
Abstract
Autophagy is involved in the pathogenesis of neurodegenerative diseases including Parkinson disease (PD). However, little is known about the regulation of autophagy in neurodegenerative process. In this study, we characterized aberrant activation of autophagy induced by neurotoxin 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) and demonstrated that melatonin has a protective effect on neurotoxicity. We found an excessive activation of autophagy in monkey brain tissues and C6 cells, induced by MPTP, which is mediated by CDK5 (cyclin-dependent kinase 5). MPTP treatment significantly reduced total dendritic length and dendritic complexity of cultured primary cortical neurons and melatonin could reverse this effect. Decreased TH (tyrosine hydroxylase)-positive cells and dendrites of dopaminergic neurons in the substantia nigra pars compacta (SNc) were observed in MPTP-treated monkeys and mice. Along with decreased TH protein level, we observed an upregulation of CDK5 and enhanced autophagic activity in the striatum of mice with MPTP injection. These changes could be salvaged by melatonin treatment or knockdown of CDK5. Importantly, melatonin or knockdown of CDK5 reduced MPTP-induced SNCA/α-synuclein aggregation in mice, which is widely thought to trigger the pathogenesis of PD. Finally, melatonin or knockdown of CDK5 counteracted the PD phenotype in mice induced by MPTP. Our findings uncover a potent role of CDK5-mediated autophagy in the pathogenesis of PD, and suggest that control of autophagic pathways may provide an important clue for exploring potential target for novel therapeutics of PD.
Abbreviations
| ATG7 | = | autophagy-related 7 |
| BAFA1 | = | bafilomycin A1 |
| CDK5 | = | cyclin-dependent kinase 5 |
| CDK5R1/p35/p25 | = | cyclin-dependent kinase 5, regulatory subunit 1 (p35) |
| DA | = | dopamine |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| i.p | = | intraperitoneally |
| s.c | = | subcutaneously |
| ICV | = | intracerebroventricular |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 β |
| MAP2 | = | microtube-associated protein 2 |
| MPTP | = | 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine |
| PD | = | Parkinson disease |
| PEBP1 | = | phosphatidylethanolamine binding protein 1 |
| PI | = | propidium iodide |
| SNc | = | substantia nigra pars compacta |
| SNCA/α-synuclein | = | synuclein, α (non A4 component of amyloid precursor) |
| TH | = | tyrosine hydroxylase. |
Introduction
Parkinson disease (PD) is the second most common neurodegenerative disease characterized by bradykinesia, tremors, and rigidity.Citation1,2 It is caused by the degeneration of dopaminergic (DAergic) neurons in the substantia nigra pars compacta (SNc), which results in a decreased dopamine level in the striatumCitation3,4 and a formation of Lewy bodies that are predominantly composed of aggregated SNCA/α-synuclein.Citation5 However, molecular mechanisms underlying PD have not been fully understood. The neurotoxin 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP), an inhibitor of mitochondrial complex I, has been commonly used as an agent to develop PD animal models for inducing specific loss of DAergic neurons in the SNc.Citation6-8 The neurotoxicity of MPTP depends on its active metabolite 1-methyl-4-phenylpyridinium (MPP+), which is specifically absorbed into the DAergic neurons through SLC6A3/DAT (solute carrier family 6 [neurotransmitter transporter], member 3).Citation9 Several studies have shown that CDK5 (cyclin-dependent kinase 5), a key regulator of cell-cycle progression, plays a key role in dopamine loss after MPTP treatment and in the pathogenesis of PD.Citation10-12 Activation of CDK5 requires its regulatory partner CDK5R1/p35/p25 (cyclin-dependent kinase 5, regulatory subunit 1 [p35]).Citation13 Accumulating evidence indicates that MPTP increases CDK5 activityCitation11,12 and triggers neuronal loss through phosphorylation and inhibition of PEBP1/RKIP (phosphatidylethanolamine-binding protein 1),Citation14 survival factor MEF2 (myocyte enhancer factor 2),Citation15 antioxidant enzyme PRDX2/Prx2 (peroxiredoxin 2),Citation16 and APEX1/Ape-1 (APEX nuclease [multifunctional DNA repair enzyme] 1).Citation11
Autophagy is a lysosomal degradation pathway that controls the turnover of cytoplasmic contents and organelles through the engulfment of cargo into double-membrane autophagosomes. It is characterized by increased MAP1LC3B-II/LC3B-II (microtubule-associated protein 1 light chain 3 β-II) and decreased SQSTM1/p62.Citation17-19 Autophagy is essential for survival, differentiation, development, and homeostasis.Citation17,18,20 Recent studies have reported that deregulation of autophagy was implicated in PD.Citation21,22 Autophagosome accumulation is evident in the brain tissues of PD patientsCitation23,24 and in MPTP-induced PD animal models.Citation25 Moreover, PINK1 (PTEN induced putative kinase 1)- and PARK2/parkin (parkin RBR E3 ubiquitin protein ligase)-mediated autophagy is actively involved in selective clearance of damaged mitochondria and protection of the neurons from death.Citation26 In addition, basal levels of autophagy and chaperone-mediated autophagy are necessary for the clearance of abnormal SNCA.Citation27,28 Excessive autophagy has also been associated with neuronal loss.Citation20,29,30 The CDK5-mediated autophagy contributes to neuronal loss in PD animal models.Citation14,31
Melatonin, the main secretory product of the pineal gland, can easily cross the blood-brain barrier and has plentiful neuroprotective properties such as regulating circadian rhythms and providing anti-inflammatory and neuronal protection.Citation32-34 As a naturally occurring compound, melatonin has potential effects on inhibiting autophagy through a redox-mediated scavenge of free radicals and lipid peroxidation,Citation35 as well as its well-known antioxidant properties.Citation36 Emerging evidence suggests that melatonin can be used as a neuroprotective agent in the rodent models of PDCitation32,37,38 by ameliorating the dysfunction of mitochondria,Citation39-42 blocking CASP3 (caspase 3, apoptosis-related cysteine peptidase) activation and cellular apoptosis,Citation43 and protecting TH (tyrosine hydroxylase)-positive nerve terminals.Citation44 However, the molecular underpinning of protective effect of melatonin on the loss of DAergic neurons in PD mouse model is still unknown. Our recent study suggests that melatonin can restore the mitochondrial DNA copy number to normal level by inhibiting autophagy induced by morphine.Citation45
Based on these lines of evidence, we hypothesized that aberrant activation of autophagy may contribute to MPTP-induced neuronal cell death, which will lead to dopamine depletion in the striatum and motor symptoms of the PD animal models. We asked whether melatonin can reduce autophagic neuronal cell death induced by MPTP and which factor is mediating the process of dopaminergic neuronal cell loss and the bradykinesia of the PD animal models. Our results showed that melatonin attenuates MPTP-induced neurotoxicity via preventing CDK5-mediated autophagy and SNCA aggregation.
Results
MPTP induced autophagy in monkey brain tissues
Male rhesus monkeys received MPTP injection (0.3 mg/kg, N = 2) and saline (N = 2) twice a wk for 3 mo and were then sacrificed for collection of brain tissues. We observed apparent parkinsonian symptoms in monkeys with MPTP treatment (). Meanwhile, the number of TH-positive cells and dendrites of dopaminergic neurons were reduced in the SNc of monkeys with MPTP injection (), along with the decreased TH level in different brain tissues (), suggesting that the monkey PD model was successfully established. An increased protein level of LC3B-II () and a decreased protein level of SQSTM1 () were observed in different brain tissues from monkeys with MPTP injection, indicating induced autophagy by this agent.
Figure 1. MPTP induces autophagy in the brain tissues of monkeys with MPTP treatment. Monkeys had a higher Parkinson disease (PD) score after MPTP injection compared to their status before the drug treatment (A). The number of TH-positive cells and dendrites of dopaminergic neurons was reduced in the SNc (B and C) and the protein levels of TH (D and E) were decreased in different brain tissues of monkeys treated with MPTP. There were increased protein levels of LC3B-II (F and G) and a decreased level of SQSTM1 (F, H). CDK5 levels were elevated (I and J), accompanied with increased levels of CDK5R1 (I, K), in different brain tissues of monkeys with MPTP treatment. *, P < 0.05; **, P < 0.01; ***, P < 0.001; Student t test. Bars represent mean ±SEM.
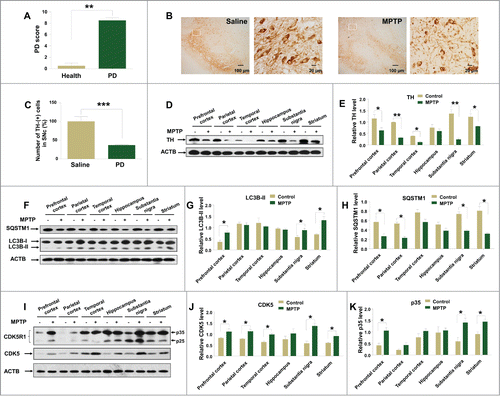
Previous studies have demonstrated that CDK5 participates in the regulation of autophagy to mediate neurotoxicity and neuronal cell death.Citation14,31,46 We investigated whether CDK5 was changed during the development of the monkey PD model. We found that MPTP could induce higher CDK5 level in the monkey brain tissues, with the most striking effect in the substantia nigra region (). In order to explore whether the increased CDK5 level was associated with the altered CDK5 kinase activity, the levels of CDK5R1that contribute to CDK5 activityCitation47 were evaluated. We found that CDK5R1 levels were significantly upregulated in several brain regions after MPTP administration (). These data showed that MPTP treatment could induce an increase of CDK5 protein level and its kinase activity in monkey brain tissues.
MPTP induced CDK5-mediated autophagy, leading to death of C6 cells
To explore whether the elevated CDK5 level participates in MPTP-induced autophagy, we measured the protein levels of CDK5 and the autophagic marker, LC3B-II, as well as SQSTM1, in MPTP-treated C6 cells. An increased protein level of CDK5 was observed in MPTP-treated C6 cells, in a dose-dependent manner (). Accordingly, we observed an increased protein level of LC3B-II and a decreased protein level of SQSTM1 (). Along with the elevated LC3B-II and the decreased SQSTM1, we observed increased EGFP-LC3 puncta () upon MPTP treatment in C6 cells transfected with the N-terminally tagged pEGFP-C1-LC3 vector. Pretreatment with bafilomycin A1 (BAFA1), an inhibitor of the vacuolar (V)-type ATPase that results in blockage of autophagosome-lysosome fusion, induced more LC3B-II accumulation compared to the control cells with or without MPTP treatment (). However, the lysosomal function was unchanged in C6 cells treated with MPTP and/or melatonin (Fig. S1), suggesting that MPTP indeed increased autophagic flux. Treatment with MPP+, a metabolite of MPTP, similarly induced autophagy and upregulated CDK5 protein levels (Fig. S2).
Figure 2. MPTP treatment induces autophagy and cell death in cultured C6 cells and this effect is CDK5-mediated. An increased protein level of CDK5 and LC3B-II and a decreased level of SQSTM1 (A and B) were observed in C6 cells treated with MPTP. EGFP-LC3 puncta were induced by MPTP treatment (C). A treatment of 100 nM BAFA1 blocked the fusion of autophagosomes and lysosomes and led to accumulation of LC3B-II (D and E). Knockdown of the Cdk5 gene by siRNA abolished the formation of EGFP-LC3 puncta (C) and the altered protein levels of LC3B-II and SQSTM1 (F and G) in C6 cells with MPTP treatment. Overexpression of CDK5 increased autophagosome formation (C), LC3B-II accumulation (H and I) and cell death (K) in C6 cells with and without MPTP treatment. MPTP induced cell death in C6 cells in a dose-dependent manner (J). Knockdown of the mRNA expression of the Atg7 gene or the Cdk5 gene blocked MPTP induced cell death (K). Relative protein abundance was normalized to ACTB. Data are representative of 3 independent experiments with similar results. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA with the Tukey post-hoc test. Bars represent mean ±SEM.
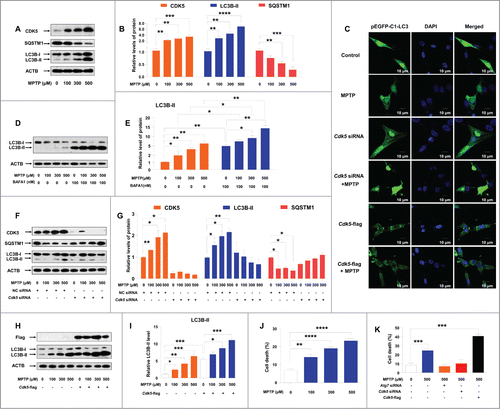
To further identify the potential roles of CDK5 in regulation of MPTP-induced autophagy, we performed experiments using siRNAs targeted to the Cdk5 gene and also, overexpression of CDK5. Among the 3 siRNAs for the mRNA of the Cdk5 gene, siRNA-1 had the best inhibitory effect (Fig. S3A and B) and was used in the following knockdown assays. Concordant with our expectation, knockdown of the mRNA of the Cdk5 gene suppressed MPTP-induced formation of autophagosomes () and salvaged the increased level of LC3B-II and the decreased level of SQSTM1 (). We got similar rescue results in a cell line stably expressing Cdk5 shRNA (Fig. S3C and D and Fig. S4A and B). MPTP could induce autophagy in C6 cells with knockdown of CDK5 in the presence of BAFA1 (Fig. S5A and B). Overexpression of CDK5 in C6 cells with or without MPTP resulted in increased autophagosome formation () and LC3B-II accumulation (). These results provided direct evidence that the autophagy induced by MPTP was mediated by CDK5. Consistent with previous reports,Citation14,46 we found that MPTP induced cell death in C6 cells in a dose-dependent manner (). Inhibition of autophagy by using Atg7 siRNA prevented cell death progression induced by MPTP (). Knockdown of the Cdk5 gene had a similar blocking effect on cell death trigged by MPTP (). In contrast, overexpression of CDK5 had a synergetic effect and significantly increased cell death induced by MPTP (). Taken together, the CDK5-mediated autophagy contributed to MPTP-induced cell death.
Melatonin restored CDK5-mediated autophagy and cell death in C6 cells, induced by MPTP
Melatonin is a well-known antioxidant molecule playing important roles against oxidative stress caused by MPTP.Citation42 It also has protective effects on methamphetamine-induced autophagic cell death in vitro.Citation48 We tested whether exogenous adding of melatonin could prevent MPTP-induced autophagy and cell death. As shown in , melatonin could restore the elevated CDK5 level induced by MPTP in C6 cells. In addition, melatonin reversed the increase of LC3B-II level, the decrease of SQSTM1 level (), as well as formation of autophagosomes induced by MPTP (). Pretreatment with melatonin could inhibit the autophagy induced by MPTP in C6 cells in the presence of BAFA1 (Fig. S5C and D). Melatonin alone had no effect on CDK5 protein level and autophagy (Fig. S6).
Figure 3. Melatonin inhibits CDK5-mediated autophagy and cell death induced by MPTP in cultured C6 cells. The increased protein levels of CDK5 and LC3B-II and the decreased protein level of SQSTM1 (A and B) and formation of EGFP-LC3 puncta (C) in C6 cells induced by MPTP could be suppressed by melatonin treatment. Knockdown of the Cdk5 gene abolished the effect of melatonin on the altered LC3B-II and SQSTM1 levels (D and E) in C6 cells with MPTP treatment. Melatonin failed to restore the altered protein level of LC3B-II (D–G) in C6 cells with knockdown or overexpression of the Cdk5 gene. Knockdown of ATG7 and CDK5 or overexpression of CDK5 abrogated the protective effect of melatonin on cell death induced by MPTP (H). Data are representative of 3 independent experiments with similar results. *, P < 0.05; **, P < 0.01; ***, P < 0.001, **** P < 0.0001; one-way ANOVA with the Tukey post-hoc test. Bars represent mean ±SEM.
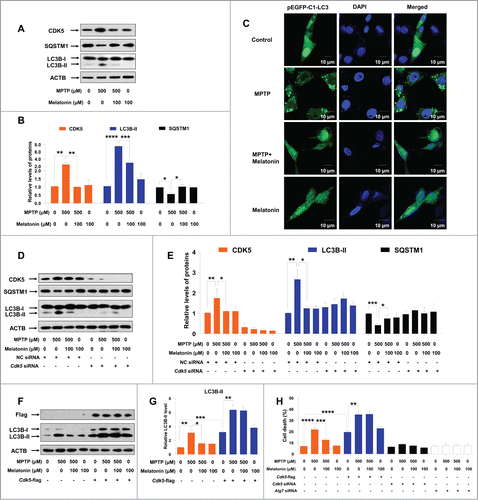
We further performed experiments using siRNA targeted to the Cdk5 gene or overexpressing Cdk5 to test whether the effect of melatonin on regulation of autophagy was CDK5-dependent. We found that melatonin had no effect on autophagy in cells with knockdown of the Cdk5 gene by using siRNA (), (partially) because CDK5 knockdown itself could inhibit the autophagy induced by MPTP. A similar result was observed in the stable cell line expressing Cdk5 shRNA (Fig. S4C and D). Overexpression of CDK5 in C6 cells transfected with the Cdk5-flag vector abolished the protective effect of melatonin on MPTP-induced autophagy (). Intriguingly, we found that melatonin blocked cell death induced by MPTP ( and Fig. S7). Overexpression of CDK5 abolished the protective effect of melatonin on MPTP-induced cell death (). Note that knockdown of the Atg7 and Cdk5 genes could also salvage the MPTP-induced cell death (). These results suggested an active role of melatonin on protecting autophagy and cell death induced by MPTP and CDK5 was involved in this process.
Melatonin reversed the dendritic morphological changes of neurons and autophagy induced by MPTP
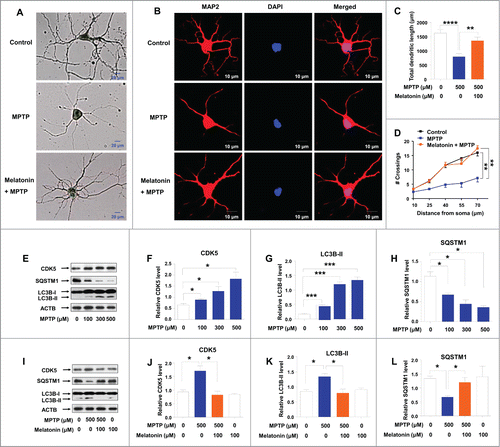
To confirm the importance of MPTP-induced autophagy in regulating neuronal cell survival, mouse primary cortical neurons were treated with 500 μM MPTP for 24 h followed by staining with Golgi-Cox or TUBB3 (tubulin, β 3 class III) to visualize neurons. MPTP treatment reduced the length of Golgi-Cox or TUBB3-positive axons and dendrites compared to untreated neurons, and this effect could be reversed by melatonin pretreatment ( and Fig. S8). We next explored the impact of MPTP treatment on the structure of neuron by evaluating the change of total dendritic length and dendritic complexity, which was demonstrated by using immunostaining for the dendritic MAP2 (microtubule-associated protein 2) (). We found that both total dendritic length () and dendritic complexity () were significantly reduced in MPTP-treated neurons relative to untreated neurons. Pretreatment with melatonin reversed the neuronal morphological changes triggered by MPTP (). These results indicated an active role of melatonin in regulating the neuro-morphological changes and counteracting the deleterious effect of MPTP, which would eventually prevent the dysfunction of the neurons. Similarly, MPTP treatment significantly elevated protein level of CDK5 with a dose-dependent manner in primary cortical neurons (), accompanied with an increased protein level of LC3B-II () and a decreased level of SQSTM1 (). Pretreatment with melatonin could restore the altered protein level of CDK5 (), LC3B-II () and SQSTM1 () to the normal levels.
Figure 4. Effect of melatonin on primary neuron dendritic morphogenesis and autophagy induced by MPTP. Mouse primary cortical neurons were cultured in Neurobasal® Medium containing 2% B27. Neurons were pretreated with melatonin (100 μM) for 1 h, followed by treatment with or without MPTP (500 μM) for 24 h. The morphological structure (A) and dendrites (B) of neurons were demonstrated by Golgi-Cox and MAP2 staining, respectively. Upon MPTP treatment, dendritic length (C) and dendritic complexity (D) were reduced. These changes could be reversed by a pretreatment with melatonin (A–D). MPTP treatment increased the protein levels of CDK5 (E and F), LC3B-II (E, G) and decreased the protein level of SQSTM1/p62 (E, H) in mouse cortical neurons. Pretreatment with melatonin restored the increased levels of CDK5 (I and J) and LC3B-II (I, K) and the decreased level of SQSTM1 (I, L) induced by MPTP. Data are representative of 2 independent experiments with similar results. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA with the Tukey post-hoc test. Bars represent mean ±SEM.
Melatonin ameliorated symptoms in the mouse PD model through preventing CDK5-mediated autophagy
A mouse PD model was used to discern whether melatonin salvaged the MPTP toxic effect in vivo. The induced dyskinesia by chronic MPTP treatment was measured using a rota rod apparatus after MPTP injection at 3 time points (). The retention time on the rota rod was significantly decreased in mice subjected to 30 mg/kg MPTP treatment (subcutaneously [s.c.]) once per d for 2 wk compared to saline controls. Pretreatment with melatonin (intraperitoneally [i.p.]; 5 mg/kg and 0.5 mg/kg) before MPTP injection in mice significantly prevented dyskinesia induced by MPTP, with a dosage-dependent effect ().
Figure 5. Pretreatment with melatonin or knockdown of CDK5 counteracts the effect of MPTP on behavioral dyskinesias and dopaminergic neuron loss in mouse models. Mice were divided into different groups (A). MPTP-treated mice (Vehicle-MPTP group) exhibited a significantly decreased retention time on the rota rod apparatus compared to control mice without any treatments (Vehicle-Saline group). Pretreatment with melatonin (0.5 mg/kg [0.5 mg/kg Melatonin-MPTP group] and 5 mg/kg [5 mg/kg Melatonin-MPTP group]) significantly improved the retention time compared to the MPTP group (B). Knockdown of CDK5 increased the retention time on the rota rod of mice with and without MPTP injection (C). Shown are photomicrographs of TH immunoreactivity in the SNc central region of different mouse groups (D): TH-positive cell number and dendrites of DA neurons were reduced in the SNc of mice with MPTP injection, which could be rescued by injection of 5 mg/kg melatonin or knockdown of the Cdk5 gene. The differences of TH-positive cell numbers between the control group and the MPTP group, between the MPTP group and the MPTP plus 5 mg/kg melatonin group (5 mg/kg Melatonin-MPTP group) were statistically significant (E). *, P < 0.05; **, P < 0.01; ***, P < 0.001; one-way ANOVA with the Tukey post-hoc test. Bars represent mean ±SEM.
![Figure 5. Pretreatment with melatonin or knockdown of CDK5 counteracts the effect of MPTP on behavioral dyskinesias and dopaminergic neuron loss in mouse models. Mice were divided into different groups (A). MPTP-treated mice (Vehicle-MPTP group) exhibited a significantly decreased retention time on the rota rod apparatus compared to control mice without any treatments (Vehicle-Saline group). Pretreatment with melatonin (0.5 mg/kg [0.5 mg/kg Melatonin-MPTP group] and 5 mg/kg [5 mg/kg Melatonin-MPTP group]) significantly improved the retention time compared to the MPTP group (B). Knockdown of CDK5 increased the retention time on the rota rod of mice with and without MPTP injection (C). Shown are photomicrographs of TH immunoreactivity in the SNc central region of different mouse groups (D): TH-positive cell number and dendrites of DA neurons were reduced in the SNc of mice with MPTP injection, which could be rescued by injection of 5 mg/kg melatonin or knockdown of the Cdk5 gene. The differences of TH-positive cell numbers between the control group and the MPTP group, between the MPTP group and the MPTP plus 5 mg/kg melatonin group (5 mg/kg Melatonin-MPTP group) were statistically significant (E). *, P < 0.05; **, P < 0.01; ***, P < 0.001; one-way ANOVA with the Tukey post-hoc test. Bars represent mean ±SEM.](/cms/asset/a7710141-6ff3-4c64-9a8a-3eda64a76cab/kaup_a_1082020_f0005_c.gif)
To test whether the CDK5 was involved in the pathogenesis of mouse PD model, we evaluated the effect of CDK5 inhibition by using intracerebroventricular (ICV) injection of siRNA oligonucleotide that targeted the Cdk5 gene. Three d after ICV injection of Cdk5 siRNA, we observed a significant decrease of CDK5 protein levels in different mouse brain regions, especially in the midbrain (Fig. S9). As expected, inhibition of CDK5 in brain tissues increased the retention time on the rota rod of mice with and without MPTP injection compared to those without Cdk5 siRNA injection (). These results indicated that CDK5 was involved in MPTP-induced dyskinesia of mice.
We detected the number and morphology of dopaminergic neurons in the midbrain of mice by using TH immunoreactivity. In control mice with saline injection, TH immunoreactivity was observed in cells with rounded somata and immunoreactive dendrites formed a dense lattice network within the SNc area (). However, the number of TH-positive cells and dendrites of dopaminergic neurons was significantly reduced in the SNc area of mice with MPTP injection (). Pretreatment with 5 mg/kg melatonin (i.p.) or knockdown of CDK5 in mice significantly attenuated the dopaminergic deficit including the reduced number and altered morphology of dopaminergic neurons induced by MPTP (). Pretreatment with a lower concentration of melatonin (0.5 mg/kg) also had a salvaging effect, albeit the effect was not significant. Treatment with melatonin alone had no effect (). We observed an increased TH-positive cells and dendrites of dopaminergic neurons in the SNc of the mice in the presence of Cdk5 siRNA (). MPTP and melatonin had no significant effect on the TH-positive cells and dendrites of dopaminergic neurons in mice with Cdk5 siRNA injection (). In addition, MPTP treatment significantly decreased the protein level of TH, as compared to the control group (). Pretreatment with 5 mg/kg melatonin (i.p.) in mice restored the decreased TH level compared to the MPTP-treated group (). In the mice with Cdk5 siRNA injection, MPTP treatment decreased the protein level of TH. However, pretreatment with melatonin had no significant effect on the level of TH in the striatum of these mice (Fig. S10).
Figure 6. Protective effect of melatonin on MPTP-induced autophagy mediated by CDK5 in the mouse model of PD. TH protein level was significantly decreased in the MPTP group relative to the control group (Vehicle-Saline group) and this change could be reversed by pretreatment with 5 mg/kg melatonin (A and B). Pretreatment of 5 mg/kg melatonin restored the elevated protein levels of CDK5 and LC3B-II (C and D) and the decreased protein level of SQSTM1 (E and F) induced by MPTP in mouse striatum. The relative protein levels of CDK5, LC3B-II and SQSTM1 were statistically significant between the control group and the MPTP group, and between the MPTP group and the MPTP plus 5 mg/kg melatonin group (5 mg/kg Melatonin-MPTP group). MPTP failed to induce autophagy and melatonin had no salvaging effect on autophagy induced by MPTP in the striatum of mice after knockdown of the Cdk5 gene (G and H). Data are expressed as mean ±SEM. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; one-way ANOVA with the Tukey post-hoc test. Bars represent mean ±SEM.
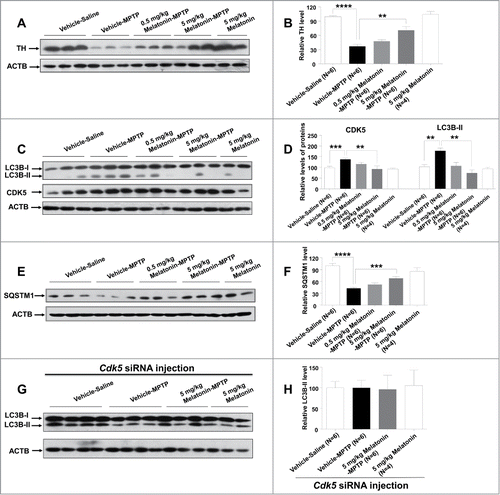
To investigate whether melatonin exerted its neuroprotective role via inhibiting the CDK5-mediated autophagy, we analyzed the effect of melatonin on protein levels of CDK5, LC3B-II, and SQSTM1 in the striatum of treated mice. In accordance with the results observed in vitro, we found increased levels of CDK5 and LC3B-II () and a decreased level of SQSTM1 () in the striatum of mice with MPTP treatment. Melatonin treatment (5 mg/kg) could restore the elevated CDK5 and LC3B-II levels and the decreased SQSTM1 level to normal levels in mouse striatum induced by chronic MPTP treatment (). MPTP could not induce autophagy in the striatum in mice with Cdk5 siRNA injection (). Collectively, our results indicated that melatonin salvaged the dopaminergic neuronal loss and decreased neurite of neuron through inhibition of MPTP-induced autophagy mediated by CDK5.
Melatonin treatment and CDK5 knockdown ameliorated SNCA aggregation
Previous studies suggest that SNCA aggregates are neurotoxic and that reducing SNCA aggregation can be used as a neuroprotective strategy against central nervous system neurodegeneration.Citation49,50 We investigated the involvement of SNCA aggregation induced by MPTP in the brain tissues of monkey and mouse. Our immunohistochemical staining results showed that MPTP increased SNCA aggregates in the SNc region of monkeys (). Western blot assay further demonstrated an increased accumulation of SNCA (51 kD) in different brain tissues of monkeys injected with MPTP (). However, MPTP did not significantly alter the SNCA monomer (17 kD) levels in these brain tissues ().
Figure 7. MPTP induced SNCA aggregation in monkey and mouse brain. MPTP increased SNCA aggregation in the monkey brain tissues, assessed by immunohistochemical staining, followed by hematoxylin counterstaining (A) and western blot assay (B–D). Mice with chronic MPTP administration (30 mg/kg once daily) showed an increased aggregation of SNCA in the SNc (E) and striatum (F). Pretreatment with 5 mg/kg melatonin or knockdown of CDK5 inhibited the SNCA aggregation induced by MPTP in mouse SNc (E) and striatum (F–H) compared to the control group. MPTP and melatonin had no effect on the SNCA aggregation in the presence of Cdk5 siRNA (E, I–K). *, P < 0.05; ****, P < 0.0001; one-way ANOVA with the Tukey post-hoc test. Bars represent mean ±SEM.
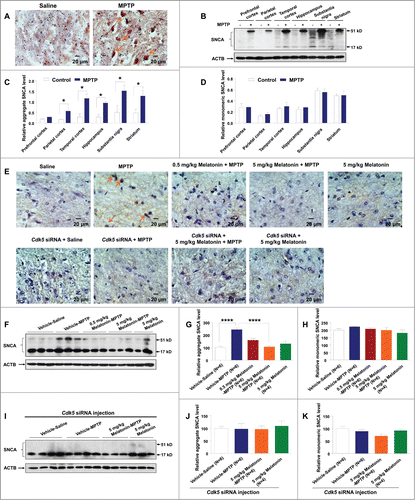
We validated the observations in monkeys relative to MPTP treatment in mouse. An increased level of SNCA aggregates was found in the SNc region and striatum of mice injected with MPTP (). Pretreatment with melatonin significantly reduced MPTP induced SNCA aggregation (), but had no effect on the SNCA monomer level (). Few SNCA aggregates were observed in the SNc region and striatum of mice in the presence of Cdk5 siRNA (). MPTP and melatonin had no effect on the aggregates of SNCA in these mice with Cdk5 siRNA injection (). It should be mentioned that melatonin enacted its protective effect only before the MPTP treatment. In the mice that were treated with MPTP and developed PD symptoms, melatonin treatment had no significant salvaging effect on behavioral change (Fig. S11A), deficiency of dopaminergic neurons (Fig. S11B and C), decrease of TH (Fig. S11D and E), upregulation of CDK5 (Fig. S12A and B), increased autophagic activity (Fig. S12A and B) and SNCA aggregation (Fig. S13). These results suggested that melatonin may not be used as a therapeutic drug, but its use as a food additive for people susceptible to PD is promising.
Discussion
PD is a progressive, debilitating neurodegenerative disease characterized by Lewy body accumulation and dopaminergic neuron death in the SNc region.Citation5,51 The development of PD is affected by a growing number of genetic and environmental risk factors and synaptic dysfunction, which would be regulated by autophagyCitation52 and SNCA aggregates.Citation50 Autophagy-mediated self-digestion of cytoplasmic inclusions may be protective against the PD.Citation53,54 However, excessive autophagic activation evokes autophagic programmed cell death.Citation14,55 Recent studies report that CDK5-mediated phosphorylation and autophagy of PEBP1 regulates neuronal death in PD.Citation14 Overexpression of the leucine-rich repeat kinase 2 (LRRK2) mutant G2019S, which is associated with autosomal dominant and sporadic forms of PD in human neuroblastoma cells, causes activation of autophagy and significantly decreases dendritic length.Citation56,57 Similarly, overexpression of mutant SNCA increases cellular autophagosomes,Citation58 which actively influences and impacts synaptic functions. Previous studies also demonstrate that both autophagy inducers and inhibitors serve as neuroprotectors against PD. For example, the autophagy inducer rapamycin (Rap) has neuroprotective effects on cellular and mouse models of PD.Citation54 The autophagy inhibitor 3-methyladenine can block 6-hydroxydopamine induced toxicity.Citation59 These findings suggest that maintaining balanced autophagy is critical for neuronal health and function.Citation60 In this study, we aimed to answer whether interference of autophagy could be of potential use in counteracting the progression of PD and whether melatonin, a readily available substance that is already widely in use in humans for other indications, could be potentially translated into human clinical trials for curing PD given melatonin's good safety profile and availability.
We presented direct evidence that MPTP could induce autophagic flux at the cellular level and this neurotoxicity effect was mediated by CDK5. These findings are in agreement with previous reports that the CDK5-mediated autophagy is actively involved in PD.Citation14,31 We next tested whether melatonin, with its antioxidant properties, could restore the abnormal autophagic activity induced by MPTP. Consistent with previous observations that melatonin can restore morphine-induced autophagyCitation45 and kainic acid-induced autophagy,Citation61 we found that melatonin inhibited MPTP-induced autophagy and cell death in vitro. This salvaging effect of melatonin was CDK5-dependent, and knockdown or overexpression of CDK5 abolished the protective effect of melatonin on MPTP-induced autophagy and cell death. Taken all these results together, it is evident that melatonin restored autophagy through inhibition of CDK5 upregulation at the cellular level.
Maintenance of axon and dendrite length is critical for neuron function and autophagy is actively involved in regulation of pathological remodeling.Citation60 Therefore, a direct test of melatonin's effect on neurons in vitro and in vivo, rather than on cell lines, is essential for further defining its role in the development of PD. Consistent with a previous finding that melatonin can save dopaminergic cells in MPTP-treated mice,Citation62 our study showed that MPTP treatment reduced the length of axons and dendrites in primary neurons and decreased the number and dendrites of dopaminergic neurons in the SNc region of monkeys and mice with MPTP injection. Furthermore, MPTP injection increased SNCA aggregation in the mouse PD model. Melatonin could reverse all these aberrant effects, including decreased number of dopaminergic neurons, increased SNCA aggregation and altered morphologic changes of neurons induced by MPTP. These results indicated an active role of melatonin in regulating the neuromorphological changes and counteracting the deleterious effect of MPTP, which might eventually prevent the dysfunction of the neurons.
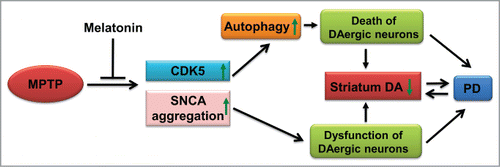
Although previous studies show that melatonin ameliorates the symptomatic features of Maneb and Paraquat-induced PD in a mouse model,Citation63 the exact mechanism of the PD or PD-like symptoms induced by these drugs has not been fully understood. We were able to show that pretreatment with melatonin significantly prevented MPTP-induced dyskinesia, and salvaged the reduction of the number of dopaminergic neurons in mice with MPTP treatment. This neuroprotective role of melatonin against MPTP-induced neurotoxicity in vivo was mediated by CDK5. First, pretreatment of melatonin reversed the increased CDK5 protein level, SNCA aggregation, and autophagy induced by MPTP in the mouse PD model. Second, injection of Cdk5 siRNA ameliorated the behavioral dyskinesia of mice with MPTP injection and MPTP failed to induce autophagy in the striatum of treated mice. These lines of evidence suggested that melatonin could prevent MPTP-induced CDK5-mediated autophagy and SNCA aggregation, and subsequently protected the substantia nigra dopaminergic neurons cells from autophagic cell death, and finally prevented the symptomatic features of PD (). Note that there are other more relevant PD models to study the impact of SNCA aggregation instead of the MPTP models,Citation3 and it might be rewarding to validate the above results in these PD models and to more thoroughly characterize the nature of the aggregates.
Figure 8. A schematic diagram of potential mechanism of MPTP-induced Parkinsonism and the reversing effect of melatonin. DA, dopamine; up arrow symbol, upregulation or activation; down arrow symbol, downregulation.
An interesting finding in this study is that injection with melatonin after MPTP treatment could not reverse PD symptoms and related deleterious effects such as dopaminergic neuronal loss, CDK5 upregulation, autophagy, as well as SNCA aggregation. These results suggested that melatonin has a protective effect only in the early stage of PD. Our study raises further questions for future investigations: What is the exact biological mechanism of CDK5 upregulation in MPTP-induced neurotoxicity and autophagy? Does it share the same pathway, i.e. CDK5-mediated phosphorylation of endophilin B1, during the induction of autophagy in PD models?Citation31 Mitochondrial dysfunction and excess reactive oxygen species have been strongly implicated in the pathogenesis of PD.Citation64 Consistent with previous studies,Citation65 we found that MPTP treatment increased the reactive oxygen species level (Fig. S14A) and decreased the mitochondrial Complex I activity (Fig. S14B and C), and these effects could be salvaged by melatonin (Fig. S14), suggesting that mitochondrial dysfunction might be actively involved in CDK5 upregulation. Conversely, deregulation of cellular Ca2+ homeostasis would lead to synaptic dysfunction, which is a hallmark of several neurodegenerative diseases.Citation66 The Ca2+-dependent protease CAPN/calpain regulates CDK5R1-CDK5 in dopaminergic neuron death.Citation67 Is CDK5 upregulation during MPTP treatment caused by the Ca2+-dependent CAPN/calpain activation? Finally, might the increased activation of autophagy be compensatory, in response to MPTP, and perhaps even protective? In this case, cells induce autophagy as a coping response, and while it is correlated with cell stress and cell death, it is not itself a primary cause of cell death. Further studies are essential to answer these intriguing and important questions.
In summary, we found excessive activation of autophagy induced by MPTP, which is mediated by CDK5 in both cellular and animal models. The detrimental effects of MPTP-induced autophagic activity could be counteracted by melatonin or CDK5 knockdown, which finally protected neuron function and prevented the behavioral abnormality (). It should be mentioned melatonin that itself could not affect autophagy under the dosage used in this study; this would exclude its potential toxicity associated with usage in other neurodegenerative disease models where autophagy induction is (apparently) beneficial. Importantly, the protective effect of melatonin occurred before the activation of CDK5. Taken together, our results suggested that pretreatment with melatonin salvaged the dopaminergic neuronal loss, decreased axon and dendritic length of neuron and dyskinesias induced by MPTP via inhibition of CDK5-mediated autophagy and SNCA aggregation. Control of autophagic pathways may provide important insights for exploring potential targets for PD therapy.
Materials and Methods
Reagents, chemicals, and cells
The following antibodies and chemicals were used in this study: mouse monoclonal anti-CDK5 (Santa Cruz Biotechnology, sc-6247), rabbit monoclonal anti-LC3B (Cell Signaling Technology, 3868), mouse monoclonal anti-SQSTM1/p62 (Merck Millipore, MABC32), rabbit polyclonal anti-TH (Merck Millipore, AB152), rabbit polyclonal anti-SNCA/α-synuclein (Merck Millipore, AB5038), rabbit polyclonal anti-MAP2 (Merck Millipore, AB5622), mouse monoclonal anti-ACTB/β-actin (Beijing Zhong Shan-Golden Bridge Biological Technology CO., LTD, TA-09), FITC-conjugated anti-rabbit antibody (KPL, 172-1506). MPTP (Sigma, M0896), melatonin (Sigma, M5250), poly-L-ornithine (Sigma, P4957) and DAPI (Roche, 10236276001), propidium iodide (PtdIns; Beyotime Institute of Biotechnology, C1502-2), pentobarbital (Sigma, P3761), procaine hydrochloride (Sigma, P9879), were purchased as indicated.
Rat C6 astroglial cells were introduced from Kunming Cell Bank, Kunming Institute of Zoology. Cells were maintained in DMEM medium (Gibco-BRL, 11965-092) with 10% fetal bovine serum (Gibco-BRL, 10099-141) at 37°C in a humidified atmosphere incubator with 5% CO2. Mouse cortical neurons were prepared and cultured using a previously described method.Citation45 Briefly, primary cortical neurons were isolated from mouse E15 embryos and were cultured in Neurobasal® Medium supplement with 2% B27 (Gibco-BRL, 17504).
Establishment of a chronic PD model in monkeys
Four male rhesus monkeys (Macaca mulatta) were obtained from the Primate Research Center of Kunming Institute of Zoology, Chinese Academy of Sciences. Captive male rhesus monkeys (mean weight: 5.2 kg; mean age: 6.1 y) were housed individually in primate cages under a 12-h light/dark cycle with free access to water and food. Monkeys were divided into 2 groups: MPTP group (N = 2) and Control group (N = 2). MPTP was dissolved in 0.9% saline. Monkeys received intravenously (i.v.) injection of low doses of MPTP (0.3 mg/kg; Sigma, M0896) or saline twice weekly for 3 mo until obvious Parkinsonism was detected.Citation68 The behavioral changes were assessed as previously described.Citation69,70 Briefly, the severity of PD was scored by using an established rating scale for (1) tremor, (2) posture, (3) gait, (4) bradykinesia, (5) balance, (6) gross motor skills, and (7) defense reaction. The minimal PD score was 0 and the maximum score was 20. All animals were euthanized by sodium pentobarbital overdose (150 mg/kg, i.v.), and the brains were removed quickly after death. Each brain was bisected along the midline. Different brain tissues were obtained and immediately frozen in liquid nitrogen and then stored at −80°C.
Establishment of a chronic PD model in mice
Experiments were performed on 8-wk-old male C57BL/6 mice (body weight: 25 to 30 g) that were obtained from the animal core facility of the Kunming Medical University. Animals were group-housed under a 12-h light/dark cycle with free access to water and food. Mice were divided into the following groups: (1) Control group (N = 11), injected with vehicle and saline; (2) MPTP group (N = 16), injected with vehicle and MPTP; (3) MPTP + 5 mg/kg Melatonin group (N = 13), injected with melatonin and MPTP. (4) MPTP + 0.5 mg/kg Melatonin group (N = 14), injected with melatonin and MPTP. (5) Melatonin group (N = 12), injected with melatonin and saline (cf. ). MPTP was dissolved in 0.9% saline; melatonin was dissolved in 0.5% ethanol saline; 0.5% ethanol saline (v/v) is regarded as vehicle. Mice were subcutaneously (s.c.) injected with MPTP (30 mg/kg) once a day for 2 wk following a previously described procedure.Citation36 Animals in the control group received an equivalent volume of 0.9% saline. Each mouse received an injection of MPTP or saline 30 min after intraperitoneal (i.p) injections of melatonin (0.5 or 5 mg/kg) or vehicle (10 mL/kg, i.p.). For the treatment group (compare in the main text), animals in the control group and the MPTP group received the same treatment as described above. Starting from d 14, 5 mice from the control group and 5 mice from the MPTP group were injected with vehicle only, another 5 mice from the MPTP group were injected with 5 mg/kg melatonin once a d for 3 consecutive d, followed by the behavioral test.
Behavioral analysis: rota-rod task
We created a mouse PD model and assessed it on a rota-rod apparatus (Panlab, LE8500) following a previously described procedure.Citation71,72 The instrument has a 30-mm diameter rotating rod and was divided into 5 compartments, thus permitting tests for 5 mice at a time. The rotation speed of the rod was fixed at 12 rpm and mice were tested for the total duration of 180 sec. Mice were trained to maintain themselves in a neutral position on a stationary rod. After the rota-rod was switched on for 3 sec, mice were timed until they fell off the rota-rod (or a maximum cutoff time of 180 sec). Mice that had a score of 180 sec were removed from the rota-rod and returned to their home cages. The animals had a total of 3 consecutive trials per d. Mice that could not hold on the stationary cylinder for 3 trials were scored as zero. The highest individual score of each animal every day was used in the analysis. In order to avoid potential data misinterpretation due to acute melatonin residual effects, we performed behavioral measurements at least 24 h after the last injection of melatonin. For all experiments in vivo, data acquisition and analysis were performed using a double-blind approach.
Surgery and intracerebroventricular (ICV) siRNA administration
The ICV cannulation was carried out as previously described.Citation73 In brief, mice were anesthetized with a pentobarbital (60 mg/kg, i.p.) and procaine hydrochloride (60 mg/kg, s.c.) combination. A guide cannula (RWD Life Science Co., 62003) was stereotaxically implanted with stereotaxic coordinates related to bregma (anterior-posterior −0.82 mm; medial-lateral +1.5 mm; dorsal-ventral +2.0 mm) according to a previous study.Citation74 After surgery, animals were allowed to recover for a wk. A total of 5.0 μL of Cdk5 siRNA (20 μM) was injected over a period of 10 min into the right lateral ventricle with a microinjection pump (World Precision Instruments, Inc.., UMP3-4) connected to an internal cannula via polyethylene tubing. The internal cannula was left in place for at least 2 min before being slowly withdrawn to avoid back flow. Drugs were administered 24 h later after siRNA injection.
All above animal study and procedures were approved by the institutional review board of Kunming Institute of Zoology.
RNA extraction, real-time quantitative PCR (RT-qPCR) assay, and western blot
Total RNA was isolated from tissues and cell lines using TRIZOL (Invitrogen, 15596-018) and cDNA was synthesized by using the M-MLV Reverse Transcriptase (Promega, M170A) according to the manufacturer's instructions. The relative mRNA expression level of Cdk5 was quantified by RT-qPCR, with normalization to the Actb/β-actin gene.
Western blot assays for the target proteins were performed using the common approach. Cell lysates of different monkey brain tissues (prefrontal cortex, parietal cortex, temporal cortex, hippocampus, substantia nigra and striatum), mouse striatum, and rat glioma C6 cells were prepared using the protein lysis buffer (Beyotime Institute of Biotechnology, P0013). Protein concentration was determined using BCA protein assay kit (Beyotime Institute of Biotechnology, P0012). A total of 25 μg protein was separated by 15% SDS PAGE, and was transferred to a polyvinylidene difluoride membrane (Roche Diagnostics, IPVH00010). The membrane was soaked with 5% (w/v) skim milk for 2 h at room temperature. The membrane was incubated with primary antibodies against LC3B (Cell Signaling Technology, 3868; 1:1000), CDK5 (Santa Cruz Biotechnology, sc-6247; 1:1000), SQSTM1/p62 (Merck Millipore, MABC32; 1:1000), TH (Merck Millipore, AB152; 1:1000), SNCA (Merck Millipore, AB5038; 1:1000) and ACTB (Beijing Zhong Shan-Golden Bridge Biological Technology CO., LTD, TA-09; 1:1000) overnight at 4°C. The membranes were washed 3 times with TBST (Tris Buffered Saline [Cell Signaling Technology, #9997] with Tween 20 [0.1%; Sigma, P1379]), each time 5 min, followed by incubation with the peroxidase-conjugated anti-mouse (lot number 474-1806) or anti-rabbit (474-1506) IgG (1:5000; KPL) for 1 h at room temperature. The epitope was visualized using an ECL western blot detection kit (Millipore, WBKLS0500). ImageJ (National Institutes of Health, Bethesda, Maryland, USA) was used to evaluate the densitometry. Western blot for ACTB was used a loading control to measure the densitometry of CDK5, SQSTM1, TH and SNCA. The LC3B-II densitometric signal is determined by the ratio of LC3B-II to LC3B-I before normalized to ACTB.
Measurement of cell death in C6 cells
C6 cells were cultured in DMEM supplemented with 5% fetal bovine serum in a humidified atmosphere incubator with 5 % CO2 at 37°C. After 24 h of drug (MPTP and/or melatonin) treatment, cells were harvested and washed once with PBS. Cell death was quantified by staining with propidium iodide (1 μg/ml). Briefly, cells were incubated with the dye at 37°C for 20 min, then washed twice with PBS, resuspended in PBS and kept on ice for an immediate detection on the FACScan (Becton Dickinson, USA).
Immunohistochemistry, confocal microscopy imaging, Golgi staining, and quantification
For immunohistochemical analysis, mice were anesthetized with pentobarbital immediately at the end of the cycle of drug treatment, and were intracardially perfused with saline, followed by 4% paraformaldehyde. The ventral midbrain was sectioned coronally at 10-μm thickness on a cryostat (Leica, CM1850UV-1-1, Amtzell, Germany). Sections were collected on slides and were incubated for 24 h with primary anti-mouse TH (1:1000; Merck Millipore, AB152) or anti-rabbit SNCA (1:1000). After 3 washes with PBST (Phosphate Buffer Saline [Beyotime Institute of Biotechnology, C0221A] with Triton™ X-100 [0.1%]), each time 5 min, sections were then incubated with a biotinylated secondary antibody (Fuzhou Maixin Biotech. Co., Ltd. SP KIT-C1) and a streptavidin horseradish peroxidase-conjugated tertiary antibody (Fuzhou Maixin Biotech. Co., Ltd., SP KIT-D1) and were visualized using a 3, 3′-diaminobenzidine/glucose oxidase reaction.Citation75 Loss of dopamine neurons was assessed by immunohistochemical analysis of TH-positive neurons in the anatomical region of the SNc of middle brain.
Cultured C6 cells were transfected with N-terminally tagged pEGFP-C1-LC3 (a kind gift from Dr. Quan Chen, Institute of Zoology, Chinese Academy of Sciences). Intact cells were imaged, using an Olympus FluoView™ 1000 confocal microscope (Olympus, Melville, NY, USA) after being treated with MPTP and/or melatonin. Primary neurons were fixed and stained with antibodies against MAP2 (Merck Millipore, AB5622; 1:500). Immunoreactivity was detected with a FITC-conjugated secondary antibody (KPL, 172-1506; 1:50) and nuclei were counterstained with DAPI and the slides were visualized under an Olympus FluoView™ 1000 confocal microscope (Olympus, America). Golgi-Cox staining (FD NeuroTechnologies, PK401) was used to visualize neurons. Primary neurons were impregnated for 2 wk and processed according to the manufacturer's specifications. Dendrites of neurons were recognized by staining with the dendritic marker MAP2. ImageJ (National Institutes of Health, Bethesda, Maryland, USA) and Image-Pro Plus (IPP; Media Cybernetics, Inc., Washington, Maryland, USA) were used to evaluate the total dendritic length and dendritic complexity.
Statistical analysis
Comparisons of relative protein levels of LC3B-II, SQSTM1, CDK5, TH, and SNCA from animals of multiple groups and cells with different treatments were conducted by one-way analysis of variance (ANOVA) with the Tukey post-hoc test using the PRISM software (GraphPad Software, Inc., La Jolla, CA, USA). Differences in the animal rota rod task were determined by ANOVA, followed by LSD (Least Significant Difference) for post-hoc comparisons. Data were represented as mean ±SEM. A P value of <0.05 was considered to be significantly different.
Disclosure of Potential Conflicts of Interest
There were no potential conflicts of interest to be disclosed.
1082020_Online_Methods_and_Data_Supplement.docx
Download MS Word (10.5 MB)Acknowledgments
We thank Dr. Quan Chen for sharing the pEGFP-C1-LC3 vector and Dr. Jia-Gui Qu for technical assistance. We are grateful to the 4 reviewers and the editor for their constructive comments that essentially improved the work. The study was done at Kunming Institute of Zoology, Chinese Academy of Sciences.
Funding
This study was supported by the Strategic Priority Research Program (B) of the Chinese Academy of Sciences (XDB02020003, XDB02020001), the National Natural Science Foundation of China (31171225), and the Ministry of Science and Technology of China (2011CB910900).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Ferrer I, Martinez A, Blanco R, Dalfo E, Carmona M. Neuropathology of sporadic Parkinson disease before the appearance of parkinsonism: preclinical Parkinson disease. J Neural Transm 2011; 118:821-39; PMID:20862500; http://dx.doi.org/10.1007/s00702-010-0482-8
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson disease. Science 2003; 302:819-22; PMID:14593166; http://dx.doi.org/10.1126/science.1087753
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson disease. Nat Neurosci 2000; 3:1301-6; PMID:11100151; http://dx.doi.org/10.1038/81834
- Collier TJ, Kanaan NM, Kordower JH. Ageing as a primary risk factor for Parkinson disease: evidence from studies of non-human primates. Nature reviews. Neuroscience 2011; 12:359-66; PMID:21587290; http://dx.doi.org/10.1038/nrn3039
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson disease. Neurobiol Aging 2003; 24:197-211; PMID:12498954; http://dx.doi.org/10.1016/S0197-4580(02)00065-9
- Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci U S A 1983; 80:4546-50; PMID:6192438; http://dx.doi.org/10.1073/pnas.80.14.4546
- Bandopadhyay R, de Belleroche J. Pathogenesis of Parkinson disease: emerging role of molecular chaperones. Trends Mol Med 2009; 16:27-36; PMID:20036196; http://dx.doi.org/10.1016/j.molmed.2009.11.004
- Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, Akram M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem 2001; 76:1265-74; PMID:11238711; http://dx.doi.org/10.1046/j.1471-4159.2001.00183.x
- Lang AE, Lozano AM. Parkinson disease. First of two parts. N Engl J Med 1998; 339:1044-53; PMID:9761807; http://dx.doi.org/10.1056/NEJM199810083391506
- Gong X, Tang X, Wiedmann M, Wang X, Peng J, Zheng D, Blair LA, Marshall J, Mao Z. Cdk5-mediated inhibition of the protective effects of transcription factor MEF2 in neurotoxicity-induced apoptosis. Neuron 2003; 38:33-46; PMID:12691662; http://dx.doi.org/10.1016/S0896-6273(03)00191-0
- Huang E, Qu D, Zhang Y, Venderova K, Haque ME, Rousseaux MW, Slack RS, Woulfe JM, Park DS. The role of Cdk5-mediated apurinic/apyrimidinic endonuclease 1 phosphorylation in neuronal death. Nat Cell Biol 2010; 12:563-71; PMID:20473298; http://dx.doi.org/10.1038/ncb2058
- Smith PD, Crocker SJ, Jackson-Lewis V, Jordan-Sciutto KL, Hayley S, Mount MP, O'Hare MJ, Callaghan S, Slack RS, Przedborski S, et al. Cyclin-dependent kinase 5 is a mediator of dopaminergic neuron loss in a mouse model of Parkinson disease. Proc Natl Acad Sci U S A 2003; 100:13650-5; PMID:14595022; http://dx.doi.org/10.1073/pnas.2232515100
- Tsai LH, Delalle I, Caviness VS Jr, Chae T, Harlow E. p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 1994; 371:419-23; PMID:8090221; http://dx.doi.org/10.1038/371419a0
- Wen Z, Shu Y, Gao C, Wang X, Qi G, Zhang P, Li M, Shi J, Tian B. CDK5-mediated phosphorylation and autophagy of RKIP regulate neuronal death in Parkinson disease. Neurobiol Aging 2014; 35:2870-80; PMID:25104559; http://dx.doi.org/10.1016/j.neurobiolaging.2014.05.034
- Tang X, Wang X, Gong X, Tong M, Park D, Xia Z, Mao Z. Cyclin-dependent kinase 5 mediates neurotoxin-induced degradation of the transcription factor myocyte enhancer factor 2. J Neurosci 2005; 25:4823-34; PMID:15888658; http://dx.doi.org/10.1523/JNEUROSCI.1331-05.2005
- Qu D, Rashidian J, Mount MP, Aleyasin H, Parsanejad M, Lira A, Haque E, Zhang Y, Callaghan S, Daigle M, et al. Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson disease. Neuron 2007; 55:37-52; PMID:17610816; http://dx.doi.org/10.1016/j.neuron.2007.05.033
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell 2010; 140:313-26; PMID:20144757; http://dx.doi.org/10.1016/j.cell.2010.01.028
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007; 3:542-5; PMID:17611390; http://dx.doi.org/10.4161/auto.4600
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Cheung ZH, Ip NY. Autophagy deregulation in neurodegenerative diseases - recent advances and future perspectives. J Neurochem 2011; 118:317-25; PMID:21599666; http://dx.doi.org/10.1111/j.1471-4159.2011.07314.x
- Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, Tolkovsky A. Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 2005; 1:11-22; PMID:16874045; http://dx.doi.org/10.4161/auto.1.1.1513
- Zhu JH, Guo F, Shelburne J, Watkins S, Chu CT. Localization of phosphorylated ERK/MAP kinases to mitochondria and autophagosomes in Lewy body diseases. Brain Pathol 2003; 13:473-81; PMID:14655753; http://dx.doi.org/10.1111/j.1750-3639.2003.tb00478.x
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson disease. Histol Histopathol 1997; 12:25-31; PMID:9046040
- Dehay B, Bove J, Rodriguez-Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson disease. J Neurosci 2010; 30:12535-44; PMID:20844148; http://dx.doi.org/10.1523/JNEUROSCI.1920-10.2010
- Koh H, Chung J. PINK1 and Parkin to control mitochondria remodeling. Anat Cell Biol 2010; 43:179-84; PMID:21212857; http://dx.doi.org/10.5115/acb.2010.43.3.179
- Cuervo AM, Stefanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004; 305:1292-5; PMID:15333840; http://dx.doi.org/10.1126/science.1101738
- Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 2008; 283:23542-56; PMID:18566453; http://dx.doi.org/10.1074/jbc.M801992200
- Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, Mao Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science 2009; 323:124-7; PMID:19119233; http://dx.doi.org/10.1126/science.1166088
- Nopparat C, Porter JE, Ebadi M, Govitrapong P. One-Methyl-4-phenylpyridinium-induced cell death via autophagy through a Bcl−2/Beclin 1 complex-dependent pathway. Neurochem Res 2014; 39:225-32; PMID:24326530; http://dx.doi.org/10.1007/s11064-013-1208-8
- Wong AS, Lee RH, Cheung AY, Yeung PK, Chung SK, Cheung ZH, Ip NY. Cdk5-mediated phosphorylation of endophilin B1 is required for induced autophagy in models of Parkinson disease. Nat Cell Biol 2011; 13:568-79; PMID:21499257; http://dx.doi.org/10.1038/ncb2217
- Singhal NK, Srivastava G, Agrawal S, Jain SK, Singh MP. Melatonin as a neuroprotective agent in the rodent models of Parkinson disease: is it all set to irrefutable clinical translation? Mol Neurobiol 2012; 45:186-99; PMID:22198804; http://dx.doi.org/10.1007/s12035-011-8225-x
- Hardeland R. Melatonin and the theories of aging: a critical appraisal of melatonin's role in antiaging mechanisms. J Pineal Res 2013; 55:325-56; PMID:24112071; http://dx.doi.org/10.1111/jpi.12090
- Rodriguez C, Mayo JC, Sainz RM, Antolin I, Herrera F, Martin V, Reiter RJ. Regulation of antioxidant enzymes: a significant role for melatonin. J Pineal Res 2004; 36:1-9; PMID:14675124; http://dx.doi.org/10.1046/j.1600-079X.2003.00092.x
- Vega-Naredo I, Caballero B, Sierra V, Garcia-Macia M, de Gonzalo-Calvo D, Oliveira PJ, Rodriguez-Colunga MJ, Coto-Montes A. Melatonin modulates autophagy through a redox-mediated action in female Syrian hamster Harderian gland controlling cell types and gland activity. J Pineal Res 2011; 52:80-92; PMID:21771054; http://dx.doi.org/10.1111/j.1600-079X.2011.00922.x
- Khaldy H, Escames G, Leon J, Bikjdaouene L, Acuna-Castroviejo D. Synergistic effects of melatonin and deprenyl against MPTP-induced mitochondrial damage and DA depletion. Neurobiol Aging 2003; 24:491-500; PMID:12600724; http://dx.doi.org/10.1016/S0197-4580(02)00133-1
- Chen ST, Chuang JI, Hong MH, Li EI. Melatonin attenuates MPP+-induced neurodegeneration and glutathione impairment in the nigrostriatal dopaminergic pathway. J Pineal Res 2002; 32:262-9; PMID:11982797; http://dx.doi.org/10.1034/j.1600-079X.2002.01871.x
- Li XJ, Gu J, Lu SD, Sun FY. Melatonin attenuates MPTP-induced dopaminergic neuronal injury associated with scavenging hydroxyl radical. J Pineal Res 2002; 32:47-52; PMID:11841600; http://dx.doi.org/10.1034/j.1600-079x.2002.10831.x
- Absi E, Ayala A, Machado A, Parrado J. Protective effect of melatonin against the 1-methyl-4-phenylpyridinium-induced inhibition of complex I of the mitochondrial respiratory chain. J Pineal Res 2000; 29:40-7; PMID:10949539; http://dx.doi.org/10.1034/j.1600-079X.2000.290106.x
- Jin BK, Shin DY, Jeong MY, Gwag MR, Baik HW, Yoon KS, Cho YH, Joo WS, Kim YS, Baik HH. Melatonin protects nigral dopaminergic neurons from 1-methyl-4-phenylpyridinium (MPP+) neurotoxicity in rats. Neurosci Lett 1998; 245:61-4; PMID:9605485; http://dx.doi.org/10.1016/S0304-3940(98)00170-0
- Chuang JI, Chen TH. Effect of melatonin on temporal changes of reactive oxygen species and glutathione after MPP(+) treatment in human astrocytoma U373MG cells. J Pineal Res 2004; 36:117-25; PMID:14962063; http://dx.doi.org/10.1046/j.1600-079X.2003.00107.x
- Thomas B, Mohanakumar KP. Melatonin protects against oxidative stress caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in the mouse nigrostriatum. J Pineal Res 2004; 36:25-32; PMID:14675127; http://dx.doi.org/10.1046/j.1600-079X.2003.00096.x
- Ortiz GG, Crespo-Lopez ME, Moran-Moguel C, Garcia JJ, Reiter RJ, Acuna-Castroviejo D. Protective role of melatonin against MPTP-induced mouse brain cell DNA fragmentation and apoptosis in vivo. Neuro Endocrinol Lett 2001; 22:101-8; PMID:11335886; http://dx.doi.org/NEL220201A04.
- Capitelli C, Sereniki A, Lima MM, Reksidler AB, Tufik S, Vital MA. Melatonin attenuates tyrosine hydroxylase loss and hypolocomotion in MPTP-lesioned rats. Eur J Pharmacol 2008; 594:101-8; PMID:18674531; http://dx.doi.org/10.1016/j.ejphar.2008.07.022
- Feng YM, Jia YF, Su LY, Wang D, Lv L, Xu L, Yao YG. Decreased mitochondrial DNA copy number in the hippocampus and peripheral blood during opiate addiction is mediated by autophagy and can be salvaged by melatonin. Autophagy 2013; 9:1395-406; PMID:23800874; http://dx.doi.org/10.4161/auto.25468
- Alvira D, Tajes M, Verdaguer E, Acuna-Castroviejo D, Folch J, Camins A, Pallas M. Inhibition of the cdk5/p25 fragment formation may explain the antiapoptotic effects of melatonin in an experimental model of Parkinson disease. J Pineal Res 2006; 40:251-8; PMID:16499562; http://dx.doi.org/10.1111/j.1600-079X.2005.00308.x
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999; 402:615-22; PMID:10604467; http://dx.doi.org/10.1038/45159
- Nopparat C, Porter JE, Ebadi M, Govitrapong P. The mechanism for the neuroprotective effect of melatonin against methamphetamine-induced autophagy. J Pineal Res 2010; 49:382-9; PMID:20738755; http://dx.doi.org/10.1111/j.1600-079X.2010.00805.x
- Stefanis L. α-Synuclein in Parkinson disease. Cold Spring Harb Perspect Med 2012; 2:a009399; PMID:22355802; http://dx.doi.org/10.1101/cshperspect.a009399
- Vekrellis K, Stefanis L. Targeting intracellular and extracellular α-synuclein as a therapeutic strategy in Parkinson disease and other synucleinopathies. Expert Opin Ther Targets 2012; 16:421-32; PMID:22480256; http://dx.doi.org/10.1517/14728222.2012.674111
- Jakowec MW, Petzinger GM. One-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned model of parkinson's disease, with emphasis on mice and nonhuman primates. Comp Med 2004; 54:497-513; PMID:15575363
- Xilouri M, Stefanis L. Autophagic pathways in Parkinson disease and related disorders. Expert Rev Mol Med 2011; 13:e8; PMID:21418705; http://dx.doi.org/10.1017/S1462399411001803
- Hollerhage M, Goebel JN, de Andrade A, Hildebrandt T, Dolga A, Culmsee C, Oertel WH, Hengerer B, Hoglinger GU. Trifluoperazine rescues human dopaminergic cells from wild-type α-synuclein-induced toxicity. Neurobiol Aging 2014; 35:1700-11; PMID:24559643; http://dx.doi.org/10.1016/j.neurobiolaging.2014.01.027
- Pan T, Kondo S, Zhu W, Xie W, Jankovic J, Le W. Neuroprotection of rapamycin in lactacystin-induced neurodegeneration via autophagy enhancement. Neurobiol Dis 2008; 32:16-25; PMID:18640276; http://dx.doi.org/10.1016/j.nbd.2008.06.003
- Xiong N, Xiong J, Jia M, Liu L, Zhang X, Chen Z, Huang J, Zhang Z, Hou L, Luo Z, et al. The role of autophagy in Parkinson disease: rotenone-based modeling. Behav Brain Funct 2013; 9:13; PMID:23497442; http://dx.doi.org/10.1186/1744-9081-9-13
- Plowey ED, Cherra SJ 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem 2008; 105:1048-56; PMID:18182054; http://dx.doi.org/10.1111/j.1471-4159.2008.05217.x
- Cherra SJ 3rd, Steer E, Gusdon AM, Kiselyov K, Chu CT. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol 2013; 182:474-84; PMID:23231918; http://dx.doi.org/10.1016/j.ajpath.2012.10.027
- Choubey V, Safiulina D, Vaarmann A, Cagalinec M, Wareski P, Kuum M, Zharkovsky A, Kaasik A. Mutant A53T α-synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 2011; 286:10814-24; PMID:21252228; http://dx.doi.org/10.1074/jbc.M110.132514
- Li L, Wang X, Fei X, Xia L, Qin Z, Liang Z. Parkinson disease involves autophagy and abnormal distribution of cathepsin L. Neurosci Lett 2011; 489:62-7; PMID:21134415; http://dx.doi.org/10.1016/j.neulet.2010.11.068
- Chu CT, Plowey ED, Dagda RK, Hickey RW, Cherra SJ 3rd, Clark RS. Autophagy in neurite injury and neurodegeneration: in vitro and in vivo models. Methods Enzymol 2009; 453:217-49; PMID:19216909; http://dx.doi.org/10.1016/S0076-6879(08)04011-1
- Chang CF, Huang HJ, Lee HC, Hung KC, Wu RT, Lin AM. Melatonin attenuates kainic acid-induced neurotoxicity in mouse hippocampus via inhibition of autophagy and α-synuclein aggregation. J Pineal Res 2012; 52:312-21; PMID:22212051; http://dx.doi.org/10.1111/j.1600-079X.2011.00945.x
- Ma J, Shaw VE, Mitrofanis J. Does melatonin help save dopaminergic cells in MPTP-treated mice? Parkinsonism & Related Disorders. 2009; 15:307-14; PMID:18793863; http://dx.doi.org/10.1016/j.parkreldis.2008.07.008
- Singhal NK, Srivastava G, Patel DK, Jain SK, Singh MP. Melatonin or silymarin reduces maneb- and paraquat-induced Parkinson disease phenotype in the mouse. J Pineal Res 2011; 50:97-109; PMID:20964710; http://dx.doi.org/10.1111/j.1600-079X.2010.00819.x
- Jenner P. Oxidative mechanisms in nigral cell death in Parkinson disease. Mov Disord 1998; 13 Suppl 1:24-34; PMID:9613715
- Danielson SR, Andersen JK. Oxidative and nitrative protein modifications in Parkinson disease. Free Radic Biol Med 2008; 44:1787-94; PMID:18395015; http://dx.doi.org/10.1016/j.freeradbiomed.2008.03.005
- Vicencio JM, Lavandero S, Szabadkai G. Ca2+, autophagy and protein degradation: thrown off balance in neurodegenerative disease. Cell Calcium 2010; 47:112-21; PMID:20097418; http://dx.doi.org/10.1016/j.ceca.2009.12.013
- Smith PD, Mount MP, Shree R, Callaghan S, Slack RS, Anisman H, Vincent I, Wang X, Mao Z, Park DS. Calpain-regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J Neurosci 2006; 26:440-7; PMID:16407541; http://dx.doi.org/10.1523/JNEUROSCI.2875-05.2006
- Graham WC, Robertson RG, Sambrook MA, Crossman AR. Injection of excitatory amino acid antagonists into the medial pallidal segment of a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) treated primate reverses motor symptoms of parkinsonism. Life Sci 1990; 47:PL91-7; PMID:2250573; http://dx.doi.org/10.1016/0024-3205(90)90376-3
- Smith RD, Zhang Z, Kurlan R, McDermott M, Gash DM. Developing a stable bilateral model of parkinsonism in rhesus monkeys. Neuroscience 1993; 52:7-16; PMID:8433810; http://dx.doi.org/10.1016/0306-4522(93)90176-G
- Ko WK, Pioli E, Li Q, McGuire S, Dufour A, Sherer TB, Bezard E, Facheris MF. Combined fenobam and amantadine treatment promotes robust antidyskinetic effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-lesioned primate model of Parkinson disease. Mov Disord 2014; 29:772-9; PMID:24610195; http://dx.doi.org/10.1002/mds.25859
- Kelly MA, Rubinstein M, Phillips TJ, Lessov CN, Burkhart-Kasch S, Zhang G, Bunzow JR, Fang Y, Gerhardt GA, Grandy DK, et al. Locomotor activity in D2 dopamine receptor-deficient mice is determined by gene dosage, genetic background, and developmental adaptations. J Neurosci 1998; 18:3470-9; PMID:9547254
- Ojha RP, Rastogi M, Devi BP, Agrawal A, Dubey GP. Neuroprotective effect of curcuminoids against inflammation-mediated dopaminergic neurodegeneration in the MPTP model of Parkinson disease. J Neuroimmune Pharmacol 2012; 7:609-18; PMID:22527634; http://dx.doi.org/10.1007/s11481-012-9363-2
- Manna SS, Umathe SN. Involvement of transient receptor potential vanilloid type 1 channels in the pro-convulsant effect of anandamide in pentylenetetrazole-induced seizures. Epilepsy Res 2012; 100:113-24; PMID:22386872; http://dx.doi.org/10.1016/j.eplepsyres.2012.02.003
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd ed (Academic Press, 2001).
- Schacht V, Kern JS. Basics of immunohistochemistry. J Invest Dermatol 2015; 135:e30; PMID:25666678; http://dx.doi.org/10.1038/jid.2014.541