
ABSTRACT
The intestinal mucosa of Crohn disease (CD) patients is abnormally colonized by adherent-invasive E. coli (AIEC). Upon AIEC infection, autophagy is induced in host cells to restrain bacterial intracellular replication. The underlying mechanism, however, remains unknown. Here, we investigated the role of the EIF2AK4-EIF2A/eIF2α-ATF4 pathway in the autophagic response to AIEC infection. We showed that infection of human intestinal epithelial T84 cells with the AIEC reference strain LF82 activated the EIF2AK4-EIF2A-ATF4 pathway, as evidenced by increased phospho-EIF2AK4, phospho-EIF2A and ATF4 levels. EIF2AK4 depletion inhibited autophagy activation in response to LF82 infection, leading to increased LF82 intracellular replication and elevated pro-inflammatory cytokine production. Mechanistically, EIF2AK4 depletion suppressed the LF82-induced ATF4 binding to promoters of several autophagy genes including MAP1LC3B, BECN1, SQSTM1, ATG3 and ATG7, and this subsequently inhibited transcription of these genes. LF82 infection of wild-type (WT), but not eif2ak4−/−, mice activated the EIF2AK4-EIF2A-ATF4 pathway, inducing autophagy gene transcription and autophagy response in enterocytes. Consequently, eif2ak4−/− mice exhibited increased intestinal colonization by LF82 bacteria and aggravated inflammation compared to WT mice. Activation of the EIF2AK4-EIF2A-ATF4 pathway was observed in ileal biopsies from patients with noninflamed CD, and this was suppressed in inflamed CD, suggesting that a defect in the activation of this pathway could be one of the mechanisms contributing to active disease. In conclusion, we show that activation of the EIF2AK4-EIF2A-ATF4 pathway upon AIEC infection serves as a host defense mechanism to induce functional autophagy to control AIEC intracellular replication.
Introduction
Evidence has suggested that the etiology of inflammatory bowel disease (IBD), including Crohn disease (CD) and ulcerative colitis, involves environmental, genetic, immunological and infectious factors.Citation1 An alteration in the intestinal microbiota, called dysbiosis, has been associated with CD, characterized by a decrease in Firmicutes, in particular Faecalibacterium prausnitzii, and an increase in Enterobacteriaceae, especially Escherichia coli.Citation2
Our group and others have shown a high prevalence of the adherent-invasive Escherichia coli (AIEC) pathotype in the ileal mucosa of CD patients.Citation3–5 Characterization of the AIEC strains show they are able to adhere to and to invade human intestinal epithelial cells (IECs),Citation4,6 survive and replicate within macrophages,Citation7,8 induce high production of proinflammatory cytokines and chemokines,Citation7,9,10 and exacerbate intestinal inflammation in a genetically susceptible mouse model.Citation9 Our recent studies showed that autophagy is induced in host cells upon AIEC infection, and a functional autophagy is required to restrain the intracellular replication of AIEC.Citation6,8,10,11 However, the mechanism involved in autophagy induction in response to AIEC infection remains largely unknown.
Autophagy is a tightly regulated homeostatic process responsible for the elimination of damaged cytoplasmic components via the lysosomal pathway.Citation12 This process plays a key role in the regulation of numerous physiological functions including cell development, differentiation and survival, and in the control of the innate and adaptive immune responses. In response to multiple stresses such as nutrient starvation, damaged organelles and unfolded protein aggregation, or infection with pathogens, autophagy is induced, and this is a crucial host defense mechanism. Dysregulated autophagy has been linked to numerous human pathologies including CD.Citation13 Genome-wide association studies have discovered the association with CD susceptibility of polymorphisms in the genes involved in autophagy, such as ATG16L1 (autophagy-related 16 like 1), IRGM (immunity-related GTPase family, M), XBP1 (X-box binding protein 1), LRRK2 (leucine-rich repeat kinase 2), PTPN2 (protein tyrosine phosphatase, non-receptor type 2) and ULK1 (unc-51 like autophagy activating kinase 1).Citation14 The known CD-associated NOD2 (nucleotide-binding oligomerization domain-containing 2) mutations have been linked to autophagy via its physical interaction with ATG16L1 in response to bacterial infection.Citation15–17 Recently, significant advances have been achieved in understanding the functional consequences of the autophagy-related gene defects in CD. Evidence has shown that the autophagy-related risk polymorphisms lead to impaired sensing and handling of intracellular bacteria by innate immunity, in particular the inappropriate stimulation of antimicrobial and inflammasome pathways eventually result in uncontrolled inflammation.Citation14,18 These studies bring autophagy to light as a key regulator with the capacity to integrate several aspects of CD pathogenesis.
Activation of the EIF2AK4-EIF2A-ATF4 pathway is necessary to activate autophagy gene expression in response to cellular stress.Citation19 EIF2AK4 (eukaryotic translation initiation factor 2 alpha kinase 4) belongs to a family of protein kinases that phosphorylate the α subunit of EIF2 (eukaryotic translation initiation factor 2) in response to various stress stimuli. Phosphorylation of EIF2A in response to EIF2AK4 activation subsequently increases translation of specific mRNAs that are important for stress remediation such as ATF4 (activating transcription factor 4).Citation20 Recent evidence demonstrates that the EIF2AK4-EIF2A-ATF4 pathway is activated in epithelial cells upon infection with Shigella and Salmonella.Citation21 However, to our knowledge, no data are available on the link between this pathway and CD and its involvement in host responses to infection with pathogenic bacteria associated with the disease.
Here, we aimed at investigating (i) the ability of AIEC to modulate the EIF2AK4-EIF2A-ATF4 pathway and (ii) the role of this pathway in regulating autophagy to control the intracellular multiplication of AIEC.
Results
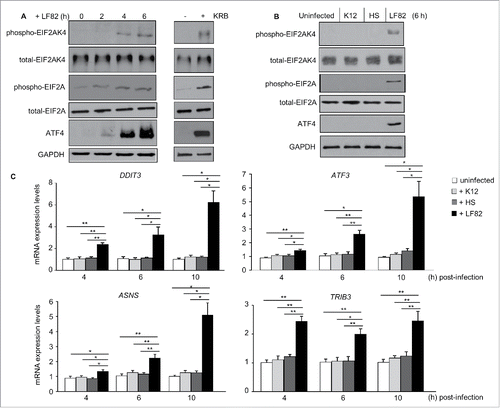
AIEC infection induces activation of the EIF2AK4-EIF2A-ATF4 pathway in IECs
We first investigated activation of the EIF2AK4-EIF2A-ATF4 pathway in human intestinal epithelial T84 cells in response to infection with the AIEC LF82 reference strain. Western blot analysis showed that LF82 infection increased the levels of phospho-EIF2AK4, phospho-EIF2A and ATF4 in T84 cells (). This effect was almost as profound as when cells were incubated for 1 h with amino acid-starvation Krebs-Ringer buffer (KRB; ). In contrast, infection with the nonpathogenic E. coli K12 MG1655 or the commensal E. coli HS strain did not significantly affect the levels of phospho-EIF2AK4, phospho-EIF2A and ATF4 (). qRT-PCR analysis showed that mRNA expression levels of the known target genes of ATF4, including TRIB3 (tribbles pseudokinase 3), ATF3 (activating transcription factor 3), DDIT3 (DNA damage inducible transcript 3) and ASNS (asparagine synthetase), were significantly increased in LF82-infected cells compared to uninfected cells or cells infected with either the K12 or HS strain (); mRNA expression levels of these genes were not significantly changed upon infection with the K12 or HS strain. These results demonstrate that the EIF2AK4-EIF2A-ATF4 pathway is activated upon AIEC infection in host IECs.
Figure 1. AIEC infection induces activation of the EIF2AK4-EIF2A-ATF4 pathway in T84 cells. T84 cells were infected with AIEC LF82 strain or the E. coli K12 MG1655 and the commensal E. coli HS strain. In parallel, cells were treated with KRB for 1 h. (A, B) Western blot analysis for the activation of the EIF2AK4-EIF2A-ATF4 pathway. (C) qRT-PCR analysis for mRNA expression levels of known ATF4 target genes, including TRIB3, ATF3, DDIT3 and ASNS. Data are means ± SEM of 6 replicates and are representative of 3 independent experiments. *, P < 0.05; **, P ≤ 0.005.
EIF2AK4 is essential to restrain AIEC intracellular replication and AIEC-induced inflammation
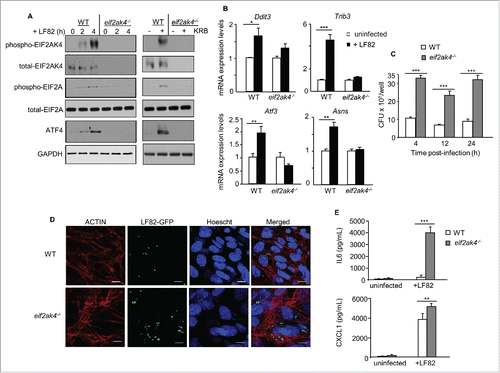
To explore the role of the EIF2AK4-EIF2A-ATF4 pathway in host response to AIEC infection, mouse embryonic fibroblasts (MEFs) harboring a deletion for the Eif2ak4 gene (eif2ak4−/− MEFs) were used. Infection with AIEC LF82 induced activation of EIF2AK4 and EIF2A, enhanced ATF4 protein levels, and increased mRNA expression levels of ATF4 target genes in wild-type (WT) MEFs (). These results were not observed in eif2ak4−/− MEFs (). Incubation of WT MEFs with KRB for 4 h, used as a positive control, also activated the EIF2AK4-EIF2A-ATF4 pathway in WT MEFs but not in eif2ak4−/− MEFs (). To examine whether the EIF2AK4-EIF2A-ATF4 pathway plays a role in the control of AIEC intracellular replication, WT and eif2ak4−/− MEFs were infected with AIEC LF82, and the number of intracellular bacteria was counted on Luria-Bertani (LB) agar plates. At different time points post-infection, there was a marked increase in the number of intracellular LF82 bacteria in eif2ak4−/− MEFs compared to WT MEFs (). Confocal microscopy analysis of MEFs infected with a LF82-GFP strain consistently showed that invalidation of Eif2ak4 led to an increased number of intracellular LF82 (). This was accompanied by an increase in AIEC-induced production of the pro-inflammatory cytokine IL6 (interleukin 6) and chemokine CXCL1 (chemokine [C-X-C motif] ligand 1), respectively, in eif2ak4−/− MEFs compared to WT MEFs (). These results show that activation of the EIF2AK4-EIF2A-ATF4 pathway in response to AIEC infection is essential to limit AIEC intracellular replication and AIEC-induced inflammation.
Figure 2. Eif2ak4 depletion results in increased AIEC intracellular replication and enhanced AIEC-induced production of cytokines and chemokines. Wild-type (WT) MEFs and MEFs depleted for Eif2ak4 (eif2ak4−/−) were infected with AIEC LF82 for 6 h (B), 12 h (D), 24 h (E) or the indicated time (A, C). In parallel, MEFs were treated with KRB for 4 h. (A) Western blot analysis for the activation of the EIF2AK4-EIF2A-ATF4 pathway. (B) qRT-PCR analysis for mRNA expression levels of ATF4 target genes, including Trib3, Atf3, Ddit3 and Asns. (C) Intracellular LF82 number counted on LB agar plates. (D) Representative confocal micrographs of cells infected with LF82-GFP. Actin cytoskeleton was stained with Rhodamine-phalloidin (red). Nuclei were stained with Hoechst (blue). Bars: 5 μm. (E) Secreted IL6 and CXCL1 amounts in cell culture supernatant quantified by ELISA. Data are means ± SEM of 6 replicates and are representative of 3 independent experiments. *, P < 0.05; **, P ≤ 0.005; ***, P ≤ 0.001.
EIF2AK4 is necessary for autophagy activation in response to AIEC infection
Given that a functional autophagy is required to restrain AIEC intracellular replication,Citation6,8,10,11 the potential involvement of the EIF2AK4-EIF2A-ATF4 pathway in autophagy response to AIEC infection was next examined. Analysis for the shift of LC3-I (microtubule associated protein 1 light chain 3; the free cytosolic form) to LC3-II (the phagophore and autophagosomal form) by western blot, an indication of autophagy induction, showed that autophagy was induced in WT MEFs upon LF82 infection (). Induction of a functional and degradative autophagy flux in LF82-infected WT MEFs was confirmed by the concomitant decrease in SQSTM1 (sequestosome 1), a receptor protein incorporated into the autophagosome and degraded inside autolysosomes (). In contrast, autophagy was not induced in LF82-infected eif2ak4−/− MEFs (). However, treatment of both WT and eif2ak4−/− MEFs with 40 μg/ml rapamycin, an inducer of autophagy and used as a positive control, led to increased LC3-II levels and SQSTM1 degradation ().
Figure 3. Eif2ak4 depletion blocks autophagy activation in response to AIEC infection by inhibiting the ATF4-mediated transcription of autophagy genes. Wild-type (WT) MEFs and MEFs depleted for Eif2ak4 (eif2ak4−/−) were infected with AIEC LF82 or treated with 40 μg/ml rapamycin for 6 h. (A) Representative western blot analysis for LC3 and SQSTM1 levels. Quantification of band intensity of LC3-II relative to GAPDH from 3 independent blots is shown in the bottom panel. Values represent means ± SEM **, P ≤ 0.005. (B) Representative confocal micrographs of cells infected with LF82-GFP (green) and immunolabeled for LC3 (red). Nuclei were stained with Hoechst (blue). Bars: 5 μm. The graph to the right shows the percentage of LF82-GFP in LC3-positive vacuoles determined from 3 independent experiments, counting 20 cells/experiment. Values represent means ± SEM ***, P ≤ 0.001. (C) Representative confocal micrographs of WT, eif2ak4−/− and atg5−/− MEFs pre-transfected with the pEGFP-LC3 plasmid and infected with LF82 or treated with rapamycin. Bars: 5 μm. The graph to the right shows the number of GFP-LC3 puncta per cell determined from 3 independent experiments, counting 10 cells/experiment. Values represent means ± SEM ***, P ≤ 0.001. (D) qRT-PCR analysis for mRNA expression levels of the autophagy genes Map1lc3b, Atg7, Atg3, Becn1 and Sqstm1. Data are means ± SEM of 6 replicates and are representative of 3 independent experiments. *, P < 0.05. (E, F) ChIP analysis for the binding of ATF4 to the autophagy gene promoters. The protein-DNA complexes were immunoprecipitated with anti-ATF4 or a negative control IgG. (E) Representative agarose gels for the ATF4-binding regions in the autophagy gene promoters or Gapdh DNA in the input amplified by semiquantitative PCR. (F) Increase in the binding of ATF4 to the autophagy gene promoters, normalized to Gapdh DNA in the input, upon LF82 infection analyzed by qPCR. Data are means ± SEM of 4 replicates and are representative of 2 independent experiments. *, P < 0.05.
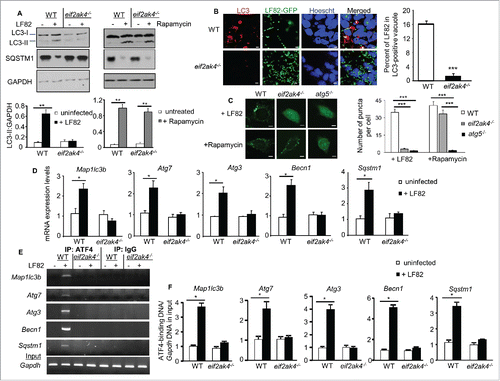
Confocal microscopy analysis showed that depletion of Eif2ak4 suppressed LC3 aggregation and therefore autophagic response to LF82 infection (). WT, eif2ak4−/− and atg5−/− MEFs were transfected with a pEGFP-LC3 plasmid and infected with LF82, and the numbers of puncta per cell, which allow estimation of autophagosome formation, were quantified. As shown in , the numbers of puncta were markedly increased in WT MEFs compared to eif2ak4−/− and atg5−/− MEFs upon LF82 infection. Rapamycin treatment, used as a positive control, increased the numbers of puncta in both WT and eif2ak4−/− MEFs, but not in atg5−/− MEFs (). These data show that EIF2AK4 is necessary for autophagy induction in response to AIEC infection.
Activation of the EIF2AK4-EIF2A-ATF4 pathway in response to AIEC infection is essential for ATF4-mediated transcription of autophagy genes
It has been recently shown that activation of the EIF2AK4-EIF2A-ATF4 pathway is required for stress-induced transcription of several autophagy genes.Citation19 In an effort to demonstrate the mechanism underlying the involvement of this pathway in autophagy response to AIEC infection, we analyzed the transcriptional levels of several genes that are involved in autophagy. Among the autophagy genes tested, mRNA expression levels of Sqstm1, Map1lc3b (microtubule-associated protein 1 light chain 3 beta), Becn1/Beclin 1, Atg3 (autophagy-related 3) and Atg7 (autophagy-related 7) were significantly increased in response to AIEC infection in WT MEFs, and this was inhibited in eif2ak4−/− MEFs ().
To investigate whether the transcription factor ATF4, activated in response to AIEC infection, can bind to promoters of these autophagy genes, a ChIP assay was performed using an anti-ATF4 antibody and a negative control IgG. In WT MEFs, AIEC infection induced binding of ATF4 to the promoter of the Sqstm1, Map1lc3b, Becn1, Atg3 and Atg7 genes as analyzed by semi-quantitative () and quantitative PCR (). This was not observed in eif2ak4−/− MEFs (). No binding was detected for the negative control IgG, indicating specificity of the ChIP assay ().
Together, these data show that EIF2AK4 is essential to induce transcription of autophagy genes via ATF4 binding, and thereby activation of autophagy in response to AIEC infection.
siRNA-mediated depletion of EIF2AK4 in IECs inhibits ATF4-mediated transcription of autophagy genes and autophagy activation in response to AIEC infection, increasing AIEC intracellular replication and AIEC-induced inflammation
To verify the results obtained with eif2ak4−/− MEFs, we examined the effect of depleting EIF2AK4 expression in T84 cells on autophagy-mediated control of AIEC intracellular replication and AIEC-induced inflammation. Transfection of T84 cells with EIF2AK4 siRNA inhibited EIF2AK4 and EIF2A activation, and ATF4 expression induced by LF82 infection compared with control siRNA (). KRB treatment, used as a positive control, also activated the EIF2AK4-EIF2A-ATF4 pathway in control siRNA-transfected cells, and this was not observed in EIF2AK4 siRNA-transfected cells ().
Figure 4. SiRNA-mediated depletion of EIF2AK4 in T84 cells inhibits autophagy activation in response to AIEC infection, increasing AIEC intracellular replication and AIEC-induced inflammation. T84 cells were transfected with control or EIF2AK4-specific siRNA for 36 h and then infected or not with the AIEC LF82 strain for 6 h or incubated with KRB for 1 h (A) or treated with 40 μg/ml rapamycin for 6 h (B). (A) Western blot analysis for the activation of the EIF2AK4-EIF2A-ATF4 pathway. (B) Representative western blot analysis for LC3 and SQSTM1 levels. Quantification of band intensity of LC3-II relative to GAPDH from 3 independent blots was shown in the bottom panel. Values are means ± SEM *, P < 0.05; **, P ≤ 0.005. (C) qRT-PCR analysis for mRNA expression levels of the autophagy genes MAP1LC3B, ATG3, ATG7, BECN1 and SQSTM1. (D) Cells were infected with AIEC LF82 in the presence or absence of 40 μg/ml rapamycin. Intracellular LF82 number was counted on LB agar plates. (E) Secreted IL8 amounts in cell culture supernatant quantified by ELISA. Data are means ± SEM of 6 replicates and are representative of 3 independent experiments. *, P < 0.05; **, P ≤ 0.005; ***, P ≤ 0.001 (C-E).
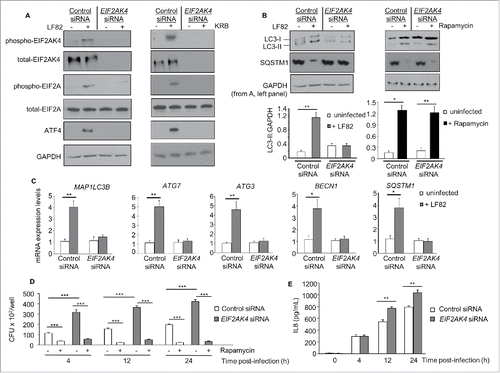
In control siRNA-transfected cells, autophagy was induced upon LF82 infection as shown by increased LC3-II levels and SQSTM1 protein degradation (). This was not observed in EIF2AK4 siRNA-transfected cells upon LF82 infection (). Rapamycin treatment, however, activated autophagy in both control siRNA- and EIF2AK4 siRNA-transfected cells (). The suppression of autophagy activation in EIF2AK4 siRNA-transfected cells was correlated with the inhibition of LF82-induced transcription of the SQSTM1, MAP1LC3B, BECN1, ATG3 and ATG7 genes (). The defect in autophagy response to LF82 infection in EIF2AK4 siRNA-transfected T84 cells consequently led to an increased number of intracellular LF82 () and enhanced IL8 (interleukin 8) production (). Rapamycin treatment prevented the increased number of intracellular LF82 bacteria in both control siRNA- and EIF2AK4 siRNA-transfected cells (). These results, which are consistent with the results obtained with eif2ak4−/− MEFs, show that EIF2AK4 is essential to induce a functional autophagy necessary to control AIEC intracellular replication and AIEC-induced inflammation.
The EIF2AK4-EIF2A-ATF4 pathway is activated in IECs from AIEC-infected mice, inducing transcription of autophagy genes and thereby the autophagy process
To gain insights into the role of the EIF2AK4-EIF2A-ATF4 pathway in autophagy response to in vivo infection, we used eif2ak4−/− mice and a model of AIEC LF82 infection. In agreement with the in vitro data, LF82 infection of WT mice activated the EIF2AK4-EIF2A-ATF4 pathway in ileal epithelial cells as shown by increased phospho-EIF2AK4, phospho-EIF2A and ATF4 protein levels (), as well as enhanced transcriptional levels of ATF4 target genes Trib3, Atf3, Ddit3 and Asns (Fig. S1). In eif2ak4−/− mice, AIEC infection did not affect the levels of phospho-EIF2AK4, phospho-EIF2A or ATF4, as well as the transcriptional levels of the ATF4 target genes ( and S1), indicating the important role of EIF2AK4 in activation of the EIF2A-ATF4 axis in response to AIEC infection. In contrast, this pathway was not activated in the ileum of mice infected with the K12 MG1655 strain (Fig. S2). As the EIF2AK4-EIF2A-ATF4 pathway is activated in IECs in response to AIEC infection, we questioned whether this could be a consequence of inflammation. We then investigated whether intestinal inflammation induces activation of this pathway using a mouse model of dextran sodium sulfate (DSS)-induced colitis. As shown in Figure S3, DSS treatment did not increase the levels of intestinal phospho-EIF2AK4 in WT mice. Together, these data suggest that activation of the EIF2AK4-EIF2A-ATF4 pathway is specific to AIEC infection and is not a consequence of inflammation.
Figure 5. Activation of the EIF2AK4-EIF2A-ATF4 pathway in IECs from AIEC-infected mice, inducing an autophagy response. Wild-type (WT) and eif2ak4−/− mice were infected with LF82 by gavage and ileal enterocytes were extracted as described in Materials and methods. (A) Representative immunoblots for the analysis of the EIF2AK4-EIF2A-ATF4 pathway. The graphs to the right represent quantification of immunoblots of phospho-EIF2AK4, phospho-EIF2A and ATF4 relative to GAPDH from n = 9 mice/group. (B) Representative western blot analysis for LC3 and SQSTM1 levels. Quantification of band intensity of LC3-II relative to GAPDH from n = 9 mice/group was shown in the bottom panel. (C) qRT-PCR analysis for mRNA expression levels of the autophagy genes Map1lc3b, Atg3, Atg7, Becn1 and Sqstm1. (D) ChIP analysis for the binding of ATF4 to the autophagy gene promoters. The protein-DNA complexes were immunoprecipitated with anti-ATF4 antibody or a negative control IgG. Representative agarose gels for the ATF4-binding regions in the autophagy gene promoters or Gapdh DNA in the input amplified by semiquantitative PCR are shown. Data are means ± SEM from n = 9 mice/group. **, P ≤ 0.005; ***, P ≤ 0.001.
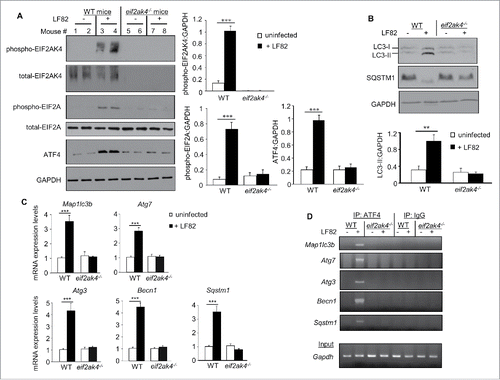
In agreement with the in vitro data, autophagy was induced in response to LF82 infection in ileal epithelial cells from WT mice, but not eif2ak4−/− mice as shown by increased LC3-II levels and SQSTM1 degradation (). LF82 infection of WT mice increased the transcriptional levels of the autophagy genes Sqstm1, Map1lc3b, Becn1, Atg3 and Atg7 in IECs (). Amplification of ATF4-binding regions in the promoters of these genes by semi-quantitative and quantitative PCR showed that LF82 infection induced binding of ATF4 to the promoters of the Sqstm1, Map1lc3b, Becn1, Atg3 and Atg7 genes in IECs from WT mice ( and S4). No binding was detected for the negative control IgG, indicating specificity of ATF4 binding (). Invalidation of Eif2ak4 suppressed the LF82-induced transcription of these genes in IECs () by inhibiting ATF4 binding to their promoters ( and S4).
Together, these data show that activation of the EIF2AK4-EIF2A-ATF4 pathway in mouse enterocytes is essential for ATF4-mediated transcription of autophagy genes, thereby activating autophagy in response to AIEC infection.
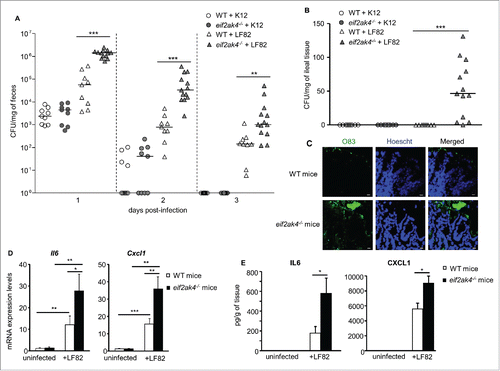
EIF2AK4 is essential to restrain AIEC intestinal colonization and AIEC-induced inflammation in mice
We next investigated the role of the EIF2AK4-EIF2A-ATF4 pathway in the control of AIEC colonization of the gut. LF82 persistence in the gut of eif2ak4−/− mice was increased compared to WT mice as shown by marked increased LF82 number counted in the feces at different days post-infection (). No difference in the number of K12 in the feces between WT and eif2ak4−/− mice was observed (). At day of sacrifice, the presence of LF82 bacteria was observed only in the ileum of eif2ak4−/− mice (). No K12 bacteria were detected in the ileal mucosa from either WT or eif2ak4−/− mice (). Confocal microscopy analysis using rabbit antiserum against E. coli lipopolysaccharide O83 consistently showed a huge number of LF82 bacteria (green) associated with the ileal mucosa of eif2ak4−/− mice, but not WT mice (). This was accompanied by increased production of the pro-inflammatory cytokine IL6 and chemokine CXCL1 at mRNA and protein levels in the intestine of eif2ak4−/− mice compared to WT mice (). These data, which are in agreement with the in vitro data, confirm the role of the EIF2AK4-EIF2A-ATF4 pathway in the control of AIEC replication and AIEC-induced inflammation.
Figure 6. Eif2ak4 depletion increases AIEC intestinal colonization and AIEC-induced inflammation in mice. Wild type (WT) and eif2ak4−/− mice were infected with 109 CFU of LF82 bacteria or the E. coli K12 MG1655 strain by gavage for 3 d. Quantification of LF82 and K12 bacteria in the feces collected every day post-infection (A) or associated with the intestinal mucosa determined at day of sacrifice (B). The quantification for each mouse (symbols) and the median (bars) are shown (A, B). (C) Representative confocal micrographs of ileal sections immunolabeled for E. coli lipopolysaccharide O83 (green) from LF82-infected WT and eif2ak4−/− mice. Nuclei were stained with Hoechst (blue). Bars: 10 μm. (D) mRNA expression levels of Cxcl1 and Il6 in mouse ileal tissues were assessed by qRT-PCR. (E) Secreted CXCL1 and IL6 amount in ileal tissue culture supernatant were quantified by ELISA. Data are means ± SEM from n = 9 mice/group. *, P < 0.05; **, P ≤ 0.005; ***, P ≤ 0.001.
We then sought to determine whether the increase in AIEC-induced intestinal inflammation in eif2ak4−/− mice compared to WT mice is because EIF2AK4 plays a role in inflammation or because its role is specific to autophagy response during AIEC infection. For that, we assessed the effect of Eif2ak4 depletion on intestinal inflammation induced by DSS treatment in mice. During the course of DSS treatment, both WT and eif2ak4−/− mice displayed severe body weight loss, rectal bleeding, and diarrhea with no significant difference in clinical score (Fig. S5A and B). In addition, after DSS treatment, WT and eif2ak4−/− mice exhibited severe inflammation with loss of entire crypts and surface epithelium in the colon and massive infiltration of inflammatory cells into the mucosa and submucosa with similar levels (Fig. S5C). As shown in Fig. S5D, WT and eif2ak4−/− mice showed a similar increase in DSS-induced colonic production of proinflammatory cytokines TNF (tumor necrosis factor), IL1B (interleukin 1 beta), and IL6 and chemokine CXCL1. These results demonstrate that depletion of Eif2ak4 did not affect susceptibility of mice to DSS-induced colitis.
Together, these data suggest that the role of the EIF2AK4-EIF2A-ATF4 pathway is specific to autophagy modulation during AIEC infection but not to inflammatory responses.
The EIF2AK4-EIF2A-ATF4 pathway is activated in noninflamed CD and deactivated in inflamed CD
We finally investigated whether there is a link between the EIF2AK4-EIF2A-ATF4 pathway and CD. Western blot analysis showed increased levels of phospho-EIF2AK4, phospho-EIF2A and ATF4, indicating activation of this pathway, in ileal biopsies from patients with noninflamed CD compared with the normal control group (). However, compared to the noninflamed CD group, activation of this pathway was suppressed in inflamed CD. In addition, mRNA expression levels of the known target genes of ATF4 (TRIB3, ATF3, DDIT3 and ASNS) and of the autophagy genes shown in this study to be regulated by ATF4 (SQSTM1, MAP1LC3B, BECN1, ATG3 and ATG7) were markedly increased in the noninflamed CD group, and were suppressed in the inflamed CD group (). These data suggest that the EIF2AK4-EIF2A-ATF4 pathway is activated in the pre-onset of CD, and a defect in the activation of this pathway could be one of the mechanisms contributing to active disease.
Figure 7. The EIF2AK4-EIF2A-ATF4 pathway is activated in noninflamed CD and deactivated in inflamed CD. Total proteins and RNA were extracted from the terminal ileal biopsy specimens from normal controls or patients with CD (with normal appearance [noninflamed CD] or with macroscopic inflammation [inflamed CD]). (A) Western blot analysis for the activation of the EIF2AK4-EIF2A-ATF4 pathway is shown in the left panel. Quantification of band intensity is shown in the right graphs. (B) qRT-PCR analysis for mRNA expression levels of the known ATF4 target genes (TRIB3, ATF3, DDIT3 and ASNS) and the autophagy genes shown to be regulated by ATF4 in this study (SQSTM1, MAP1LC3B, BECN1, ATG3 and ATG7). Data are represented as scatter dot plots; each dot shows one case number and the bars show the median. Data were analyzed by the nonparametric Mann-Whitney test. *, P < 0.05; **, P ≤ 0.005; ***, P ≤ 0.001.
![Figure 7. The EIF2AK4-EIF2A-ATF4 pathway is activated in noninflamed CD and deactivated in inflamed CD. Total proteins and RNA were extracted from the terminal ileal biopsy specimens from normal controls or patients with CD (with normal appearance [noninflamed CD] or with macroscopic inflammation [inflamed CD]). (A) Western blot analysis for the activation of the EIF2AK4-EIF2A-ATF4 pathway is shown in the left panel. Quantification of band intensity is shown in the right graphs. (B) qRT-PCR analysis for mRNA expression levels of the known ATF4 target genes (TRIB3, ATF3, DDIT3 and ASNS) and the autophagy genes shown to be regulated by ATF4 in this study (SQSTM1, MAP1LC3B, BECN1, ATG3 and ATG7). Data are represented as scatter dot plots; each dot shows one case number and the bars show the median. Data were analyzed by the nonparametric Mann-Whitney test. *, P < 0.05; **, P ≤ 0.005; ***, P ≤ 0.001.](/cms/asset/44d6e897-c927-43e5-900f-5e091af604e9/kaup_a_1156823_f0007_b.gif)
Discussion
Exciting recent progress has been made in understanding the mechanistic links between defects in autophagy-mediated pathogen handling, innate immune activation, and CD pathogenesis. A specific pathogenic group of E. coli, called adherent-invasive E. coli (AIEC), which abnormally colonize the intestinal mucosa of CD patients,Citation3–5 has been extensively used to investigate the role of autophagy in CD etiology. AIEC bacteria are able to adhere to and invade human intestinal epithelial cells (IECs),Citation4,6 survive and replicate within macrophages,Citation7,8 induce high production of proinflammatory cytokines and chemokines,Citation7,9,10 and exacerbate intestinal inflammation in a genetically susceptible mouse model.Citation9 AIEC bacteria can also target M cells to interact with Peyer patches and macrophages within the lamina propria.Citation22 We have shown that in response to AIEC infection, autophagy is induced in host cells to control the AIEC bacterial intracellular replication.Citation6,8,10,11 CD-associated risk alleles in autophagy genes ATG16L1 and IRGM result in impaired efficacy of autophagy defense with a substantial abnormal persistence of AIEC and intestinal inflammation.Citation6,11 The signaling pathways implicated in the induction of autophagy in host cells infected with AIEC remain to be investigated.
Evidence has suggested that the EIF2A-ATF4 axis could be implicated in autophagy regulation. The transcription factor ATF4 plays a key role in regulating transcription of the essential autophagy gene MAP1LC3B in response to endoplasmic reticulum stress and severe hypoxia.Citation23,24 The EIF2AK4-EIF2A-ATF4 pathway was reported to be required to induce autophagy in yeast and in mammals in response to nutrient deprivation.Citation25,26 We recently demonstrated that activation of the EIF2AK4-EIF2A-ATF4 pathway in response to cellular stress is essential for autophagy induction by increasing the transcription of a set of genes implicated in the formation, elongation and function of the phagophore and/or autophagosome.Citation19 This pathway is also induced in host cells upon infection with the intracellular bacterial pathogens Shigella and Salmonella.Citation21 However, to our knowledge, no data are available on the link between this pathway and CD and its involvement in host responses to infection with pathogenic bacteria associated with the disease.
In the present study, we investigated whether this pathway is involved in autophagy activation in host cells in response to AIEC infection. We show that activation of the EIF2AK4-EIF2A-ATF4 pathway in IECs in response to AIEC infection induces binding of the transcription factor ATF4 to promoters of several autophagy genes including MAP1LC3B, BECN1, SQSTM1, ATG3 and ATG7, activating their transcription. This confers host cells with highly active autophagy to efficiently restrain AIEC intracellular multiplication and to suppress subsequent inflammatory responses. B'Chir et al. reported that transcription of the ATG5 and ATG16L1 genes is also activated by the EIF2A-ATF4 signaling pathway in response to cellular stress.Citation19 However, under AIEC LF82 infection, we previously reported that mRNA and protein expression levels of ATG5 and ATG16L1 are decreased since a post-transcriptional regulation mediated by microRNAs is implicated.Citation10 We reason that AIEC can upregulate the levels of several host miRNAs to subvert autophagy, while host cells induce a functional autophagy as a defense mechanism to control bacterial replication. These are 2 arms of host-AIEC interaction, and depending on the physio-pathological status, one could dominate the other. We hypothesize that host cells are normally able to control the intracellular replication of AIEC by inducing an autophagy response. However, high levels of MIR30C and MIR130A under pathological conditions as observed in CD ileal mucosaCitation10 could impair the autophagy response of the host by inhibiting expression of ATG5 and ATG16L1.
Depletion of EIF2AK4 blocks the autophagy response to AIEC infection, leading to abnormal AIEC persistence in host cells and aggravated inflammation. The in vivo results using eif2ak4−/− mice were consistent with the in vitro data showing an important role for the EIF2AK4-mediated autophagy response to AIEC infection. Importantly, we demonstrate that the EIF2AK4-EIF2A-ATF4 pathway was not activated in IECs in a mouse model of DSS-induced colitis, and that depletion of Eif2ak4 did not affect susceptibility of mice to DSS treatment. This suggests that the role of the EIF2AK4-EIF2A-ATF4 pathway is specific to autophagy modulation during AIEC infection but not to inflammatory responses.
Importantly, we showed that the EIF2AK4-EIF2A-ATF4 pathway was activated in noninflamed CD compared with the normal control group, and was suppressed in inflamed CD. These data suggest that in the pre-onset of CD, the EIF2AK4-EIF2A-ATF4 pathway is activated, and a defect in the activation of this pathway could be one of the mechanisms contributing to the active disease. This is the first study showing a potential implication of the EIF2AK4-EIF2A-ATF4 pathway in CD etiology. We propose that one of the crucial events in the pathogenesis of CD might be the loss of tightly regulated autophagy caused by dysregulation of autophagy-regulating signaling pathways.
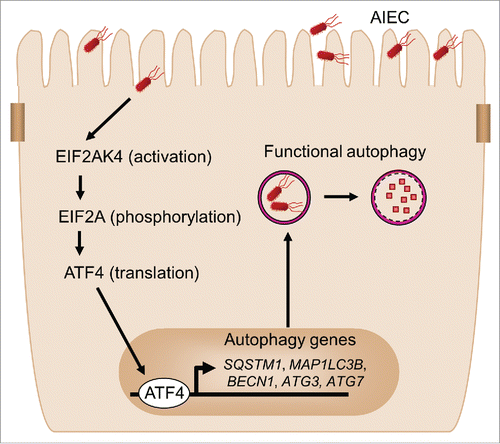
In summary, we have uncovered the critical role of the EIF2AK4-EIF2A-ATF4 signaling pathway in host autophagy response to CD-associated AIEC infection. The EIF2AK4-EIF2A-ATF4 pathway is activated in host cells upon AIEC infection, inducing transcription of autophagy genes and thereby a functional autophagy. This serves as a host defense mechanism to eliminate intracellular AIEC and to inhibit AIEC-induced intestinal inflammation (). This study highlights how the modulation of host metabolic pathways affects the cellular innate defense against CD-associated bacteria. More generally, this study should provide a better understanding of the dialogue between AIEC infection and host autophagy response, and therefore may contribute to the development of therapeutic strategies to control CD-associated intracellular bacterial pathogens.
Figure 8. Proposed model for the involvement of the EIF2AK4-EIF2A-ATF4 pathway in autophagy response to AIEC infection in intestinal epithelial cells. Upon AIEC infection, the EIF2AK4-EIF2A-ATF4 signaling pathway is activated in host cells, inducing transcription of several autophagy genes including SQSTM1, MAP1LC3B, BECN1, ATG3 and ATG7 and thereby a functional autophagic response. This serves as a host defense mechanism to eliminate intracellular AIEC and to inhibit AIEC-induced intestinal inflammation.
Materials and methods
Cell culture
The intestinal epithelial cell line T84 (ATCC, CCL-248) was maintained in an atmosphere containing 5% CO2 at 37°C in the culture medium recommended by ATCC. Eif2ak4-deficient (eif2ak4−/−) and the corresponding wild-type MEFsCitation27 were a gift from Dr. David Ron (Institute of Metabolic Science, Cambridge, UK). atg5−/− MEFsCitation28 were kindly provided by Dr. Noboru Mizushima (Graduate School of Medicine, The University of Tokyo, Tokyo, Japan). MEFs were cultured in DMEM (Gibco, 11960-044) supplemented with 10% fetal bovine serum (Dominique Dutscher, S1900-500), 2 mM L-glutamine (Gibco, 25030-024), 1 mM sodium pyruvate (PAA laboratories, SH30239.01) and 1% MEM nonessential amino acids (Dominique Dutscher, X0556-100).
Bacterial strains
The AIEC reference strain LF82 isolated from a chronic ileal lesion of a CD patient,Citation29 the nonpathogenic E. coli K12 MG1655 strain and the commensal E. coli HS strain were used. The plasmid pFPV25.1, which harbors the green fluorescent protein (GFP), was used to visualize the LF82 bacteria by confocal microscopy (LF82-GFP strain). Bacteria were grown in Luria-Bertani (LB) broth (Laboratorios Conda, 1551.0) or on LB agar (Conda, 1800.00) plates overnight at 37°C.
Human samples
Biopsies from the terminal ileum of patients with CD (with normal appearance [noninflamed CD] or with macroscopic inflammation [inflamed CD]) and normal controls were collected during endoscopic examination. Non-IBD individuals that had surveillance colonoscopies were considered “normal controls.” Biopsies were immediately soaked in RNAlater (Qiagen, 76104) and stored at -80°C until further processing. Characteristics of human biopsies are presented in Table S1.
siRNA, plamid and in vitro transfection
Control siRNA-A (Ambion, sc-37007) and EIF2AK4 siRNA (Ambion, sc-45644) were used. The pEGFP-LC3 plasmid was kindly provided by Dr. Tamotsu Yoshimori (Osaka University, Osaka, Japan).Citation30 T84 cells cultured on 12-well and 24-well plastic plates were transfected with 50 nM of EIF2AK4 siRNA or control siRNA using Lipofectamine 2000 (Invitrogen, 11668027) and Opti-MEM I reduced serum medium (Invitrogen, 319885062). WT, eif2ak4−/− and atg5−/− MEFs seeded on coverslips were transfected with 500 ng of pEGFP-LC3 plasmid using Lipofectamine 2000 and Opti-MEM I reduced serum medium.
In vitro infection and invasion assay
Cells were seeded on 24-well plates and infected at a multiplicity of infection (MOI) of 10 bacteria per cell. Invasion assays were performed as previously described.Citation6 Briefly, after 3 h of incubation in the culture medium without antibiotics, cells were washed with phosphate-buffered saline (PBS; Gibco, 14190-094). To determine the numbers of intracellular bacteria, culture medium containing 100 µg/ml gentamicin was added for the indicated time. Cells were lysed with 1% Triton X-100 (Sigma-Aldrich, G-1264) in deionized water. Samples were serially diluted and plated onto LB agar plates, and the colony-forming units (CFU) were determined.
Buffer for amino acid starvation
Cells were amino acid-starved by incubation with Krebs Ringer buffer (KRB; 118.5 mM NaCl [Euromedex, 1112-A], 4.74 mM KCl [Euromedex, P017-A], 1.18 mM KH2PO4[Euromedex, 2018], 23.4 mM NaHCO3[Euromedex, 6885-1], 5 mM glucose [Sigma-Aldrich, G8270], 2.5 mM CaCl2[Euromedex, T885], and 1.18 mM MgSO4 [Euromedex, P027-A], adjusted to pH 7.6 by titration with 1 N NaOH [Euromedex, EU2-9183-5]) for the indicated time.
Colitis induction
Colitis was induced in age- and sex-matched wild-type (WT) and eif2ak4−/− littermates by oral administration of 3% (w/v) DSS (36-50 kDa; MP Biomedicals, 160110) in the drinking water. eif2ak4−/− miceCitation27 were kindly provided by Dr. D. Ron (Institute of Metabolic Science, Cambridge, UK). Mice were weighed daily. Clinical score was assessed at the end of DSS treatment as described previously.Citation31
In vivo infection and bacterial colonization evaluation
WT and eif2ak4−/− littermates were treated with an antibiotic cocktail containing 500 mg/l metronidazole (Sigma-Aldrich, M3761), 500 mg/l vancomycin (Sigma-Aldrich, 861987), 1 g/l neomycin (Sigma-Aldrich, N6386) and 1 g/l ampicillin (Euromedex, EU0400-D) for 7 d as described,Citation32 and 24 h later were orally challenged with 109 CFU of LF82 or K12 MG1655 bacteria for 3 d. Fresh fecal samples (100-200 mg) were collected from individual mice every day post-infection and resuspended in PBS to determine bacterial persistence in the gut. At d 3 post-infection, mice were sacrificed, and 2-cm segments of ileal tissues were removed, opened longitudinally and homogenized in PBS to determine the number of bacteria associated with the intestinal mucosa. After serial dilution, fecal or ileal samples were plated on LB agar plates containing 50 µg/ml ampicillin and 20 µg/ml erythromycin (Euromedex, E002) to isolate LF82 bacteria, or containing 300 µg/ml rifampicin (Euromedex, 1059-B) to isolate K12 MG1655 bacteria, and incubated at 37°C overnight.
Isolation of intestinal epithelial cells (IECs)
Ileal epithelial cells were extracted as described previously.Citation10 Briefly, mouse ileum was dissected and flushed with a solution containing 154 mM NaCl and 1 mM DTT (Euromedex, EU0006-B) to remove fecal contents. The ileal segments were ligated, filled with PBS, and incubated in PBS at 37°C. After 15 min, the PBS was substituted with PBS supplemented with 1.5 mM EDTA (Euromedex, EU0084-A) and 0.5 mM DTT. After 30 min at 37°C, one ligature was removed and contents were collected. The recovered IECs were washed twice in PBS by centrifugation at 1,300 rpm for 5 min and were used to examine activation of the EIF2AK4-EIF2A-ATF4 pathway and autophagic activity.
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed using the Pierce Agarose ChIP kit following the manufacturer's instructions (Pierce, 1862281). Briefly, 2 × 106 uninfected T84 cells or cells infected with LF82 bacteria at a MOI of 10 for 4 h were fixed with 1% formaldehyde, washed with cold PBS and lysed. Nuclei were digested with micrococcal nuclease (supplied in the kit) to shear crosslinked chromatin, and the lysates were pelleted and precleared. The protein-DNA complexes were incubated with 4 μg of rabbit anti-ATF4 antibody (Cell Signaling Technology, 11845) or a nonspecific negative control rabbit IgG (supplied in the kit) overnight and then incubated with protein A/G agarose resin (supplied in the kit) followed by an elution, and cross-links were reversed. After recovery, DNA was subjected to semi-quantitative or quantitative PCR analysis. Specific primers used to amplify the ATF4-binding region in the promoters of autophagy genes are indicated in Table S2. The inputs were analyzed in parallel using ChIP control primers, which amplify a region of the GAPDH promoter, supplied in the Pierce ChIP kit. Fold increase in the binding of ATF4 to the autophagy gene promoter quantified by quantitative PCR was calculated using the Ct method as follows: ΔΔCt = (CtATF4-binding DNA- CtGAPDH DNA in input)infected - (CtATF4-binding DNA- CtGAPDH DNA in input)uninfected, and the final data were derived from 2−ΔΔCT.
Protein extraction and western blot analysis
T84 and MEF cells and human biopsies were lysed in radioimmune precipitation assay buffer (150 mM NaCl, 0.5% sodium deoxycholate [Sigma-Aldrich, D6750], 50 mM Tris-HCl [Euromedex, EU0011-C] pH 8, 0.1% SDS [Euromedex, EU0660-A], 0.1% Nonidet P-40 [Fluka BioChemeka, 74385]) supplemented with protease inhibitors (Roche, 11836145001). Enterocytes extracted from mouse ileum were homogenized in the same buffer and cell suspensions were sonicated. The homogenates were centrifuged at 16,000 × g for 20 min at 4°C. Proteins were separated on SDS-PAGE gels, transferred to nitrocellulose membranes, and blocked with 5% nonfat milk in PBS containing 0.1% Tween-20 (Euromedex, 2001-A). Membranes were then probed with the relevant primary antibodies: anti-phospho-EIF2AK4 (Abcam, ab75836), anti-EIF2AK4 (Cell Signaling Technology, 3302), anti-phospho-EIF2A/eIF2α (Cell Signaling Technology, 3597), anti-EIF2A/eIF2α (Cell Signaling Technology, 9722), anti-ATF4 (Cell Signaling Technology, 11845), anti-LC3 (Sigma-Aldrich, L8918), anti-SQSTM1 (Santa Cruz Biotechnology, sc-28359) and anti-GAPDH (Cell Signaling Technology, 2118). After washes, membranes were incubated with HRP-conjugated goat anti-rabbit (Cell Signaling Technology, 7074) or rabbit anti-mouse (Cell Signaling Technology, 7076) secondary antibody, and blots were detected using the Enhanced Chemiluminescence Detection kit (Amersham Biosciences, RPN2108).
RNA extraction and quantitative RT-PCR (qRT-PCR)
Total RNAs were extracted using TRIzol reagent (Invitrogen, 15-596-026) or the RNeasy Mini Kit (Qiagen, 74104), and were reverse transcribed using the PrimeScript™ RT reagent kit (Takara Bio, RR037A) to quantify mRNA expression levels. qRT-PCR was performed using SYBR Green qPCR Master Mix (Takara Bio, RR820A) on a Mastercycler Realplex (Eppendorf, Montesson, France) using the specific primers indicated in Table S3. Human RNA18S and mouse Rplp0/36B4 were used as internal controls. Fold-induction was calculated using the Ct method as follows: ΔΔCt = (Cttarget gene- Cthousekeeping gene)treatment - (Cttarget gene- Cthousekeeping gene)nontreatment, and the final data were derived from 2−ΔΔCT.
Fluorescence microscopy
T84 cells seeded on coverslips were fixed with 4% paraformaldehyde (Thermo Fisher scientific, 28906), permeabilized with 0.5% Triton X-100 for 20 min, blocked with PBS containing 0.025% Triton X-100, 3% BSA (ID-Bio, 1005-70) and 5% fetal bovine serum. Mouse ileal tissues were embedded in OCT (Tissue Tek, 4583) and cut into 5-μm sections at −20°C using a cryostat. Cryostat sections were then fixed, permeabilized and blocked similarly. Cells or cryostat sections were immunostained at room temperature for 1 h or overnight at 4°C, respectively, with the indicated primary antibodies: rabbit antiserum against E. coli lipopolysaccharide O83 generously provided by Dr. Lothar Beutin (Department of Biological Safety, Robert Koch Institut, Berlin, Germany), or anti-LC3 (MBL, M115-3). After washes with PBS, slides were incubated with Alexa Fluor 488 donkey anti-rabbit (Molecular Probes, A21206) or Cy3 goat anti-mouse (Molecular Probes, A10521) secondary antibody. Actin cytoskeleton was stained for 15 min using TRITC-phalloidin (Sigma-Aldrich, P1951) or FITC-phalloidin (Sigma-Aldrich, P8582). Nuclei were stained with Hoechst 33342 (Sigma-Aldrich, B2261). Coverslips and tissue sections were mounted with a Mowiol solution (Calbiochem, 475904) and Mountex Mounting Medium (Cell Path, SEA0504-00A), respectively. Slides were examined with a Zeiss LSM 510 Meta confocal microscope. Each confocal microscopy image is representative of 3 independent experiments.
Enzyme-linked immunosorbent assays (ELISA)
The amount of IL8, CXCL1, IL6, IL1B and TNF secreted in the supernatants from cell culture or mouse ileal tissues cultured for 24 h in RPMI medium (Gibco, 31870-025) containing penicillin/streptomycin (Hyclone, SV30079.01) and 100 µg/ml gentamicin (Euromedex, EU0540-A) in an atmosphere containing 5% CO2 at 37°C, respectively, was determined by ELISA (R&D Systems, human CXCL8/IL-8 Quantikine ELISA kit, D8000C; Mouse CXCL1/KC DuoSet ELISA, DY453; Mouse IL6 DuoSet ELISA, DY406; Mouse IL1 β DuoSet ELISA, DY401; Mouse TNF-alpha DuoSet ELISA, DY410) according to the manufacturer's instructions.
Ethics statement
Animal protocols were approved by the Ethics Committee CEMEAA (CEMEA CE16-09, Clermont-Ferrand, France). Protocols with human biopsies were approved by the local ethics commission of the Canton of Bern, Switzerland.
Statistical analysis
Results are presented as means ± SEM or median. Statistical analysis was performed using the paired t tests for in vitro data and the nonparametric Mann-Whitney test for in vivo data with GraphPad Prism version 5.01 software (GraphPad Software). A P value less than 0.05 was considered statistically significant. *, P < 0.05; **, P ≤ 0.005; ***, P ≤ 0.001.
ABBREVIATIONS
| AIEC | = | adherent-invasive Escherichia coli |
| ASNS | = | asparagine synthetase (glutamine-hydrolyzing) |
| ATF3 | = | activating transcription factor 3 |
| ATF4 | = | activating transcription factor 4 |
| ATG16L1 | = | autophagy-related 16 like 1 |
| ATG3 | = | autophagy-related 3 |
| ATG5 | = | autophagy-related 5 |
| ATG7 | = | autophagy-related 7 |
| BECN1 | = | Beclin 1, autophagy-related |
| CD | = | Crohn disease |
| CFU | = | colony-forming units |
| ChIP | = | chromatin immunoprecipitation |
| CXCL1 | = | chemokine (C-X-C motif) ligand 1 (melanoma growth stimulating activity, alpha) |
| DDIT3 | = | DNA damage inducible transcript 3 |
| DSS | = | dextran sodium sulfate |
| EIF2 | = | eukaryotic translation initiation factor 2 |
| EIF2AK4 | = | eukaryotic translation initiation factor 2 alpha kinase 4 |
| ELISA | = | enzyme-linked immunosorbent assay |
| IBD | = | inflammatory bowel diseases |
| IEC | = | intestinal epithelial cell |
| IL | = | interleukin |
| IL1B | = | interleukin 1 beta |
| IRGM | = | immunity-related GTPase family, M |
| KRB | = | Krebs-Ringer buffer |
| LRRK2 | = | leucine-rich repeat kinase 2 |
| MAP1LC3B | = | microtubule-associated protein 1 light chain 3 beta |
| MOI | = | multiplicity of infection |
| mRNA | = | messenger RNA |
| PTPN2 | = | protein tyrosine phosphatase, non-receptor type 2 |
| qRT-PCR | = | quantitative reverse-transcription polymerase chain reaction |
| SQSTM1 | = | sequestosome 1 |
| TNF | = | tumor necrosis factor |
| TRIB3 | = | tribbles pseudokinase 3 |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| WT | = | wild type |
| XBP1 | = | X-box binding protein 1 |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
KAUP_A_1156823_Supplemental.pdf
Download PDF (2.1 MB)Acknowledgments
We dedicate this article to the memory of Prof. Arlette Darfeuille-Michaud, who sadly passed away on June 28, 2014. The authors thank the Imagerie Confocale Clermont-Ferrand (ICCF) platform (IFR79 Santé Université d'Auvergne, Clermont-Ferrand, France) for confocal microscopy and the Centre Imagerie Cellulaire Santé (Facultés de Médecine et Pharmacie) for tissue embedding and sectioning. We thank Drs. Emilie Vazeille, Marie-Agnès Bringer and Pierre Lapaquette for valuable discussion.
Funding
This work was supported by the Ministère de la Recherche et de la Technologie, Inserm (UMR1071), INRA (USC 2018), grant from the Association F. Aupetit (AFA), grant from Agence nationale de la recherche (ANR) “Jeune Chercheuse Jeune Chercheur” Nutribiote SVSE 1 Edition 2013 (to N.B.), the European Union FP7 People Marie Curie International Incoming Fellowship (to H.N.) and grant from Région Auvergne (“Nouveau chercheur” grant to H.N.).
References
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol 2010; 28:573-621; PMID:20192811; http://dx.doi.org/10.1146/annurev-immunol-030409-101225
- Carriere J, Darfeuille-Michaud A, Nguyen HT. Infectious etiopathogenesis of Crohn's disease. World J Gastroenterol 2014; 20:12102-17; PMID:25232246; http://dx.doi.org/10.3748/wjg.v20.i34.12102
- Martinez-Medina M, Aldeguer X, Lopez-Siles M, Gonzalez-Huix F, Lopez-Oliu C, Dahbi G, Blanco JE, Blanco J, Garcia-Gil LJ, Darfeuille-Michaud A. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn's disease. Inflamm Bowel Dis 2009; 15:872-82; PMID:19235912; http://dx.doi.org/10.1002/ibd.20860
- Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser AL, Barnich N, Bringer MA, Swidsinski A, Beaugerie L, Colombel JF. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology 2004; 127:412-21; PMID:15300573; http://dx.doi.org/10.1053/j.gastro.2004.04.061
- Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM. Enhanced Escherichia coli adherence and invasion in Crohn's disease and colon cancer. Gastroenterology 2004; 127:80-93; PMID:15236175; http://dx.doi.org/10.1053/j.gastro.2004.03.054
- Lapaquette P, Glasser AL, Huett A, Xavier RJ, Darfeuille-Michaud A. Crohn's disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell Microbiol 2010; 12:99-113; PMID:19747213; http://dx.doi.org/10.1111/j.1462-5822.2009.01381.x
- Glasser AL, Boudeau J, Barnich N, Perruchot MH, Colombel JF, Darfeuille-Michaud A. Adherent invasive Escherichia coli strains from patients with Crohn's disease survive and replicate within macrophages without inducing host cell death. Infect Immun 2001; 69:5529-37; PMID:11500426; http://dx.doi.org/10.1128/IAI.69.9.5529-5537.2001
- Lapaquette P, Bringer M-A, Darfeuille-Michaud A. Defects in autophagy favour adherent-invasive Escherichia coli persistence within macrophages leading to increased pro-inflammatory response. Cell Microbiol 2012; 14:791-807; PMID:22309232; http://dx.doi.org/10.1111/j.1462-5822.2012.01768.x
- Carvalho FA, Barnich N, Sivignon A, Darcha C, Chan CH, Stanners CP, Darfeuille-Michaud A. Crohn's disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J Exp Med 2009; 206:2179-89; PMID:19737864; http://dx.doi.org/10.1084/jem.20090741
- Nguyen HT, Dalmasso G, Muller S, Carriere J, Seibold F, Darfeuille-Michaud A. Crohn's disease-associated adherent invasive Escherichia coli modulate levels of microRNAs in intestinal epithelial cells to reduce autophagy. Gastroenterology 2014; 146:508-19; PMID:24148619; http://dx.doi.org/10.1053/j.gastro.2013.10.021
- Brest P, Lapaquette P, Souidi M, Lebrigand K, Cesaro A, Vouret-Craviari V, Mari B, Barbry P, Mosnier JF, Hebuterne X, et al. A synonymous variant in IRGM alters a binding site for miR-196 and causes deregulation of IRGM-dependent xenophagy in Crohn's disease. Nat Genet 2011; 43:242-5; PMID:21278745; http://dx.doi.org/10.1038/ng.762
- Boya P, Reggiori F, Codogno P. Emerging regulation and functions of autophagy. Nat Cell Biol 2013; 15:713-20; PMID:23817233; http://dx.doi.org/10.1038/ncb2788
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:1845-6; PMID:23656658; http://dx.doi.org/10.1056/NEJMra1205406
- Nguyen HT, Lapaquette P, Bringer MA, Darfeuille-Michaud A. Autophagy and Crohn's disease. J Innate Immun 2013; 5:434-43; PMID:23328432; http://dx.doi.org/10.1159/000345129
- Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol 2010; 11:55-62; PMID:19898471; http://dx.doi.org/10.1038/ni.1823
- Homer CR, Richmond AL, Rebert NA, Achkar JP, McDonald C. ATG16L1 and NOD2 interact in an autophagy-dependent antibacterial pathway implicated in Crohn's disease pathogenesis. Gastroenterology 2010; 139(1630–41): 1641 e1-2; PMID:20637199
- Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 2010; 16:90-7; PMID:19966812; http://dx.doi.org/10.1038/nm.2069
- Fritz T, Niederreiter L, Adolph T, Blumberg RS, Kaser A. Crohn's disease: NOD2, autophagy and ER stress converge. Gut 2011; 60:1580-8; PMID:21252204; http://dx.doi.org/10.1136/gut.2009.206466
- B'Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 2013; 41:7683-99; PMID:Can't; http://dx.doi.org/10.1093/nar/gkt563
- Tsalikis J, Croitoru DO, Philpott DJ, Girardin SE. Nutrient sensing and metabolic stress pathways in innate immunity. Cell Microbiol 2013; 15:1632-41; PMID:23834352
- Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, Yang C, Emili A, Philpott DJ, Girardin SE. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 2012; 11:563-75; PMID:22704617; http://dx.doi.org/10.1016/j.chom.2012.04.012
- Chassaing B, Rolhion N, de Vallee A, Salim SY, Prorok-Hamon M, Neut C, Campbell BJ, Soderholm JD, Hugot JP, Colombel JF, et al. Crohn disease–associated adherent-invasive E. coli bacteria target mouse and human Peyer's patches via long polar fimbriae. J Clin Invest 2011; 121:966-75; PMID:21339647; http://dx.doi.org/10.1172/JCI44632
- Rzymski T, Milani M, Singleton DC, Harris AL. Role of ATF4 in regulation of autophagy and resistance to drugs and hypoxia. Cell Cycle 2009; 8:3838-47; PMID:19887912; http://dx.doi.org/10.4161/cc.8.23.10086
- Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL. Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 2010; 29:4424-35; PMID:20514020; http://dx.doi.org/10.1038/onc.2010.191
- Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, Bobrovnikova-Marjon E, Diehl JA, Ron D, Koumenis C. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 2010; 29:2082-96; PMID:20473272; http://dx.doi.org/10.1038/emboj.2010.81
- Talloczy Z, Jiang W, Virgin HW t., Leib DA, Scheuner D, Kaufman RJ, Eskelinen EL, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2alpha kinase signaling pathway. Proc Natl Acad Sci U S A 2002; 99:190-5; PMID:11756670; http://dx.doi.org/10.1073/pnas.012485299
- Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell 2000; 6:1099-108; PMID:11106749; http://dx.doi.org/10.1016/S1097-2765(00)00108-8
- Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguchi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, et al. Autophagy defends cells against invading group A Streptococcus. Science 2004; 306:1037-40; PMID:15528445; http://dx.doi.org/10.1126/science.1103966
- Darfeuille-Michaud A, Neut C, Barnich N, Lederman E, Di Martino P, Desreumaux P, Gambiez L, Joly B, Cortot A, Colombel JF. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn's disease. Gastroenterology 1998; 115:1405-13; PMID:9834268; http://dx.doi.org/10.1016/S0016-5085(98)70019-8
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 2000; 19:5720-8; PMID:11060023; http://dx.doi.org/10.1093/emboj/19.21.5720
- Nguyen HT, Dalmasso G, Torkvist L, Halfvarson J, Yan Y, Laroui H, Shmerling D, Tallone T, D'Amato M, Sitaraman SV, et al. CD98 expression modulates intestinal homeostasis, inflammation, and colitis-associated cancer in mice. J Clin Invest 2011; 121:1733-47; PMID:21490400; http://dx.doi.org/10.1172/JCI44631
- Dalmasso G, Nguyen HT, Ingersoll SA, Ayyadurai S, Laroui H, Charania MA, Yan Y, Sitaraman SV, Merlin D. The PepT1-NOD2 signaling pathway aggravates induced colitis in mice. Gastroenterology 2011; 141:1334-45; PMID:21762661; http://dx.doi.org/10.1053/j.gastro.2011.06.080