mice were injected with solution media to exclude the potential of artificial effects in luminescence observations.
Real time in vivo bioluminescence imaging of liver stage development
Luciferase activity in all mouse groups was determined by full-body imaging of mice using an IVIS Lumina II imager (Caliper Life Sciences, USA). Infected mice under the various fasting and treatment conditions were anaesthetized using the isofluorane anesthesia system (XGI-8, Xenogen, Caliper Life Sciences, USA). Measurements were performed at various time points post infection including 12, 24, 36, 44, 56 and 60 h, and d 3 and/or 6 post sporozoite injection. For firefly luciferase quantification, anaesthetized mice were injected with 100 µl of RediJect D-Luciferin (30mg/ml; Perkin Elmer, 760504) intraperitoneally. Measurements were performed 7 to 10 min after injection of the substrate. Imaging was acquired with a 10 cm FOV, medium binning factor, and exposure time of 3 min. Quantitative analysis was performed using the Living Image 4.1 software.
Staining and histology of liver tissue sections
Histology sections were generated for measurement of size and numbers of parasites at 40 hpi in the following sets of mice: control, fasting for 24 hpi, fasting for 36 hpi, prefasting for 12 hpi and 24 hpi, rapamycin-treated, and chloroquine-treated. Three Balb/c and 3 C57B/6 mice were used per condition, and infected with varying doses of sporozoites, ranging from 5 × 104 to 2 × 105. At 40 hpi, mice were euthanized, perfused with 5 ml of 4% PFA in PBS, and livers removed. Both complete livers lobes were further stored in 2% PFA in PBS for a further 12 h, and then paraffin-embedded. 10 × 50 μm sections of both lobes of each animal from various tissue depths were obtained, and stained with hematoxylin and eosin (H&E). Sections were thoroughly analyzed using a Leica DM 5500 B Upright widefield epifluorescence microsope using a 40× objective for general imaging, and a 100 x1.6 oil objective for detailed analysis. Images were obtained using the Leica LAS AF Software, and size measurements performed in ImageJ/Fiji.
Statistical analysis
Two groups were compared by using a 2-tailed, unpaired Student t test. All statistical analyses were performed by using Prism version 4.0a for Macintosh, GraphPad Software, San Diego, California, USA. P values less than 0.05 were considered to indicate statistical significance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1067361_supplemental_movies__10__and_files.zip
Download Zip (19.3 MB)Acknowledgments
Jonathan Howard, François Darchen, and Felix Randow are acknowledged for providing plasmid EGFP-LC3, CTSD-mRFP, and all galectin constructs, respectively. The LAMP1-GFP construct was a kind gift from John Brumel. Anti-UIS4 antiserum was kindly provided by Photini Sinnis. Stefanie Graewe is thanked for providing the Movie S10 and Janic Teutsch for Movie S6. The Microscopy Imaging Centre (MIC) in Bern is thanked for technical support.
Funding
We thank the German Academic Exchange Service (DAAD) for supporting MP, the Swiss National Foundation for supporting VH (grant 310030_140691/1), the Indo-Swiss consortium and the EviMalaR EU consortium for financial support.
Related Research Data
References
- Amino R, Thiberge S, Martin B, Celli S, Shorte S, Frischknecht F, Ménard R. Quantitative imaging of Plasmodium transmission from mosquito to mammal. Nat Med 2006; 12:220-4; PMID:16429144; http://dx.doi.org/10.1038/nm1350
- Ingmundson A, Alano P, Matuschewski K, Silvestrini F. Feeling at home from arrival to departure: protein export and host cell remodelling during Plasmodium liver stage and gametocyte maturation. Cell Microbiol 2014; 16:324-33; PMID:24330249; http://dx.doi.org/10.1111/cmi.12251
- Sturm A, Amino R, van de Sand C, Regen T, Retzlaff S, Rennenberg A, Krueger A, Pollok JM, Menard R, Heussler VT. Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science 2006; 313: 1287-90; PMID:16888102; http://dx.doi.org/10.1126/science.1129720
- Baer K, Klotz C, Kappe SH, Schnieder T, Frevert U. Release of hepatic Plasmodium yoelii merozoites into the pulmonary microvasculature. PLoS Pathog 2007; 3:e171; PMID:17997605; http://dx.doi.org/10.1371/journal.ppat.0030171
- Grutzke J, Rindte K, Goosmann C, Silvie O, Rauch C, Heuer D, Lehmann MJ, Mueller AK, Brinkmann V, Matuschewski K, et al. The spatiotemporal dynamics and membranous features of the Plasmodium liver stage tubovesicular network. Traffic 2014; 15:362-82; PMID:24423236; http://dx.doi.org/10.1111/tra.12151
- Jo EK, Yuk JM, Shin DM, Sasakawa C. Roles of autophagy in elimination of intracellular bacterial pathogens. Front Immunol 2013; 4:97; PMID:23653625; http://dx.doi.org/10.3389/fimmu.2013.00097
- Yano T, Kurata S. Intracellular recognition of pathogens and autophagy as an innate immune host defence. J Biochem 2011; 150:143-9; PMID:21729928; http://dx.doi.org/10.1093/jb/mvr083
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Cemma M, Brumell JH. Interactions of pathogenic bacteria with autophagy systems. Curr Biol 2012; 22:R540-5; PMID:22790007; http://dx.doi.org/10.1016/j.cub.2012.06.001
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Atg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/10.1093/emboj/19.21.5720
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/10.1091/mbc.E03-09-0704
- Deretic V, Levine B. Autophagy, immunity and microbial adaptations. Cell Host Microbe 2009; 5:527-49; PMID:19527881; http://dx.doi.org/10.1016/j.chom.2009.05.016
- Wang Y, Weiss L, Orlofsky A. Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J Biol Chem 2009; 284:1694-701; PMID:19028680; http://dx.doi.org/10.1074/jbc.M807890200
- Muniz-Feliciano L, Van Grol J, Portillo JA, Liew L, Liu B, Carlin CR, Carruthers VB, Matthews S, Subauste CS. Toxoplasma gondii-induced activation of EGFR prevents autophagy protein-mediated killing of the parasite. PLoS Pathog 2013; 9:e1003809; PMID:24367261; http://dx.doi.org/10.1371/journal.ppat.1003809
- Meis JF, Verhave JP, Jap PH, Hess F, Meuwissen JH. An ultrastructural study of developing stages of exo-erythrocytic Plasmodium berghei in rat hepatocytes. Parasitology 1981; 82:195-204; PMID:7012764; http://dx.doi.org/10.1017/S0031182000056936
- Meis JF, Verhave JP, Jap PH, Meuwissen JH. Transformation of sporozoites of Plasmodium berghei into exoerythrocytic forms in the liver of its mammalian host. Cell Tissue Res 1985; 241:353-60; PMID:3896506; http://dx.doi.org/10.1007/BF00217180
- Meis JF, Verhave JP, Jap PH, Meuwissen JH. Fine structure of exoerythrocytic merozoite formation of Plasmodium berghei in rat liver. J Protozool 1985; 32:694-9; PMID:3906102; http://dx.doi.org/10.1111/j.1550-7408.1985.tb03104.x
- Eickel N, Kaiser G, Prado M, Burda P, Roelli M, Stanway R, Heussler VT. Features of autophagic cell death in Plasmodium liver-stage parasites. Autophagy 2013; 9:568-80; PMID:23388496; http://dx.doi.org/10.4161/auto.23689
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Lopes da Silva M, Thieleke-Matos C, Cabrita-Santos L, Ramalho JS, Wavre-Shapton ST, Futter CE, Barral DC, Seabra MC. The Host Endocytic Pathway is Essential for Plasmodium berghei Late Liver Stage Development. Traffic 2012; 13:1351-63; PMID:22780869; http://dx.doi.org/10.1111/j.1600-0854.2012.01398.x
- Obermuller S, Kiecke C, von Figura K, Honing S. The tyrosine motifs of Lamp 1 and LAP determine their direct and indirect targetting to lysosomes. J Cell Sci 2002; 115:185-94; PMID:11801736
- Yuseff MI, Reversat A, Lankar D, Diaz J, Fanget I, Pierobon P, Randrian V, Larochette N, Vascotto F, Desdouets C, et al. Polarized secretion of lysosomes at the B cell synapse couples antigen extraction to processing and presentation. Immunity 2011; 35:361-74; PMID:21820334; http://dx.doi.org/10.1016/j.immuni.2011.07.008
- Florey O, Overholtzer M. Autophagy proteins in macroendocytic engulfment. Trends Cell Biol 2012; 22:374-80; PMID:22608991; http://dx.doi.org/10.1016/j.tcb.2012.04.005
- Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol 2014; 12:101-14; PMID:24384599; http://dx.doi.org/10.1038/nrmicro3160
- Jiang X, Chen ZJ. The role of ubiquitylation in immune defence and pathogen evasion. Nat Rev Immunol 2012; 12:35-48; PMID:22158412
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID:21189453; http://dx.doi.org/10.4161/auto.7.3.14487
- Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 2010; 12:836-41; PMID:20811356; http://dx.doi.org/10.1038/ncb0910-836
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/10.1038/nature03029
- Thurston TL, Wandel MP, von Muhlinen N, Foeglein A, Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature 2012; 482:414-8; PMID:22246324; http://dx.doi.org/10.1038/nature10744
- Puleston DJ, Simon AK. Autophagy in the immune system. Immunology 2014; 141:1-8; PMID:23991647; http://dx.doi.org/10.1111/imm.12165
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nate Cell Biol 2013; 15:713-20; PMID:23817233; http://dx.doi.org/10.1038/ncb2788
- Mehta P, Henault J, Kolbeck R, Sanjuan MA. Noncanonical autophagy: one small step for LC3, one giant leap for immunity. Curr Opin Immunol 2014; 26:69-75; PMID:24556403; http://dx.doi.org/10.1016/j.coi.2013.10.012
- Birmingham CL, Brumell JH. Autophagy recognizes intracellular Salmonella enterica serovar Typhimurium in damaged vacuoles. Autophagy 2006; 2:156-8; PMID:16874057; http://dx.doi.org/10.4161/auto.2825
- Dupont N, Lacas-Gervais S, Bertout J, Paz I, Freche B, Van Nhieu GT, van der Goot FG, Sansonetti PJ, Lafont F. Shigella phagocytic vacuolar membrane remnants participate in the cellular response to pathogen invasion and are regulated by autophagy. Cell Host Microbe 2009; 6:137-49; PMID:19683680; http://dx.doi.org/10.1016/j.chom.2009.07.005
- Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol 2011; 7:9-17; PMID:21164513; http://dx.doi.org/10.1038/nchembio.500
- Lai SC, Devenish, R. J. LC3-associated phagocytosis (LAP): connections with host autophagy. Cells 2012 1:396-408; PMID:24710482; http://dx.doi.org/10.3390/cells1030396
- Nakatogawa H, Ishii J, Asai E, Ohsumi Y. Atg4 recycles inappropriately lipidated Atg8 to promote autophagosome biogenesis. Autophagy 2012 8:177-86; PMID:22240591; http://dx.doi.org/10.4161/auto.8.2.18373
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO reports 2008; 9 859-64; PMID:18704115; http://dx.doi.org/10.1038/embor.2008.163
- Bano N, Romano JD, Jayabalasingham B, Coppens I. Cellular interactions of Plasmodium liver stage with its host mammalian cell. Int J Parasitol 2007; 37:1329-41; PMID:17537443; http://dx.doi.org/10.1016/j.ijpara.2007.04.005
- Kaushansky A, Ye A, Austin L, Mikolajczak S, Vaughan A, Camargo N, Metzger PG, Douglass AN, MacBeath G, Kappe SH. Suppression of host p53 is critical for Plasmodium liver-stage infection. Cell Rep 2013; 3:630-7; PMID:23478020; http://dx.doi.org/10.1016/j.celrep.2013.02.010
- Tarun A, Peng X, Dumpit R, Ogata Y, Silva-Rivera H, Camargo N, Daly TM, Bergman LW, Kappe SH. A combined transcriptome and proteome survey of malaria parasite liver stages. PNAS 2008; 105:305; PMID:18172196; http://dx.doi.org/10.1073/pnas.0710780104
- Albuquerque S, Carret C, Grosso A, Tarun A, Peng X, Kappe S, Prudêncio M, Mota MM. Host cell transcriptional profiling during malaria liver stage infection reveals a coordinated and sequential set of biological events. BMC Genomics 2009; 10:270; PMID:19534804; http://dx.doi.org/10.1186/1471-2164-10-270
- Graewe S, Retzlaff S, Struck N, Janse CJ, Heussler VT. Going live: a comparative analysis of the suitability of the RFP derivatives RedStar, mCherry and tdTomato for intravital and in vitro live imaging of Plasmodium parasites. Biotechnol J 2009; 4:895-902; PMID:19492329; http://dx.doi.org/10.1002/biot.200900035
- Franke-Fayard B, Trueman H, Ramesar J, Mendoza J, van der Keur M, van der Linden R, Sinden RE, Waters AP, Janse CJ. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol Biochem Parasitol 2004; 137:23-33; PMID:15279948; http://dx.doi.org/10.1016/j.molbiopara.2004.04.007
- Graewe S, Rankin KE, Lehmann C, Deschermeier C, Hecht L, Froehlke U, Stanway RR, Heussler V. Hostile takeover by Plasmodium: reorganization of parasite and host cell membranes during liver stage egress. PLoS Pathog 2011; 7:e1002224; PMID:21909271; http://dx.doi.org/10.1371/journal.ppat.1002224
- Lin JW, Annoura T, Sajid M, Chevalley-Maurel S, Ramesar J, Klop O, Franke-Fayard BM, Janse CJ, Khan SM. A novel ‘gene insertion/marker out’ (GIMO) method for transgene expression and gene complementation in rodent malaria parasites. PloS One 2011; 6:e29289; PMID:22216235
- Orr RY, Philip N, Waters AP. Improved negative selection protocol for Plasmodium berghei in the rodent malarial model. Malar J 2012; 11:103; PMID:22463060; http://dx.doi.org/10.1186/1475-2875-11-103
- Menard R, Janse C. Gene targeting in malaria parasites. Methods 1997; 13:148-57; PMID:9405198; http://dx.doi.org/10.1006/meth.1997.0507
- Janse CJ, Ramesar J, Waters AP. High-efficiency transfection and drug selection of genetically transformed blood stages of the rodent malaria parasite Plasmodium berghei. Nat Protoc 2006; 1:346-56; PMID:17406255; http://dx.doi.org/10.1038/nprot.2006.53
- Annoura T, van Schaijk BC, Ploemen IH, Sajid M, Lin JW, Vos MW, Dinmohamed AG, Inaoka DK, Rijpma SR, van Gemert GJ, et al. Two Plasmodium 6-Cys family-related proteins have distinct and critical roles in liver-stage development. FASEBJ 2014; 28:2158-70; PMID:24509910; http://dx.doi.org/10.1096/fj.13-241570
- van Dijk MR, van Schaijk BC, Khan SM, van Dooren MW, Ramesar J, Kaczanowski S, van Gemert GJ, Kroeze H, Stunnenberg HG, Eling WM, et al. Three members of the 6-cys protein family of Plasmodium play a role in gamete fertility. PLoS Pathog 2010; 6:e1000853; PMID:20386715; http://dx.doi.org/10.1371/journal.ppat.1000853
- Kimura S, Noda T, Yoshimori T. Dissection of the Autophagosome Maturation Process by a Novel Reporter Protein, Tandem Fluorescent-Tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/10.4161/auto.4451
- Dantuma NP, Groothuis TA, Salomons FA, Neefjes J. A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol 2006; 173:19-26; PMID:16606690; http://dx.doi.org/10.1083/jcb.200510071
- Thiberge S, Blazquez S, Baldacci P, Renaud O, Shorte S, Menard R, Amino R. In vivo imaging of malaria parasites in the murine liver. Nat Protoc 2007; 2:1811-8; PMID:17641649; http://dx.doi.org/10.1038/nprot.2007.257
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012; 9:676-82; PMID:22743772; http://dx.doi.org/10.1038/nmeth.2019