
ABSTRACT
Autophagy plays an essential role in cellular homeostasis through the quality control of proteins and organelles. Although a time-dependent decline in autophagic activity is believed to be involved in the aging process, the issue remains controversial. We previously demonstrated that autophagy maintains proximal tubular cell homeostasis and protects against kidney injury. Here, we extend that study and examine how autophagy is involved in kidney aging. Unexpectedly, the basal autophagic activity was higher in the aged kidney than that in young kidney; short-term cessation of autophagy in tamoxifen-inducible proximal tubule-specific autophagy-deficient mice increased the accumulation of SQSTM1/p62- and ubiquitin-positive aggregates in the aged kidney. By contrast, autophagic flux in response to metabolic stress was blunted with aging, as demonstrated by the observation that transgenic mice expressing a green fluorescent protein (GFP)-microtubule-associated protein 1 light chain 3B fusion construct, showed a drastic increase of GFP-positive puncta in response to starvation in young mice compared to a slight increase observed in aged mice. Finally, proximal tubule-specific autophagy-deficient mice at 24 mo of age exhibited a significant deterioration in kidney function and fibrosis concomitant with mitochondrial dysfunction as well as mitochondrial DNA abnormalities and nuclear DNA damage, all of which are hallmark characteristics of cellular senescence. These results suggest that age-dependent high basal autophagy plays a crucial role in counteracting kidney aging through mitochondrial quality control. Furthermore, a reduced capacity for upregulation of autophagic flux in response to metabolic stress may be associated with age-related kidney diseases.
Introduction
The kidney is typical of organs exhibiting age-associated tissue damage, and the increased incidence of chronic kidney disease (CKD) in the elderly is a health problem worldwide.Citation1,2 An increased prevalence of CKD with age is often associated with an increased risk of end-stage kidney disease, several comorbid conditions, such as cardiovascular disease, and most importantly, adverse outcomes in the elderly population.Citation3 Age-related structural kidney changes include loss of podocytes, glomerulosclerosis, vascular changes (arteriolosclerosis and intimal and medial hypertrophy), and tubule-interstitial changes (fibrosis and tubular atrophy).Citation4 Of note, the age-related decline in renal mass (and presumably, subsequent kidney dysfunction) is dependent primarily on tubule-interstitial changes rather than glomerular change.Citation5,6 Thus, illuminating the mechanism of kidney aging, especially that of age-associated tubular dysfunction, in this highly vulnerable population is of paramount importance.
Recently, aging research has advanced remarkably with the discovery that the aging process is not a passive or random process involving the loss of molecular fidelity and subsequent accumulation of waste products; rather, it is regulated by several key signaling pathways and the rate of aging can be controlled by modulating these pathways.Citation7,8 Elucidating and modulating the “coordinating centers” of aging may allow us to develop new therapeutic interventions that take aim at aging-associated diseases including kidney diseases. Accumulating evidence indicates that macroautophagy, hereafter referred to as autophagy, is a candidate “coordinating center” in aging.Citation9 Autophagy is one of the major degradation pathways in the cell, along with the ubiquitin–proteasome system.Citation10,11 A key function of autophagy is to remove damaged organelles (including mitochondria) and aberrant macromolecules, thereby preventing further injury and cellular dysfunction. Given that the time-dependent accumulation of cellular damage is the general cause of aging, it is easy to infer that a gradual, age-related decline in autophagic activity contributes to various aspects of the aging process.Citation12 Indeed, the age-related “decline” in autophagic activity has been reported.Citation13,14 However, most of these findings contain several misinterpretations and omissions presumably due to the difficulty in the assessment of autophagic flux especially in the parenchymal organs.
Regardless of the time-dependent alterations in relative autophagic activity, another important question with clinical implications is whether autophagy slows aging. Evidence from lower organisms has confirmed the tight connection between autophagy and aging: functional autophagy is required to attain the maximal life-span extension mediated by these genetic manipulations.Citation15 In mammals, many studies have revealed a close association between autophagy and aging using organ-specific conditional autophagy-deficient mice.Citation9,16 Most autophagy-deficient mice mimic several characteristics of aging, including sarcopenia, cardiac dysfunction, dysfunction of hematopoietic stem cells, and tumorigenesis, suggesting that autophagy counteracts aging stress.
Regarding the kidney, to determine basal autophagy function in adult mouse kidney proximal tubules, we recently generated KAP (kidney androgen regulated protein)-Cre/floxed Atg5 (atg5F/F-KAP) mice, in which autophagy is deficient in proximal tubular cells.Citation17-20 We observed slight hypertrophy with an accumulation of cytosolic amorphous substrates in the proximal tubular cells of the kidney of atg5F/F-KAP mice after the age of 6 mo. Kidney function is comparable in atg5F/F-KAP mice and littermate controls up to 9 mo of age. These data clearly highlight the importance of basal constitutive autophagy as a key homeostatic mechanism to maintain proximal tubule integrity; however, the long-term effects of defects in autophagy have not been analyzed.
Based on this background information, we hypothesized that the alteration of autophagic activity with age would be more complex than previously described and that the alteration could influence the aging process in the kidney. Thus, in this study, we examined 1) age-dependent alteration in autophagic activity (as a relative balance between substrate levels and degradation) under basal conditions and metabolic stress using transgenic mice with green fluorescent protein conjugated to microtubule-associated protein 1 light chain 3B (GFP-MAP1LC3B) and drug-inducible proximal tubule-specific autophagy-deficient mice, and 2) the long-term consequences of autophagy deficiency in the kidney with a focus on the hallmarks of aging.
Results
Aged kidney is more reliant on autophagy for the degradation of increasing substrates
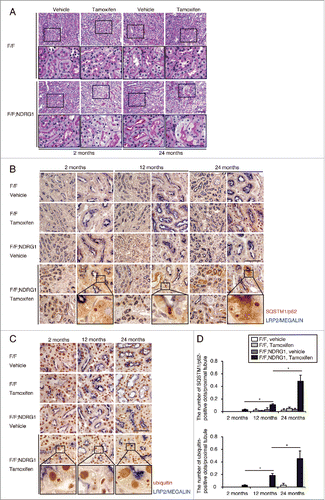
Age-dependent changes in demand and activity of basal autophagy in the proximal tubules were evaluated in NDRG1 (N-myc downstream regulated gene 1)-Cre/floxed Atg5 mice (Atg5F/F-NDRG1), in which autophagy deficiency is induced in the proximal tubule by tamoxifen treatment.Citation21 Two-, 12-, and 24-mo-old Atg5F/F-NDRG1 mice and age-matched Atg5F/F control littermates received tamoxifen-inducible genetic ablation of Atg5. Two wk after tamoxifen treatment, histological analysis and immunostaining for SQSTM1/p62 and ubiquitin were performed. The accumulation of SQSTM1 and ubiquitin represents the amount of substrate requiring degradation that accumulated over the 2-wk period, because SQSTM1 is the ubiquitin- and MAP1LC3-binding protein that regulates the formation of protein aggregates and is removed by autophagy.Citation22,23 Autophagy deficiency was confirmed by the reduction of ATG5 expression, blockage of conversion of MAP1LC3-I to MAP1LC3-II, and the accumulation of SQSTM1 (Fig. S1A and S1B). Periodic-acid Schiff (PAS) staining demonstrated that cytoplasmic swelling of the tubular cells was comparably observed in 2- and 24-mo-old Atg5F/F-NDRG1 mice (). By contrast, 2-wk ablation of autophagy triggered more accumulation of SQSTM1- and ubiquitin-positive protein aggregates in the kidneys of aged Atg5F/F-NDRG1 mice, indicating that aged kidneys utilize autophagy for the degradation of increasing these substrates (; Fig. S1A and S1B).
Figure 1. Aged kidney is more reliant on autophagy for the degradation of increasing substrates. (A) Representative images of PAS-stained kidney cortical regions of vehicle- or tamoxifen-treated 2- and 24-mo-old Atg5F/F and Atg5F/F-NDRG1 mice (n = 4 or 5 in each group). Bar: 50 μm. Magnified images are presented in the insets. (Band C) Immunostaining for SQSTM1 (B) and ubiquitin (C) in the kidneys of 2-, 12-, and 24-mo-old Atg5F/F and Atg5F/F-NDRG1 mice treated with vehicle or tamoxifen 2 wk before euthanasia. Bars: 50 μm. Images are representative of multiple experiments. Magnified images from tamoxifen-treated Atg5F/F-NDRG1 mice are presented in the insets. (D) The number of SQSTM1- or ubiquitin-positive dots was counted in at least 10 high-power fields (×400). Data are provided as mean ± SE. Statistically significant differences (*, P < 0.05) are indicated. F/F, Atg5F/F mice; F/F;NDRG1, Atg5F/F-NDRG1 mice.
Age-dependent alteration in autophagic flux in response to starvation in the proximal tubules
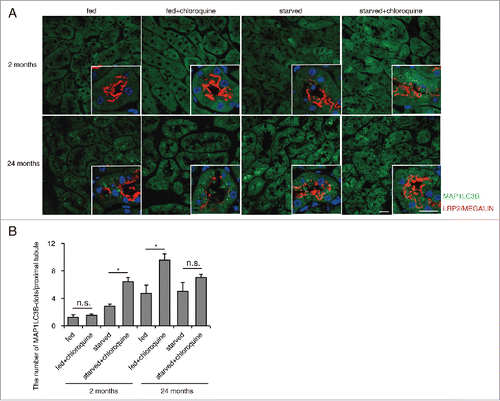
Transgenic mice expressing GFP-MAP1LC3B, in which GFP-positive puncta reflect autophagosomes,Citation24 were used to assess age-dependent alterations in autophagic flux in proximal tubules at 2- and 24-mo of age (hereafter, young refers to 2-mo of age and aged to 24 mo of age, unless otherwise indicated). To measure autophagic flux under basal and stressed conditions in vivo, we administered chloroquine, a lysosomotropic reagent that inhibits intralysosomal acidification, 6 h before euthanasia.Citation25 GFP-positive puncta were rarely observed in the fed, chloroquine-free young mice, whereas a substantial number of GFP-positive puncta were observed in the LRP2/MEGALIN-positive proximal tubular cells of the fed, chloroquine-free aged mice (). Chloroquine administration significantly increased the number of GFP-positive puncta in the aged kidney, while the number of puncta remained unchanged in young kidney. This age-dependent upregulation of basal autophagic activity confirms the data obtained from Atg5F/F-NDRG1 mice (). Next, autophagic flux in young and aged mice was assessed in response to 24 h of starvation. Chloroquine administration significantly increased GFP-positive puncta in starved young mice, whereas the increase was blunted in aged mice (). Western blot analysis (ATG12–ATG5, MAP1LC3-I, MAP1LC3-II and SQSTM1) using whole kidney lysates (Fig. S2A and S2B) and quantification of the number of autophagosome-like structures on electron microscopy images (Fig. S3A and S3B) confirmed this fluorescent study. As expected, the number of autophagosome-like structures markedly increased in the chloroquine-treated aged mice (Fig. S3A and S3B). In contrast to the proximal tubular cells, both young and aged mice exhibited comparable numbers of autophagosomes or autolysosomes in podocytes, as assessed by fluorescence in GFP-MAP1LC3B transgenic mice and electron microscopy analysis (Fig. S3C and S3D). These results demonstrate that the proximal tubular cells in young mice exhibit low basal autophagic activity and high starvation-induced autophagic flux, while those in aged mice exhibit high basal flux and blunted induction by starvation.
Figure 2. Age-dependent alterations in autophagic flux in response to starvation in the proximal tubules. GFP-positive puncta formation was assessed in the proximal tubules of young or aged GFP-MAP1LC3B transgenic mice that were either fed or subjected to 24 h of starvation, with or without chloroquine administration (n = 3 to 6 in each group). (A) Kidney sections were immunostained for LRP2/MEGALIN, a marker of proximal tubules (red), and counterstained with DAPI (blue). Bars: 20 μm. Images are representative of multiple experiments. (B) The number of GFP-positive puncta per proximal tubule under each condition was counted in at least 10 high-power fields (×600). Data are provided as mean ± SE. Statistically significant differences (*, P < 0.05) are indicated.
Long-term autophagy deficiency accelerates kidney dysfunction and atrophy
To investigate the long-term consequences of autophagy deficiency in the kidney, we observed proximal tubule-specific autophagy-deficient mice (atg5F/F-KAP) for up to 24 mo. Almost all atg5F/F-KAP mice and Atg5F/F control littermates survived during the observational period, with no tumorigenesis observed in the kidney (data not shown). Physiological parameters in young and aged atg5F/F-KAP mice and age-matched control littermates are shown in . The kidney-to-body weight ratio was comparable between young atg5F/F-KAP mice and age-matched controls, while it was significantly decreased in aged atg5F/F-KAP mice compared with young atg5F/F-KAP mice, suggesting the progression of kidney atrophy in aged atg5F/F-KAP mice (). There were no significant differences in body weight and heart-to-body, or lung-to-body ratios between atg5F/F-KAP mice and age-matched Atg5F/F control littermates, whereas the liver-to-body weight ratio and total cholesterol level were significantly decreased in aged atg5F/F-KAP mice compared with aged Atg5F/F controls (). Urinary ALB/albumin excretion and kidney function in atg5F/F-KAP mice were unchanged up to 9 mo;Citation17 however, a significant increase in urinary ALB/albumin excretion, the mRNA levels of 2 tubular injury markers Havcr1/Kim-1 and Lcn2/Ngal, and deterioration of kidney function were observed in aged atg5F/F-KAP mice compared with young atg5F/F-KAP mice or aged Atg5F/F controls (). PAS staining demonstrated that the proximal tubules of aged atg5F/F-KAP mice showed a disruption of lumen structure and an irregular arrangement of tubular epithelial cell nuclei, as well as a massive accumulation of cytosolic amorphous substrates compared with younger atg5F/F-KAP mice, while no morphologic abnormalities were observed using light microscopy in the kidney in aged Atg5F/F controls (). These findings suggest that constitutive autophagy is required for maintaining renal epithelial cell polarity and renal tubular structure in adult kidneys. Furthermore, Masson trichrome staining, immunostaining for COL1A1 (collagen, type I, alpha 1), and Col1a1 mRNA levels indicated that aged atg5F/F-KAP mice exhibited enhanced interstitial fibrosis of the kidney (). Electron microscopy analysis confirmed a disruption of lumen structure and an irregular arrangement of tubular epithelial cell nuclei (). Moreover, it showed the presence of intracellular vacuolation and aggregation, and various sizes of deformed mitochondria lacking the normal mitochondrial network and intramitochondrial structures in aged atg5F/F-KAP mice, whereas mitophagy was occasionally observed in the kidney proximal tubule in aged Atg5F/F controls (). Collectively, long-term autophagy deficiency accelerates kidney dysfunction and atrophy.
Figure 3. Long-term autophagy deficiency accelerates kidney dysfunction and atrophy. (Ato D) Age-dependent changes in the kidney-to-body weight ratio (A), urinary ALB/albumin levels (B), mRNA levels of Havcr1/Kim-1 and Lcn2/Ngal in kidney cortical regions (C) and kidney function (D) in young and aged Atg5F/Fand atg5F/F-KAP mice are shown (n = 7 to 10 in each group). Data were normalized with 18s-rRNA and presented as the ratio relative to young control kidney. (Eto G) Representative images of PAS-staining (E), Masson trichrome staining (F), and immunostaining for collagen type I (G) in kidney cortical regions of young and aged Atg5F/F and atg5F/F-KAP mice (n = 7 to 10 in each group). Bars: 50 μm (E) and 100 μm (F and G). Magnified images from 24-mo-old mice are presented in the insets. (H) The Col1a1 mRNA levels in kidney cortical regions of young and aged Atg5F/F and atg5F/F-KAP mice (n = 7 to 10 in each group). Data were normalized with 18s-rRNA and presented as the ratio relative to young control kidney. (Ito R) Electron micrographs of aged Atg5F/F (Ito M) and atg5F/F-KAP mice (Nto R) (n = 3 in each group). BM, basement membrane; TL, tubular lumen; *, nucleus. Bars: 10 μm (I, J, N, and Q), 5 μm (Kand P), 2 μm (Land Q) and 500 nm (Mand R). Representative magnified image of mitophagy is presented in the inset (L). Data are provided as mean ± SE. Statistically significant differences (*, P < 0.05 vs. age-matched Atg5F/F control littermates; #, P < 0.05 vs. young mice) are indicated. F/F, Atg5F/F mice; F/F;KAP, atg5F/F-KAP mice.
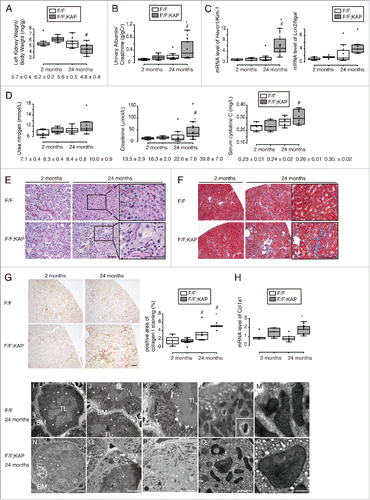
Table 1. Physiological parameters of proximal tubule-specific autophagy-deficient mice at 2 mo and 24 mo of age.
Long-term autophagy deficiency accelerates kidney aging with oxidative stress
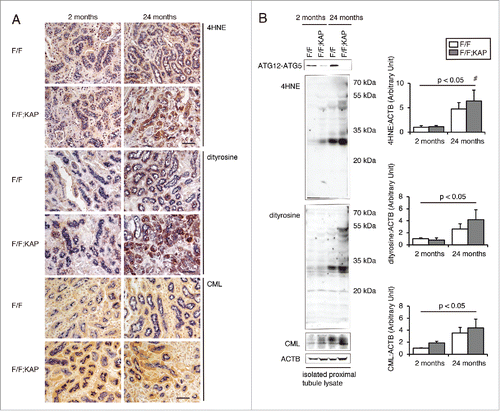
Senescence-associated (SA) GLB1/β-gal (SA-GLB1) activity is a well-defined marker of senescence representing GLB1 activity at pH 6.0, which is preferentially detectable in senescent cells and tissues.Citation26 Cells positive for SA-GLB1 staining were rarely found in kidneys from young atg5F/F-KAP mice and Atg5F/F controls. Patchy staining was observed in tubules of aged Atg5F/F mice, whereas more intense staining was found in tubules of aged atg5F/F-KAP mice (Fig. S4A). The observation of abnormally deformed mitochondria in atg5F/F-KAP mice at 24 mo of age () led to the speculation that the kidneys of older atg5F/F-KAP mice may be affected by reactive oxygen species (ROS) produced by abnormal mitochondria. Urinary excretion of the oxidative stress marker 8-OHdGCitation27 was significantly increased in aged atg5F/F-KAP mice (Fig. S4B). Furthermore, we found increased other oxidative stress markers, including 4-hydroxy-2-nonenal (HNE), dityrosine, and N-carboxymethyllysine (CML), in aged atg5F/F-KAP mouse kidneys (), which is in agreement with the previous report.Citation28
Figure 4. Long-term autophagy deficiency accelerates kidney aging with oxidative stress. (A) Representative images of immunostaining of oxidative stress indicators, including 4HNE adduct proteins, dityrosine, and AGE (CML-protein adduct), in kidney cortical regions of young and aged Atg5F/F and atg5F/F-KAP mice (n = 7 in each group). Bars: 50 μm. Kidney sections were immunostained for the proximal tubule marker LRP2/MEGALIN in blue. (B) Representative immunoblots of ATG5, 4HNE, dityrosine, and CML using isolated kidney proximal tubules of young or aged Atg5F/F and atg5F/F-KAP mice and quantification by densitometry are shown (n = 4 to 5 in each group). The mean value of young control mice was adjusted to “1” as a reference. Data are provided as mean ± SE. Statistically significant differences (*, P < 0.05 vs. age-matched Atg5F/F control littermates; #, P < 0.05 vs. young mice) are indicated. F/F, Atg5F/F mice; F/F;KAP, atg5F/F-KAP mice.
Long-term autophagy deficiency exaggerates mitochondrial dysfunction, as well as mitochondrial and nuclear DNA damage
COX (cytochrome c oxidase) and succinate dehydrogenase (SDH) staining was performed to evaluate age-related alterations in mitochondrial respiration activity in the kidney (). Young atg5F/F-KAP mice showed less intense COX staining than age-matched controls (). Tubular COX staining was somewhat decreased in aged Atg5F/F mice compared with young Atg5F/F mice, however, a more prominent decline in COX staining was observed in aged atg5F/F-KAP mice. Similarly SDH staining intensity was decreased in young atg5F/F-KAP and aged Atg5F/F mice, with the most prominent decrease observed in aged atg5F/F-KAP mice (). We also demonstrated that protein level of COX4I1 (cytochrome c oxidase subunit IV isoform 1) decreased with age, which was prominent in aged atg5F/F-KAP mice (Fig. S5A and S5B).
Figure 5. Long-term autophagy deficiency exaggerates mitochondrial dysfunction and mtDNA and nDNA abnormalities. (Aand B) Representative images of COX (A) and SDH (B) staining in the kidney cortical regions of young and aged Atg5F/F and atg5F/F-KAP mice (n = 5 in each group). Bars: 50 μm. The relative staining intensity is shown in the bar graphs. The mean value of young Atg5F/F mice is expressed as 1. (Cand D) Relative mtDNA copy number (C) and relative damage of mtDNA and nDNA (D) are shown. mtDNA and nDNA were extracted from isolated kidney proximal tubules of young or aged Atg5F/F and atg5F/F-KAP mice (n = 7 to 10 in each group). Data are expressed as the fold change relative to the mean value of young Atg5F/F mice (C). (E) Representative images of immunostaining for phospho-H2AFX, a DNA damage marker, in kidney cortical regions of Atg5F/F and atg5F/F-KAP mice (n = 7 to 9 in each group). Bar: 50 μm. Kidney sections were immunostained for the proximal tubule marker LRP2/MEGALIN in blue. The number of phospho-H2AFX-positive proximal tubular cells was counted in at least 10 high-power fields (400). Statistically significant differences (*, P < 0.05 vs. age-matched Atg5F/F control littermates; #, P < 0.05 vs. young mice; †, P < 0.05 mtDNA vs. nDNA) are indicated. F/F, Atg5F/F mice; F/F;KAP, atg5F/F-KAP mice.
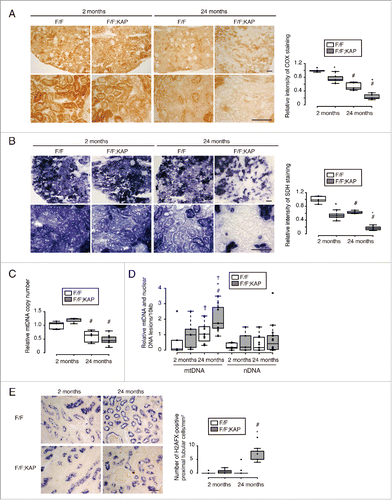
Next, the number of mitochondria in the proximal tubular cells was determined. Total DNA was extracted from isolated proximal tubules and the copy number ratio of mitochondrial DNA (mtDNA) (mt-Co1 [cytochrome oxidase subunit I]) to that of a nuclear gene (Ndufv1 [NADH dehydrogenase {ubiquinone} flavoprotein 1]) was determined using quantitative PCR (). In young mice, the mtDNA:nuclear DNA (nDNA) ratios were similar regardless of autophagy competency. However, we found significantly lower mtDNA:nDNA ratios in proximal tubules from aged mice compared with young mice. The decrease was more pronounced in atg5F/F-KAP mice (59.4% reduction) compared with Atg5F/F mice (38.8% reduction), suggesting age-related mitochondrial depletion was exaggerated by autophagy deficiency. This trend is in agreement with findings from the ultrastructural analysis (), COX and SDH staining () and protein level of COX4I1 (Fig. S5A and S5B).
Furthermore, the relative levels of damaged mtDNA and damaged nDNA in isolated kidney proximal tubules were determined using long-amplicon quantitative PCR (). We observed a significant increase in mtDNA damage in aged atg5F/F-KAP mice compared with young atg5F/F-KAP mice and aged Atg5F/F mice; however, no significant changes in nDNA damage were observed. Of note, the levels of mtDNA damage were higher than those of nDNA damage in aged atg5F/F-KAP mice, suggesting that mtDNA is more vulnerable to long-term autophagy-deficiency-induced-damage compared to nDNA. Immunostaining for phospho-H2AFX/γ-H2AX (H2A histone family, member X), a marker for nDNA damage, demonstrated that nuclear staining was observed only in the proximal tubular cells of aged atg5F/F-KAP mice (), which was confirmed by western blot analysis using isolated proximal tubule lysates (Fig. S5B). Thus, these findings suggest that mitochondrial dysfunction and mtDNA and nDNA abnormalities are exaggerated in proximal tubules from aged atg5F/F-KAP mice.
Discussion
In this study, we found a close association between dysregulation of autophagic activity and kidney aging. Specifically, age-dependent high basal autophagy was found to play a crucial role in counteracting kidney aging primarily through mitochondrial quality control. Furthermore, a lack of autophagic flux upregulation in response to metabolic stress with age may lead to age-related kidney diseases.
To overcome several drawbacks present in prior studies, we directly analyzed age-dependent alteration of autophagic activity in the kidney. Halting autophagy for 2 wk in young and aged animals, using tamoxifen-inducible proximal tubule-specific autophagy-deficient mice, demonstrated that autophagy is substantially upregulated in the aged kidney, and that the aged kidney is more dependent on autophagy than young kidneys. We speculate that basal autophagic activity increases with age to cope with the time-dependent accumulation of cellular ‘garbage,' and/or increased levels of ROS and hypoxia, which are major stimulators of autophagy. In addition to inconspicuous aging stress, aged organisms could be sporadically challenged by additional stresses such as ischemia and metabolic stress. In reference to a recent study,Citation25 we assessed the alteration of autophagic flux in the kidney in response to starvation, a canonical autophagy stimulator. We found that autophagic flux in response to starvation is markedly blunted in the proximal tubules of aged GFP-MAP1LC3B transgenic mice. Failure of cellular autophagic adaptation to metabolic stress may account for the age-dependent vulnerability to CKD. Although these findings are contrary to the dogma that autophagic activity downregulates with aging (which appears to be derived from a simple extrapolation from the expression study of autophagy-related proteins or the number of autophagosomes), we believe that we newly assessed the autophagic activity as strictly as possible. Indeed, another study reports the similar finding that old hematopoietic stem cells have higher basal autophagic flux than young hematopoietic stem cells.Citation29
Although the mechanism underlying decreased sensitivity to metabolic stress in aged kidney is largely unknown, it is possible that aged kidney cannot afford to boost autophagic activity because age-dependent increases in constitutive stress (such as ROS) lead to a reduced capacity for additional autophagy stimulation. Another explanation is age-dependent dysregulation of nutrient sensing, including the MTOR (mechanistic target of rapamycin [serine/threonine kinase])-, AMP-activated protein kinase (AMPK)-, and SIRT1 (sirtuin 1)-related pathways. On the one hand, SIRT1 expression declined with aging, which does not appear to explain our findings (Fig S1 and S2). On the other hand, the levels of phosphorylated AMPK relative to total AMPK upregulated with aging; however, a lack of further induction in response to starvation in the aged proximal tubules was observed (Fig S1 and S2). Indeed, increased phosphorylation of ACAC (acetyl-CoA carboxylase), which is regulated by AMPK activation, was observed in the aged proximal tubules (Fig S6). In contrast, hyperactivation of the MTORC1 signal, as determined by positive staining for phosphorylated (p-) RPS6 (ribosomal protein S6) on Ser235/236, was observed only in the aged proximal tubules. The aged proximal tubules showing positive p-RPS6 (Ser235/236) staining tend to exhibit suppression of autophagic activity, as assessed by the reduced number of GFP-MAP1LC3B dots, and 24 h of starvation did not reduce the MTORC1 signal in the aged mice (Fig. S6). These observations may explain a lack of further induction of the autophagic activity in response to starvation. Alternatively, a lack of autophagic flux in response to starvation in the aged kidney may be attributed to defective lysosomal function, which has been reported in almost all tissues of aging organisms from nematode worms to mammals. Accumulation of undigested material (e.g., lipofuscin) inside secondary lysosomes may interfere with their ability to fuse with autophagosomes and to degrade their cargo.Citation30
It is intriguing to investigate whether the aforementioned age-dependent dysregulation in autophagic activity contributes to the aging process in the kidney. Diverse findings observed in the kidney of aged proximal tubule-specific Atg5-knockout mice could be summarized as indicating that autophagy counteracts kidney aging through mitochondrial homeostasis and maintenance of genome stability.
Here, it was observed that autophagy deficiency in the kidney exaggerates age-dependent mitochondrial dysfunction, reduces mtDNA copy number and increases mtDNA mutations. Although many studies have demonstrated damaged mitochondria in autophagy-deficient cells, we, for the first time, comprehensively analyzed the mitochondrial dysfunction and mtDNA abnormalities due to the long-term autophagy deficiency. These findings indicate that autophagy counteracts the aging process by improving mitochondrial homeostasis, although causality between aging and a decline in mitochondrial function has long been debated.Citation31 The mitochondrial free-radical theory of aging proposes that progressive mitochondrial dysfunction occurring with aging results in increased ROS production, which in turn causes further mitochondrial dysfunction and global cellular damage.Citation32,33 Recent studies have forced the reevaluation of this theory. Of particular impact has been the unexpected observation that genetic manipulations in mice that increase mitochondrial ROS and oxidative damage do not accelerate aging,Citation34,35 while genetic manipulations that impair mitochondrial function, but do not increase ROS, accelerate aging.Citation36-39 Accumulating evidence suggests that ROS plays a compensatory role in triggering proliferation and survival in response to physiological signals and stress conditions; however, beyond a certain threshold, ROS aggravates age-associated damage.Citation33 The present observation that increased staining for oxidative stress markers, including HNE and CML, is concomitant with an increased cellular proliferation rate of proximal tubule cells in aged atg5F/F-KAP mice (Fig. S7), support this new conceptual framework regarding the dual roles of ROS on aging. Given that mtDNA mutations and mitochondrial dysfunction could affect aging regardless of ROS production, it is possible that depletion of mtDNA could contribute to cell death during aging. Indeed increased apoptosis and/or cell death has now been reported in a variety of mtDNA-depleted mouse tissues including the heart,Citation40 pyramidal neurons,Citation41 and pancreatic β cells.Citation42 The observed increase in apoptosis in proximal tubule cells of aged atg5F/F-KAP mice in this study support this possibility (Fig. S8). The fact that the mitochondria do not accumulate (dysfunctional or not) even when autophagy is inhibited is counterintuitive, given the role of autophagy and mitophagy in mitochondrial clearance. Although the mechanism remains to be elucidated, one possibility is that Atg5- and Atg7-independent alternative autophagy could compensate for the clearance of the damaged mitochondria as indicated by a recent paper.Citation43 Another possibility is that mitochondrial biogenesis could be suppressed with aging.
Another important finding is that DNA damage, as assessed by phospho-H2AFX/γ-H2AX staining, was increased in proximal tubular cells of aged atg5F/F-KAP mice. The genetic links between deficient autophagy and tumorigenesis have been highlighted recently.Citation44-46 Although the molecular mechanisms by which autophagy functions in tumor suppression are poorly defined, autophagy deficiency may promote genomic instability in metabolically stressed cells, leading to oncogene activation and tumor progression.Citation47 Insufficient generation of ATP required for DNA repair, damage to critical cellular proteins that control mitosis or centrosome function, or excessive generation of ROS due to inefficient removal of damaged mitochondria in autophagy-deficient cells are potential causes of chromosomal instability. The accumulation of genomic damage throughout life is a common dominator in aging.Citation8 Various forms of DNA alteration are a consequence of the age-related decline in autophagic activity and may affect essential genes and transcriptional pathways, thereby resulting in cellular dysfunction that may jeopardize kidney homeostasis.
Pharmacological approaches to modulate autophagy could hold promise for treating age-related kidney disease. One class of candidate drugs that upregulates autophagy is the MTOR inhibitors. First-generation MTOR inhibitors including rapamycin and its derivatives, ATP competitive small-molecule MTOR inhibitors (such as PP242 and Torin1), and catalytic inhibitors that selectively block the MTOR kinase (such as AZD8055) have proven effective in inducing autophagy in yeast and mammalian cell lines.Citation48 Indeed, Harrison et al. have recently reported that rapamycin extends the life span of both male and female mice when fed beginning at 600 or 270 d of age.Citation49 In addition, numerous other compounds have been described that induce autophagy, including AMPK activators, inhibitors of the phosphatidylinositol signaling pathway and others. It should be investigated whether these drugs could improve decreased sensitivity to metabolic stress in aged kidney in the future study.
In conclusion, we report that an age-dependent elevation in basal autophagy plays a crucial role in counteracting kidney aging through mitochondrial quality control and maintenance of genome stability, as well as a reduced capacity for upregulation of autophagic flux in response to metabolic stress may be associated with age-related kidney diseases. Strategies aimed at modulating autophagy hold promise for treating age-related kidney disease.
Materials and methods
Mice
To generate tamoxifen-inducible proximal tubule-specific Atg5 knockout mice (Atg5F/F-NDRG1), Atg5-floxed mice were crossed with NDRG1-Cre mice.Citation21 For the induction of Atg5 deletion, we intraperitoneally injected tamoxifen (Sigma-Aldrich, T5648; 1 mg/10 g body weight) dissolved in corn oil (Sigma-Aldrich, C8267) at a concentration of 10 mg/mL 3 d a wk. To assess Cre recombinase activity, we performed LacZ staining in the kidneys of double transgenic heterozygote mice established by crossing CAG-CAT-Z mice (a gift from Jun-ichi Miyazaki) expressing LacZ after Cre-mediated recombination with NDRG1-Cre transgenic mice (Fig. S9). GFP-MAP1LC3B transgenic mice and atg5F/F-KAP with C57BL/6 background have been described previously.Citation17,24 Chloroquine (Sigma-Aldrich, C6628; 50 μg/g body weight) was injected intraperitoneally and mice sacrificed 6 h following the injection. Cisplatin (Sigma-Aldrich, P4394; 15 μg/g body weight) was injected intraperitoneally as previously described.Citation18 All animal experiments were approved by the institutional committee of the Animal Research Committee of Osaka University and the Japanese Animal Protection and Management Law (No. 25).
Antibodies
We used the following antibodies: antibodies for LRP2/MEGALIN (a gift from T. Michigami, Department of Bone and Mineral Research, Osaka Medical Center and Research Institute for Maternal and Child Health, Japan), COL1A1 (Abcam, ab34710), ATG5 (MBL, M153-3), SQSTM1 (MBL, PM045), MAP1LC3 (Cell Signaling Technology, 2755), ubiquitin (Cell Signaling Technology, 3936), SIRT1 (Millipore, 07-131), phosphorylated PRKAA/AMPKα (Thr172; Cell Signaling Technology, 2535), PRKAA/AMPKα (Cell Signaling Technology, 5831), phosphorylated ACAC (Ser79; Cell Signaling Technology, 11818), phosphorylated RPS6 (Ser235/236; Cell Signaling Technology, 2211), ACTB (Sigma-Aldrich, A5316), phospho-H2AFX/γ-H2AX (Ser139; Millipore, 05-636-I), 4HNE (Japan Institute for the Control of Aging, MHN-020P), dityrosine (Japan Institute for the Control of Aging, MDT-020P), CML (Transgenic, KH024), MKI67 (BD Pharmingen, 550609), COX4I1 (MBL, PM063), biotinylated secondary antibodies (Vector Laboratories, BA-1000 [anti-rabbit IgG], BA-2001 [anti-mouse IgG]), horseradish peroxidase-conjugated secondary antibodies (DAKO, P0448 [anti-rabbit IgG], P0447 [anti-mouse IgG]), and Alexa Fluor 555-conjugated secondary antibody (Invitrogen, A31572).
Histological analysis
Histological analysis was performed as previously described with modification.Citation18 To assess dots positive for GFP or monitoring the levels of the post-translational modifications of MTORC1 and AMPK substrates (p-RPS6 and p-ACAC), post-fixed frozen tissue was sectioned and immunostained for LRP2 (proximal tubular cell marker) or p-RPS6 and p-ACAC. GFP-MAP1LC3B dots per proximal tubule in at least 10 high-power fields (×600) were counted. The fluorescence images were collected using confocal microscopy (FV1000-D [Olympus, Tokyo, Japan]). The intensity of the positive staining area was measured using a digital image-analyzing software, ImageJ (available at http://rsbweb.nih.gov/ij/index.html; National Institutes of Health, Bethesda, MD). For each kidney, at least 10 high-power fields (×600) were analyzed. Immunohistochemical staining for SQSTM1, ubiquitin, COL1A/collagen type I, HNE, dityrosine, CML, COX4I1, and MKI67 was performed on paraffin-embedded sections after antigen retrieval via autoclaving in 0.01 mmol/L citrate buffer (pH 6.0) for 10 min at 120°C and blocking with 1.5% bovine serum albumin (Sigma-Aldrich, A3059-100G) in phosphate-buffered saline (composed of 137 mM NaCl (Nacalai, 31320-05), 8 mM Na2HPO4 (Nacalai, 31723-35), 2.7mM KCl (Nacalai, 28514-75), 1.5 mM KH2PO4 (Wako, 169-04245), pH 7.4) for 60 min. For double staining for SQSTM1 (or ubiquitin, HNE, dityrosine, CML, COX4I1, and MKI67) and LRP2/MEGALIN, each molecule was first visualized using a horseradish peroxidase–diaminobenzidine compound (Nichirei, 415172), followed by detection of LRP2/MEGALIN using alkaline phosphatase and NBT/BCIP Stock Solution (Roche, 11681451001). The number of SQSTM1- or ubiquitin-positive dots was quantified per individual LRP2/MEGALIN-positive proximal tubule. The percentage of the immune-positive area for COL1A was calculated using the ImageJ imaging software. For each kidney, at least 10 high-power fields (×400) were analyzed. For electron microscopy, kidney specimens were fixed with 2.5% glutaraldehyde (Wako, 071-01931) and observed using a Hitachi H-7650 transmission electron microscope (Hitachi, Tokyo, Japan). Autophagosomes and autolysosomes in podocytes or proximal tubules of GFP-MAP1LC3B transgenic mice were counted on electron micrographs. For each kidney, at least 10 podocytes or tubules were analyzed by 2 nephrologists (T.Y. and T.N.) in a blinded manner.
Biochemical parameters
Plasma urea nitrogen, creatinine, cystatin C, total cholesterol, and triglycerides were measured using the BUN-Test-Wako (Wako, 279-36201), the CRE-EN Kainos (Kainos, TKA7500), cystatin C (mouse) ELISA kit (BioVendor, RD291009200R), the cholesterol E-test (WAKO, 439-17501), and the TG E-test (WAKO, 432-40201) respectively. Urinary ALB/albumin excretion and urinary 8OHdG excretion were measured with the MicrofluoralTM microalbumin test (Progen, PR2005) and the Highly Sensitive ELISA kit for 8OHdG (Japan Institute for the Control of Aging, KOG-HS10/E), respectively. They were adjusted by urinary creatinine measured using the CRE-EN Kainos.
Senescence-associated GLB1/β-galactosidase (SA-GLB1) staining
Senescent cells were detected using Senescence Cells Histochemical Staining Kit (Sigma-Aldrich, CS0030) following the manufacturer's instructions. Tissue sections were counterstained with eosin and examined under a microscope. The intensity of the positive staining area was measured using the ImageJ imaging software. For each kidney, at least 10 high-power fields (×400) were analyzed.
COX and SDH staining
COX and SDH staining were performed as previously described.Citation18 We used fresh cryosections (5-μm thick) of kidney tissues. For COX staining, the sections were incubated in the solution composed of 2 μg/ml of catalase (Sigma-Aldrich, C1345), 1 mg/ml of cytochrome c (Sigma-Aldrich, C3131), 0.5 mg/ml of diaminobenzidine (Wako, 040-27001) in 0.1 M of phosphate buffer (pH 7.4) for 1 h at 37°C. The SDH staining was performed according to the modified method of Martin et al.Citation50 In brief, we prepared NBT stock composed of KCN 6.5 mg (Nacalai, 28529-82), EDTA 185 mg (Sigma-Aldrich, ED2SS), nitroblue tetrazolium 100 mg (Nacalai, 24720-56) in 100 ml of 0.1 M phosphate buffer (pH 7.6) and 500 mM of sodium succinate solution (Nacalai, 32406-65) diluted in distilled water. Then the sections were incubated in a solution mixed with 2 ml of NBT stock solution, 0.2 ml of succinate solution, and 0.7 mg of phenazine methosulfate (Nacalai, 26712-51) for 15 min at 37°C. The intensity of the positive staining area was measured using the ImageJ imaging software. For each kidney, at least 10 high-power fields (×400) were analyzed.
Isolation of mouse kidney proximal tubule cells
Kidney proximal tubule cells were isolated from mice using the CELLection Biotin Binder Kit (Invitrogen, 11533D) and biotinylated Lotus Tetragonolobus Lectin (Vector Laboratories, B-1325) as described previously.Citation17 Isolated proximal tubule cells were snap-frozen in liquid nitrogen and stored at −80°C before further process of extraction of total DNA and mRNA.
Relative quantification of mtDNA copy numbers
Total DNA (mtDNA and nDNA) was extracted from isolated proximal tubules with Wizard® Genomic DNA purification kit according to the manufacturer's instructions (Promega, A1120). Real-time quantitative PCR of the mtDNA encoded gene mt-Co1 (GenBank accession no. NC_001807) and the single-copy nuclear gene Ndufv1 (GenBank accession number NM_133666) was carried out with an ABI PRISM 7900HT sequence detection system (Applied Biosystems, Foster City, CA, USA). Relative quantification of mtDNA copy numbers were depicted as the ratio of the relative concentrations of mtDNA:nDNA. The sequences of primers for the mt-Co1 and Ndufv1 gene are as follows: mt-Co1-F 5′-tgctagccgcaggcattac-3′; mt-Co1-R 5′-gggtgcccaaagaatcagaac-3′; Ndufv1-F 5′-cttccccactggcctcaag-3′; Ndufv1-R 5′-ccaaaacccagtgatccagc-3′.
Analysis of mtDNA and nDNA damage using long-amplicon quantitative real-time PCR
Analysis of mtDNA and nDNA damage was performed as previously described with modification.Citation51 Using the comparative Ct method, DNA damage was quantified by comparing the relative amplification of large fragments of DNA from each total DNA sample with controls and normalizing this to the amplification of small fragments. In this assay, we set the total DNA extracted from the proximal tubule cells of 3-week-old Atg5F/F mice as nondamaged control samples and those of cisplatin-treated Atg5F/F mice as damaged control samples (Fig. S5C). The long-amplicon quantitative PCR was carried out on an ABI PRISM 7900HT sequence detection system with the PrimeSTAR® GXL DNA Polymerase kit (TaKaRa, R050A) with properly diluted SYBR® Green I Nucleic Acid Stain (Lonza, 50513). Representative amplifications of large fragments of DNA are shown (Fig. S5D). The short fragments of single-copy nuclear gene Ndufv1 and the mouse mitochondrial genome were quantified using a usual real-time PCR method for normalization to nDNA and mtDNA copy number. The sequences of the primers used were as follows: for the 8.7-kb fragment of the Hbb/β-globin gene (GenBank accession no. X14061), 5′-ttgagactgtgattggcaatgcct-3′ and 5′-cctttaatgcccatcccggact-3′; for the 10-kb fragment of the mouse mitochondrial genome, 5′-gccagcctgacccatagccataatat-3′ and 5′-gagagattttatgggtgtaatgcgg-3′; for the 121-bp fragment of Ndufv1 (as described above), and for the 117-bp fragment of the mouse mitochondrial genome, 5′-cccagctactaccatcattcaagt-3′ and 5′-gatggtttgggagattggttgatgt-3′. DNA lesion frequencies were calculated using the Poisson transformation as previously described.Citation51 We determined the relative DNA damage by combining the results from 2 separate qPCR runs.
Quantitative RT-PCR and western blot analysis
Quantitative RT-PCR and western blot analyses were performed as previously described.Citation52 The sequences of the primers used were as follows: Havcr1-F, 5′-tcagctcgggaatgcaca-3′; Havcr1-R, 5′-tggttgccttccgtgtct-3′; Lcn2-F, 5′-ctacaaccagttcgccatgg-3′; Lcn2-R, 5′-acactcaccacccattcagt-3′; Col1a1-F, 5′-acgccatcaaggtctactgc-3′; Col1a1-R, 5′-actcgaacgggaatccatcg-3′; 5′-ggcttcagactggtacacat-3′; 18s-rRNA-F, 5′-aaacggctaccacatccaag-3′; 18s-rRNA-R, 5′-cctccaatggatcctcgtta-3′.
Tunel staining
Apoptotic cells were detected using the TUNEL assay with an in situ apoptosis detection kit (Takara, MK-500). Tissue sections were counterstained with Methyl Green Solution (Wako, 138-12701) and examined under a microscope.
Statistics
We used box plots to illustrate the spread and differences of samples (n > 5). Center lines show the medians; box limits indicate the 25th and 75th percentiles as determined by R software; whiskers extend 1.5 times the interquartile range from the 25th and 75th percentiles, outliers are represented by dots. n = 7 to 10 sample points. For very small samples (n < 5), we used bar plots. All results are presented as mean ± SE. Statistical analyses were conducted using JMP software (SAS Institute). Multiple-group comparisons were performed using analysis of variance with post-testing using the Tukey-Kramer test. Two-tailed unpaired Student t tests were used for comparison between 2 groups when appropriate. Statistical significance was defined as P < 0.05.
Abbreviations
| ACTB | = | actin, beta |
| ACAC | = | acetyl-CoA carboxylase |
| AMPK | = | AMP-activated protein kinase |
| Atg | = | autophagy-related |
| CKD | = | chronic kidney disease |
| CML | = | N-carboxymethyllysine |
| COL1A1 | = | collagen type I alpha 1 |
| COX | = | cytochrome c oxidase |
| COX4I1 | = | cytochrome c oxidase subunit IV isoform 1 |
| GFP | = | green fluorescent protein |
| HAVCR1/Kim-1 | = | hepatitis A virus cellular receptor 1 |
| H2AFX | = | H2A histone family, member X |
| HNE | = | 4-hydroxy-2-nonenal |
| KAP | = | kidney androgen regulated protein |
| LCN2/Ngal | = | lipocalin 2 |
| LRP2 | = | low density lipoprotein receptor-related protein 2 |
| MAP1LC3B/LC3 | = | microtubule-associated protein 1 light chain 3B |
| MKI67 | = | antigen identified by monoclonal antibody Ki67 |
| MT-CO1 | = | mitochondrially encoded cytochrome c oxidase I |
| mtDNA | = | mitochondrial DNA |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| NDRG1 | = | N-myc downstream regulated gene 1 |
| NDUFV1 | = | NADH:ubiquinone oxidoreductase core subunit V1 (formerly also termed NADH dehydrogenase (ubiquinone) flavoprotein 1) |
| nDNA | = | nuclear DNA |
| PAS | = | periodic-acid Schiff |
| ROS | = | reactive oxygen species |
| RPS6 | = | ribosomal protein S6 |
| SA | = | senescence-associated |
| SDH | = | succinate dehydrogenase complex |
| SIRT1 | = | sirtuin 1 |
| SQSTM1 | = | sequestosome 1 |
Disclosure of potential conflicts of interest
The authors declare that they have no conflict of interest.
KAUP_A_1159376_Supplementary_Fig1-9_correction.pdf
Download PDF (1.2 MB)Acknowledgments
We thank N. Mizushima, University of Tokyo, for Atg5F/F and GFP-MAP1LC3B transgenic mice; T. Michigami, Osaka Medical Center and Research Institute, for LRP2/MEGALIN antibody; and N. Horimoto, K. Shibayama, M. Kameda, and M. Nishio for technical and secretary assistance.
Funding
This work was supported by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology in Japan (22590890 [to H.K.], 24591196 [to Y.T.], and 24659416 [to Y.I.]), Takeda Medical Research Foundation [to Y.T.], Manpei Suzuki Diabetes Foundation [to T.K.] and Uehara Memorial Foundation [to T.K.].
References
- Zhou X, Rakheja D, Yu X, Saxena R. The aging kidney. Kidney Int 2008; 74:710-20; PMID:18614996; http://dx.doi.org/10.1038/ki.2008.319
- Imai E, Horio M, Watanabe T, Iseki K, Yamagata K, Hara S, Ura N, Kiyohara Y, Moriyama T, Ando Y, et al. Prevalence of chronic kidney disease in the Japanese general population. Clin Exp Nephrol 2009; 13:621-30; PMID:19513802; http://dx.doi.org/10.1007/s10157-009-0199-x
- Astor BC, Matsushita K, Gansevoort RT, van der Velde M, Woodward M, Levey AS, Jong PE de, Coresh J, de Jong PE, El-Nahas M, et al. Lower estimated glomerular filtration rate and higher albuminuria are associated with mortality and end-stage renal disease. A collaborative meta-analysis of kidney disease population cohorts. Kidney Int 2011; 79:1331-40; PMID:21289598; http://dx.doi.org/10.1038/ki.2010.550
- Bolignano D, Mattace-Raso F, Sijbrands EJG, Zoccali C. The aging kidney revisited: A systematic review. Ageing Res Rev 2014; 14C:65-80; http://dx.doi.org/10.1016/j.arr.2014.02.003
- Tauchi H, Tsuboi K, Okutomi J. Age changes in the human kidney of the different races. Gerontologia 1971; 17:87-97; PMID:5093734; http://dx.doi.org/10.1159/000211811
- Martin JE, Sheaff MT. Renal ageing. J. Pathol 2007; 211:198-205
- Kenyon CJ. The genetics of ageing. Nature 2010; 464:504-12; PMID:20336132; http://dx.doi.org/10.1038/nature08980
- López-Otín C, Blasco M A, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153:1194-217; PMID:Can't; http://dx.doi.org/10.1016/j.cell.2013.05.039
- Rubinsztein D, Mariño G, Kroemer G. Autophagy and aging. Cell 2011; 146:682-95; PMID:21884931; http://dx.doi.org/10.1016/j.cell.2011.07.030
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27-42; PMID:18191218; http://dx.doi.org/10.1016/j.cell.2007.12.018
- Choi AMK, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:651-62; PMID:23406030; http://dx.doi.org/10.1056/NEJMra1205406
- Cuervo A. Autophagy and aging: keeping that old broom working. Trends Genet 2008; 24:604-12; PMID:18992957; http://dx.doi.org/10.1016/j.tig.2008.10.002
- Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner B A, Scherzer CR, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A 2010; 107:14164-9; PMID:20660724; http://dx.doi.org/10.1073/pnas.1009485107
- Cui J, Bai X-Y, Shi S, Cui S, Hong Q, Cai G, Chen X. Age-related changes in the function of autophagy in rat kidneys. Age (Dordr) 2012; 34:329-39; PMID:21455601; http://dx.doi.org/10.1007/s11357-011-9237-1
- Lionaki E, Markaki M, Tavernarakis N. Autophagy and ageing: insights from invertebrate model organisms. Ageing Res Rev 2013; 12:413-28; PMID:22634332; http://dx.doi.org/10.1016/j.arr.2012.05.001
- Takabatake Y, Kimura T, Takahashi A, Isaka Y. Autophagy and the kidney: health and disease. Nephrol Dial Transplant 2014; 29(9):1-9
- Kimura T, Takabatake Y, Takahashi A, Kaimori J, Matsui I, Namba T, Kitamura H, Niimura F, Matsusaka T, Soga T, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol 2011; 22:902-13; PMID:21493778; http://dx.doi.org/10.1681/ASN.2010070705
- Takahashi A, Kimura T, Takabatake Y, Namba T, Kaimori J, Kitamura H, Matsui I, Niimura F, Matsusaka T, Fujita N, et al. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol 2012; 180:517-25; PMID:22265049; http://dx.doi.org/10.1016/j.ajpath.2011.11.001
- Maejima I, Takahashi A, Omori H, Kimura T, Takabatake Y, Saitoh T, Yamamoto A, Hamasaki M, Noda T, Isaka Y, et al. Autophagy sequesters damaged lysosomes to control lysosomal biogenesis and kidney injury. EMBO J 2013; 7:1-12
- Namba T, Takabatake Y, Kimura T, Takahashi A, Yamamoto T, Matsuda J, Kitamura H, Niimura F, Matsusaka T, Iwatani H, et al. Autophagic Clearance of Mitochondria in the Kidney Copes with Metabolic Acidosis. J Am Soc Nephrol 2014; 25(10):1-13
- Endo T, Nakamura J, Sato Y, Asada M, Yamada R, Takase M, Takaori K, Oguchi A, Iguchi T, Higashi AY, et al. Exploring the origin and limitations of kidney regeneration. J Pathol 2015; 236:251-63; PMID:25664690; http://dx.doi.org/10.1002/path.4514
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Øvervatn A, Stenmark H, Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol 2005; 171:603-14; PMID:16286508; http://dx.doi.org/10.1083/jcb.200507002
- Komatsu M, Waguri S, Koike M, Sou Y shin, Ueno T, Hara T, Mizushima N, Iwata J ichi, Ezaki J, Murata S, et al. Homeostatic Levels of p62 Control Cytoplasmic Inclusion Body Formation in Autophagy-Deficient Mice. Cell 2007; 131:1149-63; PMID:18083104; http://dx.doi.org/10.1016/j.cell.2007.10.035
- Mizushima N, Yamamoto A. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 2004; 15:1101-11; PMID:14699058; http://dx.doi.org/10.1091/mbc.E03-09-0704
- Zois CE, Giatromanolaki A, Sivridis E, Papaiakovou M, Kainulainen H, Koukourakis MI. “Autophagic flux” in normal mouse tissues: focus on endogenous LC3A processing. Autophagy 2011; 7:1371-8; PMID:21997374; http://dx.doi.org/10.4161/auto.7.11.16664
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995; 92:9363-7; PMID:7568133; http://dx.doi.org/10.1073/pnas.92.20.9363
- Kasai H, Nishimura S. Hydroxylation of deoxy guanosine at the C-8 position by polyphenols and aminophenols in the presence of hydrogen peroxide and ferric ion. Gann 1984; 75:565-6; PMID:6468841
- Liu S, Hartleben B, Kretz O, Wiech T. Autophagy plays a critical role in kidney tubule maintenance, aging and ischemia-reperfusion injury. Autophagy 2012; 1:230-5
- Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, Passegué E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013; 494:323-7; PMID:23389440; http://dx.doi.org/10.1038/nature11895
- Rajawat YS, Hilioti Z, Bossis I. Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev 2009; 8:199-213; PMID:19427410; http://dx.doi.org/10.1016/j.arr.2009.05.001
- Bratic A, Larsson N. The role of mitochondria in aging. J Clin Invest 2013; 123:951-7; PMID:23454757; http://dx.doi.org/10.1172/JCI64125
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 1956; 11:298-300; PMID:13332224; http://dx.doi.org/10.1093/geronj/11.3.298
- Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012; 48:158-67; PMID:23102266; http://dx.doi.org/10.1016/j.molcel.2012.09.025
- Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, Thorpe SR, Alderson NL, Baynes JW, Epstein CJ, Huang T-T, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics 2003; 16:29-37; PMID:14679299; http://dx.doi.org/10.1152/physiolgenomics.00122.2003
- Zhang Y, Ikeno Y, Qi W, Chaudhuri A, Li Y, Bokov A, Thorpe SR, Baynes JW, Epstein C, Richardson A, et al. Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J Gerontol A Biol Sci Med Sci 2009; 64:1212-20; PMID:19776219; http://dx.doi.org/10.1093/gerona/glp132
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004; 429:417-23; PMID:15164064; http://dx.doi.org/10.1038/nature02517
- Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 2005; 309:481-4; PMID:16020738; http://dx.doi.org/10.1126/science.1112125
- Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA, Nijtmans L, Nedergaard J, Cannon B, Larsson N-G, Trifunovic A. Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab 2009; 10:131-8; PMID:19656491; http://dx.doi.org/10.1016/j.cmet.2009.06.010
- Hiona A, Sanz A, Kujoth GC, Pamplona R, Seo AY, Hofer T, Someya S, Miyakawa T, Nakayama C, Samhan-Arias AK, et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One 2010; 5:e11468
- Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG. Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci U S A 2001; 98:4038-43; PMID:11259653; http://dx.doi.org/10.1073/pnas.061038798
- Sörensen L, Ekstrand M, Silva JP, Lindqvist E, Xu B, Rustin P, Olson L, Larsson NG. Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J Neurosci 2001; 21:8082-90; PMID:11588181
- Silva JP, Köhler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat Genet 2000; 26:336-40; PMID:11062475; http://dx.doi.org/10.1038/81649
- Hirota Y, Yamashita S, Kurihara Y, Jin X, Aihara M, Saigusa T, Kang D, Kanki T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 2015; 11:332-43; PMID:25831013; http://dx.doi.org/10.1080/15548627.2015.1023047
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672-6; PMID:10604474; http://dx.doi.org/10.1038/45257
- Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev 2011; 25:795-800; PMID:21498569; http://dx.doi.org/10.1101/gad.2016211
- Guo JY, Xia B, White E. Autophagy-mediated tumor promotion. Cell 2013; 155:1216-9; PMID:24315093; http://dx.doi.org/10.1016/j.cell.2013.11.019
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21:1367-81; PMID:17510285; http://dx.doi.org/10.1101/gad.1545107
- Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol 2011; 7:9-17; PMID:21164513; http://dx.doi.org/10.1038/nchembio.500
- Harrison D, Strong R, Sharp Z, Nelson J. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460:392-5; PMID:19587680
- Martin TP, Vailas AC, Durivage JB, Edgerton VR, Castleman KR. Quantitative histochemical determination of muscle enzymes: biochemical verification. J Histochem Cytochem 1985; 33:1053-9; PMID:4045183; http://dx.doi.org/10.1177/33.10.4045183
- Furda AM, Bess AS, Meyer JN, Van Houten B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol Biol 2012; 920:111-32; PMID:22941600; http://dx.doi.org/10.1007/978-1-61779-998-3_9
- Matsui I, Ito T, Kurihara H, Imai E, Ogihara T, Hori M. Snail, a transcriptional regulator, represses nephrin expression in glomerular epithelial cells of nephrotic rats. Lab Invest 2007; 87:273-83; PMID:17260001; http://dx.doi.org/10.1038/labinvest.3700518