
ABSTRACT
The secretion of proteins that lack a signal sequence to the extracellular milieu is regulated by their transition through the unconventional secretory pathway. IDE (insulin-degrading enzyme) is one of the major proteases of amyloid beta peptide (Aβ), a presumed causative molecule in Alzheimer disease (AD) pathogenesis. IDE acts in the extracellular space despite having no signal sequence, but the underlying mechanism of IDE secretion extracellularly is still unknown. In this study, we found that IDE levels were reduced in the cerebrospinal fluid (CSF) of patients with AD and in pathology-bearing AD-model mice. Since astrocytes are the main cell types for IDE secretion, astrocytes were treated with Aβ. Aβ increased the IDE levels in a time- and concentration-dependent manner. Moreover, IDE secretion was associated with an autophagy-based unconventional secretory pathway, and depended on the activity of RAB8A and GORASP (Golgi reassembly stacking protein). Finally, mice with global haploinsufficiency of an essential autophagy gene, showed decreased IDE levels in the CSF in response to an intracerebroventricular (i.c.v.) injection of Aβ. These results indicate that IDE is secreted from astrocytes through an autophagy-based unconventional secretory pathway in AD conditions, and that the regulation of autophagy is a potential therapeutic target in addressing Aβ pathology.
Introduction
Alzheimer disease (AD) is characterized by senile plaques, neurofibrillary tangles, and neuronal cell death.Citation1 Abnormal aggregates of the amyloid-beta peptide (Aβ) are deposited in the extracellular senile plaques and are associated with neurodegeneration in AD.Citation2 In AD brains, intracellular and extracellular Aβ accumulation occurs because of the imbalance between the production of Aβ and its removal (clearance) from the brain. IDE (insulin-degrading enzyme) is a major endogenous Aβ degrading enzyme that mediates Aβ clearance, and its levels and enzymatic activity are negatively correlated with the size of the amyloid plaques and AD pathology.Citation3 In AD brains, IDE is mainly detected in astrocytes,Citation4 and located in the cytosol and intracellular compartments such as mitochondria and peroxisomes.Citation5 However, it is also known to play a role in the extracellular milieu via the secretory pathway.Citation6 IDE has no signal peptide sequence for secretion through the conventional secretory pathway, and despite many studies demonstrating that IDE is secreted and/or associated with the cell surface, little is known about the underlying secretory mechanism.
Macroautophagy (hereafter referred to as autophagy) is a fundamental biological process in eukaryotes,Citation7 and has an impact on essential biological processes in human health including aging, cancer, neurodegenerative disease, immunity, and metabolic disorders.Citation8,9 Autophagy is currently best known as a degradative pathway that delivers cytoplasmic material and organelles to the lysosomes for degradation.Citation10 All autophagy-related processes include the formation of double-membrane structures called the autophagosomes, and are induced by the inhibition of MTOR (mechanistic target of rapamycin [serine/threonine kinase]) signaling pathway. Autophagosomes and their contents undergo clearance upon fusion with endosomes (amphisomes) or lysosomes (autolysosomes) for degradation and recycling (autophagic flux).Citation7,10 However, recent studies show that autophagy also has a role in nonautophagic processes, especially in the secretory pathway.Citation11 In eukaryotic cells, the autophagy-based secretory pathway (autosecretionCitation12) regulates the unconventional secretion of several cytosolic proteins such as IL1B (interleukin 1, beta), IL18, HMGB1 (high mobility group box 1), and VWF (von Willebrand factor), as well as ATP.Citation13-15 There are 3 principal features defining autophagy-based secretion: 1) proteins lacking a signal sequence, 2) contribution of autophagy-related (ATG) genes, and 3) association with GORASP (Golgi reassembly and stacking protein).Citation16
In the brains of patients with AD and animal models, pathological autophagic vacuoles (AVs) are accumulated through increased induction via the AMPK-MTOR pathway and dysfunction in the autophagy-lysosomal degradation.Citation17-19 Maintaining autophagic flux is important for cell survival, and dysregulated autophagy is expected to accelerate AD progression; however, the mechanism is not yet fully understood. In this study, we found that Aβ increased IDE secretion from astrocytes in a time- and concentration-dependent manner, and Aβ-induced IDE secretion was associated with autophagy. In addition, in vitro and in vivo data suggests that autophagic flux is important in IDE secretion. GORASP and RAB8A also had a role in IDE secretion. To determine the IDE secretion levels in vivo, the cerebrospinal fluid (CSF) was analyzed because the CSF has more physical contact with the brain than any other fluid in the body, as it is not separated from the brain by the tightly regulated blood brain barrier (BBB). We found that the mice with global haploinsufficiency of an ATG7 (Atg7+/− mice) showed decreased IDE activities and expression levels in CSF in response to an intracerebroventricular (i.c.v.) injection of Aβ, indicating that this Aβ-induced IDE secretion is mediated by autophagy. Investigating the role of autophagy on IDE secretion in the AD brain revealed that 1.5-mo-old Tg6799 mice, an AD mouse model,Citation20 showed increased IDE secretion in the CSF compared to the littermate control mice. The CSF of 7-mo-old Tg6799, however, showed reduced IDE level, and dysfunction in the lysosome. Finally, CSF from patients with AD showed decreased IDE activity and level, and this reduction was correlated with cognitive impairment in AD patients using clinical dementia rating (CDR) and mini-mental state examination (MMSE) scores. Overall, these results indicate that IDE is secreted from astrocytes through autophagy-based secretory pathway in AD, and regulation of autophagy is a potential therapeutic target in Aβ pathology.
Results
Aβ induces extracellular secretion of functional IDE from astrocytes
Previous studies have reported astrocytes as a main source for IDE in AD pathology;Citation4 therefore, we determined whether Aβ regulates IDE levels in the extracellular space of astrocytes. Exogenous Aβ1-42, but not Aβ42-1 (reverse form of Aβ1-42; revAβ), increased IDE secretion from the mouse primary astrocytes and human astrocytoma cells, U373MG, in a time- and dose-dependent manner ( to C for primary astrocytes; Fig. S1A and B for U373MG cells). To determine whether the IDE secretion is regulated by Aβ, Ide siRNA was transfected into astrocytes. Aβ increased IDE secretion in the scrambled siRNA-transfected cells, but not the Ide siRNA-transfected cells (Fig. S1C and D). To examine whether the IDE secreted by Aβ treatment has a function as insulysin (an insulin-degrading function), an IDE enzymatic activity assay was adopted. Increased fluorescence intensity generated by the cleavage of fluorometric IDE substrates in the Aβ-treated, astrocyte-conditioned media (Aβ-ACM) was detected (). When bacitracin A, an IDE inhibitor, was added to the Aβ-ACM, the fluorescence intensity was decreased, while thiorphan, a MME (membrane metallo-endopeptidase) inhibitor, did not alter the increased fluorescence intensity by Aβ (), indicating that Aβ-ACM contains functional IDE that degrades insulin. In an alternative approach, Aβ degradation assay was performed. When the Aβ-ACM was incubated with recombinant Aβ peptide, the level of the remaining Aβ peptide was reduced (). Furthermore, the reduced Aβ levels after incubation with the Aβ-ACM were restored when either bacitracin A or insulin (competing partner of Aβ to IDE) was added (). These data demonstrate that Aβ-induced IDE, secreted from astrocytes, functions as a protease.
Figure 1. Aβ induces extracellular secretion of functional IDE from primary astrocytes. (A, B) Increased IDE levels secreted from the primary astrocytes with Aβ treatment, in a concentration-(A) and time-(B) dependent manner. Treatments were 0.5, 1 or 2 μM of Aβ for 24 h (A), and 1 µM of Aβ for 1, 3, 12 or 24 h (B). Blots are representative of at least 3 independent experiments. (C) Measurement of IDE levels in the media from primary astrocytes by ELISA. (D) IDE enzymatic activity in media containing 1 µM Aβ and/or several inhibitors (Thiorphan [10 µM], Bacitracin A [10 µM]) for 24 h. Baci. A, Bacitracin A. (E) Cell-free Aβ degradation assay. rIDE, recombinant IDE protein (0.3 ng/μL). The arrowhead indicates remaining Aβ levels. Data were obtained from at least 3 replicates for each group (N = 3 experiments). (F) Aβ degradation assay with several drugs. Ins, insulin (0.5 μM); Baci. A, bacitracin A (10 µM). Data were obtained from at least 3 replicates for each group (N = 3 experiments). Values are mean ± SEM *, P < 0.05; **, P < 0.01; and ***, P < 0.001 vs. vehicle-treated cells; #, P < 0.05 vs. cells treated with Aβ.
![Figure 1. Aβ induces extracellular secretion of functional IDE from primary astrocytes. (A, B) Increased IDE levels secreted from the primary astrocytes with Aβ treatment, in a concentration-(A) and time-(B) dependent manner. Treatments were 0.5, 1 or 2 μM of Aβ for 24 h (A), and 1 µM of Aβ for 1, 3, 12 or 24 h (B). Blots are representative of at least 3 independent experiments. (C) Measurement of IDE levels in the media from primary astrocytes by ELISA. (D) IDE enzymatic activity in media containing 1 µM Aβ and/or several inhibitors (Thiorphan [10 µM], Bacitracin A [10 µM]) for 24 h. Baci. A, Bacitracin A. (E) Cell-free Aβ degradation assay. rIDE, recombinant IDE protein (0.3 ng/μL). The arrowhead indicates remaining Aβ levels. Data were obtained from at least 3 replicates for each group (N = 3 experiments). (F) Aβ degradation assay with several drugs. Ins, insulin (0.5 μM); Baci. A, bacitracin A (10 µM). Data were obtained from at least 3 replicates for each group (N = 3 experiments). Values are mean ± SEM *, P < 0.05; **, P < 0.01; and ***, P < 0.001 vs. vehicle-treated cells; #, P < 0.05 vs. cells treated with Aβ.](/cms/asset/0cbfb9cc-ed68-48e1-87ac-73289f278fbb/kaup_a_1159375_f0001_b.gif)
Aβ-induced IDE secretion is associated with an autophagy-based unconventional secretory pathway
To examine the mechanism of Aβ-induced IDE secretion, transcript levels of IDE after Aβ treatment were measured by RT-PCR and qPCR (quantitative real-time PCR). Aβ did not alter Ide mRNA levels in astrocytes (). Since IDE has no signal sequence for secretion, we further investigated the secretory mechanism of IDE. To examine the IDE secretion via an unconventional secretory pathway, BFA (brefeldin A), a blocker of conventional secretion, was added along with Aβ. BFA failed to inhibit Aβ-induced IDE secretion, and even increased IDE secretion (Fig. S1E and F). Because MMP9 (matrix metallopeptidase 9), another Aβ-degrading enzyme, has a signal sequence, BFA inhibited MMP9 secretion in the Aβ treatment (Fig. S1G). Our results also confirmed that the secretion of HMGB1, a previously known protein secreted through unconventional secretory pathway,Citation13 increased after BFA treatment, similarly to IDE (Fig. S1H). Because BFA induced the endoplasmic reticulum (ER) stress (indicated by increased DDIT3 levels; Fig. S1E and I) and increased IDE secretion (Fig. S1E and F), we investigated the role of ER stress on IDE secretion. We found that treatment with TUDCA, a chemical chaperone which can reduce ER stress,Citation21 inhibited Aβ-induced IDE secretion from primary astrocytes (Fig. S1J). Recent reports have shown that autophagy plays a role in unconventional protein secretion in mammalian cells.Citation11,13,14,22 To examine whether Aβ-induced IDE secretion is associated with autophagy, we utilized several autophagic modulators. We found that Aβ-induced autophagy is activated in astrocytes (; Fig. S2A and B), and that Aβ modulated the AMP-activated protein kinase (PRKA)-MTOR signal pathway, which in turn can regulate autophagy induction (Fig. S2C).Citation17 When 3-methyladenine (3MA), an autophagy inhibitor, was added along with Aβ, Aβ-induced IDE secretion was significantly inhibited by 3MA (; Fig. S2A), but not MMP9 (Fig. S1G). These data were confirmed by inhibiting autophagy with Atg5 siRNA (). To activate autophagy, rapamycin, a functional autophagy inducer that inhibits MTOR, was applied to astrocytes, resulting in increased IDE secretion from astrocytes (). To investigate whether IDE exists in the autophagosomes, IDE and LC3 were stained with their specific antibodies and then visualized using confocal microscopy and super resolution microscopy. Immunocytochemistry data showed that IDE was located in the autophagosomes (). Taken together, these data imply that Aβ-induced IDE secretion is tightly regulated by the autophagy pathway.
Figure 2. Aβ-induced IDE secretion from primary astrocytes is associated with the autophagy-mediated unconventional secretion pathway. (A) RT-PCR analysis of Ide mRNA levels under Aβ treatment conditions. (B) Quantification of Ide mRNA levels by qPCR. (C) Increased LC3-II levels in astrocytes by Aβ treatment demonstrate the role of autophagy in Aβ-induced IDE secretion; 3-MA (1 mM) was administered to vehicle- or Aβ-treated cells. (D) Quantitative analysis of (N = 4 experiments). (E) Western blot analysis of IDE levels in the media from Atg5 knockdown primary astrocytes. Representative images are shown, and data are represented as mean ± SEM from 4 independent experiments (N = 4 experiments). (F) Western blot analysis of secreted IDE levels from astrocytes after the treatment with inhibitors and/or activators in the presence of Aβ (1 µM for 24 h). Rap, rapamycin (2 µM for 6 h); MG, MG132 (5 µM for 6 h). Representative images are shown, and data are represented as mean ± SEM from 3 independent experiments (N = 3 experiments). (G) 3D-SIM reconstruction images of IDE expression in the autophagosomes. The 3D-SIM image was taken in a z-direction with a thickness of 0.150 μm, reconstructed into a 3D volume image. Scale bar represents 10 µm in the image with confocal microscopy. Low panels (a1, a2, and a3) show enlarged images using 3D-SIM. The b1 and b2 panels show lines of fluorescence tracing from images in a3. (H) The Pearson colocalization coefficient for IDE and LC3. The Pearson coefficient was derived from 3 independent experiments with 3 fields per experiment, for a total of 9 fields contributing to the cumulative result. *, P < 0.05; and **, P < 0.01 vs. vehicle-treated cells; #, P < 0.05; and ##, P < 0.01 vs. cells treated with Aβ. n.s. indicates not significant.
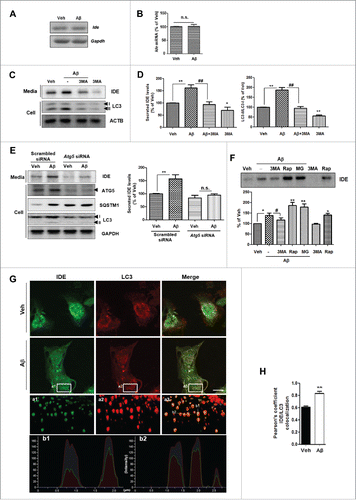
IDE is degraded through the proteasome pathway
Since the treatment with proteasome inhibitor, MG132, increased the IDE levels (), IDE might be a substrate for the proteasome. To examine this possibility, astrocytes were cotreated with MG132 and Aβ, resulting in increased IDE levels both intracellularly and extracellularly (Fig. S3A and B). Moreover, treatment with MG132 alone also increased IDE levels compared to vehicle treatment (Fig. S3A and B), indicating that basal IDE levels were regulated by the proteasome pathway.
Autophagic flux is important for Aβ-induced IDE secretion
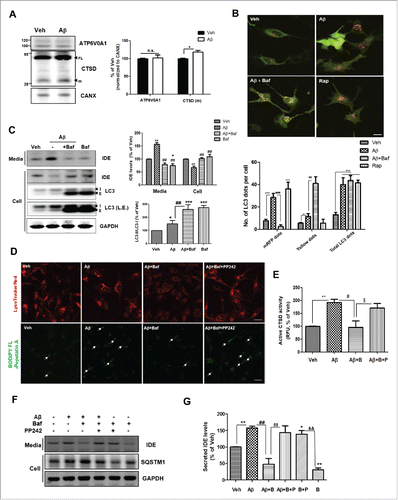
To determine the effect of the autophagy-lysosomal pathway on Aβ-induced IDE secretion, we investigated the change in lysosomal function following treatment with Aβ. When levels of ATP6V0A1 (ATPase, H+ transporting, lysosomal V0 subunit a1) and the mature form of CTSD (cathepsin D) were analyzed by western blotting (WB), we found that treatment with 1µM Aβ resulted in increased astrocytic expression of the mature form of CTSD (). Dupont et al. (2011)Citation13 reported that the secretion of IL1B, a substrate for autophagy-based secretion, was inhibited under conditions of lysosomal dysfunction, as induced by treatment with bafilomycin A1 (Baf; an inhibitor of vacuolar ATPase, which prevents lysosomal acidification and autophagosomal cargo degradation). To further examine the Aβ-induced IDE secretory pathway, Baf was used to block fusion between the autophagosome and the lysosome. If IDE is a substrate for autophagy-lysosomal degradation, Baf may increase total IDE levels intracellularly and extracellularly. When a monomeric red fluorescent protein-green fluorescent protein (mRFP-GFP) tandem fluorescence-tagged LC3 (Tf-LC3) construct was transfected to U373MG cells, Baf treatment increased the yellow signal (produced by the mRFP-GFP fusion form), indicating that the fusion of autophagosomes with lysosomes is blocked (). Baf treatment decreased IDE secretion from astrocytes (), suggesting that autophagic flux is important for IDE secretion and IDE is not a substrate for the autophagy-lysosomal pathway (). In addition, by live-cell imaging, we observed that IDE colocalized with the autophagosome (MDC-positive) and lysosome (LysoTracker Red-positive), and that Aβ accelerated the colocalization of IDE with the lysosome (Fig. S4A and B). To determine the involvement of lysosome function in Aβ-induced IDE secretion, restoration of the lysosome function was challenged by PP242, a well-known ATP-competitive MTOR inhibitor that activates the lysosomal function.Citation23 When PP242 was added together with Aβ, the signal intensity of BODIPY FL pepstatin A, a fluorescent probe for CTSD,Citation24 increased (), suggesting that PP242-induced lysosome activation in astrocytes. PP242 restored IDE secretion, which was decreased by Baf treatment (). Considered together, these data indicate that autophagic flux is important for Aβ-induced IDE secretion.
Figure 3. Autophagic flux is important for Aβ-induced IDE secretion. (A) Change in lysosomal proteins in primary astrocytes following treatment with Aβ (1 µM for 24 h). FL, full-length; m, mature form; CTSD, cathepsin D. CANX is a loading control for the membrane fraction. Representative images are shown, and data are represented as mean ± SEM from 3 independent experiments (N = 3 experiments). (B) Determination of autophagic flux using the Tf-LC3 construct. U373MG cells were preincubated with or without 10 nM Baf for 2 h, then exposed to 1 µM Aβ for 24 h. Cells were treated with rapamycin (Rap) for 6 h. The mRFP-positive dots indicate only mRFP-LC3 signals (autolysosome), and the yellow signal indicates colocalization of GFP and mRFP signals (defects in autophagic maturation). Scale bar represents 10 µm. Values represent the number of positive dots for red signals and yellow signals, and are mean ± SEM of 50 different cells in each independent experiment (N = 3). (C) Western blot analysis of IDE secretion after treatment with the lysosome inhibitor, Baf. L.E. indicates long exposure. (D) Lysosomal activity was determined with LysoTracker Red probe or BODIPY-FL-Pepstatin probe (for detection of active CTSD). The white arrows represent active CTSD signals, and Aβ-induced lysosomal clustering. Scale bar represents 10 µm. (E) Quantitative analysis of (N = 3 experiments). (F) Secreted IDE levels under the lysosome-modulating conditions (Baf, PP242 or Baf + PP242). Baf (10 nM for 2 h), PP242 (5 µM for 2 h) (G) Quantitative analysis of (N = 3 experiments). B, Baf; P, PP242. *, P < 0.05; **, P < 0.01; and ***, P < 0.001 vs. vehicle-treated cells; #, P < 0.05; ##, P < 0.01; and ###, P < 0.001 vs. cells treated with Aβ; §, P < 0.05; and §§, P < 0.01 vs. cells treated with Aβ + Baf; &&, P < 0.01 vs. cells treated with Baf.
The SlyX domain of IDE regulates IDE secretion by protecting degradation through the lysosome
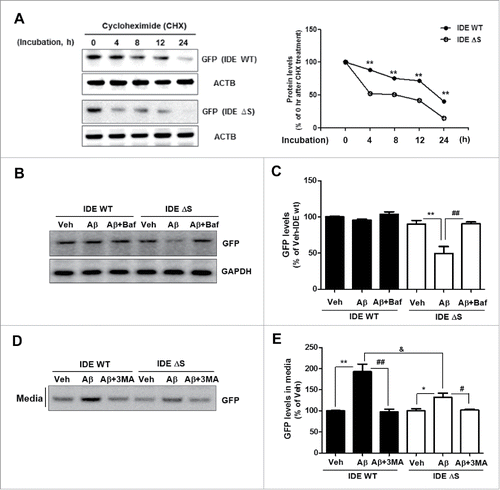
Then, how can IDE protein escape from the lysosome without degradation? Previous studies demonstrate that the SlyX motif, a novel amino acid motif (853EKPPHY858) close to the C terminus of IDE, determines IDE secretion –and yet, the mechanism by which it does so remains elusive.Citation25 To examine whether the SlyX domain of IDE has a role in protection from lysosomal degradation, protein stability was measured with IDE wild type (IDE WT) or SlyX domain-deletion mutant (IDE ΔS)-transfected U373MG cells. When cycloheximide (CHX) was added into U373MG cells, IDE ΔS degraded faster than IDE WT (). Moreover, we found that following Aβ treatment, IDE ΔS levels in the cells decreased—this can be reversed by inhibition of the lysosomes (). Secretion of IDE ΔS decreased more than that of IDE WT under both basal and Aβ treatment conditions (). These data indicate that the SlyX domain of IDE regulates Aβ-induced IDE secretion by preventing its lysosomal degradation.
Figure 4. The SlyX domain of IDE regulates IDE degradation through lysosomes. (A) Protein stability in IDE wild-type (IDE WT) and SlyX domain-deletion mutant (IDE ΔS)-transfected U373MG cells after cycloheximide (CHX) treatment. **, P < 0.01 vs. IDE protein levels immediately (0 h) after CHX treatment. (B) The increased degradation of IDE ΔS through lysosomes in the Aβ treatment (1 µM for 24 h). Baf (10 nM for 2 h). (C) Quantitative analysis of (N = 3 experiments). **, P < 0.01 vs. vehicle-treated, IDE ΔS-GFP-transfected cells; ##, P < 0.01 vs. IDE ΔS-GFP-transfected cells treated with Aβ. (D) The reduced secretion of IDE ΔS compared to IDE WT protein. (E) Quantitative analysis of (N = 3 experiments). *, P < 0.05; and **, P < 0.01 vs. vehicle-treated cells; #, P < 0.05; and ##, P < 0.01 vs. cells treated with Aβ; &, P < 0.05 vs. IDE WT-GFP-transfected cells, treated with Aβ.
Aβ-induced IDE secretion is mediated by RAB8A activity
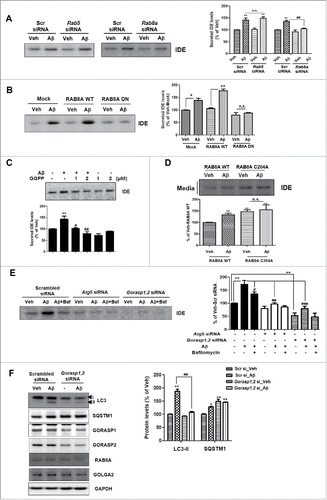
Several factors regulate the secretion of IDE. A previous study reports multiple roles of small GTPase RAB proteins in the secretory pathway.Citation26 In particular, RAB8A is known as one of the main regulators of membrane fusion and exocytosis.Citation13,27 To determine whether RAB8A regulates IDE secretion from astrocytes, we treated the Rab8a and Rab5 (as control) siRNA-transfected cells with Aβ ( and S5A). In Rab8a, but not Rab5, knockdown cells, IDE secretion was reduced significantly under Aβ treatment conditions (), suggesting that RAB8A was required for the enhanced IDE secretion caused by Aβ treatment. To investigate the association of RAB8A protein with Aβ-induced IDE secretion, the protein levels and the cellular localization of RAB8A were examined. When astrocytes were treated with Aβ, the RAB8A protein level was unchanged (Fig. S5B). However, RAB8A protein clustered in the cytoplasm following Aβ treatment (Fig. S5C). In this condition, RAB8A surprisingly colocalized with LC3-positive organelles, the autophagosomes (Fig. S5D). Furthermore, RAB8A dominant negative (DN) construct (RAB8AS22N) inhibited Aβ-induced IDE secretion (), indicating that RAB8A plays an important role in Aβ-induced IDE secretion. Then, how does Aβ affect RAB8A to modulate IDE secretion? Hooff et al. (2010) report that RAB prenylation, which is the addition of GGPP (geranylgeranylpyrophosphate) to a RAB protein, is necessary to regulate the activity of RAB proteins because RAB protein should be inserted into membranes to activate various biochemical pathways.Citation28 Since Aβ leads to the inhibition of prenylation in the neurons according to a previous study,Citation29 we examined whether Aβ regulates prenylation of RAB8A in astrocytes. When Aβ was applied to astrocytes, unprenylated RAB8A was increased, and conversely, prenylated RAB8A was decreased (Fig. S5E). This effect was restored by GGPP treatment (Fig. S5E). In addition, Aβ-induced IDE secretion was reduced by cotreatment with GGPP (), suggesting that the increased unprenylation of RAB8A induced IDE secretion. To confirm these results, we established RAB8A prenylation-deficient mutant cells (RAB8AC204A) (Fig. S5F and G). When the prenylation-deficient mutant of RAB8A was expressed, IDE secretion increased over that in RAB8A wild type (WT)-expressing cells (). These data indicate that IDE secretion is mediated by RAB8A activity, and that Aβ increases IDE secretion from astrocytes by the inhibition of RAB8A prenylation.
Figure 5. Aβ-induced IDE secretion is mediated by RAB8A activity and GORASP. (A) WB analysis of secreted IDE levels in the media from Rab5 or Rab8a knockdown primary astrocytes under treatment with Aβ or vehicle. Representative images are shown, and data are represented as mean ± SEM from 3 independent experiments (N = 3 experiments). (B) Association of Aβ-induced IDE secretion with RAB8A activity in primary astrocytes. RAB8A DN indicates a dominant-negative mutant form of RAB8A (RAB8AS22N) (N = 3). (C) Effect of prenylated proteins on Aβ-induced IDE secretion (1 µM for 24 h). (D) Effect of RAB8A on Aβ-induced IDE secretion, with a RAB8A prenylation-deficient mutant. Aβ (1 µM) treatment was applied to RAB8A WT or RAB8AC204A-transfected astrocytes for 24 h (N = 3). (E) WB analysis of IDE secretion from Atg5 knockdown and Gorasp (Gorasp1 and Gorasp2) double-knockdown astrocytes. Aβ (1 µM for 24 h); Baf (10 nM for 2 h) (N = 4). (F) Inhibition of autophagy in Gorasp1,2 double-knockdown astrocytes (N = 3). GOLGA2 was used as a marker for Golgi apparatus. *, P < 0.05; and **, P < 0.01 vs. vehicle-treated cells; #, P < 0.05; ##, P < 0.01; and ###, P < 0.001 vs. cells treated with Aβ.
GORASP regulates Aβ-induced IDE secretion by regulating autophagy
Since previous studies have reported that GORASP proteins control unconventional secretion of Acb1 in yeast and IL1B in macrophages,Citation13,30,31 we examined whether GORASP (GORASP1 and GORASP2 in mammals) regulate Aβ-induced IDE secretion. Double knockdown of Gorasp1 and Gorasp2 inhibited IDE secretion from astrocytes (). To determine if any particular GORASP is required for IDE secretion, siRNA for Gorasp1 or Gorasp2 was transfected into astrocytes. Both GORASP1 and GORASP2 reduced Aβ-induced IDE secretion, indicating that both are important for IDE secretion (Fig. S6A and B). To examine how the GORASP protein regulates IDE secretion, involvement of autophagy was examined because previous studies reported that GORASP2 controls autophagy initiation in macrophages.Citation13 When the effect of knockdown of Gorasp1 and Gorasp2 in astrocytes was examined, the LC3-II level was reduced, and the SQSTM1 (sequestosome 1) level increased in the Gorasp1, 2 double-knockdown astrocytes (). In addition, when the Tf-LC3 construct was transfected to scrambled siRNA- or Gorasp1, 2 siRNA-transfected cells, Gorasp1, 2 double-knockdown cells showed decreased mRFP and yellow dot numbers with Aβ and/or Baf treatments (Fig. S6C and D). When the autophagy inducers such as rapamycin were applied to Gorasp1, 2 double-knockdown cells, we found that Gorasp1, 2 double-knockdown cells showed decreased LC3-II levels compared to scrambled siRNA-transfected cells (Fig. S6E). These data indicate that GORASP regulates IDE secretion by modulating autophagy.
Autophagy-insufficient mouse brains show decreased IDE levels in the CSF, with Aβ-injection
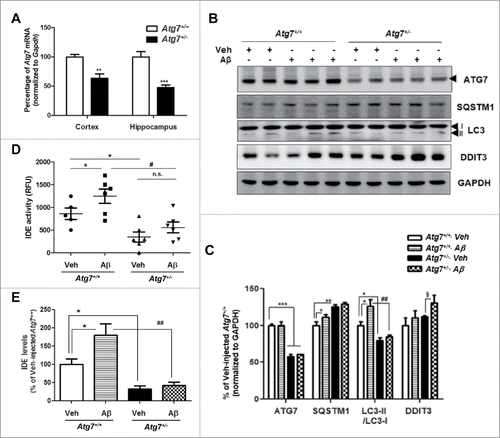
The above in vitro data showed increased IDE secretion from Aβ-treated astrocytes, mediated by an autophagy-based unconventional secretory pathway. Consistent with the in vitro data, we examined whether Aβ-induced IDE secretion occurs via the autophagy-based secretory pathway in vivo using mice with global haploinsufficiency of an essential autophagy gene (Atg7+/− mice). The brains from Atg7+/− mice showed reduced Atg7 mRNA and protein levels (), decreased LC3-II levels, and increased SQSTM1 levels, a substrates for autophagic protein degradation, as previously reported ().Citation32 With the i.c.v. injection of Aβ into the 2- to 3-mo-old- Atg7+/+ and Atg7+/− mice for 2 wk, increased autophagy (represented by an increase in LC3-II levels) in the brains of Atg7+/+ mice, but not Atg7+/− mice were detected (). IDE levels (and activity) were detected in the CSF because CSF has more physical contact with the brain than any other fluid. By ELISA and enzymatic activity assay for IDE in the CSF, Aβ-injected Atg7+/− mice showed a significant reduction in the IDE level and activity as compared to Atg7+/+ mice (). These data suggest that Aβ-induced IDE secretion is mediated by autophagy in vivo.
Figure 6. Autophagy-insufficient mouse brains show decreased IDE levels in the CSF under Aβ-injection conditions. (A) The analysis of Atg7 mRNA level by qPCR in the cortex and hippocampus of vehicle-injected Atg7+/+ and Atg7+/− mice (2- to 3-mo-old, N = 6 each group). **, P < 0.01; ***, P < 0.001 vs. vehicle-injected Atg7+/+ mice. (B) WB analysis of LC3, SQSTM1, or ATG7 expression in vehicle or Aβ-injected Atg7+/+ and Atg7+/− mice. DDIT3 was used to measure the level of ER stress. (C) Quantitative analysis of . (D) IDE enzymatic activities in the CSF of vehicle or Aβ-injected Atg7+/+ and Atg7+/− mice. (E) Results of quantification of IDE levels detected with ELISA in the CSF of vehicle or Aβ-injected Atg7+/+ and Atg7+/− mice. (N = 6 each group) *, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. vehicle-injected Atg7+/+ mice; #, P < 0.05; ##, P < 0.01 vs. Aβ-injected Atg7+/+ mice; §, P < 0.05 vs. vehicle-injected Atg7+/− mice. n.s. indicates not significant.
IDE activity is inversely correlated with age in AD mice
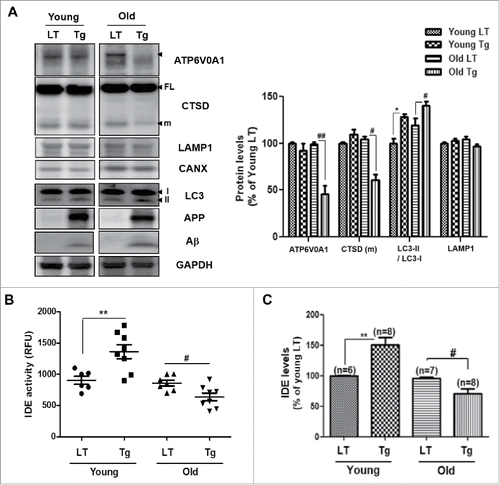
Previous studies have demonstrated accumulated autophagy in AD brains, and suggested that dysregulated autophagy regulates the progression of AD.Citation17,33 Because IDE secretion is associated with Aβ-induced autophagy from the aforementioned data, the regulation of IDE secretion in Tg6799 mice, an AD mouse model,Citation20 was examined. Since the Tg6799 mice are known to overexpress Aβ, have amyloid plaque deposition in the subiculum at 2 mo of age, and exhibit reduced synaptic markers, neuronal loss, and memory impairment at 6 mo of age,Citation20 1.5-mo-old (before plaque formation) and 7-mo-old (after plaque formation) Tg6799 mice were used in this study. Both 1.5-mo-old (young) and 7-mo-old (old) Tg6799 mice showed increased Aβ and autophagy levels (), but only the old Tg6799 mice showed defects in the lysosomes (decreased ATP6V0A1 and CTSD mature form) (). Electron microscopy analysis showed increased AVs in the old Tg6799 mice (Fig. S7A). To examine the secreted IDE levels, ELISA and enzymatic activity assays for the IDE protein were performed on the CSF from the Tg6799 mice and littermate control mice. IDE activity and protein levels increased in the CSF of the young Tg6799 mice, however, the old Tg6799 mice showed decreased IDE activity and levels in the CSF (). Based on in this study, IDE secretion is increased when the lysosomal function is intact to form autolysosomes in the young Tg6799 mice (). As the lysosomal function is defective in the old Tg6799 mice (), autophagic flux is inhibited, and Aβ-induced IDE secretion is reduced, followed by accumulation of Aβ and AVs in the brain (Fig. S7A).
Figure 7. IDE activity is inversely correlated with age in AD mice. (A) Lysosomal dysfunction in the brain of the 7-mo-old Tg6799 mice (old Tg), and in 1.5-mo-old Tg6799 (young Tg) mice. LT, age matched littermate controls; Tg, Tg6799; CTSD, cathepsin D. CANX is a loading control for the membrane fraction of the brain tissue, and GAPDH is a loading control for the total lysates. (B) IDE enzymatic activities in the CSF of 1.5-mo-old and 7-mo-old Tg6799 mice (and littermate controls). (C) Results of quantification of IDE levels detected with ELISA in the CSF of LT and Tg mice. Values are mean ± SEM *, P < 0.05; and **, P < 0.01 vs. young LT; #, P < 0.05; and ##, P < 0.01 vs. old LT. RFU, relative fluorescence unit.
Decreased enzymatic activity of IDE in the CSF of patients with AD
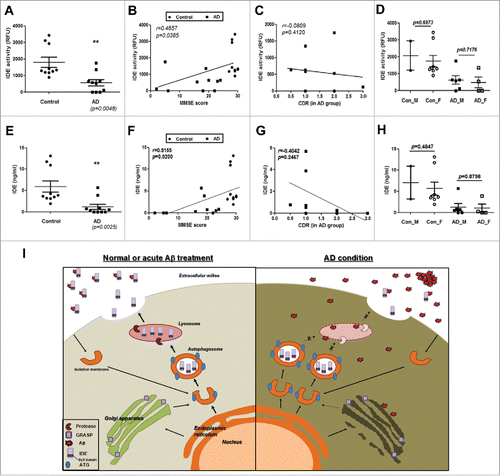
Since previous reports show accumulated autophagosomes and dysfunction in lysosomes in the AD-afflicted brain,Citation33,34 IDE activity and levels in the CSF of patients with AD were measured. When the IDE activity and levels in the CSF from AD patients (Table S1A and B) were measured, both activity and levels of IDE decreased in the CSF of AD patients (). These results showed a significant positive correlation with MMSE scores (), and, although not significant, a negative correlation tendency with CDR in the AD groups (). There was no difference between genders (). These results indicate that AD patients have reduced IDE protein levels and decreased enzymatic activity of the same, in their CSF, and that these correlate with cognitive impairment.
Figure 8. Decreased enzymatic activity and protein levels of IDE in the CSF of patients with AD. (A) IDE enzymatic activities in the CSF of control subjects and patients with AD (N = 10 in each group). RFU, relative fluorescence unit. (B) IDE activity data from the CSF show a correlation with the MMSE score (r = 0.4657, P value = 0.0385). (C) IDE activities in the CSF of AD patients show a tendency of negative correlation with AD group CDR scores (r = −0.0809, P = 0.4120). (D) Gender difference in IDE enzymatic activities. (E) IDE expression levels in the CSF of control subjects and patients with AD (N = 10 in each group). (F) The levels of IDE proteins in the CSF correlate with the MMSE score (r = 0.5155, P value = 0.0200). (G) Like IDE activity, IDE protein levels in the CSF of AD patients show a negative correlation with CDR scores calculated in the AD groups (r = −0.4042, P = 0.2467). (H) Gender difference in IDE levels in the CSF. Values are mean ± SEM *, P < 0.05; **, P < 0.01. ‘n.s.’ means not significant. (I) Schematic diagram of this study. Aβ increases IDE secretion from astrocytes via an autophagy-based unconventional secretory pathway, and Aβ-induced IDE secretion depends on the activity of RAB8A and GORASP. In the AD brain, lysosomal dysfunction occurs and, as a result, Aβ-induced IDE secretion is inhibited because autophagic flux is important for IDE secretion. Reduced IDE levels may result in increased levels of extracellular Aβ and contribute to the progression of AD pathogenesis.
Discussion
In this study, we found that IDE secretion from astrocytes under conditions of AD pathology is regulated by autophagy (). IDE is well known as a major protease for Aβ degradation, and the regulation of functional IDE secretion is important in AD progression. Prior to our work, it was known that, as IDE has no signal sequences, it is secreted from the microglia via an exosome-associated unconventional secretory pathway under statin treatment.Citation35 To determine whether Aβ-induced IDE secretion is associated with exosomes in astrocytes, both exosomes and nonexosome fractions were isolated from vehicle- or Aβ-treated ACM. Aβ increased the IDE secretion both in the exosomes and nonexosome fractions (Fig. S8A and B), indicating that the secretory pathway for IDE is mediated by both exosome- and nonexosome- associated pathways. Next, to determine whether Aβ-induced IDE secretion is caused by cell death, cell death assays were performed. 3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) and calcein-AM assays were performed (Fig. S9A and B). We found that treatment with 1 or 2 µM of Aβ for 24 h did not induce cell death in astrocytes. The mitochondrial uncoupling agent, CCCP, induced cell death as indicated by a positive control cell death assay. To confirm this result, TdT-mediated dUTP nick-end labeling (TUNEL) assay was performed (Fig. S9C). Consistent with the results of MTT and calcein-AM assay, Aβ did not induce cell death in this study. These results indicate that Aβ-induced IDE secretion is not caused by cell death. We examined the mechanisms underlying IDE secretion, especially under AD conditions, and found that Aβ increased functional IDE secretion in a time- and dose-dependent manner, and that this Aβ-induced IDE secretion was blocked by autophagy inhibition. Because a fundamental role of autophagy is the clearance of protein aggregates and pathogens, we investigated whether IDE secretion is regulated by the autophagy-lysosome pathway. When lysosomes were disrupted by the lysosomal inhibitor Baf, IDE secretion was blocked, and inversely, cotreatment with a lysosome activator, PP242, restored IDE secretion. These data indicate that autophagic flux is important for IDE secretion. From our in vivo data using AD-model mice, we found that the IDE activity and levels increased in the CSF of the young (1.5-mo-old) Tg6799 mice, while the old (7-mo-old) Tg6799 mice showed decreased IDE activity and protein levels in the CSF. These were consistent with the in vitro data because the old Tg6799 mice brains showed lysosomal dysfunction (decreased ATP6V0A1 and matured form of CTSD levels or increased AVs shown in EM images based on previous studies) (; Fig. S7).Citation36 In addition, patients with AD showed decreased IDE activity and levels in the CSF, and this reduction is correlated with cognitive impairment (). These data suggest that because the defect in the lysosome is a causative risk factor for AD, modulation of the lysosomal activity may serve as a novel therapeutic target in AD treatment by regulating IDE secretion.
It is surprising that IDE can be secreted from astrocytes via the autophagy-lysosome pathway. How can IDE proteins escape the acidic and harsh environment of the lysosome without degradation? The lysosomal lumen contains approximately 60 different soluble hydrolases, which are active in the acidic environment of the lumen (pH 4.5–5.0).Citation37 Many lysosomal proteins, such as lysosomal-associated membrane protein 1 (LAMP1) and cathepsins, are protected from lysosomal degradation by post-translational modifications such as mannose-6-phosphate tagging or hyper-glycosylation. Since IDE does not have these modification sites, we investigated mechanisms other than lysosomal degradation. We found that the SlyX domain (EKPPHY) of IDE contributes to the Aβ-induced IDE secretion by preventing its lysosomal degradation (), although the mechanism by which it does so is not yet known. Indeed, the SlyX domain is present in other proteins (HS3ST/heparan sulfate proteoglycans and PLSCR1 [phospholipid scramblase 1]), and these proteins are reportedly secreted via the unconventional secretory pathways.Citation38,39 We will, in a future study, try to identify the binding partner on the SlyX domain of IDE, and establish whether such a structure may confer a protective effect to IDE during lysosomal degradation. In this study, we found that IDE might be a substrate for the proteasome. Treatment with MG132, a proteasome inhibitor, increased both intracellular and extracellular IDE levels, as well as LC3-II levels (Fig. S3A and B). An association between the proteasome and autophagy has been noted in previous studies,Citation40 and we further found that MG132 treatment increased LC3-II levels in astrocytes. These results suggest that MG132 treatment increased IDE secretion both by increasing IDE protein levels and by autophagy induction. In a future study, we will check whether MG132 regulates key proteins of secretory autophagy (such as RAB8A or GORASP), as well as total autophagy levels. Results of this study have shown that application of 1 µM Aβ reduces proteasome activity by approximately 20% (data now shown) and activates autophagy in astrocytes; thus, IDE may escape degradation by the proteasome and autophagic secretion from astrocytes treated with Aβ.
What are the regulatory factors for the autophagy-based secretion of IDE? We focused on the RAB proteins because they play multiple roles in the secretory pathway.Citation26 According to our data, among the RAB proteins, RAB8A was important for IDE secretion. Specifically, IDE secretion was mediated by RAB8A activity, and Aβ increased IDE secretion from astrocytes by the inhibition of RAB8A prenylation. Although it is indirect evidence, previous studies have shown that statin treatment increases IDE secretion, and inhibits protein prenylation.Citation35,41 Whether IDE secretion by statin treatment occurs through same pathway as Aβ treatment should be investigated. Further, how IDE inside the lysosome is transported to the extracellular medium needs to be determined. Previous studies reported the topological inversion properties of autophagy, ferrying molecules from the cytosol to the lumen of secretory vesicles, however, this wanes quickly with time.Citation42,43 In addition, the core secretory machinery for vesicle exocytosis consists of SNAREs (N-ethylmaleimide-sensitive factor attachment protein receptors) and sec1/STXBP/Munc18-like proteins.Citation44 The marker protein of secretory lysosomes as the major vesicular compartment for Ca2+-regulated exocytosis from astrocytesCitation45 is VAMP7/TI-VAMP (vesicle associated membrane protein 7).Citation46 Verderio et al. (2012)Citation47 report that VAMP7 is the SNARE of secretory lysosomes contributing to ATP secretion from astrocytes. Considering that ATP is a cytosolic protein, which can be secreted from astrocytes following Aβ treatment,Citation48 and that ATP is known as a protein secreted via an autophagy-based unconventional secretory pathway,Citation49 VAMP7 might play a role as a regulatory factor for Aβ-induced autophagy-based secretion of IDE. In a future study, we will investigate the effect of possible regulatory factors on IDE secretion from astrocytes.
Astrocytes play a critical role in maintaining neuronal homeostasis by providing energy, eliminating waste, and regulating ionic flux.Citation50 Furthermore, astrocytes are well-positioned both metabolically and anatomically to play an important homeostatic role under basal conditions, as well as in pathological conditions such as AD. Astrocytes are activated to degrade Aβ at an early stage of AD. For the removal of extracellular Aβ, astrocytes take up Aβ bound to membrane receptors via endocytosis,Citation51 and degrade Aβ in the lysosomes. In a recent study, enhancing astrocytic lysosome biogenesis facilitates Aβ clearance, and attenuates AD pathogenesis.Citation52 Our results suggest another mechanism for the blockade of Aβ toxicity by astrocytes. In other words, we found that Aβ could be degraded by inducing IDE secretion from astrocytes via autophagy-based secretory pathway under AD conditions. In previous studies, IDE is expressed in several cell types in brain such as neurons and endothelial cells as well as astrocytes,Citation53,54 however, Dorfman et al.Citation4 show that astrocytes are the main cell types for IDE in AD pathology. In addition, when neurons, astrocytes or endothelial cells were treated with Aβ, we found that only astrocytes increased IDE secretion (Fig. S10). For detection of secreted IDE in vivo, we used the CSF of humans and mice as previously described.Citation55 CSF is a translucent body fluid that occupies the subarachnoid space and the ventricular system around the brain.Citation56 CSF protects the brain by providing mechanical and immunological defense. CSF can be obtained via a lumbar puncture, and is probably the most informative biomarker for the prognosis of neurodegenerative diseases. As a result, proteins or peptides that may be directly reflective of brain-specific activities, as well as of disease pathology, would most likely diffuse into the CSF, rather than into any other bodily fluid. These proteins and metabolites can serve as excellent biomarkers of AD as well as other neurodegenerative diseases.Citation56
The regulation of Aβ levels is of great interest for its therapeutic potential in AD. Apart from the interference with Aβ generation, a promising alternative may be the enhancement of Aβ degradation by targeting Aβ-degrading enzymes.Citation57 Many reports highlight the impact of decreased Aβ clearance from the central nervous system in late-onset AD.Citation58 Thus, regulating autophagy-based secretion of IDE by modulating lysosomal activity in astrocytes may lead to new therapeutic approaches for sporadic AD.
Materials and methods
Study participants
The subject population for this study included patients from the Soonchunhyang University Bucheon Hospital, South Korea, with a diagnosis of probable AD along with healthy controls. Participants were diagnosed by National Institute of Neurological and Communicative Disorders and Stroke–Alzheimer Disease and Related Disorders Association criteria and CDR of 0.5 (mild AD) – 3.0 (severe AD). Clinical evaluation included medical history, physical examination, MMSE, and CDR. All those with available biochemical measures were included in the study, resulting in 10 subjects with AD and 10 healthy controls (IRB No. SCHBC_IRB_2012-124) (Table S1A and B).
CSF preparation
For preparation of the CSF samples from AD and control subjects, CSF was obtained through lumbar punctures, which were performed between 8 and 10 a.m. subsequent to an overnight fast. After collection of 5 mL in a polypropylene tube, the CSF was centrifuged, and aliquoted into 1.0-mL polypropylene tubes. The tubes were stored at -80°C until analysis was performed. Isolation of the CSF from mice was performed as described previously.Citation59
Animals
The Tg6799 mice (formerly JAX Stock No. 008730) overexpress the mutant human APP (amyloid β precursor protein) form with the Swedish (K670N, M671L), Florida (I716V), and London (V717I) familial AD (FAD) mutations, and human PS1 harboring 2 FAD mutations, M146L and L286V.Citation20 Nontransgenic littermate controls (B6SJL) were used for the experiments. All mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA), and the animals were treated and maintained in the Seoul National University's mouse facility. Atg7+/− and Atg7+/+ mice (N = 12 each group) were kindly provided by Dr. Myung-Shik Lee (Sungkyunkwan University School of Medicine, Korea).Citation32 Aβ or phosphate-buffered saline (PBS) was administered via i.c.v. injections into 2- to 3-mo-old-Atg7+/− and Atg7+/+ mice as previously described with some modification.Citation60 Briefly, the mice received a single i.c.v. injection into the lateral ventricles using a Hamilton syringe (Fisher Scientific Co, Pittsburgh, Pennsylvania, USA) 2 weeks before the experiments were performed, while being secured in a stereotactic apparatus (Harvard Apparatus, Holliston, MA). The coordinates according to bregma were −0.9 mm lateral, −0.1 mm posterior, and −3.1 mm below. All experiments involving animals were performed according to protocols approved by the Seoul National University Institutional Animal Care and Use Committee (SNU IACUC) guidelines.
Cell cultures, drug treatments, and transfection
The human astroglioma U373MG cells (ATCC, HTB-17), human neuroblastoma SH-SY5Y cells (ATCC, CRL-2266) and mouse brain endothelial cell line BEND3 (ATCC, CRL-2299) were cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma-Aldrich, D6546) containing 10% fetal bovine serum (FBS), 0.1 mg/mL penicillin/streptomycin and 2 mM L-glutamine (Sigma-Aldrich, G7513) at 37°C under humidified 5% CO2 air. The U373MG cells (5 × 105 cells) were plated onto 6-well plates. Primary neurons were prepared from E18 ICR mouse embryos, and primary astrocytes were prepared from newborn (P1) ICR mice as described previously.Citation48 Astrocytes underwent 2 passages for the experiments. Cells were treated with several reagents alone, or cotreated with Aβ followed by a 24 h incubation. The reagents used in this study were the following: thiorphan (T6031), bacitracin A (B0125), insulin (I2643), 3MA (M9281), MG132 (M7449), GGPP (G6025), PP242 (P0037), bafilomycin A1/Baf (B1793), chloroquine (C6628) from Sigma-Aldrich; and rapamycin (LC Laboratories, R-5000). The cDNAs in this study were purchased from Origene (IDE-GFP; RG220700) or Addgene (24898 for Rab8a; 24899 for Rab8aS22N) and the siRNAs were purchased from Bioneer Inc. (Daejeon, Korea). cDNA and siRNA were transfected into the cells using Lipofectamine 2000 (11668027) and RNAimax (13778) from Invitrogen according to the manufacturer's instructions or by electroporation (ECM 830 Square Wave Electroporation System, Harvard Apparatus).
Western blot analysis
Harvested cell pellets and mouse brain lysates were prepared as described elsewhere.Citation61 The antibodies for the western blot analysis were: anti-LC3B antibody (1:2,000; MBL, M152-3; and Cell Signaling Technology, 2775); anti-Beta-amyloid (Aβ) 1-16 (6E10) antibody (1:5000; Biolegend, 803016); anti-RAB8A (6975), anti-ATG5 (12994), anti-ATG7 (8558), anti-CANX (calnexin) (2433), anti-p-PRKAA1/2 (T172) (2535), anti-PRKAA1/2 (2532), anti-p-RPS6KB (T389) (9205) and anti-RPS6KB (2708) (1:2000, Cell Signaling Technology); anti-DDIT3 (DNA damage-inducible transcript 3) (sc-575), anti-MMP9 (sc-6840), anti-GORASP2 (sc-271840), anti-GORASP1 (sc-19481), anti-ATP6V0A1 (sc-28801), anti-CTSD (sc-6486), anti-RAB5A (sc-309) and anti-GFP antibody (sc-9996) (1:1,000; Santa Cruz Biotechnologies Inc.); anti-IDE (ab32216), anti-GAPDH (ab9485), anti-TSG101 (ab83), anti-LAMP1 (ab25245), and anti-HMGB1 antibody (ab18256) (1:2,000; Abcam); anti-GOLGA2 (golgin A2) (1:4,000; BD BioScience, 610822); anti-SQSTM1 (P0067), and anti-ACTB (actin, beta) (A1978) (1:4,000; Sigma-Aldrich). Immunoreactivity was determined by chemiluminescence (GE Healthcare, RPN2108). The chemiluminescence signal was detected with a digital image analyzer (LAS-3000; Fuji, Tokyo, Japan).
Preparation and quantification of Aβ
Aβ was prepared as previously described.Citation17 Most of the Aβ used exists as oligomers and some monomers. For quantification of Aβ, sandwich enzyme-linked immunosorbent assay (ELISA) was performed as described for Aβ quantification.Citation61
Aβ degradation assays
Aβ degradation assays were performed as previously described, with some modification.Citation35 Briefly, primary astrocytes or U373MG cells grown in a 6-well plate to 70% confluency were incubated with Aβ42 or DMSO (vehicle) for 24 h. After PBS (Sigma-Aldrich, P4417) washing, cells were incubated for an additional 6 h in serum-free OPTI-MEM (GIBCO, 31985-070). The supernatant fraction (vehicle- or Aβ-ACM) was then collected after centrifugation and incubated with 1 μM Aβ for 12 h at 37°C in the absence or presence of 10 μg/mL bacitracin A, 0.3 ng/μL recombinant IDE (rIDE) (R&D Systems, 31985-070), or 0.5 μM insulin. The remaining Aβ levels were quantified by western blot analysis with 6E10 antibody, which can detect both Aβ40 and Aβ42 peptides.
Trichloroacetic acid (TCA) precipitation
For analyzing protein in the medium, we performed TCA precipitation as previously described.Citation61 Briefly, cell medium was centrifuged at 5,000 rpm for 5 min to remove cell debris and subjected to TCA precipitation (up to 10%) (Sigma-Aldrich, T6399). By TCA precipitation with media, 40X concentrated samples were loaded on the SDS-PAGE gels.
IDE enzymatic activity assay and IDE level measurement
The IDE enzymatic activity in the CSF or media was determined per the manufacturer's protocol (Calbiochem, CBA079). Briefly, 5 μL of the CSF from mouse and 50 μL of the CSF from human were diluted with PBS, and loaded into in a 96-well plate, which contained an affinity-purified polyclonal antibody that recognizes IDE (Calbiochem, CBA079). Following 1-h incubation, substrate was added and incubated for 2 to 4 h at 37°C in the dark. The fluorescence was measured using an excitation wavelength of 320 nm and an emission wavelength of 405 nm. The relative fluorescence unit represents total IDE activity in the media or CSF without normalizing to IDE protein levels. Cell media were concentrated with Amicon Ultra filter (UFC510024, Millipore, Billerica, MA). To determine the protein level of IDE in the media and CSF, ELISA was performed according to the manufacturer's protocol (Mybiosource, MBS018913 and MBS725082).
Partial purification of lysosomes
Partial isolation of lysosomes was performed as previously described.Citation62 Briefly, cells were resuspended in a hypotonic buffer (10 mM KCl, 30 mM Tris, pH 7.5, 5 mM MgOAc, 1 mM β-mercaptoethanol) and dissociated by piston strokes. Homogenates were centrifuged at 1,000 X g to precipitate the nuclei, then the supernatant fractions were collected and centrifuged at 100,000 X g for 1 h at 4°C.
Measurement of lysosomal activity
The lysosomal activity was determined as per the manufacturer's protocol (pepstatin A-BODIPY-conjugate [Molecular Probes, P12271] and LysoTracker Red [Molecular Probes, L7528]).
Isolation and characterization of exosomes
Exosomes and nonexosome fractions in the media from astrocytes were prepared as described earlier.Citation63
Cell viability assay
To measure cell viability, we performed MTT, calcein-AM and TUNEL assays (Promega, G7130) as previously described.Citation64 To analyze the TUNEL assay results, similar cell numbers from each group were analyzed (n = 300). Data are represented as the mean ± SEM from 3 independent experiments (N = 3).
Separation of prenylated and unprenylated RAB protein
RAB protein prenylation was examined by extraction of prenylated proteins with Triton X-114 (Sigma-Aldrich, X114) as previously described.Citation29
Examination of LC3-II translocation
To analyze green fluorescent protein (GFP)-LC3, red fluorescent protein (RFP)-LC3, and tandem-fluorescence LC3 (Tf-LC3), plasmids encoding GFP-LC3, RFP-LC3, and Tf-LC3 (a gift of Dr. Lee, Sungkyunkwan University, Korea) were transfected into astrocytes. The appearance of RFP-LC3 and GFP-LC3 puncta was visualized on a confocal laser-scanning microscope (FV10i-w; Olympus, Tokyo, Japan).
RNA isolation and reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from astrocytes treated with vehicle or Aβ using TRIzol reagent (Life Technologies, 15596-026). The following sense and antisense primers were used for mouse Ide: 5- TCTCCTTCCAAGTCAGCTGGT -3 (sense), 5- TTGGAAGGCCTCTTCTGTCAT -3 (antisense).
Quantitative real-time PCR (qPCR)
To examine the levels of mouse Ide mRNA, qPCR was performed as previously described.Citation64 The following sense and antisense primers were used: 5′-TACCTCCGCTTGCTGATGACTG-3′ (sense), 5′-ACAGGAGCTGAGGTATGAAGGC-3′ (antisense) for Ide; 5′-ACAGCCGCATCTTCTTGTGCAGTG-3′(sense), 5′-GGCCTTGACTGTGCCGTTGAATTT -3′ (antisense) for Gapdh.
3D-SIM (super resolution structured illumination microscopy)
To check the colocalization of IDE and autophagosomes, we used the super-resolution structured illumination microscopy (SIM; Nikon N-SIM). 3D-SIM images were taken for each fixed cell by moving the stage in the z-direction with a step size of 0.150 μm. The sequential z-sections were reconstructed to a 3D-SIM image (z-axis; brain slice thickness ∼5.0 ± 0.4 μm) and the 3D-deconvolution with the α blending function using NIS-E software (Nikon, Tokyo, Japan) was created. Images were taken by an Eclipse Ti-E research inverted microscope (Nikon, Tokyo, Japan) with Nikon's CFI Apo TIRF 100× oil objective lens (NA 1.49) and iXon DU-897 EMCCD camera (Andor Technology, Tokyo, Japan) at 512 × 512 pixel resolution. Multicolor fluorescence was acquired using a diode laser (488 nm, 561 nm) and exposure times of 40 ms, electron microscopic (EM) gain of 150, conversion gain of 1× were applied. The image was processed with NIS-E software and later exported to the Adobe Photoshop program.
Immunostaining
Immunocytochemical staining was performed as described previously.Citation21 Briefly, the fixed cells were incubated with mouse anti-IDE (1:300) or anti-LC3B (1:300) or anti-RAB8A (1:300) primary antibodies in PBST (PBS with 0.2% Triton X-100 [Sigma-Aldrich, T8787]) buffer overnight at 4°C. After several washes, the cells were incubated with secondary antibody, and images were taken on a confocal laser scanning microscope (FV10i-w).
Electron microscopy (EM)
Brain tissues of Tg6799 mice and the littermate control mice were fixed overnight in a mixture of cold 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2) and 2% paraformaldehyde in 0.1 M phosphate or cacodylate buffer (pH 7.2) and embedded with epoxy resin. The epoxy resin-mixed samples were loaded into capsules and polymerized at 38°C for 12 h and 60°C for 48 h. Thin sections were sliced on an ultramicrotome (RMC MT-XL; RMC Products, Tucson, AZ, USA) and collected on a copper grid. Appropriate areas for thin sectioning were cut at 65 nm and stained with saturated 4% uranyl acetate and 4% lead citrate before examination on a transmission electron microscope (JEM-1400; JEOL, Tokyo, Japan) at 80 kV.
Data analysis and statistics
For western blots, protein levels were normalized to pan forms or a housekeeping protein, such as ACTB or GAPDH. All data were expressed as means ± standard error of the mean (SEM). Statistical analysis was performed using GraphPad Prism5 (San Diego, CA, USA). The Student t test was used for 2-group comparisons, and one-way analysis of variance (ANOVA) followed by the Tukey-Kramer multiple comparison test was used to compare 3 or more groups. P values of < 0.05 were considered statistically significant.
Abbreviations
| 3-MA | = | 3-methyladenine |
| AD | = | Alzheimer disease |
| Aβ | = | amyloid beta |
| AMPK | = | AMP-activated protein kinase |
| APP | = | amyloid beta precursor protein |
| ATG | = | autophagy related |
| AVs | = | autophagic vacuoles |
| ATP6V0A1 | = | ATPase, H+ transporting, lysosomal V0 subunit a1 |
| ACM | = | astrocyte-conditioned media |
| ACTB | = | actin, beta |
| BBB | = | blood brain barrier |
| BFA | = | brefeldin A |
| Baf | = | bafilomycin A1 |
| CANX | = | calnexin; |
| CSF | = | cerebrospinal fluid |
| CDR | = | clinical dementia rating |
| CQ | = | chloroquine |
| CHX | = | cycloheximide |
| CTSD | = | cathepsin D |
| DDIT3 | = | DNA damage inducible transcript 3 |
| DMEM | = | Dulbecco's modified Eagle's medium |
| ELISA | = | enzyme-linked immunosorbent assay |
| EM | = | electron microscopy |
| FBS | = | fetal bovine serum |
| GORASP | = | Golgi reassembly stacking protein |
| GGPP | = | geranylgeranylpyrophosphate |
| GORASP KD | = | GORASP1 and GORASP2 double-knockdown cell |
| GAPDH, | = | glyceraldehyde-3-phosphate dehydrogenase |
| GOLGA2 | = | golgin A2 |
| HMGB1 | = | high mobility group box 1 |
| IDE | = | insulin-degrading enzyme |
| i.c.v. | = | intracerebroventricular |
| IL1B | = | interleukin 1, beta |
| IL18 | = | interleukin 18 |
| LC3A/B | = | microtubule associated protein 1 light chain 3 alpha/beta |
| LRP1 | = | LDL receptor related protein 1 |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| MMSE | = | mini-mental state examination |
| MMP9 | = | matrix metalloproteinase 9 |
| MDC | = | monodansylcadaverine |
| PBS | = | phosphate-buffered saline |
| RPS6KB | = | ribosomal protein S6 kinase 70kDa |
| RT-PCR | = | reverse transcription polymerase chain reaction |
| qPCR | = | quantitative real-time PCR |
| SIM | = | super-resolution structured illumination microscopy |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| Tf-LC3 | = | monomeric RFP (mRFP)-GFP tandem fluorescence tagged LC3 |
| TCA | = | trichloroacetic acid |
| TSG101 | = | tumor susceptibility 101 |
| VWF | = | von Willebrand factor |
| WB | = | western blotting |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary Files
Download Zip (11.4 MB)Funding
This work was supported by grants from the NRF (2015R1A2A1A05001794, 2014M3C7A1046047, 2015M3C7A1028790) and MRC (2012R1A5A2A44671346) for I.M-J.
References
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 2004; 44:181-93; PMID:15450169; http://dx.doi.org/10.1016/j.neuron.2004.09.010
- Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol 2004; 6:1054-61; PMID:15516999; http://dx.doi.org/10.1038/ncb1104-1054
- Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, Rosner MR, Selkoe DJ. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci 2000; 20:1657-65; PMID:10684867
- Dorfman VB, Pasquini L, Riudavets M, Lopez-Costa JJ, Villegas A, Troncoso JC, Lopera F, Castaño EM, Morelli L. Differential cerebral deposition of IDE and NEP in sporadic and familial Alzheimer's disease. Neurobiol Aging 2010; 31:1743-57; PMID:19019493; http://dx.doi.org/10.1016/j.neurobiolaging.2008.09.016
- Duckworth WC, Bennett RG, Hamel FG. Insulin degradation: progress and potential. Endocr Rev 1998; 19:608-24; PMID:9793760
- Qiu WQ, Ye Z, Kholodenko D, Seubert P, Selkoe DJ. Degradation of amyloid beta-protein by a metalloprotease secreted by microglia and other neural and non-neural cells. J Biol Chem 1997; 272:6641-6; PMID:9045694; http://dx.doi.org/10.1074/jbc.272.10.6641
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069-75; PMID:18305538; http://dx.doi.org/10.1038/nature06639
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol 2010; 12:823-30; PMID:20811354; http://dx.doi.org/10.1038/ncb0910-823
- Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009; 5:527-49; PMID:19527881; http://dx.doi.org/10.1016/j.chom.2009.05.016
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931-7; PMID:17712358; http://dx.doi.org/10.1038/nrm2245
- Subramani S, Malhotra V. Non-autophagic roles of autophagy-related proteins. EMBO Rep 2013; 14:143-51; PMID:23337627; http://dx.doi.org/10.1038/embor.2012.220
- Deretic V, Jiang S, Dupont N. Autophagy intersections with conventional and unconventional secretion in tissue development, remodeling and inflammation. Trends Cell Biol 2012; 22:397-406; PMID:22677446; http://dx.doi.org/10.1016/j.tcb.2012.04.008
- Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1beta. Embo J 2011; 30:4701-11; PMID:22068051; http://dx.doi.org/10.1038/emboj.2011.398
- Torisu T, Torisu K, Lee IH, Liu J, Malide D, Combs CA, Wu XS, Rovira II, Fergusson MM, Weigert R, et al. Autophagy regulates endothelial cell processing, maturation and secretion of von Willebrand factor. Nat Med 2013; 19:1281-7; PMID:24056772; http://dx.doi.org/10.1038/nm.3288
- Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Métivier D, Galluzzi L, Perfettini JL, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ 2014; 21:79-91; PMID:23852373; http://dx.doi.org/10.1038/cdd.2013.75
- Jiang S, Dupont N, Castillo EF, Deretic V. Secretory versus degradative autophagy: unconventional secretion of inflammatory mediators. J Innate Immun 2013; 5:471-9; PMID:23445716; http://dx.doi.org/10.1159/000346707
- Son SM, Jung ES, Shin HJ, Byun J, Mook-Jung I. Abeta-induced formation of autophagosomes is mediated by RAGE-CaMKKbeta-AMPK signaling. Neurobiol Aging 2012; 33:1006 e11-23; http://dx.doi.org/10.1016/j.neurobiolaging.2011.09.039
- Yang DS, Stavrides P, Mohan PS, Kaushik S, Kumar A, Ohno M, Schmidt SD, Wesson D, Bandyopadhyay U, Jiang Y, et al. Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain 2011; 134:258-77; PMID:21186265; http://dx.doi.org/10.1093/brain/awq341
- Boland B, Kumar A, Lee S, Platt FM, Wegiel J, Yu WH, Nixon RA. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci 2008; 28:6926-37; PMID:18596167; http://dx.doi.org/10.1523/JNEUROSCI.0800-08.2008
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006; 26:10129-40; PMID:17021169; http://dx.doi.org/10.1523/JNEUROSCI.1202-06.2006
- Son SM, Byun J, Roh SE, Kim SJ, Mook-Jung I. Reduced IRE1alpha mediates apoptotic cell death by disrupting calcium homeostasis via the InsP3 receptor. Cell Death Dis 2014; 5:e1188; PMID:24743743; http://dx.doi.org/10.1038/cddis.2014.129
- Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido TC. Abeta secretion and plaque formation depend on autophagy. Cell reports 2013; 5:61-9; PMID:24095740; http://dx.doi.org/10.1016/j.celrep.2013.08.042
- Zhou J, Tan SH, Nicolas V, Bauvy C, Yang ND, Zhang J, Xue Y, Codogno P, Shen HM. Activation of lysosomal function in the course of autophagy via mTORC1 suppression and autophagosome-lysosome fusion. Cell Res 2013; 23:508-23; PMID:23337583; http://dx.doi.org/10.1038/cr.2013.11
- Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, Wolfe DM, Martinez-Vicente M, Massey AC, Sovak G, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell 2010; 141:1146-58; PMID:20541250; http://dx.doi.org/10.1016/j.cell.2010.05.008
- Glebov K, Schutze S, Walter J. Functional relevance of a novel SlyX motif in non-conventional secretion of insulin-degrading enzyme. J Biol Chem 2011; 286:22711-5; PMID:21576244; http://dx.doi.org/10.1074/jbc.C110.217893
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol 2001; 2:107-17; PMID:11252952; http://dx.doi.org/10.1038/35052055
- Bravo-Cordero JJ, Marrero-Diaz R, Megias D, Genis L, Garcia-Grande A, Garcia MA, Arroyo AG, Montoya MC. MT1-MMP proinvasive activity is regulated by a novel Rab8-dependent exocytic pathway. Embo J 2007; 26:1499-510; PMID:17332756; http://dx.doi.org/10.1038/sj.emboj.7601606
- Hooff GP, Wood WG, Muller WE, Eckert GP. Isoprenoids, small GTPases and Alzheimer's disease. Biochimica et biophysica acta 2010; 1801:896-905; PMID:20382260; http://dx.doi.org/10.1016/j.bbalip.2010.03.014
- Mohamed A, Saavedra L, Di Pardo A, Sipione S, Posse de Chaves E. beta-amyloid inhibits protein prenylation and induces cholesterol sequestration by impairing SREBP-2 cleavage. J Neurosci 2012; 32:6490-500; PMID:22573671; http://dx.doi.org/10.1523/JNEUROSCI.0630-12.2012
- Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol 2010; 188:527-36; PMID:20156967; http://dx.doi.org/10.1083/jcb.200911154
- Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol 2010; 188:537-46; PMID:20156962; http://dx.doi.org/10.1083/jcb.200911149
- Lim YM, Lim H, Hur KY, Quan W, Lee HY, Cheon H, Ryu D, Koo SH, Kim HL, Kim J, et al. Systemic autophagy insufficiency compromises adaptation to metabolic stress and facilitates progression from obesity to diabetes. Nat Commun 2014; 5:4934; PMID:25255859; http://dx.doi.org/10.1038/ncomms5934
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med 2013; 19:983-97; PMID:23921753; http://dx.doi.org/10.1038/nm.3232
- Harris H, Rubinsztein DC. Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 2012; 8:108-17; http://dx.doi.org/10.1038/nrneurol.2011.200
- Tamboli IY, Barth E, Christian L, Siepmann M, Kumar S, Singh S, Tolksdorf K, Heneka MT, Lütjohann D, Wunderlich P, et al. Statins promote the degradation of extracellular amyloid {beta}-peptide by microglia via stimulation of exosome-associated insulin-degrading enzyme (IDE) secretion. J Biol Chem 2010; 285:37405-14; PMID:20876579; http://dx.doi.org/10.1074/jbc.M110.149468
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 2007; 120:4081-91; PMID:18032783; http://dx.doi.org/10.1242/jcs.019265
- Settembre C, Fraldi A, Medina DL, Ballabio A. Signals from the lysosome: a control centre for cellular clearance and energy metabolism. Nat Rev Mol Cell Biol 2013; 14:283-96; PMID:23609508; http://dx.doi.org/10.1038/nrm3565
- Nickel W, Rabouille C. Mechanisms of regulated unconventional protein secretion. Nat Rev Mol Cell Biol 2009; 10:148-55; PMID:19122676; http://dx.doi.org/10.1038/nrm2617
- Merregaert J, Van Langen J, Hansen U, Ponsaerts P, El Ghalbzouri A, Steenackers E, Van Ostade X, Sercu S. Phospholipid scramblase 1 is secreted by a lipid raft-dependent pathway and interacts with the extracellular matrix protein 1 in the dermal epidermal junction zone of human skin. J Biol Chem 2010; 285:37823-37; PMID:20870722; http://dx.doi.org/10.1074/jbc.M110.136408
- Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, Rockenstein E, Masliah E, Hyman BT, McLean PJ, Unni VK. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci 2011; 31:14508-20; PMID:21994367; http://dx.doi.org/10.1523/JNEUROSCI.1560-11.2011
- Morck C, Olsen L, Kurth C, Persson A, Storm NJ, Svensson E, Jansson JO, Hellqvist M, Enejder A, Faergeman NJ, et al. Statins inhibit protein lipidation and induce the unfolded protein response in the non-sterol producing nematode Caenorhabditis elegans. Proc Natl Acad Sci U S A 2009; 106:18285-90; PMID:19826081; http://dx.doi.org/10.1073/pnas.0907117106
- Deretic V. Autophagy: an emerging immunological paradigm. J Immunol 2012; 189:15-20; http://dx.doi.org/10.4049/jimmunol.1102108
- Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012; 13:255-63; PMID:22286270; http://dx.doi.org/10.1038/ni.2215
- Jahn R, Lang T, Sudhof TC. Membrane fusion. Cell 2003; 112:519-33; PMID:12600315; http://dx.doi.org/10.1016/S0092-8674(03)00112-0
- Li D, Ropert N, Koulakoff A, Giaume C, Oheim M. Lysosomes are the major vesicular compartment undergoing Ca2+-regulated exocytosis from cortical astrocytes. J Neurosci 2008; 28:7648-58; PMID:18650341; http://dx.doi.org/10.1523/JNEUROSCI.0744-08.2008
- Rao SK, Huynh C, Proux-Gillardeaux V, Galli T, Andrews NW. Identification of SNAREs involved in synaptotagmin VII-regulated lysosomal exocytosis. J Biol Chem 2004; 279:20471-9; PMID:14993220; http://dx.doi.org/10.1074/jbc.M400798200
- Verderio C, Cagnoli C, Bergami M, Francolini M, Schenk U, Colombo A, Riganti L, Frassoni C, Zuccaro E, Danglot L, et al. TI-VAMP/VAMP7 is the SNARE of secretory lysosomes contributing to ATP secretion from astrocytes. Biol Cell 2012; 104:213-28; PMID:22188132; http://dx.doi.org/10.1111/boc.201100070
- Jung ES, An K, Hong HS, Kim JH, Mook-Jung I. Astrocyte-originated ATP protects Abeta(1-42)-induced impairment of synaptic plasticity. J Neurosci 2012; 32:3081-7; PMID:22378880; http://dx.doi.org/10.1523/JNEUROSCI.6357-11.2012
- Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Métivier D, Galluzzi L, Perfettini JL, et al. Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ 2014; 21:79-91; PMID:23852373; http://dx.doi.org/10.1038/cdd.2013.75
- Eroglu C, Barres BA. Regulation of synaptic connectivity by glia. Nature 2010; 468:223-31; PMID:21068831; http://dx.doi.org/10.1038/nature09612
- Verghese PB, Castellano JM, Garai K, Wang Y, Jiang H, Shah A, Bu G, Frieden C, Holtzman DM. ApoE influences amyloid-beta (Abeta) clearance despite minimal apoE/Abeta association in physiological conditions. Proc Natl Acad Sci U S A 2013; 110:E1807-16; PMID:23620513; http://dx.doi.org/10.1073/pnas.1220484110
- Xiao Q, Yan P, Ma X, Liu H, Perez R, Zhu A, Gonzales E, Burchett JM, Schuler DR, Cirrito JR, et al. Enhancing astrocytic lysosome biogenesis facilitates Abeta clearance and attenuates amyloid plaque pathogenesis. J Neurosci 2014; 34:9607-20; PMID:25031402; http://dx.doi.org/10.1523/JNEUROSCI.3788-13.2014
- Vekrellis K, Ye Z, Qiu WQ, Walsh D, Hartley D, Chesneau V, Rosner MR, Selkoe DJ. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J Neurosci 2000; 20:1657-65; PMID:10684867
- Gao W, Eisenhauer PB, Conn K, Lynch JA, Wells JM, Ullman MD, McKee A, Thatte HS, Fine RE. Insulin degrading enzyme is expressed in the human cerebrovascular endothelium and in cultured human cerebrovascular endothelial cells. Neurosci Lett 2004; 371:6-11; PMID:15500957; http://dx.doi.org/10.1016/j.neulet.2004.07.034
- Kilger E, Buehler A, Woelfing H, Kumar S, Kaeser SA, Nagarathinam A, Walter J, Jucker M, Coomaraswamy J. BRI2 protein regulates beta-amyloid degradation by increasing levels of secreted insulin-degrading enzyme (IDE). J Biol Chem 2011; 286:37446-57; PMID:21873424; http://dx.doi.org/10.1074/jbc.M111.288373
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 2010; 6:131-44; PMID:20157306; http://dx.doi.org/10.1038/nrneurol.2010.4
- Turner AJ, Fisk L, Nalivaeva NN. Targeting amyloid-degrading enzymes as therapeutic strategies in neurodegeneration. Ann N Y Acad Sci 2004; 1035:1-20; PMID:15681797; http://dx.doi.org/10.1196/annals.1332.001
- Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 2010; 330:1774; PMID:21148344; http://dx.doi.org/10.1126/science.1197623
- Liu L, Duff K. A technique for serial collection of cerebrospinal fluid from the cisterna magna in mouse. J Vis Exp 2008; 10(21):pii: 960; PMID:19066529; http://dx.doi.org/10.3791/960
- Moon M, Choi JG, Nam DW, Hong HS, Choi YJ, Oh MS, Mook-Jung I. Ghrelin ameliorates cognitive dysfunction and neurodegeneration in intrahippocampal amyloid-beta1-42 oligomer-injected mice. Journal of Alzheimer's disease : JAD 2011; 23:147-59; PMID:20930280
- Son SM, Song H, Byun J, Park KS, Jang HC, Park YJ, Mook-Jung I. Altered APP processing in insulin-resistant conditions is mediated by autophagosome accumulation via the inhibition of mammalian target of rapamycin pathway. Diabetes 2012; 61:3126-38; PMID:22829447; http://dx.doi.org/10.2337/db11-1735
- Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates beta-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem 2013; 288:1295-306; PMID:23155049; http://dx.doi.org/10.1074/jbc.M112.409250
- Trajkovic K, Hsu C, Chiantia S, Rajendran L, Wenzel D, Wieland F, Schwille P, Brügger B, Simons M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008; 319:1244-7; PMID:18309083; http://dx.doi.org/10.1126/science.1153124
- Byun J, Son SM, Cha MY, Shong M, Hwang YJ, Kim Y, Ryu H, Moon M, Kim KS, Mook-Jung I. CR6-interacting factor 1 is a key regulator in Abeta-induced mitochondrial disruption and pathogenesis of Alzheimer's disease. Cell Death Differ 2015; 22(6):959-73. http://dx.doi.org/10.1038/cdd.2014.184