
Abstract
Metastatic renal cell carcinomas (mRCC) are highly vascularized tumors that are a paradigm for the treatment with antiangiogenesis drugs targeting the vascular endothelial growth factor (VEGF) pathway. The available drugs increase the time to progression but are not curative and the patients eventually relapse. In this study we have focused our attention on the molecular mechanisms leading to resistance to sunitinib, the first line treatment of mRCC. Because of the anarchic vascularization of tumors the core of mRCC tumors receives only suboptimal concentrations of the drug. To mimic this in vivo situation, which is encountered in a neoadjuvant setting, we exposed sunitinib-sensitive mRCC cells to concentrations of sunitinib below the concentration of the drug that gives 50% inhibition of cell proliferation (IC50). At these concentrations, sunitinib accumulated in lysosomes, which downregulated the activity of the lysosomal protease CTSB (cathepsin B) and led to incomplete autophagic flux. Amino acid deprivation initiates autophagy enhanced sunitinib resistance through the amplification of autolysosome formation. Sunitinib stimulated the expression of ABCB1 (ATP-binding cassette, sub-family B [MDR/TAP], member 1), which participates in the accumulation of the drug in autolysosomes and favor its cellular efflux. Inhibition of this transporter by elacridar or the permeabilization of lysosome membranes with Leu-Leu-O-methyl (LLOM) resensitized mRCC cells that were resistant to concentrations of sunitinib superior to the IC50. Proteasome inhibitors also induced the death of resistant cells suggesting that the ubiquitin-proteasome system compensates inhibition of autophagy to maintain a cellular homeostasis. Based on our results we propose a new therapeutic approach combining sunitinib with molecules that prevent lysosomal accumulation or inhibit the proteasome.
Abbreviations
| ABCB1 | = | ATP-binding cassette, sub-family B (MDR/TAP), member 1 |
| ATF4 | = | activating transcription factor 4 |
| CTSB | = | cathepsin B |
| CHL | = | chloroquine |
| EEA1 | = | early endosome antigen 1 |
| FACS | = | fluorescence-activated cell sorting |
| HBSS | = | Hank's balanced salt solution |
| IC50 | = | concentration of a drug that gives 50% inhibition of cell proliferation |
| LAMP1/2 | = | lysosomal-associated membrane protein 1/2 |
| MAP1LC3A/LC3A | = | microtubule-associated protein 1 light chain 3 α |
| MAP1LC3-II/LC3-II | = | lipidated forms of LC3 |
| LLOM | = | Leu-Leu-O-methyl |
| MAPK1/2/3 | = | mitogen-activated protein kinase1/2/3 |
| mRCC | = | metastatic renal cell carcinomas |
| NAA10/ARD1 | = | N(α)-acetyltransferase 10, NatA catalytic subunit |
| PSMB8/9/10 | = | proteasome (prosome macropain) subunit, β type 8/9/10 |
| PSMF1 | = | proteasome (prosome macropain) inhibitor subunit 1 (PI31) |
| SQSTM1/p62 | = | sequestosome 1 |
| V-ATPase | = | vacuolar-type ATPase |
| VEGFA/C | = | vascular endothelial growth factor A/C. |
Introduction
A large number of drugs currently used in the clinic are weak bases that make them lysosomotropic. These drugs are extensively sequestered in acidic lysosomes through an ion-trapping mechanism. Drug accumulation in lysosomes is driven by the large pH gradient that exists across the lumen of the organelle and the cytosol. Lysosomal sequestration can directly influence the activity of the drug by preventing its availability to bind with intended intracellular targets. In addition, some cancer cells have acquired an enhanced ability to sequester anticancer drugs in lysosomes, which constitutes a mechanism of drug resistance. The clear cell form of kidney cancers, the most represented one, is characterized by VHL/von Hippel-Lindau mutations and deletions which lead to stabilization of HIF1A (hypoxia inducible factor 1, α subunit [basic helix-loop-helix transcription factor]) protein and consequent overexpression of VEGFA (vascular endothelial growth factor A). This feature has rendered mRCC attractive targets for the treatment with antiangiogenesis drugs, particularly with the development of tyrosine kinase inhibitors targeting the VEGF (KDR, FLT1, FLT4) and the PDGF (PDGFRB) receptors, both implicated in proliferation of endothelial cells and pericytes. These inhibitors include notably sunitinib,Citation1 which since 2006 is considered as the standard first line treatment option for this disease. Hence, we have analyzed the tumor cell fate with chronic exposure to the drug. Such a question may seem inappropriate since sunitinib was originally defined as an antiangiogenesis compound that inhibits endothelial cell proliferation. However, we and others have recently shown that sunitinib also inhibits mRCC cell proliferation probably because they aberrantly express the tyrosine kinase receptors targeted by the drug therefore leading to the selection of resistant cells.Citation2,3 Sunitinib has been designed to disrupt major signaling pathways (HRAS-RAF1-MAP2K1/2-MAPK1/3 and MTOR pathways) that are responsible for the abnormal proliferation of cancer cells and tumor angiogenesis.Citation4 However, sunitinib has not significantly improved the overall survival of the majority of patients compared to treatment with IFNA/interferon α or IL2/interleukin 2 (median time of survival after the diagnosis of about 20 mo),Citation1,5 the standard treatments used before the development of antiangiogenesis drugs. Moreover, the fact that mRCC patients gradually become refractory to sunitinib represents an important obstacle to better outcomes for patients. Therefore, it is urgent to better understand the molecular mechanisms associated with resistance to sunitinib to improve the final outcome of the patients. In this context, we have analyzed the fate of tumor cells following chronic exposure to the drug. The selection pressure exerted by chronic exposure has led to the selection of resistant cells,Citation2,3 but the mechanisms inducing resistance are unknown.
It was previously reported that sunitinib induces autophagy in bladder cancer cells,Citation6 and that inhibition of autophagy potentiates the antiproliferative effects of sunitinib.Citation7,8 However, in these experiments, cells are exposed to high doses of sunitinib and the cells are not representative of cancers for which sunitinib is the treatment of reference.Citation7 Moreover, these reports do not investigate the molecular link between sunitinib treatment and autophagy. Lysosomal sequestration of sunitinib may be explained by the fact that it is a hydrophobic weak base (pKa 8.95).Citation9 Sequestration in lysosomes may prevent access of the drug to the kinase domain of tyrosine kinase receptors present in the cytoplasm, thus participating in the loss of efficacy of the drug. However, the effect of sunitinib on autophagy has not been described for mRCC cells. Furthermore, the implication of autophagy and lysosome trapping in the mechanisms of resistance has not been addressed. In the present study we aimed at deciphering the link between autophagy and the mechanisms of resistance to sunitinib in mRCC. To overcome resistance we propose treatment with a combination of drugs preventing lysosomal accumulation.
Results
mRCC cells showed reduced proliferation in the presence of concentrations of sunitinib below the IC50 (suboptimal concentration)
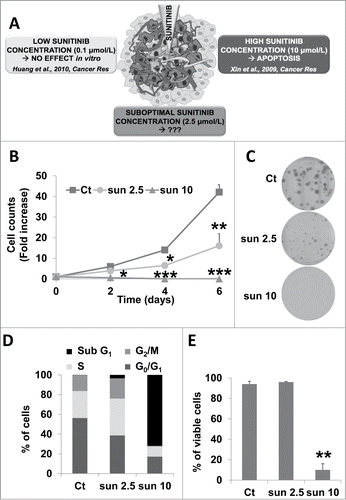
Because of the abnormal vascularization of tumors, the core of primary mRCC or metastases is not exposed to optimal concentrations of the drug (). We first determined the in vitro concentrations that resulted in progressive adaptation to sunitinib and final selection of resistant cells. The plasma concentrations of patients or mice exposed to sunitinib was low (0.1 to 1 μmol/L range) compared to the intratumor amount, which was 10 times higher (10 μmol/L range). Whereas the IC50 of endothelial cells for sunitinib was approximately 0.1 μmol/L,Citation10 the IC50 of mRCC cells was approximately 5 μmol/L.Citation2,9 In the presence of a concentration of sunitinib below the IC50 (2.5 μmol/L), mRCC cells ( and Fig. S1A, 786-O and RCC10, respectively) have a reduced proliferation rate, which was linked to prolonged S and G2/M phases of the cell cycle (). A suboptimal concentration of sunitinib (2.5 μmol/L) did not affect cell viability () whereas exposure to a higher concentration resulted in cell death, as measured by cell counting or a clonogenic assay.
Figure 1. A sunitinib concentration below the IC50 slowed down cell proliferation but did not induce cell death. (A) General schema illustrating the different concentrations to which RCC cells may be exposed to in a tumor. (B) The proliferative capacity of 786-O cells in the absence (Ct) or presence of increasing concentrations of sunitinib (sun) was evaluated by counting the cells at the indicated times. Data are the mean fold increase ±SD. The fold increase of untreated cells was taken as the reference value for statistics. Statistical significances of the results compared to untreated cells are indicated; *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) Clonal growth of 786-O cells in the absence (Ct) or presence of sunitinib (sun) (2.5 or 10 μmol/L). (D) The proportion of cells in each phase of the cell cycle was determined by DNA labeling with propidium iodide followed by FACS analysis. (E) Determination of viable cells in the absence (Ct) or presence of 2.5 μmol/L (2.5) or 10 μmol/L (10) of sunitinib (sun).
Sunitinib accumulated in lysosomes
To decipher the adaptation to suboptimal concentrations of sunitinib, the rest of the experiments were conducted at the concentration of 2.5 μmol/L.
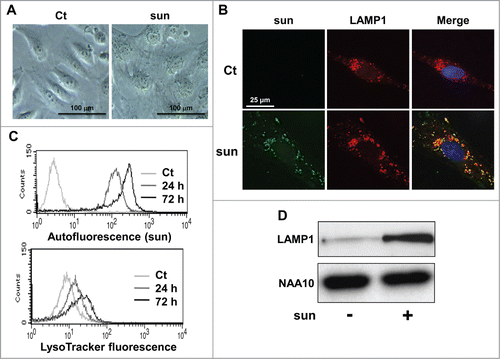
Phase contrast microscopy highlighted a modification of the cell shape and the appearance of a yellowish color inside the cells after incubation with sunitinib for 2 d (). The intracellular localization of sunitinib was confirmed by visualization of its autofluorescence. Sunitinib autofluorescence colocalized with a specific lysosomal staining (LAMP1 [lysosomal-associated membrane protein 1]; ) confirming that sunitinib accumulated in acidic lysosomal structures. Accumulation in lysosomes was also observed in 2 independent cell lines (RCC10 and A498) and 2 RCC primary cell lines (CC and TFE3) that we previously described (Fig. S1B).Citation2 However, sunitinib did not accumulate in early endosomes (no colocalization with EEA1 [early endosome antigen 1]; Fig. S2). This result suggests accumulation of sunitinib in intracellular compartments with no major consequences to cell viability. This characteristic defines sunitinib as a lysomotropic agent.Citation11 FACS analysis showed that sunitinib accumulated in lysosomes in a time-dependent manner and that there was an increase in the lysosomal mass (), which coincided with increased expression of LAMP1 (). Such accumulation of sunitinib was not dependent on the oxygen concentration since sunitinib sequestration was equivalent in normoxia or hypoxia (Fig. S3A). At the concentration of 2.5 μmol/L of sunitinib, hypoxia did not modify the cell viability (Fig. S3B). Intracellular accumulation of sunitinib was also observed in experimental RCC in mice (Fig. S3C).
Figure 2. Sunitinib accumulated in lysosomes. (A) Phase contrast microscopy showing modifications of the cell shape and accumulation of yellow granules in 786-O cell incubated with 2.5 μmol/L of sunitinib for 24 h. (B) Immunofluorescence to LAMP1 in control (Ct) or sunitinib-treated (2.5 μmol/L, sun) 786-O cells for 48 h. Sunitinib autofluorescence is shown. Merged fluorescence is also shown. (C) The autofluorescence of sunitinib and the fluorescence of the lysosomal probe (LysoTracker Red DND-99, Lyso) were followed by FACS analysis performed at the indicated times. (D) Cell extracts from control (-) or sunitinib-treated (2.5 μmol/L) 786-O cells incubated for 72 h were tested for LAMP1 expression by immunoblotting. NAA10 is shown as a loading control.
Sunitinib neutralized the pH of lysosomes and inhibited CTSB
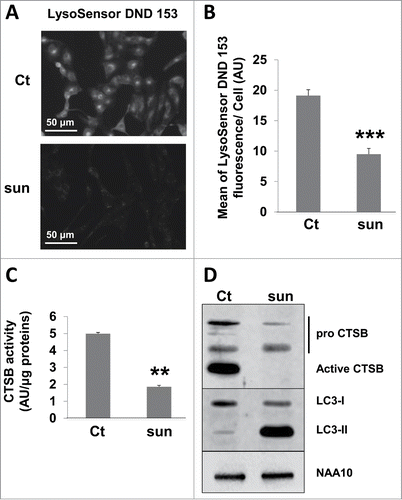
Sunitinib is a weak base (pKa 8.95), which accumulates in lysosomes where it is protonated by a pH-partitioning process.Citation11 Once ionized, sunitinib becomes membrane impermeable with the impossibility of diffusing out of the organelle, which results in lysosome trapping. Accumulation continues as long as the low pH is maintained by the vacuolar proton pump (V-type H+ATPase) but ultimately results in buffering of the acidic pH of lysosomes. The LysoSensor Green DND-153 fluorescence (pKa 7.5) was intense in control conditions (this dye fluoresces in an acidic environment) but almost disappeared in the presence of sunitinib suggesting that the acidic pH of lysosomes has been neutralized (). This correlated with decreased expression and activity of one of the major lysosome-associated proteases CTSB ().
Figure 3. Sunitinib neutralized the lysosomal acidic pH and inhibited the proteolytic activity. (A) Acid pH-dependent fluorescence to LysoSensor Green DND-153 in 786-O cells in the absence or presence of sunitinib. (B) Quantification of the fluorescence shown in A (n = 3; ***, P < 0.001). (C) Quantification of the CTSB activity in 786-O cells in the absence (Ct) or presence of sunitinib (sun) in the organelles compartment (lysosomes, mitochondria, Golgi, endoplasmic reticulum) activity as described in Materials and Methods. (D) Analysis of the presence of pro and mature CTSB by immunoblotting. NAA10 is shown as a loading control.
Suboptimal concentrations of sunitinib initiated incomplete autophagic flux
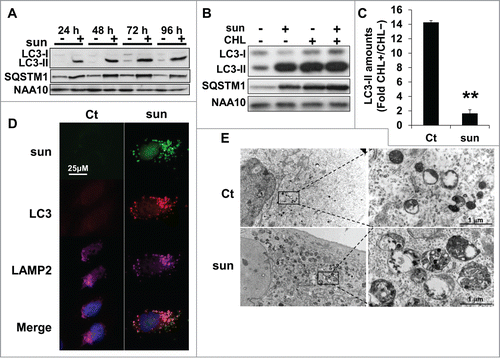
In physiological situations, autophagy is responsible for the degradation of dysfunctional organelles and proteins and allows cell survival during nutrient deprivation.Citation12 So, autophagy is important in maintaining cell homeostasis, but if exacerbated, it can lead to cell death.Citation13 Sunitinib treatment resulted in an increase in the lysosomal pH and inhibition of the lysosomal protease activity. Hence, we investigated the consequences of these modifications on autophagy. Autophagy is characterized by the accumulation of cleaved and lipidated forms of MAP1LC3A/LC3A (microtubule-associated protein 1 light chain 3 α; LC3-II) and the degradation of SQSTM1/p62 protein. Sunitinib treatment induced accumulation of LC3-II, which indicated either initiation or blockade of autophagy (in 786-O cells, and in 2 additional cell lines [RCC10 and A498] and the above mentioned primary cells [CC and TFE3], Fig. S4A and S4B, respectively). However, sustained expression of SQSTM1 strongly suggested incomplete processing. To confirm incomplete autophagic flux, we evaluated the LC3-II level in response to sunitinib in the presence or absence of chloroquine (CHL), a lysomotropic agent that increases the lysosomal pH. A higher fold change compared to control conditions reflected induction of autophagy whereas no modification or a lower fold change in the presence of CHL was indicative of inhibition of autophagy.Citation14 The fold change in LC3-II in sunitinib-treated cells after CHL treatment was equivalent compared to cells treated with sunitinib alone (), indicating an inhibition of the autophagy flux. Furthermore, the merged localization of sunitinib, LC3 and LAMP2 indicated accumulation of nonfunctional autolysosomes (), which was visualized by electron microscopy ().
Figure 4. Suboptimal concentrations of sunitinib initiated incomplete autophagic flux. (A) The autophagic flux was examined in 786-O cells in the absence (-) or presence (+) of 2.5 μmol/L of sunitinib for the indicated times. The levels of LC3-I/LC3-II and SQSTM1 were evaluated by immunoblotting. NAA10 is shown as a loading control. (B) To gauge the level of autophagic flux in 786-O cells incubated in the absence (-) or presence (+) of 2.5 or 5 μmol/L of sunitinib, the level of LC3-I/LC3-II and SQSTM1 was evaluated and compared to cells incubated with CHL, 40 μmol/L. NAA10 is shown as a loading control. (C) Quantification of LC3-II in CHL-treated and CHL-untreated cells. **, P < 0.01. (D) Sunitinib autofluorescence and immunofluorescence to LC3-II and LAMP2 in control (Ct) or sunitinib-treated (2.5 μmol/L) 786-O cells after incubation for 48 h. Merged fluorescence is shown. Pink illustrates colocalization of the 3 markers. (E) Electron micrographs showing autolysosomes in 786-OS cells incubated in the absence (Ct) or presence of sunitinib 2.5 μmol/L (sun) for 48 h.
Amino acid deprivation enhanced resistance to sunitinib
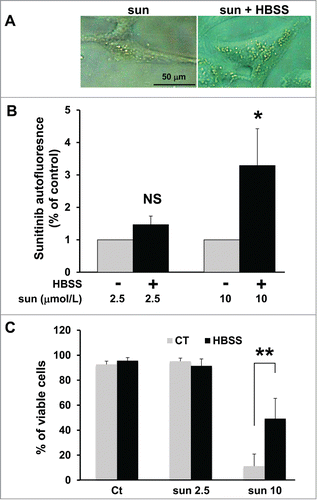
It is well known that cell starvation stimulates autophagy, which helps cells to resist this unfavorable environment. Autophagy is also used by tumor cells to survive when they have consumed all the nutrients easily accessible from the blood stream. Hence, the autolysosomes generated through this process may engulf sunitinib thereby amplifying resistance to the drug by preventing drug access to its targets. To test this hypothesis, we cultured 786-OS cells in a saline medium (Hank's balanced salt solution; HBSS) deprived of amino acids, which initiates autophagosome and lysosome formation. We questioned if this increased the capacity of the cells to store sunitinib in acidic intracellular compartments. Phase contrast microscopy clearly showed an enhanced accumulation of yellow granules when cells are cultured in HBSS medium (). Quantification by FACS confirmed this qualitative observation (). A high concentration (10 μmol/L) of sunitinib induces cell death, but at a lower concentration (2.5 μmol/L), it slows proliferation without inducing cell death, as expected. However, if the cells were first cultured in HBSS medium, cell proliferation was minimally affected by 2.5 μmol/L sunitinib and cell death was substantially decreased, even at a high concentration of the drug (10 μmol/L; ). These results were confirmed with 2 other independent cell lines (RCC10 and A498 cells; Fig. S5).
Figure 5. Amino acid starvation enhanced sunitinib resistance. (A) Phase contrast microscopy showing more accumulation of yellow granules in 786-O cells incubated with 10 μmol/L of sunitinib cultured in HBSS medium (sun + HBSS) for 24 h compared to cells cultured with 10 μmol/L of sunitinib in normal medium. (B) Determination of sunitinib autofluorescence by FACS after incubation with 2 concentrations of the drug (sun 2.5 and 10 μmol/L) during 24 h. Cells were cultured either in normal or HBSS medium; *, P < 0.05; **, P < 0.01; NS, nonsignificant. (C) Determination of the percentage of viable 786-O cells in the absence (Ct) or presence of 2.5 (sun 2.5) or 10 μmol/L (sun 10) of sunitinib during 24 h. Cells were cultured either in normal or HBSS medium.
Sunitinib-resistant cells showed exacerbated, incomplete autophagy and a more aggressive phenotype
We hypothesized that sequestration of sunitinib in lysosomes and the subsequent inhibition of the autophagy flux participated in sunitinib resistance. To test this hypothesis, we generated sunitinib-resistant cells by chronic exposure of cells to the drug (786-OR and RCC10R). Incomplete autophagy in these cells was attested by accumulation of LC3-II and sustained expression of SQSTM1 (Fig. S6). Primary sunitinib-resistant cells were also derived from a RCC removed surgically from a patient, as we previously described (TFE3 cells).Citation2 The 786-OR and RCC10R cells survive and proliferate in the presence of a high concentration of the drug, which is sufficient to induce parental cell death. TFE3 cells are highly resistant to sunitinib in vitro even at high concentrations of the drug (IC50 10 μmol/L).Citation2 The ability to accumulate sunitinib inside 786-OR was increased compared to parental cells (Fig. S7). The viability of parental (786-OS) and resistant cells (786-OR) at different concentrations of sunitinib is illustrated in . As shown previously by Gotink et al.,Citation9 resistance (maintained several weeks) is not genetically acquired since it can be reverted by culturing the cells in the absence of the drug for a few passages. Electron microscopy showed that 786-OR cells accumulate bigger vacuolar structures, identified above as autolysosomes, compared to 786-OS cells when incubated in the presence of 2.5 μmol/L of sunitinib, a finding in favor of an exacerbated incomplete autophagy ().
Figure 6. Enhanced inhibition of autophagy and aggressiveness of 786-OR cells. (A) Determination of the percentage of viable 786-OS and 786-OR cells in the absence (Ct) or presence of 2.5 (sun2.5) or 10 μmol/L (sun10) of sunitinib after incubation for 24 h. (B) Electron micrographs of 786-OS cells incubated in the absence (Ct) or presence of sunitinib 2.5 μmol/L (sun) for 48 h and of 786-OR cells showing autolysosomes (arrows). Note the presence of autolysosomes of increased size in 786-OR cells. (C) Quantification of the number (left panel) and the size (right panel) of colonies obtained for 786-OS and 786-OR cells seeded in soft agar; *, P < 0.05. (D) Cell migration in real time was analyzed with the xCELLigence RTCA. The chart shows the outcome of the kinetics analysis of the cell migration for 786-OS and 786-OR cells (left panel). The right panel shows the average cell indexes at 24 h from 3 independent experiments. **, P < 0.01.
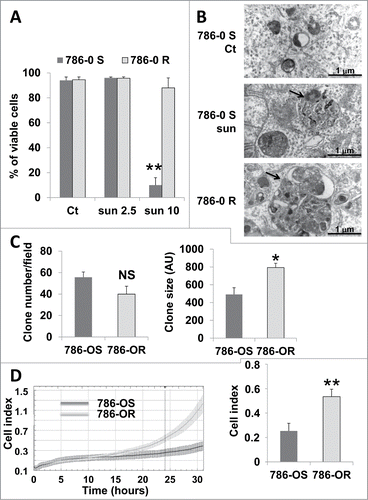
Previous reports have shown that treatment of tumor-bearing mice with sunitinib results in the selection of more aggressive cells. This has been observed in vivo, but we hypothesized that such selection did not involve cells from the tumor microenvironment but implicated an intrinsic phenotypic adaptation of tumor cells. We observed that 786-OR cells acquired greater anchorage independency illustrated by the formation of bigger colonies in soft agar (786-OR, ; and RCC10R, Fig. S8). In addition, the 786-OR cells showed an increase in their ability to migrate compared to 786-OS cells (). These results suggest that incomplete autophagy correlated with the acquisition of the more aggressive phenotype of 786-OR cells. We tested different markers implicated in epithelial/mesenchymal transition including CDH/N-cadherin, VIM/vimentin, and the transcription factors SNAI2/slug and SNAI1/snail, but no significant changes were observed between the sensitive and resistant cells.
Inhibition of the ATP binding cassette (ABC) transporter ABCB1 and lysosomal permeabilization enhanced the efficacy of sunitinib
We hypothesized that sequestration of sunitinib in lysosomes is involved in resistance to sunitinib. If so, destabilization of lysosomes with LLOM may result in increased cell death in the presence of a suboptimal concentration of sunitinib since the drug inhibits the kinase activity of target tyrosine kinase receptors located in the cytoplasm. However, we observed that LLOM reduced accumulation of sunitinib inside the cells. Moreover, in the presence of LLOM, the amount of sunitinib in the culture medium was increased. These results strongly suggest that if sunitinib is not sequestered in lysosomal compartment, it is actively exported outside the cells.
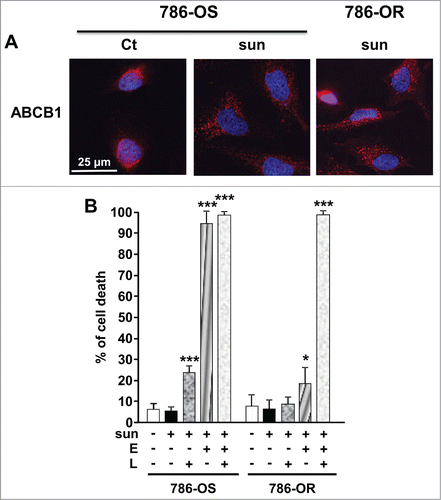
Inhibition of the ABC transporters improves sunitinib accumulation in the brain suggesting that these transporters participate in the efflux of the drug.Citation15 These transporters are present at the lysosomal membrane and could participate in sunitinib accumulation in this cellular compartment.Citation16 Moreover, sunitinib is a substrate of ABCB1.Citation17 Hence, we hypothesized that ABC transporters could be present on the plasma and/or lysosomal membranes to mediate accumulation in specific cell compartments and/or to participate in sunitinib efflux out of the cells. Sunitinib treatment of 786-OS stimulated ABCB1 expression and increased accumulation in 786-OR cells (). ABCB1 expression was also increased in A498, TFE3, and CC cells (Fig. S4). Whereas elacridar, an inhibitor of ABC transporters, induced slightly 786-OS cell death at a low concentration (1 μmol/L; Fig. S9A), it potentiated sunitinib activity on 786-OS cells (). Elacridar did not significantly mediate 786-OR cell death when alone (Fig. S9A) but potentiated sunitinib activity (). The lysomotropic agent LLOM had little effect on cell death at a low concentration (1 μmol/L). Higher concentrations are needed to induce cell death probably through the release of cathepsins and induction of lysosome membrane permeabilization leading finally to apoptosis (Fig. S9B).Citation18 A low concentration of LLOM increased the efficacy of sunitinib in 786-OS cells (), which strongly suggested that preventing drug sequestration in the lysosomes allowed a better accessibility to its targets. Finally, the triple combination of sunitinib, LLOM, and elacridar resulted in 100% cell death of 786-OS and 786-OR cells (). Of note, a mix of elacridar plus LLOM had a strong detrimental effect on 786-OS cells even at low concentrations, but had little effect on 786-OR cell viability (Fig. S9C). Although less potent, bafilomycin A1 (BAF), an inhibitor of the V-ATPase pump, which is responsible for the maintenance of the low pH of the lysosomes, exerted a comparable effect to LLOM (Fig. S9D). Hence, the triple combination sunitinib, BAF and elacridar, was less potent than the sunitinib and LLOM-elacridar mix. Equivalent results were obtained with an independent cell line (RCC10, Fig. S10). These results suggest that blockade of sunitinib trapping in the lysosomes is an efficient way to increase the potency of the drug and prevent resistance.
Figure 7. A combination of a lysosome destabilizing agent and an inhibitor of ABC transporters reverted sunitinib resistance of 786-OR cells. (A) Expression of ABCB1 was detected by immunofluorescence in control (Ct) or sunitinib-treated (2.5 μmol/L) sensitive 786-O cells (786-OS) for 48 h (sun) and in resistant 786-O cells (786-OR) in the presence of sunitinib (sun). (B) Determination of the percentage of dead 786-OS and 786-OR cells after incubation for 24 h the indicated combinations of drugs (sunitinib (sun) 2.5 μmol/L; LLOM (L) 1 μmol/L; elacridar (E) 5 μmol/L). *, P < 0.05; ***, P < 0.001.
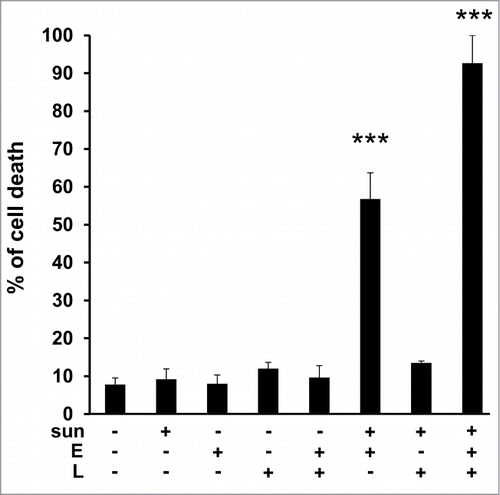
Elacridar and LLOM alone or in combination had no effect on TFE3 cell viability. Elacridar was more active in the presence of a concentration of 2.5 μmol/L sunitinib, (55% cell death) but this was not the case for the LLOM and sunitinib combination. As for 786-OR cells, massive TFE3 cell death was obtained with the triple combination ().
Figure 8. Primary tumor cells derived from a patient who progressed on sunitinib, were sensitive to sunitinib when in the presence of elacridar or elacridar and LLOM. Determination of the percentage of viable/dead TFE3 cells after incubation for 24 h with the indicated combinations of drugs (sunitinib (sun) 2.5 μmol/L; LLOM (L) 0.2 μmol/L; elacridar (E) 1 μmol/L). *, P < 0.05; ***, P < 0.001.
Proteasome inhibitors induced the death of sunitinib-resistant cells
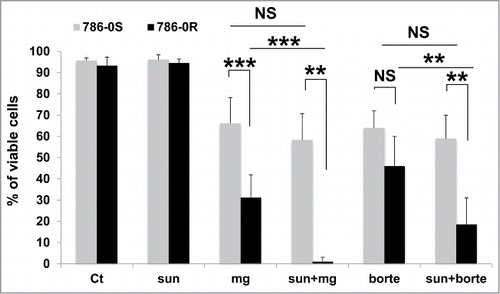
Autophagy and the ubiquitin-proteasome system are 2 linked mechanisms leading to the degradation of abnormal proteins and the recycling of amino acids. These 2 mechanisms compensate for each other when one is inhibited.Citation19 Hence, we speculate that incomplete autophagy resulted in enhanced proteasomal activity. As a consequence, sunitinib and inhibitors of the proteasome may have additional effects on cell death. We observed that MG132, a proteasome inhibitor, or bortezomib, which is approved for the treatment of multiple myeloma,Citation20 induced the death of sensitive and resistant cells. However, proteasome inhibitors combined with sunitinib induced a higher level of mortality of 786-OR cells than that induced by the individual compounds (). A higher level of mortality was also observed for RCC10S, RCC10R, and CC cells when proteasome inhibitors, and especially bortezomib, was combined with sunitinib (Fig. S11).
Figure 9. Proteasome inhibitors induced the death of cells resistant to sunitinib. The proteasome inhibitors MG132 (mg, 10 μmol/L) or bortezomib (borte, 5 μmol/L) alone or in combination with sunitinib (sun, 2.5 μmol/L) decreased the viability of 786-OS and 786-OR cells after incubation for 24 h. **, P < 0.01; ***, P < 0.001, NS, nonsignificant.
Discussion
Most of the current research in the field of antiangiogenesis drugs has focused on the adaptation of the endothelial cells to these drugs, which target VEGFA or its receptors.Citation21 However, it has been shown that other members of the VEGF family especially VEGFC are induced after exposure to anti-VEGFA antibodies.Citation22 Research has also concentrated on modifications to the genetic program of tumor cells exposed to antiangiogenesis drugs, which lead to the expression of redundant proangiogenesis factors or the ability to migrate.Citation23,24 Different reports have described the acquisition of a MET-dependent aggressive phenotype associated with sunitinib treatment, in particular in animal models.Citation25–27 However, it is difficult to address this modification in patients since sunitinib is administered mainly after radical nephrectomy to challenge metastatic sites and metastatic cells are generally not sampled for ethical purposes. Dissemination of mRCC cells via the lymphatic system has also been reported but again in animal models.Citation28,29 The role of the tumor microenvironment in the adaptation to treatments has also been addressed.Citation30
The major question was how to define the best treatment among the different ones that have been approved since 2007. A great hope was to use mouse avatars xenografted with fragments of a tumor to test the available treatments in vivo. With this experimental model, a correlation between the capacity of a tumor to develop in nude mice and tumor aggressiveness has been described. This model is also used to test the efficacy of a new compound.Citation31 The results of many groups including ours show that the growth of these tumor fragments in mice takes 3 to 6 mo. Hence, testing various treatments with this method is not compatible time-wise for a rapid therapeutic decision between surgical removal of the tumor and the beginning of the treatment.Citation32 Tumor-derived slice cultures of head and neck cancers have also been used to test the sensitivity to targeted therapies, a technique that may also be used for mRCC, but the procedure needs to be improved.Citation33 Our recent study shows a good correlation between the response of the patient and the sensitivity to a FDA (food and drug administration)-approved drug.Citation2 We postulate that the antiangiogenesis treatment equivalently targets the blood vessels constituted of normal cells from one patient to another. However, we believe that tumor cell genetic plasticity was at the origin of the variation in tumor response. Our procedure consisted of testing cell survival and death in response to available drugs on isolated tumor cells grown in a specific culture medium. This technique is rapid and the procedure can be performed within one month after surgery. It may allow assistance in a therapeutic decision considering that the response to a given treatment on primary cells that represents tumor heterogeneity will reflect the general effect on patients.Citation34
Although all the aspects that lead to acquired resistance are of major importance, to become resistant the cells require chronic exposure to the drugs. So it would take several months before genetically modified tumor cells emerge that survive antiangiogenesis treatment.
The ability of cells to compartmentalize the drug in subcellular organelles to avoid accessibility to its target has not been examined in detail. Several reports have shown that certain weakly basic compounds (i.e. daunorubicin, doxorubicin) with pKa values near neutrality are selectively sequestered into lysosomes of multidrug-resistant cell lines. Alternatively, other weakly basic compounds, also with pKa near neutrality, specifically accumulate within mitochondria (i.e., rhodamine 123).Citation35 In our case, we have observed lysosomal sequestration of sunitinib but no accumulation in mitochondria, as previously shown.Citation9
Lysosomal trapping reduces the activity of sunitinib, since its targets, the kinase domain of tyrosine kinase receptors are located in the cytoplasm. This mechanism has been described in chronic myeloid leukemia for which agents that destabilize lysosomes revert resistance to imatinib.Citation18 Among the different drugs that have obtained FDA and/or EMA (Food and Drug Administration and/or European Medicines Agency) approval for the treatment of mRCC, in addition to sunitinib, axitinib and dovitinib can be protonated at physiological pH and subsequently trapped in the lysosomes. Hence, resistance mechanisms equivalent to that described herein for sunitinib may be the cause of reduced efficacy of these drugs. Pazopanib, another ATP mimetic approved for the treatment of mRCC is the only drug that cannot be protonated and trapped in the lysosomes, hence not concerned by this mechanism of resistance. However, sunitinib and pazopanib show the same overall survival,Citation36 but pazopanib is preferred by physicians and patients mainly for its better quality of life.Citation37 Several reports have previously shown that drugs that are lysosomotropic shared certain physicochemical properties, possessing a ClogP>2 and a basic pKa between 6.5 and 11, predictably influenced their intracellular localization.Citation38 Sunitinib enters perfectly into this category with a ClogP = 5.2 and a basic pKa = 8.95. We also observed that the amine group of sunitinib, added to improve the solubility of the drug is responsible for the high pKa value. Thus, the synthesis of an analog of sunitinib devoid of this amine group may prevent its accumulation in lysosomes.
Sequestration of chemotherapeutic agents in lysosomes is largely due to their lysosomotropic properties but sequestration within lysosomes may also be dependent on the ABC transporter activity.Citation16,39 We observed an increase in the expression of the ABCB1 transporter after sunitinib treatment, but the regulatory mechanism implicated is unknown. Preliminary experiments suggest that the initiation of autophagy induces ATF4 (activating transcription factor 4) expression. ATF4 is a major transcription factor implicated in the adaptation to nutrient stress of tumor cells.Citation40 Moreover, ATF4 has also been implicated in resistance to cisplatin and cells overexpressing ATF4 showed multidrug resistance.Citation41 Hence, ATF4 may be the driver of a transcriptional program leading to expression of ABCB1, as previously shown.Citation42,43
Moreover, accumulation of ABCB1 may be due to a lack of its degradation. Several membrane proteins, including receptors and transporters, recycle to the plasma membrane through the recycling endosomal system. Some cargo proteins sort cell membranes and discarded proteins into internal luminal vesicles of multivesicular bodies (early endosomes), and mature multivesicular bodies (late endosomes) that can fuse with lysosomes for proteolysis by lysosomal enzymes. In the context of this study, the lysosomal degradation pathway is impaired because of the modification of the lysosomal pH and could explain the decrease in ABCB1 degradation and its subsequent accumulation. Similar consequences were observed with CHL treatment, which resulted in NOTCH1 accumulation due to a decrease in the activity of lysosomes.Citation44
Lysosomal sequestration is rapid, occurring as soon as the drug is in contact with the target cells, and does not modify the genetic program. Recent studies have also demonstrated that numerous cancer cells have defective acidification of their lysosomes. Hence, lysomotropic agents would be in contact with their targets in the cytoplasm of cancer cells devoid of lysosome trapping.Citation45 This elegant approach would limit toxicity to normal cell and would concentrate the cytotoxic or cytostatic effects on tumor cells. However, we showed that the acidification of the lysosomes of mRCC cells was not defective. We observed that the TFE3 cells were resistant to a high concentration of sunitinib (IC50 = 10 μmol/L). However, we did not observe an increase in the number of lysosomes (LysoTracker Red labeling) in TFE3 cells compared to 786–0 cells. The pH of lysosomes dictates the predicted degree of lysosomal accumulation of sunitinib; the greater the lysosome-to-cytosol pH gradient the greater the extent of lysosomal sequestration. As long as the pH gradient is maintained, significant accumulation of the drug is possible. The proton pump, the vacuolar-type (V-) ATPase, which is located on the lysosomal membrane, maintains acidification of lysosomes. Tumor cells with drug resistance exhibit an increase in V-ATPase activity, which may explain the resistance to sunitinib of TFE3 cells.Citation46,47
To prevent lysosomal trapping and avoid export of the drug out of the cells, we used lysosomal destabilizing agents and inhibitors of ABCB1. This combination was very efficient in promoting cell death of cancer cell lines and cancer cells derived from a patient who progressed on sunitinib. The recapitulative schema we propose to prevent sunitinib resistance is shown in . Lysosome stabilizing agents are far from entering into the clinic, because of major toxic effects whereas clinical assays using inhibitors of ABC transporters are ongoing.Citation48 Moreover, we found that proteasome inhibitors induced strong tumor cell death especially on cells resistant to sunitinib. As sunitinib and proteasome inhibitors have independent targets, the toxic effects should be manageable. In silico analysis of online-available microarrays highlighted a cluster of proteasome-associated genes that are overexpressed in primary and mRCC but also in paired pulmonary metastasis (Fig. S12A, Table S1, Supplemental Materials and Methods).Citation49,50 The proteins encoded by these genes comprised a subset of the proteasome β subunits that affect the generation of peptides to promote efficient antigen recognition (PSMB8/9/10; proteasome [prosome, macropain] subunit, β type, 8/9/10),Citation51 and a cellular regulator of proteasome formation and of proteasome-mediated antigen processing (PSMF1; proteasome [prosome, macropain] inhibitor subunit 1 [PI31]).Citation52 A slight increase in expression (1.4 [PSMB8, PSMB9, PSMB10] to fold2- [PSMF1] above the median) of each gene was associated with a decrease in overall survival (OS) with significant P values (P = 0.035 for PSMB8; 0.0006 for PSMB9; 0.018 for PSMB10; 0.036 for PSMF1) as revealed by data analysis at cbioportal.Citation53,54 Moreover, overexpression of the genes of the cluster were indicative of both disease free survival (P = 0.0008) and overall survival is much more decreased for patients that overexpressed the different genes of the cluster (P = 0.0002). Overexpression of the genes of the cluster was also indicative of disease free survival for non metastatic patients (P = 0.007) and of overall survival for metastatic patients (0.006) (Fig. S12B). This in silico analysis clearly showed the prognostic significance of specific proteasome-associated genes. It corroborated our "in cellulo" analysis for the relevance of association of sunitinib and proteasome inhibitors.
Figure 10. Description of the different phases of events justifying a combinatorial approach for treatment. (A) Maximal resistance (+++) is mediated by lysosomal trapping of sunitinib (yellow circles in lysosomes). The presence of autolysosomes (green circles) is indicative of incomplete autophagy. The proteasome degrades misfolded proteins and participates in recycling of amino acids. (B) The lysosome destabilizing agent Leu-Leu-O-Methyl (LLOM) prevents the trapping of sunitinib in lysosomes so the drug is localizes to the cytoplasm but is "taken in charge" by the ABCB1, which transport the drug out of the cell thus leading to intermediate resistance (++). (C) Maximal sensitivity to sunitinib can be obtained by 1) by destabilization of lysosomes with LLOM combined with inhibitors of ABC transporters (elacridar) or 2) with proteasome inhibitors (MG132 [mg] or bortezomib [borte]).
![Figure 10. Description of the different phases of events justifying a combinatorial approach for treatment. (A) Maximal resistance (+++) is mediated by lysosomal trapping of sunitinib (yellow circles in lysosomes). The presence of autolysosomes (green circles) is indicative of incomplete autophagy. The proteasome degrades misfolded proteins and participates in recycling of amino acids. (B) The lysosome destabilizing agent Leu-Leu-O-Methyl (LLOM) prevents the trapping of sunitinib in lysosomes so the drug is localizes to the cytoplasm but is "taken in charge" by the ABCB1, which transport the drug out of the cell thus leading to intermediate resistance (++). (C) Maximal sensitivity to sunitinib can be obtained by 1) by destabilization of lysosomes with LLOM combined with inhibitors of ABC transporters (elacridar) or 2) with proteasome inhibitors (MG132 [mg] or bortezomib [borte]).](/cms/asset/76bcf1f6-9932-460d-9a3b-202bc575d8aa/kaup_a_1085742_f0010_c.gif)
The resistance to any targeted therapies is a constant debate between the presence of mutations within the primary tumor and acquired mutations under the selection pressure induced by the treatment. Tumor heterogeneity constitutes a major problem for defining personalized treatment. For mRCC, genomic sequencing highlighted an evolution of the metastatic niche that may refine tumor progression.Citation55 As analyzed by cbioportal, proteasome genes were highly informative as prognosis markers of poor survival of nonmetastatic and metastatic patients while ATF4 and ABCB1 were not. A biopsy of metastases is rarely possible because it would threaten patients' life. However, if it is possible, it would be interesting to test in metastases that become refractory to sunitinib treatment if ABCB1, ATF4 and PSMB8, PSMB9, PSMB10, and PMSF1 are upregulated compared to the primary tumor hence defining them as markers of resistance as suggested by our study.
Taken together, our study highlighted a primary mechanism of resistance to the major antiangiogenic compound used in the treatment of mRCC. Having deciphered this mechanism, we can now propose relevant therapeutic combinations that deserve testing in preclinical models but also putatively in phase I clinical trials.
Materials and Methods
Materials
Sunitinib and bortezomib came from residual materials given to patients (Center Antoine Lacassagne, Nice, France) and prepared as 2.5 mmol/L and 6.5 mmol/L stock solutions in dimethyl sulfoxide (Sigma, 472301) and stored at -20°C. CHL (C6628), elacridar (SML0486) and LLOM (L7393) were purchased from Sigma. MG132 was purchased from Calbiochem (474790) Anti-LC3 antibody (5F10) was obtained from Nanotools (0231–100/LC3–5F10). Anti-LAMP1 (H4A3, sc-20011) and anti-EEA1 (N-19, sc-6415) were from Santa Cruz Biotechnology, anti SQSTM1 was from BD Bioscience (610833), CTSB (Ab-1, IM27L) was purchased from Merck, anti-LAMP2 was from Abcam (H4B4, ab25631), anti-NAA10/ARD1 antibodies were produced and characterized in our laboratory,Citation56 anti-actin (I-19, sc-1616) was from Santa Cruz Biotechnology, anti-phospho-AKT1 (Ser473; 9271S), anti-AKT1 (9272), anti-phospho-MAPK1/3 (Thr185/Tyr187 and Thr202/Tyr204; 4370), anti-MAPK1/3 (137F5; 4695) and anti-ABCB1 (EY7B, 133442S) antibodies were all obtained from Cell Signaling Technology. HBSS was from Life Technologies (14025092).
Cell culture
Human 786–0 cells were purchased from the American Tissue Culture Collection (ATCC→ CRL-1932™). RCC10 cells were a kind gift from W.H. Kaelin (Dana-Farber Cancer Institute, Boston, MA) and were used in one of our published studies.Citation57 RCC cells were grown in DMEM (Life Technologies, 61965–026) supplemented with 7% fetal calf serum at 37°C in a humidified atmosphere containing 5% CO2. For HBSS experiments, cells were preincubated in HBSS for 30 min before sunitinib treatment for 24 h for the determination of cell viability. For clonogenic assays, cells were incubated for 7 d in fresh medium after the same procedure. Resistant cells were obtained by chronic exposure to increasing concentrations of sunitinib up to 8 μmol/L. An INVIVO2 200 workstation (Ruskinn Technology Biotrace International Plc, Sanford, FL, USA) set at 1% oxygen, 94% nitrogen and 5% carbon dioxide was used for hypoxic conditions.
Growth curves and cell viability
Cells were seeded in 6-well dishes and transiently treated with sunitinib the following day. Cells were next detached from d 2 to 6 and counted with a Coulter counter (Beckman, Pasadena, CA, USA) in duplicate to assess cell proliferation. Cell viability and cell death was assessed using the ADAM-MC apparatus (NanoEnTek, Guro-gu, Seoul, Korea) based on fluorescent propidium iodide staining according to the manufacturer's instructions.
Colony formation assay
RCC cells (500 cells per condition) were treated or not with sunitinib. Colonies were detected after 10 d of culture. Cells were then washed, fixed at room temperature for 20 min with 3% paraformaldehyde (PFA; Electron Microscopy Sciences, 15713) and colored by crystal violet (Sigma, C3886).
Kinetics of cell migration
Cell migration in real time was monitored by using the xCELLigence Real-Time Cell Analyzer (RTCA) DP Instrument equipped with a CIM-plate 16 (Roche, Indianapolis, IN, USA).
Each well of the plate is composed of upper and lower chambers separated by a microporous membrane. Migration was measured as the relative impedance change (cell index) across microelectronic sensors integrated into the bottom side of the membrane. TenCitation4 cells were added in triplicate to the upper chambers. Migration and invasion were monitored every min for 48 h. For quantification, the cell index at the indicated time points was averaged from 3 independent measurements.
Immunoblotting
Cells treated with sunitinib and/or exposed to pharmacological inhibitors, were lysed in buffer containing 3% SDS (Euromedex, EU0660), 10% glycerol, 0.825 mM Na2HPO4. Samples (30 μg) were separated by 10% SDS-PAGE, transferred onto a PVDF membrane (Immobilon, Millipore, IPVH00010) and then exposed to the appropriate antibodies: anti-LC3, anti-SQSTM1, anti-LAMP1, anti-NAA10, anti-CTSB or anti-actin. Proteins were visualized with the ECL system using horseradish peroxidase-conjugated anti-rabbit (W4011) or anti-mouse (W4021) secondary antibodies (Promega).
Subcellular colocalization studies
Cells were incubated with sunitinib and LysoTracker Red DND-99 (Invitrogen, L7528) or LysoSensor Green DND-153 (Invitrogen, L7534). Viable cells were imaged in real time with EVOS Cell Imaging Systems (Life Technologies, Carlsbad, CA, USA).
Immunofluorescence
RCC cells seeded on glass coverslips (150,000 cells for 24 h or 60,000 cells for 48 h) in 6-well dishes were treated or not with sunitinib. Twenty-four or 48 h after, cells were then washed, fixed at room temperature for 20 min with 3% paraformaldehyde and permeabilized with phosphate-buffered saline (PBS; Euromedex, ET330-A) containing 0.2% Triton X-100 (Amresco, 0694–1L) for 2 min before being exposed to anti-LC3, anti-SQSTM1, anti-LAMP1, anti-LAMP2, anti-EEA1 or anti-ABCB1 (Sigma, P7965) for 1 h at room temperature. Cells were washed 3 times with PBS, and then incubated for 1 h at room temperature with 1:1000 dilution anti-mouse or anti-rabbit Alexa Fluor 488- (A21207) or Alexa Fluor 594-labeled (A21203) secondary antibody (Invitrogen, Life Technologies) and mounted using Mountant Permafluor (Thermo Scientific, TA-030-FM). Fluorescence images were examined and collected under a DeltaVision Microscopy Imaging System (GE Healthcare/Life Technologies, Carlsbad, CA, USA).
Subcellular fractionation
Subcellular fractionation was performed using a proteo-extract subcellular proteome extraction kit according to the manufacturer's instructions (Calbiochem, 539790).
Flow cytometry– cell cycle distribution
Cells were trypsinized, washed, and resuspended in cold 70% ethanol overnight. After 2 washes with PBS, cells were resuspended in propidium iodide (40 μg/ml; Sigma, P4170) containing ribonuclease A (10 μg/ml; Sigma, R4642) for 15 min at room temperature and were analyzed using a fluorescence-activated cell sorter (BD healthcare FACSCALIBUR, analyzer, San Jose, CA, USA).
Transmission and scanning electron microscopy
For ultrastructural analysis, cells were fixed in 1.6% glutaraldehyde (Electron Microscopy Sciences, 16210) in 0.1 M phosphate buffer, rinsed in 0.1 M cacodylate buffer, postfixed for 1 h in 1% osmium tetroxide (Electron Microscopy sciences, 19170) and 1% potassium ferrocyanide (Sigma, 14459–95–1) in 0.1 M cacodylate buffer (Sigma, 124–65–2) to enhance the staining of membranes. Cells were rinsed in distilled water, dehydrated in alcohols and lastly embedded in epoxy resin (Sigma, 45345). Contrasted ultrathin sections (70 nm) were analyzed under a JEOL 1400 transmission electron microscope (JEOL Europe, Croissy sur Seine, France) mounted with a Morada Olympus CCD camera (Olympus-SIS Europe).
CTSB activity
RCC cells treated with sunitinib for 24 h were lysed for 30 min at 4°C in lysis buffer (400 mmol/L Na-phosphate, pH 6, 150 mmol/L NaCl, 4 mmol/L ethylene-diaminetetraacetic acid, 1 mmol/L phenylmethylsulfonyl fluoride (Sigma, 78830), 10 μg/ml aprotinin (Euromedex, A162-A) and 1% Triton X-100 (Sigma, T9284) and lysates were cleared at 10,000 g for 15 min at 4°C. Each assay (in quadruplicate) was performed with 50 μg of protein prepared from control or sunitinib treated cells. Briefly, cellular extracts were incubated in a 96-well plate, with 60 μm of z-RR-AMC (7-amino-4-methylcoumarin; Peptide Institute, 3123-v) as substrate for various times at 37°C. The CTSB activity was measured by following the emission at 460 nm (excitation at 390 nm) in the presence or absence of 1 μm of CA-074Me (an inhibitor of CTSB activity; Calbiochem, 205531) Enzyme activities were expressed in arbitrary units per mg of protein.
Statistical analysis
Statistical significance and P values were determined by the 2-tailed Student t test.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
1085742_supplemental_files.zip
Download Zip (6 MB)Acknowledgment
We thank Dr M Christiane Brahimi-Horn for editorial assistance and Mr Benoit Front for the detection of sunitinib in experimental tumors.
Funding
This work was supported by the French Association for Cancer Research (ARC), the Fondation de France (SG and MD financial supports), the French National Institute for Cancer Research (INCA), the “Conseil Général des Alpes Maritimes,” the association Monégasque “Cordons de Vie” (www.cordonsdevie.com) and Novartis (Prime award for SG). This work was performed using the microscopy (PICMI) and cytometry (CYTOMED) facilities of IRCAN. The materials of CytoMed were supported by the Conseil Général 06, the FEDER, the Ministère de l'Enseignement Supérieur, the Région Provence Alpes-Côte d'Azur and the INSERM.
Related Research Data
References
- Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Oudard S, Negrier S, Szczylik C, Pili R, Bjarnason GA, et al. Overall survival and updated results for sunitinib compared with interferon alfa in patients with metastatic renal cell carcinoma. J Clin Oncol 2009; 27:3584-90; PMID:19487381; http://dx.doi.org/10.1200/JCO.2008.20.1293
- Grepin R, Ambrosetti D, Marsaud A, Gastaud L, Amiel J, Pedeutour F, Pages G. The relevance of testing the efficacy of anti-angiogenesis treatments on cells derived from primary tumors: a new method for the personalized treatment of renal cell carcinoma. PLoS ONE 2014; 9:e89449; PMID:24676409; http://dx.doi.org/10.1371/journal.pone.0089449
- Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H. Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res 2009; 69:2506-13; PMID:19244102; http://dx.doi.org/10.1158/0008-5472.CAN-08-4323
- Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis 2010; 13:1-14; PMID:20012482; http://dx.doi.org/10.1007/s10456-009-9160-6
- Escudier B, Bellmunt J, Negrier S, Bajetta E, Melichar B, Bracarda S, Ravaud A, Golding S, Jethwa S, Sneller V. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol 2010; 28:2144-50; PMID:20368553; http://dx.doi.org/10.1200/JCO.2009.26.7849
- Santoni M, Amantini C, Morelli MB, Liberati S, Farfariello V, Nabissi M, Bonfili L, Eleuteri AM, Mozzicafreddo M, Burattini L, et al. Pazopanib and sunitinib trigger autophagic and non-autophagic death of bladder tumour cells. Br J Cancer 2013; 109:1040-50; PMID:23887605; http://dx.doi.org/10.1038/bjc.2013.420
- Abdel-Aziz AK, Shouman S, El-Demerdash E, Elgendy M, Abdel-Naim AB. Chloroquine synergizes sunitinib cytotoxicity via modulating autophagic, apoptotic and angiogenic machineries. Chem Biol Interact 2014; 217:28-40; PMID:24751611; http://dx.doi.org/10.1016/j.cbi.2014.04.007
- Ikeda T, Ishii KA, Saito Y, Miura M, Otagiri A, Kawakami Y, Shimano H, Hara H, Takekoshi K. Inhibition of autophagy enhances sunitinib-induced cytotoxicity in rat pheochromocytoma PC12 cells. J Pharmacol Sci 2013; 121:67-73; PMID:23269235; http://dx.doi.org/10.1254/jphs.12158FP
- Gotink KJ, Broxterman HJ, Labots M, de Haas RR, Dekker H, Honeywell RJ, Rudek MA, Beerepoot LV, Musters RJ, Jansen G, et al. Lysosomal sequestration of sunitinib: a novel mechanism of drug resistance. Clin Cancer Res 2011; 17:7337-46; PMID:21980135; http://dx.doi.org/10.1158/1078-0432.CCR-11-1667
- Huang D, Ding Y, Li Y, Luo WM, Zhang ZF, Snider J, Vandenbeldt K, Qian CN, Teh BT. Sunitinib acts primarily on tumor endothelium rather than tumor cells to inhibit the growth of renal cell carcinoma. Cancer Res 2010; 70:1053-62; PMID:20103629; http://dx.doi.org/10.1158/0008-5472.CAN-09-3722
- Kaufmann AM, Krise JP. Lysosomal sequestration of amine-containing drugs: analysis and therapeutic implications. J Pharm Sci 2007; 96:729-46; PMID:17117426; http://dx.doi.org/10.1002/jps.20792
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Stanton MJ, Dutta S, Zhang H, Polavaram NS, Leontovich AA, Honscheid P, Sinicrope FA, Tindall DJ, Muders MH, Datta K. Autophagy control by the VEGF-C/NRP-2 axis in cancer and its implication for treatment resistance. Cancer Res 2013; 73:160-71; PMID:23149913; http://dx.doi.org/10.1158/0008-5472.CAN-11-3635
- Tang SC, Lagas JS, Lankheet NA, Poller B, Hillebrand MJ, Rosing H, Beijnen JH, Schinkel AH. Brain accumulation of sunitinib is restricted by P-glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) and can be enhanced by oral elacridar and sunitinib coadministration. Int J Cancer 2012; 130:223-33; PMID:21351087; http://dx.doi.org/10.1002/ijc.26000
- Yamagishi T, Sahni S, Sharp DM, Arvind A, Jansson PJ, Richardson DR. P-glycoprotein mediates drug resistance via a novel mechanism involving lysosomal sequestration. J Biol Chem 2013; 288:31761-71; PMID:24062304; http://dx.doi.org/10.1074/jbc.M113.514091
- Shukla S, Robey RW, Bates SE, Ambudkar SV. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos 2009; 37:359-65; PMID:18971320; http://dx.doi.org/10.1124/dmd.108.024612
- Puissant A, Dufies M, Raynaud S, Cassuto JP, Auberger P. Targeting lysosomes to eradicate imatinib-resistant chronic myelogenous leukemia cells. Leukemia 2010; 24:1099-101; PMID:20376083; http://dx.doi.org/10.1038/leu.2010.55
- Lamark T, Johansen T. Autophagy: links with the proteasome. Curr Opin Cell Biol 2010; 22:192-8.; PMID:19962293; http://dx.doi.org/10.1016/j.ceb.2009.11.002
- Einsele H. Bortezomib. Recent Results Cancer Res 2014; 201:325-45; PMID:24756802; http://dx.doi.org/10.1007/978-3-642-54490-3_20
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008; 8:592-603; PMID:18650835; http://dx.doi.org/10.1038/nrc2442
- Grau S, Thorsteinsdottir J, von Baumgarten L, Winkler F, Tonn JC, Schichor C. Bevacizumab can induce reactivity to VEGF-C and -D in human brain and tumour derived endothelial cells. J Neurooncol 2011; 104:103-12; PMID:21308397; http://dx.doi.org/10.1007/s11060-010-0480-6
- Giuliano S, Pages G. Mechanisms of resistance to anti-angiogenesis therapies. Biochimie 2013; 95:1110-9; PMID:23507428; http://dx.doi.org/10.1016/j.biochi.2013.03.002
- Ebos JM, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol 2011; 8:210-21; PMID:21364524; http://dx.doi.org/10.1038/nrclinonc.2011.21
- Sennino B, Ishiguro-Oonuma T, Wei Y, Naylor RM, Williamson CW, Bhagwandin V, Tabruyn SP, You WK, Chapman HA, Christensen JG, et al. Suppression of tumor invasion and metastasis by concurrent inhibition of c-Met and VEGF signaling in pancreatic neuroendocrine tumors. Cancer Discov 2012; 2:270-87; PMID:22585997; http://dx.doi.org/10.1158/2159-8290.CD-11-0240
- Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 2009; 15:232-9; PMID:19249681; http://dx.doi.org/10.1016/j.ccr.2009.01.021
- Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 2009; 15:220-31; PMID:19249680; http://dx.doi.org/10.1016/j.ccr.2009.01.027
- Cao Y, Hoeppner LH, Bach S, E G, Guo Y, Wang E, Wu J, Cowley MJ, Chang DK, Waddell N, et al. Neuropilin-2 promotes extravasation and metastasis by interacting with endothelial alpha5 integrin. Cancer Res 2013; 73:4579-90; PMID:23689123; http://dx.doi.org/10.1158/0008-5472.CAN-13-0529
- Sennino B, Ishiguro-Oonuma T, Schriver BJ, Christensen JG, McDonald DM. Inhibition of c-Met reduces lymphatic metastasis in RIP-Tag2 transgenic mice. Cancer Res 2013; 73:3692-703; PMID:23576559; http://dx.doi.org/10.1158/0008-5472.CAN-12-2160
- Ferrara N. Role of myeloid cells in vascular endothelial growth factor-independent tumor angiogenesis. Curr Opin Hematol 2010; 17:219-24; PMID:20308892
- Sivanand S, Pena-Llopis S, Zhao H, Kucejova B, Spence P, Pavia-Jimenez A, Yamasaki T, McBride DJ, Gillen J, Wolff NC, et al. A validated tumorgraft model reveals activity of dovitinib against renal cell carcinoma. Sci Transl Med 2012; 4:137ra75; PMID:22674553; http://dx.doi.org/10.1126/scitranslmed.3003643
- Karam JA, Zhang XY, Tamboli P, Margulis V, Wang H, Abel EJ, Culp SH, Wood CG. Development and characterization of clinically relevant tumor models from patients with renal cell carcinoma. Eur Urol 2011; 59:619-28; PMID:21167632; http://dx.doi.org/10.1016/j.eururo.2010.11.043
- Gerlach MM, Merz F, Wichmann G, Kubick C, Wittekind C, Lordick F, Dietz A, Bechmann I. Slice cultures from head and neck squamous cell carcinoma: a novel test system for drug susceptibility and mechanisms of resistance. Br J Cancer 2014; 110:479-88; PMID:24263061; http://dx.doi.org/10.1038/bjc.2013.700
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med 2012; 366:883-92; PMID:22397650; http://dx.doi.org/10.1056/NEJMoa1113205
- Duvvuri M, Gong Y, Chatterji D, Krise JP. Weak base permeability characteristics influence the intracellular sequestration site in the multidrug-resistant human leukemic cell line HL-60. J Biol Chem 2004; 279:32367-72; PMID:15181006; http://dx.doi.org/10.1074/jbc.M400735200
- Motzer RJ, Hutson TE, Cella D, Reeves J, Hawkins R, Guo J, Nathan P, Staehler M, de Souza P, Merchan JR, et al. Pazopanib versus sunitinib in metastatic renal-cell carcinoma. N Engl J Med 2013; 369:722-31; PMID:23964934; http://dx.doi.org/10.1056/NEJMoa1303989
- Escudier B, Porta C, Bono P, Powles T, Eisen T, Sternberg CN, Gschwend JE, De Giorgi U, Parikh O, Hawkins R, et al. Randomized, controlled, double-blind, cross-over trial assessing treatment preference for pazopanib versus sunitinib in patients with metastatic renal cell carcinoma: PISCES Study. J Clin Oncol 2014; 32:1412-8; PMID:24687826; http://dx.doi.org/10.1200/JCO.2013.50.8267
- Nadanaciva S, Lu S, Gebhard DF, Jessen BA, Pennie WD, Will Y. A high content screening assay for identifying lysosomotropic compounds. Toxicol In Vitro 2011; 25:715-23; PMID:21184822; http://dx.doi.org/10.1016/j.tiv.2010.12.010
- Kalayda GV, Wagner CH, Buss I, Reedijk J, Jaehde U. Altered localisation of the copper efflux transporters ATP7A and ATP7B associated with cisplatin resistance in human ovarian carcinoma cells. BMC Cancer 2008; 8:175; PMID:18565219; http://dx.doi.org/10.1186/1471-2407-8-175
- Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, Koumenis C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 2010; 29:2082-96; PMID:20473272; http://dx.doi.org/10.1038/emboj.2010.81
- Igarashi T, Izumi H, Uchiumi T, Nishio K, Arao T, Tanabe M, Uramoto H, Sugio K, Yasumoto K, Sasaguri Y, et al. Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene 2007; 26:4749-60; PMID:17297441; http://dx.doi.org/10.1038/sj.onc.1210289
- Hamdan AM, Koyanagi S, Wada E, Kusunose N, Murakami Y, Matsunaga N, Ohdo S. Intestinal expression of mouse Abcg2/breast cancer resistance protein (BCRP) gene is under control of circadian clock-activating transcription factor-4 pathway. J Biol Chem 2012; 287:17224-31; PMID:22396548; http://dx.doi.org/10.1074/jbc.M111.333377
- Zhu H, Xia L, Zhang Y, Wang H, Xu W, Hu H, Wang J, Xin J, Gang Y, Sha S, et al. Activating transcription factor 4 confers a multidrug resistance phenotype to gastric cancer cells through transactivation of SIRT1 expression. PLoS ONE 2012; 7:e31431; PMID:22363646; http://dx.doi.org/10.1371/journal.pone.0031431
- Maes H, Kuchnio A, Peric A, Moens S, Nys K, De Bock K, Quaegebeur A, Schoors S, Georgiadou M, Wouters J, et al. Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014; 26:190-206; PMID:25117709; http://dx.doi.org/10.1016/j.ccr.2014.06.025
- Duvvuri M, Konkar S, Hong KH, Blagg BS, Krise JP. A new approach for enhancing differential selectivity of drugs to cancer cells. ACS Chem Biol 2006; 1:309-15; PMID:17163760; http://dx.doi.org/10.1021/cb6001202
- Luciani F, Spada M, De Milito A, Molinari A, Rivoltini L, Montinaro A, Marra M, Lugini L, Logozzi M, Lozupone F, et al. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J Natl Cancer Inst 2004; 96:1702-13; PMID:15547183; http://dx.doi.org/10.1093/jnci/djh305
- Martinez-Zaguilan R, Lynch RM, Martinez GM, Gillies RJ. Vacuolar-type H(+)-ATPases are functionally expressed in plasma membranes of human tumor cells. Am J Physiol 1993; 265:C1015-29; PMID:8238296
- Falasca M, Linton KJ. Investigational ABC transporter inhibitors. Expert Opin Investig Drugs 2012; 21:657-66; PMID:22493979; http://dx.doi.org/10.1517/13543784.2012.679339
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005; 102:15545-50; PMID:16199517; http://dx.doi.org/10.1073/pnas.0506580102
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet 2003; 34:267-73; PMID:12808457; http://dx.doi.org/10.1038/ng1180
- Pamer E, Cresswell P. Mechanisms of MHC class I–restricted antigen processing. Annu Rev Immunol 1998; 16:323-58; PMID:9597133; http://dx.doi.org/10.1146/annurev.immunol.16.1.323
- Zaiss DM, Standera S, Kloetzel PM, Sijts AJ. PI31 is a modulator of proteasome formation and antigen processing. Proc Natl Acad Sci U S A 2002; 99:14344-9; PMID:12374861; http://dx.doi.org/10.1073/pnas.212257299
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:pl1; PMID:23550210; http://dx.doi.org/10.1126/scisignal.2004088
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; http://dx.doi.org/10.1158/2159-8290.CD-12-0095
- Gerlinger M, Horswell S, Larkin J, Rowan AJ, Salm MP, Varela I, Fisher R, McGranahan N, Matthews N, Santos CR, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet 2014; 46:225-33; PMID:24487277; http://dx.doi.org/10.1038/ng.2891
- Bilton R, Mazure N, Trottier E, Hattab M, Dery MA, Richard DE, Pouyssegur J, Brahimi-Horn MC. Arrest-defective-1 protein, an acetyltransferase, does not alter stability of hypoxia-inducible factor (HIF)-1alpha and is not induced by hypoxia or HIF. J Biol Chem 2005; 280:31132-40; PMID:15994306; http://dx.doi.org/10.1074/jbc.M504482200
- Grepin R, Guyot M, Giuliano S, Boncompagni M, Ambrosetti D, Chamorey E, Scoazec JY, Negrier S, Simonnet H, Pages G. The CXCL7/CXCR1/2 axis is a key driver in the growth of clear cell renal cell carcinoma. Cancer Res 2014; 74:873-83; PMID:24335961; http://dx.doi.org/10.1158/0008-5472.CAN-13-1267