
ABSTRACT
Most of the proteins that are specifically turned over by selective autophagy are recognized by the presence of short Atg8 interacting motifs (AIMs) that facilitate their association with the autophagy apparatus. Such AIMs can be identified by bioinformatics methods based on their defined degenerate consensus F/W/Y-X-X-L/I/V sequences in which X represents any amino acid. Achieving reliability and/or fidelity of the prediction of such AIMs on a genome-wide scale represents a major challenge. Here, we present a bioinformatics approach, high fidelity AIM (hfAIM), which uses additional sequence requirements—the presence of acidic amino acids and the absence of positively charged amino acids in certain positions—to reliably identify AIMs in proteins. We demonstrate that the use of the hfAIM method allows for in silico high fidelity prediction of AIMs in AIM-containing proteins (ACPs) on a genome-wide scale in various organisms. Furthermore, by using hfAIM to identify putative AIMs in the Arabidopsis proteome, we illustrate a potential contribution of selective autophagy to various biological processes. More specifically, we identified 9 peroxisomal PEX proteins that contain hfAIM motifs, among which AtPEX1, AtPEX6 and AtPEX10 possess evolutionary-conserved AIMs. Bimolecular fluorescence complementation (BiFC) results verified that AtPEX6 and AtPEX10 indeed interact with Atg8 in planta. In addition, we show that mutations occurring within or nearby hfAIMs in PEX1, PEX6 and PEX10 caused defects in the growth and development of various organisms. Taken together, the above results suggest that the hfAIM tool can be used to effectively perform genome-wide in silico screens of proteins that are potentially regulated by selective autophagy. The hfAIM system is a web tool that can be accessed at link: http://bioinformatics.psb.ugent.be/hfAIM/.
Introduction
Macroautophagy (hereafter referred to as autophagy) is a highly conserved biological process in eukaryotes, which mainly functions in the degradation of macromolecules in the lytic compartment.Citation1-3 One of the central core proteins of the autophagy machinery is Atg8. Atg8 binding to specific target proteins is often, though not always, mediated by a conserved motif, the Atg8-interacting motif (AIM), on the target protein.Citation4,5 The core AIM motif is comprised of 4 amino acids, defined as F/W/Y-X-X-L/I/V,Citation6 in which ‘X’ represents any amino acid. Notably, structural analysis suggested a striking bias toward negatively charged amino acids present within or upstream of the core AIM.Citation4,7,8 Therefore, it has been proposed that the acidic amino acid Asp (D) and Glu (E), and potentially also Ser (S) and Thr (T) that generate negative charges when phosphorylated, can improve the strength of binding of Atg8 to the AIMs.Citation4,9 Furthermore, it has been hypothesized that the closer the acidic or phosphorylated amino acids are to the core AIM or their presence in the 2 X positions within the core AIM, the higher is the fidelity of binding of Atg8 to the AIMs.Citation7
Based on the degenerate consensus sequence of AIMs, it is possible to use bioinformatics tools to look for potential Atg8-interacting proteins by searching for AIM motifs followed by verification of their binding to Atg8 by experimental methods, such as yeast 2-hybrid and bimolecular fluorescence complementation (BiFC). Indeed, 2 bioinformatics tools that identify consensus AIMs in proteins were previously developed, the first being reported by our laboratoryCitation10 and the second, iLIR, reported by Kalavari and associates.Citation11 Our methodCitation10 took into account the contribution of acidic amino acids to the fidelity of binding of Atg8 to the AIM. The iLIR system defines an AIM, termed xLIR, based on a regular expression pattern that is based on the sequences of a set of verified AIMs and the 2 amino acids that precede it.Citation11 Additionally, iLIR scores xLIRs against a custom position-specific scoring matrix (PSSM) and identifies potentially disordered subsequences with protein interaction potential overlapping with detected xLIR-motifs (ANCHOR).Citation12,13 Interestingly, the regular pattern of xLIR, that is based on experimentally determined Atg8-interacting regions, does not contain positively charged amino acids in the minus 1 and plus 1 positions upstream or downstream of the F/W/Y sequence of the AIM, strengthening the notion that the presence of positively charged amino acids in these 2 positions may hinder the binding of Atg8 to the AIM.
The peroxisome is a highly dynamic organelle involved in metabolism, development and response to stresses, and its homeostasis is regulated by selective autophagy.Citation14-16 Recent work indicates that peroxisomes and peroxisomal proteins accumulate in autophagy (atg) mutants.Citation17,18 Moreover, the Atg8 protein frequently colocalizes with peroxisome aggregates, indicating that peroxisomes are selectively degraded by autophagy.Citation17 Therefore, it has been proposed that autophagy apparently regulates the homeostasis of peroxisomes through the degradation of certain peroxisomal proteins.Citation16,19 However, it is still not clear which peroxisome proteins are selectively turned over by autophagy. Peroxin (PEX) proteins are peroxisomal proteins that serve multiple functions in the operation of this organelle.Citation20 Interestingly, a recent report showed that a G-to-E point mutation in the Arabidopsis PEX10 protein alters the shape of the peroxisome and that this mutant displays a dominant negative plant phenotype, which is highly similar to that of AtPEX10-knockout mutants.Citation21 These results suggest that the G-to-E mutation results in an irreversible degradation of AtPEX10 protein. Taking into account the key role of PEX10 in the peroxisome,Citation21,22 it is reasonable to speculate that PEX10 may be one of the candidate peroxisomal proteins degraded by autophagy. However, this hypothesis, as well as the identification of other peroxisomal proteins that are regulated by autophagy, still remains to be further determined.
In the present report, we present a bioinformatics approach, termed high fidelity AIM system (hfAIM), for in silico genome-wide identification of AIMs. Application of the hfAIM system facilitates a rapid identification of potentially interesting proteins that are associated with autophagy, as well as studying the network regulation of autophagy. As a test case, we utilized hfAIM to identify potential AIMs in PEX proteins from multiple model organisms. Evolutionary conservation of the predicted AIMs was further used to refine the predictions. BiFC experiments were used to validate hfAIM predictions for Arabidopsis PEX proteins. Our results suggest that PEX6, PEX10 and likely also PEX1 contain functional AIMs and interact with Atg8, suggesting that autophagy regulates the homeostasis of peroxisomes through the degradation of specific PEX proteins. The hfAIM system is a web tool (link: http://bioinformatics.psb.ugent.be/hfAIM/), which allows users to upload FASTA files to scan for our 5-hfAIM motifs (as default) and add or remove motifs as they wish. The code is deposited in a github at https://gitlab.psb.ugent.be/thpar/hfAIM/blob/master/README.md.
Results
Estimation of the contribution of acidic amino acids to functional AIMs
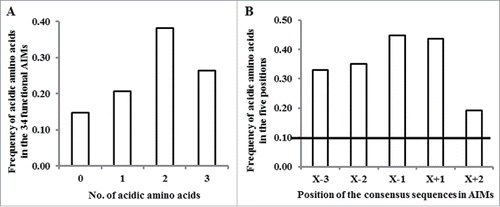
Atg8-interacting proteins often possess one or more functional Atg8-interacting motifs (AIMs), which are comprised of the core consensus sequence F/W/Y-X-X-L/I/V.Citation1,3,6 The presence of acidic amino acids either immediately upstream the F/W/Y sequence or at any of the 2 X positions between the F/W/Y and the L/I/V sequences appear to contribute to the fidelity of binding of Atg8 to the AIM.Citation7,23 Thus, it might be useful to consider a longer 6-amino acid X−2-X−1-F/W/Y-X+1-X+2-L/I/V motif for AIMs. Based on this degenerate sequence, we have previously developed a bioinformatics tool,Citation10 termed “canonical AIM” (cAIM), for identifying AIMs in a group of plant exocyst subunits, whose transport to the vacuole was suggested to require the autophagy apparatus.Citation24 Another more recently developed tool, iLIR,Citation10 also defines an AIM as a 6-amino acid motif, termed xLIR, based on the following degenerate amino acid sequence: [ADEFGLPRSK][DEGMSTV][WFY][DEILQTV][ADEFHIKLMPSTV][ILV]. As mentioned above, accumulating data suggested that the presence of acidic amino acids in any of the 2 “X” positions within the core AIM (namely X+1, X+2) or in any of the 2 “X” positions upstream to the F/W/Y sequence (namely X−2, X−1) may improve the binding efficiency of Atg8 to the AIM.Citation6,7 While looking further into the 36 verified functional AIMs collected from the literature (see detail in Table S1), we found that 29 functional AIMs contain one or more acidic amino acid (, Table S1). Of the remaining 7 AIMs, 4 AIMs contain at least one S residue, only one AIM possesses neither acidic amino acid nor S and T residues, and 2 functional AIMs are atypical AIMs (Table S1), which thereby were excluded from our following studies. Furthermore, the frequency of acidic amino acids at the degenerate 5 positions of the AIM motif (X−3, X−2, X−1, X+1 and X+2) is specifically higher than the percentage found in a random sequence of 5 amino acids (). These results suggest that introducing a requirement for negatively charged amino acids around the core consensus sequence of the AIM motif might improve the predictive power of bioinformatics tools.
Figure 1. The distribution of acidic amino acids in known functional AIMs. (A) The frequency of acidic amino acids present in the 34 functional AIMs (excluding the 2 atypical AIMs). (B) The frequency of acidic amino acids present in each of the 5 analyzed positions in the 34 functional AIMs. The horizontal line indicates the expected frequency of acidic amino acid in a random distribution (2/20 amino acids).
Generation of the hfAIM system
Based on preference for acidic amino acids in AIM motifs we developed a bioinformatics tool, hfAIM, for the prediction of AIM motifs in proteins. An hfAIM motif was defined as a motif containing at least 2 acidic amino acids in the X−3, X−2, X−1, X+1 or X+2 positions. Since the contribution of phosphorylation of S and T residues to AIM motif binding to Atg8 still needs to be verified experimentally, we did not introduce a bias toward these residues in our AIM prediction algorithm. This definition of the hfAIM motif resulted in 10 regular patterns (Fig. S1). Subsequently, we examined the distribution of these 10 regular patterns among the 19 functional AIMs in our data sets that contain at least 2 acidic amino acids (Table S1), excluding the 7 AIMs that contain only one acidic amino acid, 5 AIMs that do not contain any acidic amino acid and the 2 atypical AIMs. Interestingly, we found that 6 out of these 10 regular patterns were enough to fully cover the above 22 AIMs (A, B, C, F, H and I, see Table S1). In addition, only one functionally proved AIM, “PSHWPLI,” out of the 34 typical verified AIMs contains a positively charged amino acid His (H) at the X−1 position, and none contain positively charged amino acids at the X+1 position (Table S1), supporting the notion that positively charged amino acids have a negative effect on the binding of Atg8 to AIMs. Thus, we excluded putative AIMs containing positively charged amino acids at either the X−1 or the X+1 position (see below). Finally, according to the hypothesis that the closer the acidic amino acids are to the F/W/Y sequence of the core AIM, the higher is the fidelity of binding of Atg8 to these AIMs,Citation7 we excluded the “H” regular AIM pattern from the 6 regular expression patterns, resulting in only 5 regular expression patterns that meet the standard of an hfAIM motif ().
Figure 2. AIMs prediction by hfAIM. (A) The 5 regular patterns used for the analysis of AIMs in the hfAIM system. X, represent any amino acids. X−p, represent any amino acids except for the positively charged amino acids. (B) Comparison of the quality of the hfAIM and iLIR systems according to the statistical validation described by Kalvari et al.Citation10 “ACC,” accuracy of the AIM prediction; “Sens,” sensitivity of the AIM prediction; “Spec,” specificity of the AIM prediction; “BACC,” balanced accuracy of the AIM prediction. (C) Distribution of the number of AIMs in Arabidopsis proteins as predicted by the hfAIM and iLIR systems.
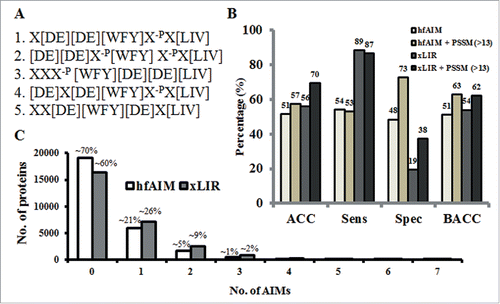
To validate the quality of our prediction of potentially functional hfAIMs, we used the statistical method reported by Kalvari and associatesCitation11 to compare our hfAIM predictions to the iLIR predictions of AIMs in the dataset of experimentally verified AIMs (Table S1). As shown in , the 2 approaches seem to give similar results in terms of both the accuracy and balanced accuracy of prediction of AIMs in this data set that compiles mostly human proteins. Nevertheless, these 2 systems displayed different sensitivity and specificity of AIM prediction. While the hfAIM system appears more powerful in specificity, the iLIR system is better at sensitivity (). Similarly to iLIR, the hfAIM algorithm is based on a regular expression pattern, a sequence of symbols and characters expressing a string or pattern to be searched for within a longer piece of sequence. Though searching for a regular expression pattern is a useful method for scanning large sequence data sets for sequences of interest, it sometimes suffers from high rate of false negative sequences. Adding a stricter criterion based on position-specific scoring matrix (PSSM) can improve the specificity of AIM prediction, as it has been previously shown for the iLIR.Citation11 PSSM is a tool that is used to score how close any sequence is to the collected sequences used to create the scoring matrix. Based on the training sequences, a score is assigned to the presence of a residue in each position in the sequence. A higher total score represents a sequence that is closer to the training sequences relative to other sequences of similar length. Thus, a genuine AIM motif is expected to have higher PSSM scores. As the hfAIM motif is defined as a 7-amino acid long motif, while the xLIR is a 6-amino acid long motif, PSSM scores calculated for the motifs predicted by each of the approaches cannot be directly compared. Therefore, PSSM values were calculated based on a 6-amino acid motif and the Kalvari et al.Citation11 custom PSSM for both the iLIR and hfAIM predictions of AIMs in our dataset (Table S1). The predictions were re-evaluated using a PSSM threshold value of 13 (i.e. only predicted sequences with PSSM value higher than 13 are taken into accountCitation11).
Although hfAIM still provides higher specificity relative to iLIR predictions, adding the PSSM predictor improves the specificity of both iLIR and hfAIM predictions leading to an improved balanced accuracy ().
The data set of verified AIMs used above (Table S1) contained mostly human proteins. To compare the predictive power of the iLIR and hfAIM approaches in plants, we first separately applied each of the 2 systems to identify putative AIMs in the entire Arabidopsis proteome (the in-house script for the hfAIM system is deposited in a github https://gitlab.psb.ugent.be/thpar/hfAIM/blob/master/README.md). Nearly 40% of the proteins of the entire Arabidopsis proteome contain AIMs according to the iLIR system, whereas about ∼30% of the Arabidopsis proteins contain AIMs as defined by the hfAIM system (). Next, we applied these 2 systems to identify AIMs in a dataset of 26 verified Arabidopsis Atg8-interacting proteins that were collected from the literature (see details in Table S2). We found differences between the predictions of the hfAIM and iLIR systems. Ten xLIR motifs derived from 9 proteins were identified by the iLIR system, while 16 hfAIM motifs derived from 9 proteins were identified by the hfAIM system. Among these Atg8-interacting proteins, 4 proteins were recognized as containing AIMs by both systems and only 2 identical AIM motifs were recognized by both systems (Table S2). When a PSSM threshold value of 13 was applied to the predictions (calculated according to Kalvari et al.Citation11), iLIR identified only 5 xLIR motifs in 4 proteins in the data set of verified Atg8-interacting proteins from Arabidopsis, while hfAIM identified 8 AIMs in 8 of the proteins. Taken together, these results suggest that hfAIM might be somewhat better suited for the prediction of AIMs in plants.
Global analysis of the role of autophagy-associated AIMs-containing proteins in multiple biological processes in plants
To broadly investigate a potential contribution of autophagy to various biological processes in plants, we performed a gene ontology (GO) enrichment analysis for the AIMs-containing proteins (hereafter termed as ACPs) identified by hfAIM in Arabidopsis plants. The GO enrichment analysis was conducted on groups of proteins containing increasing numbers of AIMs (Table S3). Since only 4 ACPs contained 7 to 8 AIMs and > 5000 proteins contained only one AIM, these 2 groups of protein genes were discarded from the GO enrichment analysis. The GO enrichment results suggest that ACPs are involved in multiple biological processes and molecular functions (See Table S3 for full list of GO enrichments). Notably, some GO terms associated with the ACPs were directly connected to autophagy-associated cellular catabolic process (GO:0044248), and proteolysis involved in cellular protein catabolic process (GO:0051603), suggesting that hfAIM is able to predict AIMs in proteins that are indeed likely to be involved in autophagy-related processes. The GO terms of other ACPs were related to metabolism, like gluconeogenesis (GO:0006094) and carbohydrate biosynthetic processes (GO:0016051), which is consistent with recent reports showing the comprehensive participation of autophagy in maintaining the homeostasis of cellular metabolism.Citation25-27 Surprisingly, we found that > 30% of the ACPs are related to the adenyl ribonucleotide binding (GO:0032559) and nucleobase-containing compound metabolic process (GO:0006139). In addition, one of the largest groups of ACPs is involved in the regulation of transcription, implying a new, relatively poorly understood role of autophagy in transcriptional control in plants. Furthermore, ACPs were also involved in signaling transduction, stress response and protein transport, as well as with other biological processes. Taken together, our results suggest that the hfAIM system enables an in-house genome-wide identification of ACPs, and thus facilitates the high-throughput analysis of the role of autophagy not only in plants, but also in various other organisms.
AtPEX10 interacts with Atg8 and this interaction requires a functional AIM motif predicted by hfAIM
To further verify the ability of the hfAIM system to identify functional AIMs, we used a group of peroxisome peroxin (PEX) proteins as a test case. The choice of PEX proteins was based on 4 independent reasons: (i) autophagy participates in the homeostasis of peroxisomes by a process termed pexophagy;Citation14-16 (ii) it has already been reported that AtPEX10-YFP fusion protein localizes to peroxisomes in tobacco leavesCitation28 and that a GFP-Atg8 fusion protein is a functional protein;Citation29 (iii) a G-to-E point mutation in the Arabidopsis AtPEX10 protein, which resulted in a peroxisome deficient phenotype,Citation21 occurs in a sequence predicted to be an AIM by the hfAIM system. The underlined G in the predicted AIM GEEYCDI sequence was mutated to E, introducing an extra acidic amino acid, that might improve the binding to Atg8 (); and (iv) additional analysis indicated that this natural GEEYCDI AIM (amino acids 93 to 99 in AtPEX10 sequence) is evolutionarily conserved (Fig. S3).
Figure 3. Identification of the functional AIM in AtPEX10. (A) Schematic representation of the AIM sequences predicted by either hfAIM or iLIR in AtPEX10 and their mutants. The referred G-to-E mutation is denoted by a star. (B to E) BiFC analysis was performed following transient coexpression of YN-Atg8f and different variants of PEX10-YC in N. benthamiana leaves. The interaction of wild-type PEX10 with Atg8f results in YFP fluorescence (in green) (B). No interaction was observed when YN-Atg8f was coexpressed with PEX10Y96A-YC (C), demonstrating that the GEEYCDI AIM motif is necessary and sufficient for Atg8f interaction. On the other hand, mutating the AIM motif identified by iLIR did not abolish PEX10 interaction with Atg8f (D), suggesting that this putative AIM is not required for the interaction. The G93E mutation in the hfAIM predicted motif retained the interaction between PEX10 and Atg8f (E). Chloroplast autofluorescence is shown in magenta. Bar: 20 µm.
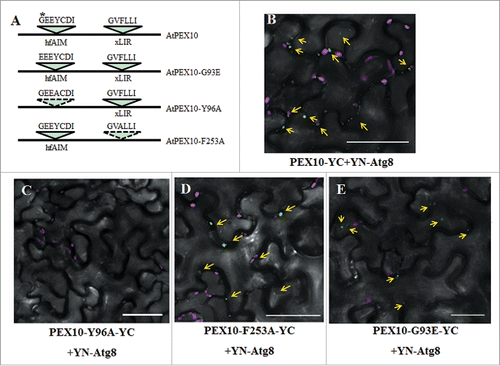
To look at the interaction of Atg8 with AtPEX10, we utilized the bimolecular fluorescence complementation (BiFC) assay as previously described.Citation30 Thus, we produced a C-terminal split YFP fusion protein with AtPEX10 (AtPEX10-YC), as well as an N-terminal split YFP fusion protein with Atg8 (YN-Atg8). Transient coexpression of AtPEX10-YC and YN-Atg8 in N. benthamiana leaves showed that AtPEX10 indeed interacts with Atg8 in vivo (), whereas the negative controls had no signals (Fig. S2A and B). Notably, the iLIR system identified a different AIM motif in AtPEX10, “GVFLLI” (amino acids 251 to 256 in AtPEX10 sequence), with a lower PSSM score compared to the predicted hfAIM motif (). Therefore, we were interested in testing which of these 2 potential AIMs in AtPEX10 is needed for the interaction with Atg8. To address this issue, we eliminated the potential AIMs in AtPEX10 by substituting the Tyr residue at position 96 (Y96) and the Phe residue at position 253 (F253) to Ala (A) (), respectively, and then generated the AtPEX10Y96A-YC and AtPEX10F253A-YC fusion proteins respectively. Using the BiFC assay, we transiently cotransformed N. benthamiana leaves with YN-Atg8 and either AtPEX10Y96A-YC or AtPEX10F253A-YC. The results indicated that while the point mutation in AtPEX10Y96A abolished its interaction with Atg8, the point mutation in AtPEX10F253A did not affect its Atg8 binding (). As a G-to-E point mutation in the hfAIM predicted AIM motif in AtPEX10 was previously shown to cause a peroxisome deficient phenotype,Citation21 we also generated an AtPEX10G93E-YC construct, and then transiently coexpressed this construct together with YN-Atg8 in tobacco leaves. The G93E mutation in AtPEX10 did not influence the interaction between AtPEX10 and Atg8 in the BiFC assay (). Together, we concluded that AtPEX10 interacts with Atg8 in vivo and that the AIM motif identified by hfAIM is the functional AIM in AtPEX10.
Table 1. Identification of putative AIMs in AtPEX proteins by hfAIM and iLIR.
Identification of putative AIMs in members of the Arabidopsis PEX family
Triggered by the above results, we employed both the hfAIM and the iLIR systems to elucidate whether additional AtPEX proteins also contain putative AIMs. The hfAIM and iLIR systems identified 20 AIMs in 13 AtPEX proteins, including the AtPEX10 protein (). Further analysis demonstrated that 9 AtPEX proteins contain 12 AIMs in total based on the hfAIM system, including AtPEX1, AtPEX3-2, AtPEX5, AtPEX6, AtPEX7, AtPEX10, AtPEX14, AtPEX17 and AtPEX19-1 (). Utilization of the iLIR system identified 12 xLIR motifs that were present in AtPEX1, AtPEX5, AtPEX10, AtPEX11C, AtPEX11D, AtPEX11E, AtPEX12 and AtPEX17 (). Among these AIMs, 3 hfAIM motifs that are present in AtPEX1 as well as one hfAIM motif in AtPEX17 were also predicted by the iLIR system. The rest of the AIMs differed between the hfAIM and iLIR systems (). AtPEX1, AtPEX5, AtPEX10 and AtPEX17 were predicted to contain AIMs by both systems, and are therefore considered as Atg8-interacting proteins with higher confidence.
Evolutionary conservation of PEX1, PEX6 and PEX10 AIMs
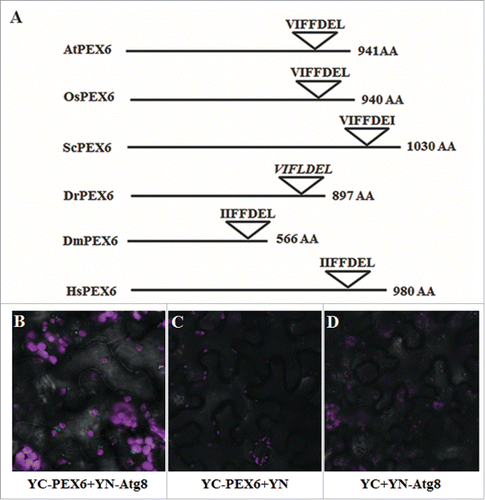
We were next interested to elucidate whether any of the 20 AIMs mentioned above have been evolutionarily conserved. To address this issue, we compared the sequences of the PEX proteins that were predicted to contain AIMs from multiple organisms represented in the Peroxisome DB 2.0 (www.peroxisomedb.org/).Citation31 Sequence alignment revealed that only PEX1, PEX6 and PEX10 contained highly conserved AIMs (Table S4). The hfAIM predicted sequence “GEEYCDI” in AtPEX10 (hfAIM pattern 1, see ) was found in the conserved PEX10 regions of 92% organisms that were analyzed (Fig. S3). The hfAIM predicted sequences “EDDWEVL” and “FEDFDSI” in AtPEX1 (hfAIM pattern 1, 2, 4 and 5, see ) were found in the conserved PEX1 regions of 76% or 89% organisms that were analyzed, respectively (Fig. S4), and the predicted hfAIM “VIFFDEL” sequence in AtPEX6 (hfAIM pattern 3, ) was found in the conserved PEX6 regions of 93% organisms that were analyzed (Fig. S5). Interestingly, 2 out of the 3 AIMs in AtPEX1 that were identified both by hfAIM and iLIR were evolutionarily conserved, while the third predicted AIM was not conserved among various species (, schematic representation of the sequences of only 6 representative organisms is presented). Moreover, the conserved AIM in AtPEX6 was only identified by hfAIM, while the iLIR detected no such motif in AtPEX6 ( and ). To determine the accuracy of the hfAIM prediction, we performed a BiFC assay to look at the potential interaction of Atg8 with AtPEX6 in vivo. As shown in , transient coexpression of YN-Atg8 and YC-AtPEX6 results in YFP fluorescence that is visualized as punctate spherical structures similar in size to plant peroxisomes, while neither the cotransformation of YC-AtPEX6 with YN nor YN-Atg8 and YC yielded any signal (). These results indicate that AtPEX6 indeed binds to Atg8 in vivo as predicted by hfAIM. In addition, a “LQLWDEL” sequence in AtPEX3-2 was also predicted by hfAIM as a potential AIM motif (hfAIM pattern 3, ), and this pattern appeared in 56% organisms that were analyzed (Fig. S6). With respect to the rest of the AIMs identified in the other PEX proteins by either the hfAIM or iLIR systems, all of these AIMs were not conserved in evolution (Fig. S7 to S11 and Table S4). Taken together, our results suggest that AtPEX6 and AtPEX10, and likely also AtPEX1 and AtPEX3-2 interact with Atg8 in planta. Furthermore, the autophagy mechanism underlying the degradation of these AtPEX proteins is apparently highly evolutionary conserved.
Figure 4. Evolutionarily conserved AIMs predicted in PEX1 by either hfAIM or iLIR. Schematic representation of the PEX1 proteins from 6 representative organisms and their AIMs sequences as predicted by either hfAIM or iLIR. The AtPEX1 AIM that was predicted only by iLIR is indicated by a dashed angle. The sequences in italics indicate nonconserved AIM sequences. At, the dicot plant Arabidopsis (Arabidopsis thaliana); Os, the monocot plant rice (Oryza sativa); Sc, yeast (Saccharomyces cerevisiae); Dm, Drosophila (Dorosophila melanogaster); Dr, Zebrafish (Danio rerio); Hs, human (Homo sapiens). AA, amino acid.
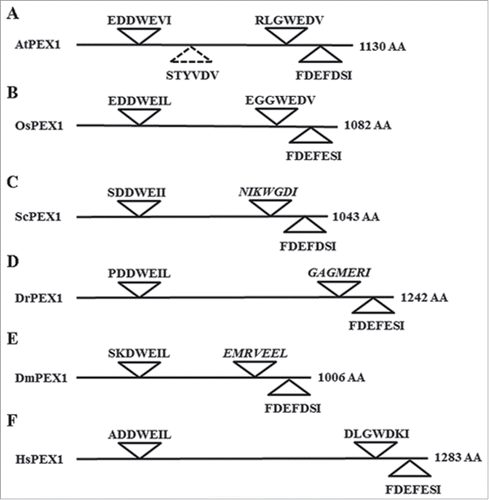
Figure 5. AtPEX6 interacts with Atg8 in vivo. (A) Schematic representation of the PEX6 proteins from 6 representative organisms and the AIMs sequences present in them as predicted by hfAIM. The sequence in italics indicates AIM that was not recognized by hfAIM. At, the dicot plant Arabidopsis (Arabidopsis thaliana); Os, the monocot plant rice (Oryza sativa); Sc, yeast (Saccharomyces cerevisiae); Dm, Drosophila (Dorosophila melanogaster); Dr, zebrafish (Danio rerio); Hs, human (Homo sapiens). AA, amino acid. (B) BiFC analysis was performed following transient coexpression of YN-Atg8f and YC-PEX6 in N. benthamiana leaves. YFP fluorescence (green bodies indicated by arrows) demonstrates the interaction of PEX6 with Atg8f. Transient coexpression of YC-PEX6 with unfused YN (C) or coexpression of YN-Atg8f with unfused YC (D) did not result in any YFP fluorescence demonstrating the specificity of the interaction. Chloroplast autofluorescence is shown in magenta. Bar: 20 µm.
Developmental phenotypes of pex1, pex6 and pex10 mutants are apparently associated with mutations occurring within or nearby their AIMs
A number of atg mutants and their corresponding genes have already been isolated and well characterized in plants.Citation3 Interestingly, recent studies revealed that peroxisome degradation is noticeably attenuated in the backgrounds of autophagy mutants,Citation15,18,32 implying that autophagy is involved in the degradation of peroxisomes, possibly through the interactions of Atg8 with peroxisome proteins possessing AIMs. Based on this assumption, we attempted to figure out whether mutations occurring in AIMs of PEX proteins influence the interaction of Atg8 with these proteins, eventually leading to abnormal peroxisome phenotypes. According to the Polyphen prediction method,Citation33 a R949W mutation in the homo sapiens PEX1 protein has been suggested to cause a “probably damaging” phenotype.Citation34 Intriguingly, this mutation is located close to an hfAIM predicted AIM (). In AtPEX6 protein, a mutation of the conserved R766 residue, also located close to an hfAIM predicted AIM motif, to Q (pex6-1) (), has been reported to cause peroxisomal targeting signaling (PTS2) processing defect in Arabidopsis.Citation35 Notably, a distinct mutation in AtPEX6 (pex6-2) results in similar physiological responses, such as resistance to inhibition by 2,4-dichlorophenoxybutyric acid (2, 4-DB) and to the promotive effects by protoauxin indole-3-butyric acid (IBA), but has no effect on peroxisomal matrix proteins import.Citation36 Thus, it is possible that the effect on matrix proteins import is related to changes in the autophagic degradation of Atpex6-1. Yet, the most striking findings were several mutations that occur within the hfAIM predicted AIM in PEX10 (). Prestele and associatesCitation21 identified a dominant negative G93E mutant in PEX10 that exhibited vermiform peroxisome shapes. Since it is proposed that the presence of acidic amino acids within or nearby the core AIM would increase the strength of binding of Atg8 to this AIM,Citation4,7 the G93E mutation might increase the Atg8-mediated turnover of AtPEX10. Indeed, the quantity of peroxisomes was reduced in the PEX10G93E mutant supporting our hypothesis that the G93E mutation enhances the binding efficiency of Atg8 to the mutated AtPEX10, thus increasing the turnover of peroxisomes by pexophagy. Additionally, 4 other mutations that occurred within or nearby this AIM cause possibly damaging phenotypes according to the Polyphen prediction.Citation34 Among these 4 mutations, an E71K mutation is expected to reduce the strength of Atg8 binding to AtPEX10 due to the conversion of the negatively charged E residue to a positively charged K residue (E71K; ). Taken together, the information described above supports our above mentioned results, suggesting that PEX1, PEX6 and PEX10 are turned over by autophagy via the interactions with Atg8.
Table 2. Mutations associated with or nearby AIMs in Arabidopsis and human PEX1, PEX6 and PEX10.
Discussion
Autophagy is an evolutionarily conserved process in eukaryotic organisms, including animals and plants. Accumulating evidence suggests that most of the proteins that are selectively turned over by autophagy contain one or multiple Atg8-interacting motif (AIM).Citation3,7,23 The core consensus of the AIM motif is F/W/Y-X-X-L/I/V.Citation4 Using this degenerate consensus sequence, it is possible to screen and identify AIM-containing proteins (ACPs) on a genome-wide scale by bioinformatics approaches.Citation10,11,24 But, as the consensus AIM motif is short and degenerate, a simple search for this motif will likely generate multiple false positive results. Thus, generating reliable, high fidelity bioinformatics tools will minimize the experimental work required to verify the predictions and therefore it is highly desirable.
The binding of Atg8 to the AIMs present in various proteins was shown to be enhanced by negative charge. Thus the presence of aspartate or glutamate, or phosphorylated serine and threonine residues, either immediately upstream or within the core AIM, strengthen the interaction between Atg8 and the AIM motif.Citation7 This finding is supported by our sequence analysis of experimentally verified AIM motifs that indicated that these motifs are highly enriched in D and E residues (). Based on this information we developed a bioinformatics tool, hfAIM, that uses a definition of an AIM motif as a sequence of 7 amino acids, X-X-X-F/W/Y-X-X-L/I/V, that contains at least 2 acidic amino acids. Although there is no direct evidence to support the negative influence of positively charged amino acids on the binding strength of Atg8 to the AIM, we still excluded these amino acids in the regular patterns of our hfAIM system in an attempt to improve the reliability of AIM prediction (). Note that in the present report, we only considered the contribution of the acidic amino acids to Atg8 binding in our prediction scheme. This restrain might enhance the fidelity of the AIM prediction with the expense of reducing the sensitivity, as the contribution of the S and T residues is not taken into account.
Another recently developed bioinformatics tool for the identification of AIM motifs in proteins, the iLIR tool,Citation11 is also based on regular expression patterns. iLIR defines an AIM motif as a sequence of 6 amino acids, X-X-F/W/Y-X-X-L/I/V, where the permitted residues at any given “X” position are based on multiple sequence alignment of verified AIM motifs. A comparison of iLIR and hfAIM predictions using a dataset of verified AIM motifs suggested that while iLIR has a better sensitivity, hfAIM is more stringent (). Furthermore, hfAIM was somewhat better at identifying AIM motifs in verified Arabidopsis Atg8-interacting proteins (Table S2). Unfortunately, experimental information about the specific sequences needed for the Atg8 interaction of the verified Atg8 interacting proteins from Arabidopsis is still scarce. However, our BiFC analysis suggested that the hfAIM motif identified in AtPEX10 is indeed a functional AIM and is necessary for the interaction with Atg8, while the iLIR identified AIM is not (Table S2 and ). Furthermore, a single AIM was identified solely by hfAIM in AtPEX6, and BiFC analysis indeed verified AtPEX6 - Atg8 interaction (Table S2 and ). It is possible that as iLIR regular expression patterns were defined using mostly non-plant verified AIMs, it does not represent well the composition of amino acids in plant AIM motifs and therefore it is not best suited to identify AIMs in plants.
Recently, the role of autophagy in multiple biological processes has been characterized in Arabidopsis by omics analysis.Citation25,26 However, the identification of the specific and/or novel components regulated by autophagy from the large-scale data is still not entirely resolved. Our hfAIM system can be used as a complementary approach to identify potential ACPs and may provide new insight into the regulation of autophagy-mediated degradation processes. For instance, differentially expressed proteins (DEPs) identified by proteomic analysis in Arabidopsis can be further analyzed by the hfAIM system to look at their potential regulation by the autophagy apparatus. Those DEPs containing hfAIM motifs could be considered as potential Atg8 interacting candidates, and then selected for further analysis. However, it is important to remember that Atg8 binding to target proteins is not always mediated by a typical AIM motif.Citation5 For example, the verified Atg8 binding proteins Calcium-binding and coiled-coil domain-containing protein 2 (UniProtKB accession: CACO2_HUMAN) and Tax1-binding protein 1 (UniProtKB accession: TAXB1_HUMAN) do not contain a typical AIM motif (Table S1). Therefore, the characterization of additional verified functional AIMs by experimental methods is essential for the development of more accurate prediction tools.
The fidelity of AIMs prediction by hfAIM can be improved by several approaches. One possible approach could be to combine hfAIM predictions with iLIR predictions, and to consider as more promising AIM motifs that were also predicted by iLIR, or had PSSM scores according to Kalvari et al. above a defined cut-off value.Citation11 As demonstrated using the verified AIM motifs data set (Table S1), the use of a PSSM cut-off value improved the specificity for both hfAIM and iLIR ( and Kalvari et al.Citation11). However, the use of this higher level of stringency can lead to false negative results, as demonstrated with AtPEX10. Though the PSSM score of the predicted hfAIM in AtPEX10 is quite low (PSSM = 9), and therefore it would be regarded as a low confidence AIM, the protein was experimentally shown to interact with Atg8 () through this predicted hfAIM.
Prediction of potential AIMs by bioinformatics tools can also be strengthened by additional lines of evidence such as evolutionary conservation of the AIMs.Citation11,24 This approach assumes that a functional AIM will be conserved across species in proteins that are involved in selective autophagy-mediated degradation processes or whose homeostasis is regulated by selective autophagy. The peroxisomal PEX proteins were used as a case study to evaluate this approach. Peroxisomes are highly dynamic organelles functioning in multiple biological processes.Citation20 Furthermore, pexophagy, the process of selective degradation of peroxisomes by autophagy, is an essential mechanism regulating the homeostasis of the peroxisomes. Although pexophagy in plants was demonstrated by several recent reports,Citation14,17,18,32 the regulatory mechanisms underlying the degradation of peroxisomes still await additional studies. Indeed, the colocalization of Atg8 and peroxisomes, specifically aggregated peroxisomes,Citation14,17,18 suggests that Atg8 interacts with certain peroxisome PEX proteins, leading to their specific autophagy-mediated transport, or the transport of the entire peroxisome to the vacuole for degradation. Therefore, we used both the hfAIM and the iLIR tools to identify potential AIMs in the Arabidopsis family of PEX proteins (), and then compared the sequences of PEX proteins from 38 different organisms and used them to analyze the evolutionary conservation of the predicted AIMs in these proteins (Fig. S3 to S11). Out of the 22 AtPEX proteins, 13 proteins contain AIM motifs according to either hfAIM or iLIR (), but only the AIM motifs in PEX1, PEX6 and PEX10 are highly conserved across species (Figs. S3 to S5). PEX6 encodes a peroxisomal AAA-ATPase, and forms a complex with PEX1 that participates in peroxisomal matrix proteins import.Citation35,37 Only a single, highly conserved AIM was detected in PEX6 by our hfAIM system, and this AIM is located in the Walker B domain of PEX6 (). Indeed, our BiFC results verified that PEX6 does interact with Atg8 in planta (), supporting the functional role of the conserved AIM motif. Therefore, we propose that this conserved amino acid sequence in PEX6 may have a dual function, either serving as a Walker B domain and/or serving as an AIM, allowing the binding of Atg8 to this protein. AtPEX1 contains 3 hfAIMs, which are also recognized by iLIR and an additional motif that is recognized solely by iLIR (). Two of these 4 motifs are highly conserved across species ( and S4) implying that PEX1 interacts with Atg8 at a reasonable confidence.
PEX10 is involved in both peroxisome formation and matrix protein import.Citation21,22 The evolutionary highly conserved amino acid sequences in PEX10 are thought to be essential for peroxisome biogenesis and plant development.Citation21 Looking further into PEX10 proteins from various organisms, we found that all of them contain a highly conserved AIM (GEEYCDI) recognized by our hfAIM method, but not by the iLIR system (). A G93E mutation in PEX10 that causes vermiform peroxisome shape Citation21 occurs in the minus 3 position of the core hfAIM (). Moreover, this mutation also leads to a lower number of peroxisomes,Citation21 as expected from a mutation that strengthen Atg8-PEX10 interaction and therefore will lead to increase turnover by autophagy. Indeed, BiFC experiments confirmed the functional role of this conserved AIM in PEX10 interaction with Atg8 in planta (). Interestingly, an additional AIM, located in another position of the PEX10 protein, is predicted by the iLIR system. Yet, this xLIR motif is not evolutionarily conserved in PEX10 proteins of the various lower and higher organisms that have been studied (Fig. S3). Moreover, our BiFC analysis also verified that, unlike the conserved AIM, this xLIR motif is not necessary for Atg8 binding to PEX10 (). The rest of the AIMs present in the other PEX proteins are not conserved in evolution (Figs. S6 to S11). Therefore, we propose that PEX6, PEX10 and possibly also PEX1, are likely to interact with Atg8 through their evolutionarily conserved AIMs, and that selective autophagy in probably involved in the turnover of these proteins.
In the present study, 9 AtPEX proteins were identified by our hfAIM approach as potential Atg8-interacting proteins (). Interestingly, the iLIR system also identified 8 AtPEX proteins as potential Atg8-interacting proteins. However, AIMs in PEX3 and PEX14, which interact with Atg8 in mammalian cells,Citation38,39 were recognized only by the hfAIM system (). Though the predicted AIM motif in AtPEX14 does not seem to be very well conserved among other organisms (Fig. S9), it was recently shown that Atg8 colocalized with AtPEX14 in peroxisome aggregates, suggesting that the interaction between Atg8 and PEX14 is conserved. Furthermore, though fission events are suggested to be involved in the degradation of yeast peroxisomes following protein aggregation,Citation40 no AIM motifs were predicted by hfAIM in AtPEX family members involved in peroxisome division-proliferation (). Interestingly, the iLIR tool did predict xLIR motifs in several division-proliferation AtPEX proteins and these motifs contained S and T residues, strengthening the notion that S and T might contribute to Atg8 binding of AIMs. Alternatively, as recent studies demonstrate that some proteins do not require a typical AIM motif to bind Atg8,Citation5,41,42 these proteins might interact with Atg8 in an AIM-independent manner.
In summary, we have generated a high-fidelity bioinformatics tool, termed hfAIM, available as a web tool (http://bioinformatics.psb.ugent.be/hfAIM/) for in silico genome-wide prediction of AIMs in proteins. Using hfAIM it is possible to perform fast and reliable genome-wide screening of AIM-containing proteins that may be regulated by autophagy, and to select candidates for further studies using experimental approaches. This bioinformatics approach can facilitate a better understanding of the contribution of autophagy to multiple biological processes in various organisms. Using PEX proteins as a test case, our investigations indicate that PEX1, PEX6 and PEX10 are selectively turned over by autophagy. More specifically, our results may also shed a new light on the regulatory mechanism(s) underlying how Atg8 coordinates the homeostasis of specific PEX proteins as well as the operation of pexophagy.
Materials and methods
Identification of AIMs within proteins
To identify AIMs that meet the standards determined in within proteins, we adapted the stand-alone version of the PatMatch software.Citation43 This program is available for download at The Arabidopsis Information Resource (TAIR) at ftp://ftp.arabidopsis.org/home/tair/Software/Patmatch/). In-house scripts were tailored to calculate the percentage of Arabidopsis proteins possessing either zero, 1, 2, 3 or more AIMs in the entire Arabidopsis proteome. Position specific scoring matrices (PSSM), were calculated using the iLIR web interface provided by Kalvari et al (http://repeat.biol.ucy.ac.cy/iLIR/).Citation11
Plasmid construction
To generate PEX6-YC, PEX10-YC and YN-Atg8, we used the pSAT vector system for BiFC assays.Citation44 Cloning was done with the In-Fusion kit (Clontech, 639649) according to the manual instructions by using the corresponding primers (Table S3). The resulting clones were finally introduced into the pPZP vectors as previously described.Citation30 To generate the PEX10E95A-YC, PEX10E95H-YC, PEX10Y96A-YC and PEX10F253A-YC, we also used the pSAT vector system (https://www.arabidopsis.org/abrc/catalog/vector_2.html) for BiFC assays.Citation44 Mutagenesis of AtPEX10 was generated via substituting the corresponding amino acids by specific primers (Table S5) and the corresponding binary vector was produced as described above.
Multiple alignments
To test the homology of the protein sequences among various organisms, PEX protein sequences derived from the PeroxisomeDB (http://www.peroxisomedb.org/home.jsp) were aligned by the ClustalW method in the MEGA 6 software with defaults settings.Citation45 To simplify the presentation, only the alignment of a window of 10 to 30 amino acids that includes the predicted AIM motifs is shown in Figs. S3 to S11.
BiFC assay and confocal microscopy
To verify the interactions of ATG8f with either AtPEX6 or AtPEX10, we used Agrobacterium strains harboring each of the following plasmids separately: YN-Atg8 with either PEX6-YC or PEX10-YC were transiently cotransformed in Nicotiana benthamiana leaves as previously described.Citation30 For analysis of the interactions of ATG8f with the AtPEX10 mutants PEX10E95A-YC, PEX10E95H-YC, PEX10Y96A-YC or PEX10F253A-YC, we employed the same approach as above. Confocal microscopy analysis was performed using Olympus Fluoview 1000 IX81 (Olympus Life Science, Tokyo, Japan) and the Nikon A1 (Nikon, Japan) systems as previously described.Citation30 Briefly, samples were put between 2 microscope glass cover slips. Images were taken from a single focal plane unless otherwise indicated. GFP fluorescence images were taken using 488-nm laser excitation and the emission was collected via the 525-nm filter. Chlorophyll autofluorescence was taken with the 640-nm laser and collected with the 700-nm filter. Acquired images were analyzed by either Olympus Fluoview 1000 viewer or the NIS-Elements AR imaging software.
Abbreviations
| ACP | = | AIM-containing proteins |
| AIM | = | Atg8-interacting motif |
| At | = | Arabidopsis thaliana |
| BiFC | = | bimolecular fluorescence complementation |
| hfAIM | = | high-fidelity AIM |
| PEX | = | peroxin |
| PSSM | = | position-specific scoring matrix |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary files
Download Zip (26.3 MB)Acknowledgements
The authors thank Omrit Zemach and Dana Averbuch for excellent technical assistance.
Funding
Our research was supported by grants from The Israeli Ministry of Agriculture, The Israel Science Foundation (grant No.395/11), and the J & R center for scientific research at the Weizmann Institute of Science. GG is an incumbent of the Bronfman Chair of Plant Science at the Weizmann Institute of Science.
References
- Avin-Wittenberg T, Honig A, Galili G. Variations on a theme: plant autophagy in comparison to yeast and mammals. Protoplasma 2012; 249:285-99; PMID:21660427; http://dx.doi.org/10.1007/s00709-011-0296-z
- Yoshimoto K, Takano Y, Sakai Y. Autophagy in plants and phytopathogens. FEBS Lett 2010; 584:1350-8; PMID:20079356; http://dx.doi.org/10.1016/j.febslet.2010.01.007
- Li F, Vierstra RD. Autophagy: a multifaceted intracellular system for bulk and selective recycling. Trends Plant Sci 2012; 17:526-37; PMID:22694835; http://dx.doi.org/10.1016/j.tplants.2012.05.006
- Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, Fujioka Y, Ohsumi Y, Inagaki F. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells 2008; 13:1211-8; PMID:19021777; http://dx.doi.org/10.1111/j.1365-2443.2008.01238.x
- Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010; 466:68-76; PMID:20562859; http://dx.doi.org/10.1038/nature09204
- Noda NN, Ohsumi Y, Inagaki F. Atg8-family interacting motif crucial for selective autophagy. FEBS Lett 2010; 584:1379-85; PMID:20083108; http://dx.doi.org/10.1016/j.febslet.2010.01.018
- Schreiber A, Peter M. Substrate recognition in selective autophagy and the ubiquitin-proteasome system. Biochim Biophys Acta 2014; 1843:163-81; PMID:23545414; http://dx.doi.org/10.1016/j.bbamcr.2013.03.019
- Kumeta H, Watanabe M, Nakatogawa H, Yamaguchi M, Ogura K, Adachi W, Fujioka Y, Noda NN, Ohsumi Y, Inagaki F. The NMR structure of the autophagy-related protein Atg8. J Biomol NMR 2010; 47:237-41; PMID:20428927; http://dx.doi.org/10.1007/s10858-010-9420-1
- Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 2011; 333:228-33; PMID:21617041; http://dx.doi.org/10.1126/science.1205405
- Tzfadia O, Galili G. The Arabidopsis exocyst subcomplex subunits involved in a golgi-independent transport into the vacuole possess consensus autophagy-associated atg8 interacting motifs. Plant signal Behav 2013; 9; PMID:24494242; http://dx.doi.org/10.4161/psb.26732
- Kalvari I, Tsompanis S, Mulakkal NC, Osgood R, Johansen T, Nezis IP, Promponas VJ. iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy 2014; 10:913-25; PMID:24589857; http://dx.doi.org/10.4161/auto.28260
- Meszaros B, Simon I, Dosztanyi Z. Prediction of protein binding regions in disordered proteins. PLoS Comput Biol 2009; 5:e1000376; PMID:19412530; http://dx.doi.org/10.1371/journal.pcbi.1000376
- Dosztanyi Z, Meszaros B, Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009; 25:2745-6; PMID:19717576; http://dx.doi.org/10.1093/bioinformatics/btp518
- Yoshimoto K, Shibata M, Kondo M, Oikawa K, Sato M, Toyooka K, Shirasu K, Nishimura M, Ohsumi Y. Organ-specific quality control of plant peroxisomes is mediated by autophagy. J cell Sci 2014; 127:1161-8; PMID:24463818; http://dx.doi.org/10.1242/jcs.139709
- Shibata M, Oikawa K, Yoshimoto K, Goto-Yamada S, Mano S, Yamada K, Kondo M, Hayashi M, Sakamoto W, Ohsumi Y, et al. Plant autophagy is responsible for peroxisomal transition and plays an important role in the maintenance of peroxisomal quality. Autophagy 2014; 10:936-7; PMID:24732712; http://dx.doi.org/10.4161/auto.28529
- Lee HN, Kim J, Chung T. Degradation of plant peroxisomes by autophagy. Front Plant Sci 2014; 5:139; PMID:24782878
- Shibata M, Oikawa K, Yoshimoto K, Kondo M, Mano S, Yamada K, Hayashi M, Sakamoto W, Ohsumi Y, Nishimura M. Highly oxidized peroxisomes are selectively degraded via autophagy in Arabidopsis. Plant Cell 2013; 25:4967-83; PMID:24368788; http://dx.doi.org/10.1105/tpc.113.116947
- Kim J, Lee H, Lee HN, Kim SH, Shin KD, Chung T. Autophagy-related proteins are required for degradation of peroxisomes in Arabidopsis hypocotyls during seedling growth. Plant Cell 2013; 25:4956-66; PMID:24368791; http://dx.doi.org/10.1105/tpc.113.117960
- Kim J, Lee HN, Chung T. Plant cell remodeling by autophagy: switching peroxisomes for green life. Autophagy 2014; 10:702-3; PMID:24492493; http://dx.doi.org/10.4161/auto.27953
- Hu J, Baker A, Bartel B, Linka N, Mullen RT, Reumann S, Zolman BK. Plant peroxisomes: biogenesis and function. Plant Cell 2012; 24:2279-303; PMID:22669882; http://dx.doi.org/10.1105/tpc.112.096586
- Prestele J, Hierl G, Scherling C, Hetkamp S, Schwechheimer C, Isono E, Weckwerth W, Wanner G, Gietl C. Different functions of the C3HC4 zinc RING finger peroxins PEX10, PEX2, and PEX12 in peroxisome formation and matrix protein import. Proc Natl Acad Sci USA 2010; 107:14915-20; PMID:20679226; http://dx.doi.org/10.1073/pnas.1009174107
- U. S, Prestele J, Henriette O, Brueggeman R, Wanner G, Gietl C. Requirement of the C3CH4 zing RING finger of the Arabidopsis PEX10 for photorespiration and leaf peroxisome contact with chloroplasts. Proc Natl Acad Sci USA 2007; 104:1069-74; PMID:17215364; http://dx.doi.org/10.1073/pnas.0610402104
- Birgisdottir AB, Lamark T, Johansen T. The LIR motif - crucial for selective autophagy. J Cell Sci 2013; 126:3237-47; PMID:23908376
- Cvrckova F, Zarsky V. Old AIMs of the exocyst: evidence for an ancestral association of exocyst subunits with autophagy-associated Atg8 proteins. Plant Signal Behav 2013; 8:e27099; PMID:24305598; http://dx.doi.org/10.4161/psb.27099
- Avin-Wittenberg T, Bajdzienko K, Wittenberg G, Alseekh S, Tohge T, Bock R, Giavalisco P, Fernie AR. Global Analysis of the Role of Autophagy in Cellular Metabolism and Energy Homeostasis in Arabidopsis Seedlings under Carbon Starvation. Plant Cell 2015; 27:306-22; PMID:25649436
- Masclaux-Daubresse C, Clement G, Anne P, Routaboul JM, Guiboileau A, Soulay F, Shirasu K, Yoshimoto K.. Stitching together the Multiple Dimensions of Autophagy Using Metabolomics and Transcriptomics Reveals Impacts on Metabolism, Development, and Plant Responses to the Environment in Arabidopsis. Plant Cell 2014; 26:1857-77; PMID:24808053; http://dx.doi.org/10.1105/tpc.114.124677
- Ren C, Liu J, Gong Q. Functions of autophagy in plant carbon and nitrogen metabolism. Front Plant Sci 2014; 5:301; PMID:25009547; http://dx.doi.org/10.3389/fpls.2014.00301
- Sparkes IA, Brandizzi F, Slocombe SP, El-Shami M, Hawes C, Baker A. An Arabidopsis pex10 null mutant is embryo lethal, implicating peroxisomes in an essential role during plant embryogenesis. Plant Physiol 2003; 133:1809-19; PMID:14576288; http://dx.doi.org/10.1104/pp.103.031252
- Contento AL, Xiong Y, Bassham DC. Visualization of autophagy in Arabidopsis using the fluorescent dye monodansylcadaverine and a GFP-AtATG8e fusion protein. Plant J 2005; 42:598-608; PMID:15860017; http://dx.doi.org/10.1111/j.1365-313X.2005.02396.x
- Michaeli S, Honig A, Levanony H, Peled-Zehavi H, Galili G. Arabidopsis ATG8-INTERACTING PROTEIN1 Is Involved in Autophagy-Dependent Vesicular Trafficking of Plastid Proteins to the Vacuole. Plant Cell 2014; 26:4084-101; PMID:25281689; http://dx.doi.org/10.1105/tpc.114.129999
- Schluter A, Real-Chicharro A, Gabaldon T, Sanchez-Jimenez F, Pujol A. PeroxisomeDB 2.0: an integrative view of the global peroxisomal metabolome. Nucleic Acids Res 2010; 38:D800-5; PMID:19892824; http://dx.doi.org/10.1093/nar/gkp935
- Farmer LM, Rinaldi MA, Young PG, Danan CH, Burkhart SE, Bartel B. Disrupting autophagy restores peroxisome function to an Arabidopsis lon2 mutant and reveals a role for the LON2 protease in peroxisomal matrix protein degradation. Plant Cell 2013; 25:4085-100; PMID:24179123; http://dx.doi.org/10.1105/tpc.113.113407
- Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics / editorial board. Jonathan L Haines [et al] 2013; Chapter 7:Unit7 20.
- Ebberink MS, Mooijer PAW, Gootjes J, Koster J, Wanders RJA, Waterham HR. Genetic classification and mutational spectrum of more than 600 patients with a zellweger syndrome spectrum disorder. Hum Mutat 2011; 32:59-69; PMID:21031596; http://dx.doi.org/10.1002/humu.21388
- Zolman BK, Bartel B. An Arabidopsis indole-3-butyric acid-response mutant defective in PEROXIN6, an apparent ATPase implicated in peroxisomal function. Proc Natl Acad Sci USA 2004; 101:1786-91; PMID:14745029; http://dx.doi.org/10.1073/pnas.0304368101
- Burkhart SE, Lingard MJ, Bartel B. Genetic dissection of peroxisome-associated matrix protein degradation in Arabidopsis thaliana. Genetics 2013; 193:125-241; PMID:23150599; http://dx.doi.org/10.1534/genetics.112.146100
- Goto S, Mano S, Nakamori C, Nishimura M. Arabidopsis ABERRANT PEROXISOME MORPHOLOGY9 is a peroxin that recruits the PEX1-PEX6 complex to peroxisomes. Plant Cell 2011; 23:1573-87; PMID:21487094; http://dx.doi.org/10.1105/tpc.110.080770
- Hara-Kuge S, Fujiki Y. The peroxin Pex14p is involved in LC3-dependent degradation of mammalian peroxisomes. Exp Cell Res 2008; 314:3531-41; PMID:18848543; http://dx.doi.org/10.1016/j.yexcr.2008.09.015
- Farre JC, Manjithaya R, Mathewson RD, Subramani S. PpAtg30 tags peroxisomes for turnover by selective autophagy. Developmental cell 2008; 14:365-76; PMID:18331717; http://dx.doi.org/10.1016/j.devcel.2007.12.011
- Manivannan S, de Boer R, Veenhuis M, van der Klei IJ. Lumenal peroxisomal protein aggregates are removed by concerted fission and autophagy events. Autophagy 2013; 9:1044-56; PMID:23614977; http://dx.doi.org/10.4161/auto.24543
- Sawa-Makarska J, Abert C, Romanov J, Zens B, Ibiricu I, Martens S. Cargo binding to Atg19 unmasks additional Atg8 binding sites to mediate membrane-cargo apposition during selective autophagy. Nat Cell Biol 2014; 16:425-33; PMID:24705553; http://dx.doi.org/10.1038/ncb2935
- Lin L, Yang P, Huang X, Zhang H, Lu Q, Zhang H. The scaffold protein EPG-7 links cargo-receptor complexes with the autophagic assembly machinery. J Cell Biol 2013; 201:113-29; PMID:23530068; http://dx.doi.org/10.1083/jcb.201209098
- Yan T, Yoo D, Berardini TZ, Mueller LA, Weems DC, Weng S, Cherry JM, Rhee SY. PatMatch: a program for finding patterns in peptide and nucleotide sequences. Nucleic Acids Res 2005; 33:W262-6; PMID:15980466; http://dx.doi.org/10.1093/nar/gki368
- Tzfira T, Tian GW, Lacroix B, Vyas S, Li J, Leitner-Dagan Y, Krichevsky A, Taylor T, Vainstein A, Citovsky V. pSAT vectors: a modular series of plasmids for autofluorescent protein tagging and expression of multiple genes in plants. Plant Mol Biol 2005; 57:503-16; PMID:15821977; http://dx.doi.org/10.1007/s11103-005-0340-5
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evolut 2013; 30:2725-9; PMID:24132122; http://dx.doi.org/10.1093/molbev/mst197
- Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Øvervatn A, Kirkin V, Johansen T.. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem 2012; 287:39275-90; PMID:23043107; http://dx.doi.org/10.1074/jbc.M112.378109
- Svenning S, Lamark T, Krause K, Johansen T. Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy 2011; 7:993-1010; PMID:21606687; http://dx.doi.org/10.4161/auto.7.9.16389
- Zhou J, Wang J, Cheng Y, Chi YJ, Fan B, Yu JQ, Chen Z.. NBR1-mediated selective autophagy targets insoluble ubiquitinated protein aggregates in plant stress responses. PLoS Genet 2013; 9:e1003196; PMID:23341779; http://dx.doi.org/10.1371/journal.pgen.1003196
- Arabidopsis Interactome Mapping Consortium. Evidence for network evolution in an Arabidopsis interactome map. Science 2011; 333:601-7; PMID:21798944; http://dx.doi.org/10.1126/science.1203877
- Chen J, Lalonde S, Obrdlik P, Noorani Vatani A, Parsa SA, Vilarino C, Revuelta JL, Frommer WB, Rhee SY. Uncovering Arabidopsis membrane protein interactome enriched in transporters using mating-based split ubiquitin assays and classification models. Front Plant Sci 2012; 3:124; PMID:22737156; http://dx.doi.org/10.3389/fpls.2012.00124
- Kim DY, Scalf M, Smith LM, Vierstra RD. Advanced proteomic analyses yield a deep catalog of ubiquitylation targets in Arabidopsis. Plant Cell 2013; 25:1523-40; PMID:23667124; http://dx.doi.org/10.1105/tpc.112.108613
- Li F, Chung T, Vierstra RD. AUTOPHAGY-RELATED11 plays a critical role in general autophagy- and senescence-induced mitophagy in Arabidopsis. Plant Cell 2014; 26:788-807; PMID:24563201; http://dx.doi.org/10.1105/tpc.113.120014
- Zhuang X, Wang H, Lam SK, Gao C, Wang X, Cai Y, Jiang L. A BAR-domain protein SH3P2, which binds to phosphatidylinositol 3-phosphate and ATG8, regulates autophagosome formation in Arabidopsis. Plant Cell 2013; 25:4596-615; PMID:24249832; http://dx.doi.org/10.1105/tpc.113.118307
- Yamaguchi M, Matoba K, Sawada R, Fujioka Y, Nakatogawa H, Yamamoto H, Kobashigawa Y, Hoshida H, Akada R, Ohsumi Y, et al. Noncanonical recognition and UBL loading of distinct E2s by autophagy-essential Atg7. Nat Struct Mol Biol 2012; 19:1250-6; PMID:23142983; http://dx.doi.org/10.1038/nsmb.2451
- Klopffleisch K, Phan N, Augustin K, Bayne RS, Booker KS, Botella JR, Carpita NC, Carr T, Chen JG, Cooke TR, et al. Arabidopsis G-protein interactome reveals connections to cell wall carbohydrates and morphogenesis. Mol Syst Biol 2011; 7:532; PMID:21952135; http://dx.doi.org/10.1038/msb.2011.66
- Honig A, Avin-Wittenberg T, Ufaz S, Galili G. A new type of compartment, defined by plant-specific Atg8-interacting proteins, is induced upon exposure of Arabidopsis plants to carbon starvation. Plant Cell 2012; 24:288-303; PMID:22253227; http://dx.doi.org/10.1105/tpc.111.093112