
ABSTRACT
Chronic lung inflammation is accepted as being associated with the development of lung cancer caused by nickel exposure. Therefore, identifying the molecular mechanisms that lead to a nickel-induced sustained inflammatory microenvironment that causes transformation of human bronchial epithelial cells is of high significance. In the current studies, we identified SQSTM1/p62 as a novel nickel-upregulated protein that is important for nickel-induced inflammatory TNF expression, subsequently resulting in transformation of human bronchial epithelial cells. We found that nickel exposure induced SQSTM1 protein upregulation in human lung epithelial cells in vitro and in mouse lung tissues in vivo. The SQSTM1 upregulation was also observed in human lung squamous cell carcinoma. Further studies revealed that the knockdown of SQSTM1 expression dramatically inhibited transformation of human lung epithelial cells upon chronic nickel exposure, whereas ectopic expression of SQSTM1 promoted such transformation. Mechanistic studies showed that the SQSTM1 upregulation by nickel was the compromised result of upregulating SQSTM1 mRNA transcription and promoting SQSTM1 protein degradation. We demonstrated that nickel-initiated SQSTM1 protein degradation is mediated by macroautophagy/autophagy via an MTOR-ULK1-BECN1 axis, whereas RELA is important for SQSTM1 transcriptional upregulation following nickel exposure. Furthermore, SQSTM1 upregulation exhibited its promotion of nickel-induced cell transformation through exerting an impetus for nickel-induced inflammatory TNF mRNA stability. Consistently, the MTOR-ULK1-BECN1 autophagic cascade acted as an inhibitory effect on nickel-induced TNF expression and cell transformation. Collectively, our results demonstrate a novel SQSTM1 regulatory network that promotes a nickel-induced tumorigenic effect in human bronchial epithelial cells, which is negatively controlled by an autophagic cascade following nickel exposure.
Introduction
Lung cancer is the most common type of cancer in the world and it is estimated that 242,550 new cases and 163,660 deaths were attributed to lung cancer in the USA in 2014.Citation1 Lung cancer includes non-small cell lung carcinoma (NSCLC) and small cell lung carcinoma (SCLC).Citation2 SCLC appears to arise from neuroendocrine cells,Citation3 whereas NSCLC, which accounts for approximately 85% of all cases, derives from pulmonary epithelial cells.Citation4 Its morbidity and mortality have increased rapidly in the past 30 years, particularly in developing countries, such as China.Citation5 Unlike many other cancers, the etiological factor of the majority of cases of lung cancer is tobacco consumption and environmental pollution.Citation6,7 Therefore, understanding the factors that predispose an individual to lung cancer is of prime importance. Among lung carcinogens, nickel is classified as group 1 carcinogen (confirmed carcinogen) for humans by International Agency for Research on Cancer in 1990.Citation8 Nickel is abundant in metallurgy waste air, burning fossil fuels, and cigarette smoke.Citation9 Increased mortality from lung malignant tumors has been found in nickel refinery workers who are exposed daily to nickel-contaminated air.Citation10 The carcinogenic effects of nickel are also supported by results observed in human epidemiological investigation, in vitro cell transformation studies and in vivo animal experiments.Citation11-13 The association between lung inflammation and lung cancer development is supported by at least 10 cohort studies and animal studies.Citation14 And although the chronic lung inflammatory microenvironment is accepted to be a major driving force for the development of lung cancers from the inflammatory process,Citation15,16 it is still unclear how chronic nickel exposure results in chronic lung inflammation and how chronic lung inflammation develops into tumors.
SQSTM1/p62 (sequestosome 1) is a multifunctional protein, and acts as a scaffold for intracellular signaling that controls bone remodeling, obesity and smooth muscle proliferation.Citation17-19 It has been reported that sustained SQSTM1 expression resulting from autophagy defects leads to NFKB activation and gene expression, which in turn promotes tumorigenesis in mouse models.Citation20 Paradoxically, SQSTM1 synergizes with autophagy for tumor growth in vivoCitation21 and the knockdown of SQSTM1 shows significant inhibitory effects on autophagy activation and tumor growth of human colon cancer cells both in vitro and in a xenograft tumor model.Citation22 Thus, the biological role of SQSTM1 in cancer is far from understood. Although SQSTM1 upregulation has been reported to be associated with poor prognosis in patients with lung adenocarcinoma,Citation23 nothing is known about the effect of exposure to environmental carcinogens on SQSTM1 expression. More importantly, nothing is known about the relationship between SQSTM1 upregulation and TNF overexpression, or the upstream regulators and/or downstream effectors that induce TNF expression and cause human bronchial epithelial cell transformation upon environmental carcinogen exposure. Thus, we explored the potential effects of nickel exposure on SQSTM1 expression, autophagy activation, and the relationship between SQSTM1 expression, autophagy activation and inflammatory TNF expression, as well as cell transformation in human bronchial epithelial cells following nickel exposure in the current studies. Moreover, our important findings were also applicable to an in vivo animal model.
Results
Upregulation of SQSTM1 expression was observed as a result of nickel exposure both in vitro and in vivo, and in human lung cancer tissues.
Although SQSTM1 overexpression has been reported in some cancer tissues,Citation23 to the best of our knowledge, its potential induction in lung carcinogenesis due to environmental lung carcinogen exposure have never been explored. Although lung inflammation involves events from inflammatory cells, such as macrophages and leukocytes, the lung epithelial cells are the cells that are first exposed and respond to nickel exposure. Therefore, the current studies, focused on the effects of nickel exposure in human lung epithelial cells. Since the Occupational Safety and Health Administration permissible exposure limit is 1 mg Ni/m3, the in vitro dose of nickel chloride at 1.0 mM is equivalent to the same alveolar dose of a human exposed to this limit for 8 h with light work.Citation24 We also performed a colony-survival assay to determine the cytotoxic effects of 1.0 mM nickel on Beas-2B cells, and the results indicated that there were no significant inhibition of colonies in Beas-2B cells following nickel exposure as shown in Fig. S1. To test the effects of nickel exposure in human lung epithelial cells, Beas-2B cells were exposed to NiCl2, and SQSTM1 protein expression was assessed by western blot. As shown in , nickel exposure (1.0 mM) resulted in a significant upregulation of SQSTM1 protein expression. We further extended our observation of nickel upregulation of SQSTM1 protein to normal human bronchial epithelial cells (NHBECs) and human bronchial epithelial BEP2D cells (Fig. S2). It has been reported that SQSTM1 exhibits its function mostly acting as a scaffold for intracellular signaling.Citation25 To understand the potential SQSTM1 cellular location following nickel exposure, GFP-SQSTM1 was stably transfected into Beas-2B cells and stable transfectant Beas-2B(GFP-SQSTM1) was established, as identified (). Beas-2B(GFP-SQSTM1) was then exposed to nickel and the dynamic distribution of GFP-SQSTM1 was observed in various time points following nickel exposure under a Leica fluorescence microcope. The results showed that GFP-SQSTM1 was mainly present in the cytoplasm in Beas-2B cells without nickel exposure, whereas nickel exposure resulted in GFP-SQSTM1 fluorescence puncta in a time-dependent manner (). These results reveal that SQSTM1 protein upregulation might be associated with its biological function in cells due to nickel exposure.
Figure 1. Upregulation of SQSTM1 expression by nickel in vitro and in vivo, as well as SQSTM1 overexpression in human lung cancer tissues. (A and B) 2×105 of Beas-2B cells were seeded into each well of 6-well plates. After the cell density reached 80∼90%, the cells were exposed to 1.0 mM NiCl2 for the various time points indicated. The cells were then extracted with an SDS-sample buffer (10 mM Tris-HCl, pH 7.4, 1% SDS, 1mM Na3VO4, and 1× complete protease inhibitor) and cell extracts were subjected to protein gel blot. ACTB was used as a control for protein loading. The data shown in (A) are representative of 3 independent experiments, and the SQSTM1 expression band was quantitatively analyzed by using prism software and presented in (B). (C) Beas-2B GFP-SQSTM1 stable transfectants were identified with western blotting; primary antibodies against GFP or SQSTM1 were used to determine exogenous GFP-SQSTM1 expression. (D) Beas-2B(GFP-SQSTM1) cells (1×104) were seeded into each well of an 8-well chamber. Following culturing at 37°C for 48 h, the cells were treated with 1.0 mM NiCl2 for the time points indicated, and the GFP-SQSTM1 distributions in Beas-2B cells were captured using fluorescence microscopy. (E-G) C57BL/6J mice were exposed to nickel nanoparticles via inhalation for 24 h or 48 h (E) or 5-months (F). Lung tissues were extracted for protein extracts and the extracts were subjected to western blot. ACTB was used as a protein loading control. (G) Seven pairs of human primary lung cancers and their adjacent normal lung tissues were obtained from the First Affiliated Hospital of Wenzhou Medical University; the tissue extracts were subjected to western blotting and TUBA was used as a protein loading control.
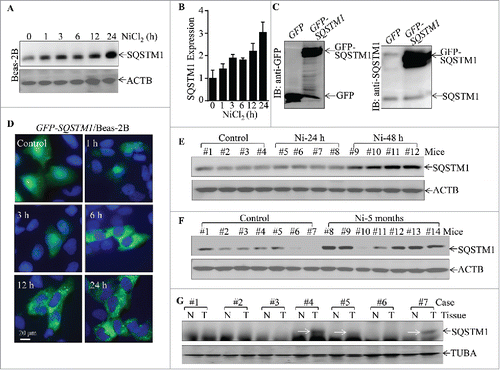
Although it is easy to use soluble nickel compound for mechanistic in vitro studies, nano-Ni-hydroxide would be more realistic in reflecting the real world exposure scenarios for in vivo experiments.Citation24 To evaluate the in vivo effect of nickel on SQSTM1 expression, we exposed C57BL/6J mice to nanoparticle nickel via inhalation for the indicated time points. The lung tissues from exposed mice were extracted for determination of SQSTM1 protein expression. The results obtained from protein gel blotting showed that SQSTM1 expression was significantly upregulated in mouse lung tissues following nickel exposure as early as 48 h post-exposure () and that there were sustained increases in mouse lung tissues (6 of 7 mice) following nickel exposure for 5 months, in comparison to the lung tissues obtained from control particle-exposed mice in all of 7 mice examined (). These results demonstrate that nickel is capable of upregulating SQSTM1 protein expression both in vitro and in vivo. To determine whether SQSTM1 upregulation is relevant to human lung carcinoma development, we extended our observation to human lung cancer tissues. Since the nickel exposure is associated with lung squamous cell carcinoma development,Citation26 we evaluated SQSTM1 expression in lung tissues from the patients with human lung squamous cell carcinoma. As shown in , SQSTM1 upregulation was observed in 3 (#4, #5, #7) of the 7 cases of lung squamous cell carcinoma patients (Table S1) as compared with that of the corresponding adjacent normal lung tissues. Collectively, our results demonstrate that SQSTM1 upregulation is not only observed in human lung epithelial cells and in mouse lung tissues following nickel exposure, but also exhibits in human lung cancer tissues.
SQSTM1 upregulation plays an important role in nickel-induced malignant transformation of human bronchial epithelial cells.
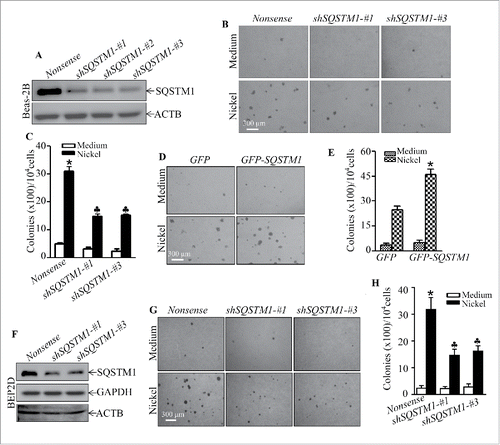
To determine whether SQSTM1 upregulation played an important role in nickel-induced carcinogenic effects, 3 shRNAs (short hairpin RNA) specifically targeting different sequences of human SQSTM1 were stably transfected into Beas-2B cells with puromycin selection, and Beas-2B shSQSTM1 stable transfectants, Beas-2B(shSQSTM1-#1), Beas-2B(shSQSTM1-#2) and Beas-2B(shSQSTM1-#3) were established. As shown in , the stable transfection of either one of 3 shRNAs that specially target different sequence of SQSTM1 mRNA dramatically knocked down SQSTM1 protein expression in Beas-2B cells in comparison to Beas-2B(Nonsense) transfectant. The repeated exposure of these transfectants to nickel resulted in Beas-2B(Nonsense) transfectant cells gaining the capability of anchorage-independent growth in soft agar, a hallmark of cellular malignant transformation. Knockdown of SQSTM1 expression led to a significant inhibition of Beas-2B cell transformation by nickel compared to the Beas-2B(Nonsense) transfectant under the same experimental conditions (). Consistently, ectopic expression of GFP-SQSTM1 in Beas-2B cells significantly increased nickel-induced cell transformation in comparison with Beas-2B(Vector) cells (). The Beas-2B cell line is a human bronchial epithelial cell line that has been immortalized by overexpression of SV40 large T antigen.Citation27 To exclude the possibility that nickel-induced cell transformation was mediated by nickel upregulation of Large T antigen gene, we evaluated the potential effect of nickel on Large T antigen expression in Beas-2B. The results showed that nickel exposure did not exhibit upregulation of Large T antigen in Beas-2B cells (Fig. S3). Moreover, we employed a second human bronchial epithelial cell line, BEP2D, which is immortalized by human papillomavirus.Citation28 The results showed a similar effect of SQSTM1 upregulation upon nickel exposure, whereas knockdown of SQSTM1 in BEP2D cells also attenuated nickel-induced cell transformation in . Taken together, our results strongly demonstrate that SQSTM1 upregulation contributes to human bronchial epithelial cell malignant transformation due to nickel exposure.
Figure 2. SQSTM1 upregulation played an important role in nickel-induced malignant transformation of human bronchial epithelial cells. (A and F) shSQSTM1-#1, shSQSTM1-#2, and shSQSTM1-#3 represent different shRNAs that specifically target 3 different sequences in SQSTM1 mRNA and its nonsense vector were stably transfected into Beas-2B cells (A) or BEP2D cells (F), respectively, and their stable transfectants were established and identified by western blot with specific anti-SQSTM1 antibody. (B and C) Beas-2B(Nonsense), Beas-2B(shSQSTM1-#1) and Beas-2B(shSQSTM1-#3) cells were repeatedly exposed to 0.5 mM NiCl2 for 6 months and then subjected to soft agar assay. The cell colonies were counted by microscopy. Each bar indicates the mean and SD from triplicate assays. The symbol (*) indicates a significant increase as compared with the medium control (p < 0.05), while the symbol (♣) indicates a significant decrease as compared with the Beas-2B (Nonsense) cells (p < 0.05). (D and E) Beas-2B(GFP) and Beas-2B(GFP-SQSTM1) cells were repeatedly exposed to 0.5 mM NiCl2 for 5 months and then subjected to the soft agar assay. Each bar indicates the mean and SD from triplicate assays. The symbol (*) indicates a significant increase in comparison to the Beas-2B(GFP) control (p < 0.05). (G and H) BEP2D(Nonsense), BEP2D(shSQSTM1-#1) and BEP2D(shSQSTM1-#3) cells were repeatedly exposed to 0.5 mM NiCl2 for 4 months and then subjected to the soft agar assay. The cell colonies were counted by microscopy. Each bar indicates the mean and SD from triplicate assays. The symbol (*) indicates a significant increase as compared with the medium control (p < 0.05), while the symbol (♣) indicates a significant inhibition as compared with the Beas-2B(Nonsense) cells (p < 0.05).
Activation of MTOR-ULK1-BECN1 autophagic cascade by nickel provides an inhibitory effect on SQSTM1 protein expression.
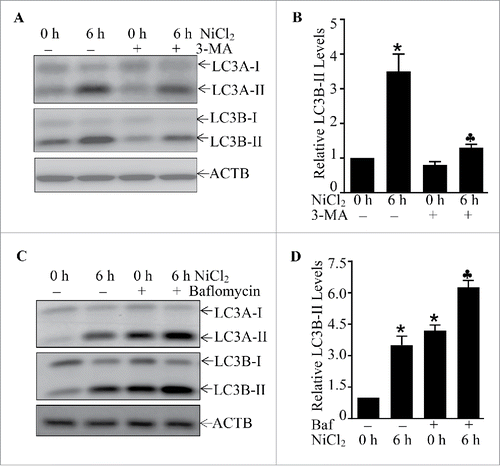
Autophagy is the basic catabolic mechanism that involves cell degradation of unnecessary or dysfunctional cellular components through the actions of lysosomes.Citation29 In addition, autophagy allows for the degradation and recycling of cellular components to promote cellular survival during starvation by maintaining cellular energy levels.Citation30 It has been reported that SQSTM1 is downregulated by an autophagy-mediated mechanism.Citation20 To elucidate the molecular mechanisms underlying SQSTM1 upregulation due to nickel exposure, the potential effect of nickel on cell autophagy was evaluated in Beas-2B cells exposed to nickel. The results unexpectedly showed that nickel exposure induced autophagy in Beas-2B cells as demonstrated by increases of accumulation of both LC3A-II and LC3B-II, 2 well-known autophagic markers (). This notion was further supported by formation of GFP-LC3B puncta in Beas-2B(GFP-LC3B) transfectants following nickel exposure (). The puncta could be observed as early as 3 h after nickel treatment, and the number of puncta constantly increased at 6 h and reached a peak at 12∼24 h (). Consistently, the results from western blotting also indicated that the increases of exogenous GFP-LC3B-II and endogenous LC3B-II were observed in Beas-2B(GFP-LC3B) transfectants upon nickel treatment (). Moreover, to assure this accumulation of LC3B-II was due to autophagy activation, the autophagy inhibitor 3-MA was employed as a pretreatment of cells prior to nickel exposure. The results revealed that 3-MA pretreatment inhibited the formation of LC3A-II and LC3B-II () and GFP-LC3B puncta (Fig. S4A) following nickel exposure in comparison to vehicle-treated cells. To confirm autophagic induction due to nickel exposure in Beas-2B, bafilomycin A1, a lysosomal inhibitor of autophagic flux,Citation31 was employed. As shown in and S4B, treatment of Beas-2B cells with bafilomycin A1 markedly increased levels of LC3A-II and LC3B-II, as well as GFP-LC3B puncta in comparison to those exposed to nickel alone. Similar autophagic induction was also observed in NHBECs and BEP2D cells following nickel exposure (Fig. S5). These results demonstrate that nickel exposure is able to efficiently induce autophagy in human bronchial epithelial cells.
Figure 3. The induction of Beas-2B cell autophagy by nickel exposure. (A and B) 2 × 105 Beas-2B cells were seeded into each well of 6-well plates. After the cell density reached 80∼90%, Beas-2B cells were treated with 1.0 mM NiCl2 for the indicated time (A) or with various concentrations of NiCl2 for 12 h (B). The cells were then extracted with SDS-sample buffer and cell extracts were subjected to western blot. (C) 1 × 104 stable Beas-2B(GFP) and Beas-2B(GFP-LC3B) transfectants were seeded into each well of an 8-well chamber. After being cultured at 37°C for 48 h, the cells were exposed to 1.0 mM NiCl2 for the time indicated. The images were captured using fluorescence microscopy. The average GFP-LC3B puncta number were quantified and presented as puncta number/cell (D). The symbol (*) indicates a significant increase as compared with the medium control (p < 0.05). (E) 2×105 Beas-2B(GFP-LC3B) transfectants were seeded into each well of 6-well plates and the cells were exposed to 1.0 mM NiCl2 for various time points, as indicated. The cell extracts were subjected to western blotting.
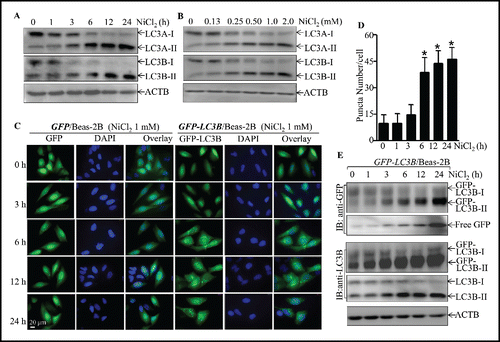
Figure 4. The effects of 3-MA and bafilomycin A1 on the nicekl-induced LC3-II formation. (A and B) 2 × 105 Beas-2B cells were seeded into each well of 6-well plates and the cells were pretreated with 3-MA (5.0 mM) for 30 min and then exposed to 1.0 mM NiCl2 for 6 h. The cell extracts were subjected to western blotting. The symbol (*) indicates a significant increase as compared with the medium control (p < 0.05), while the symbol (♣) indicates a significant inhibition as compared with nickel-treated alone (p < 0.05). (C and D) 2 × 105 Beas-2B cells were seeded into each well of 6-well plates and the cells were exposed to NiCl2 (1 mM) and bafilomycin A1 (Baf, 10 nM) for 6 h. The cell extracts were subjected to western blotting. The symbol (*) indicates a significant increase as compared with the medium control (p < 0.05), while the symbol (♣) indicates a significant increase as compared with the nickel-treated alone (p < 0.05).
We next elucidated the signaling cascade leading to autophagy of Beas-2B cells due to nickel exposure. The autophagy-related (ATG) protein family and BECN1/Beclin1 are central proteins that regulate cell autophagy in many experimental systems.Citation32 We therefore determined the effect of nickel exposure on the expression of various ATG family members, including ATG3, ATG7, ATG12–ATG5 and BECN1. The results indicated that nickel exposure did not affect the expression of any of these proteins (), suggesting that they might not be major players in nickel-induced autophagy. We further evaluated the nickel effects on activation of the MTOR cascade that regulates cell autophagy.Citation33 As shown in , MTOR phosphorylation at Ser2448, p-MTOR Ser2448, was dramatically inhibited at 3 h and thereafter following nickel exposure. Consistently, the phosphorylation of RPS6KB1/p70s6K and RPS6/S6 and EIF4EBP1/4E-BP1, which are regulated by MTOR, were also attenuated in similar patterns by nickel exposure (). To test whether MTOR inhibition mediated autophagy due to nickel exposure, the dominant active mutant of PIK3CA/p110α, phosphoinositide 3-kinase (PI3K)-DA, was stably transfected into Beas-2B cells and the stable transfectant was used to evaluate the protective effect of the PI3K-MTOR cascade on nickel-induced autophagy. As shown in , activation of MTOR by PI3K-DA dramatically inhibited LC3B-II formation, suggesting that MTOR inhibition by nickel did play a role in nickel-induced cell autophagy.
Figure 5. The important role of MTOR, ULK1, and BECN1 in mediation of cell autophagy upon nickel exposure in Beas-2B cells. (A, B and D) 2 × 105 Beas-2B cells were seeded into each well of 6-well plates. After the cell density reached 80∼90%, the cells were exposed to 1.0 mM NiCl2 for the times indicated. The cell extracts were subjected to western blotting. (C) 2 × 105 Beas-2B(PI3K-DA) and Beas-2B(Vector) transfectants were seeded into each well of 6-well plates. The cells were exposed to 1.0 mM NiCl2 for 6 h. The cell extracts were subjected to western blotting. (E and G) 2 × 105 Beas-2B transfectants, as indicated, were seeded into each well of 6-well plates and the cells were exposed to 1.0 mM NiCl2 for 6 h (E) or various concentrations of NiCl2 for 6 h (G).The cell extracts were subjected to western blot and TUBA was used as protein loading control. (F and H) 2 × 105 indicated Beas-2B transfectants were seeded into each well of 6-well plates and the cells were exposed to 1.0 mM NiCl2 and 10 nM of bafilomycin A1 (Baf) for 6 h. The cell extracts were subjected to western blot and ACTB was used as protein loading control.
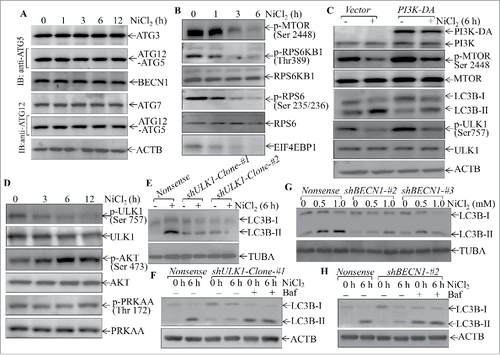
It has been reported that ULK1 activation by inhibition of phosphorylation of ULK1 at Ser757 promotes cell autophagy.Citation34 We next investigated whether ULK1 could be involved in nickel-induced autophagy. As shown in , nickel exposure resulted in a dramatic reduction of phosphorylation of ULK1 at Ser757 in a time-dependent manner, suggesting that nickel did activate ULK1. In contrast, nickel treatment led to an increase in AKT phosphorylation with no observable alteration of PRKAA/AMPKα phosphorylation (), excluding the possible association of AKT and PRKAA activation with either MTOR inhibition or ULK1 activation due to nickel exposure.
Given the ULK1 activation and cell autophagy following nickel exposure, we next evaluated the possible relationship between ULK1 activation and autophagy due to nickel exposure. To this end, the specific shRNA targeting ULK1 was transfected into Beas-2B cells and the knockdown level of ULK1 in 2 clones of Beas-2B cells were determined, as shown in Figure S6A. The knockdown of ULK1 expression impaired nickel-induced cell autophagy in Beas-2B cells (). BECN1 acts downstream of ULK1 to regulate autophagy.Citation34 To identify whether BECN1 is involved in the regulation of nickel-activated autophagy, 4 BECN1 shRNAs (#1, #2, #3 and #4) were stably transfected into Beas-2B cells (Fig. S6B). The results showed that shRNA-#2 and -#3 were able to markedly knockdown BECN1 protein expression (Fig. S6B). As expected, knockdown of BECN1 expression attenuated the nickel-induced LC3B-II level (). It was noted that nickel failed to increase the lysosome inhibitor bafilomycin A1-induced LC3B-II level in both transfectants of Beas-2B(shULK1) and Beas-2B(shBECN1), although marked LC3B-II induction was still observed in Beas-2B(shULK1) and Beas-2B(shBECN1) cells treated with bafilomycin A1 alone (), revealing the specificity of inhibition of nickel-induced autophagy in Beas-2B(shULK1) and Beas-2B(shBECN1) transfectants. Collectively, our results demonstrate that nickel-induced autophagy requires inhibition of MTOR and activation of ULK1 and BECN1.
Nickel induced SQSTM1 transcription via a NFKB RELA-dependent axis
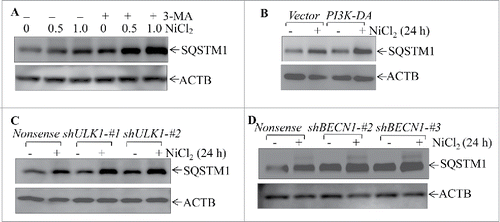
To investigate the role of the autophagy cascade in regulation of nickel-induced SQSTM1 expression, the autophagy inhibitor 3-MA was employed. The disruption of autophagy by 3-MA markedly elevated the basal level and nickel-induced levels of SQSTM1 protein abundance (). Consistently, inhibition of autophagy by either ectopic expression of PI3K-DA, or knockdown of ULK1 or BECN1 expression using their specific shRNAs also increased both basal and nickel-induced levels of SQSTM1 protein in Beas-2B cells (). Our results indicate that autophagy is a negative regulatory mechanism for SQSTM1 expression in Beas-2B cells.
Figure 6. Autophagy induced by nickel inhibited the SQSTM1 level in human Beas-2B cells. (A) 2 × 105 Beas-2B cells were seeded into each well of 6-well plates. The cells were pretreated with 5.0 mM 3-MA for 30 min and then exposed to 0.5 mM or 1.0 mM NiCl2 for 24 h. (B-D) 2 × 105 of Beas-2B stable transfectants, including Beas-2B(PI3K-DA), Beas-2B(shULK1), Beas-2B(shBECN1), and their corresponding control vector transfectants as indicated, were seeded into each well of 6-well plates. After the cell density reached 80∼90%, the cells were exposed to 1.0 mM NiCl2 for 24 h. The cells were extracted with SDS-sample buffer and western blot was carried out as described in the “Materials and Methods.” ACTB was used as a control for protein loading.
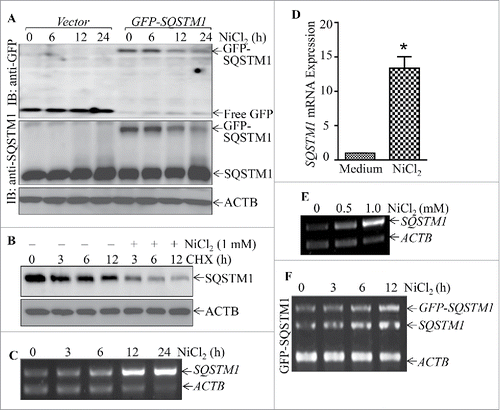
To determine the molecular mechanisms underlying nickel upregulation of SQSTM1 protein expression, we compared the effects of nickel on endogenous SQSTM1 protein and exogenous GFP-SQSTM1 protein expression. The results showed that nickel exposure specifically attenuated the exogenous GFP-SQSTM1 protein level, whereas overall it enhanced endogenous SQSTM1 protein abundance (). Moreover, nickel exposure increased the SQSTM1 protein degradation rate in the presence of cycloheximide (CHX, a protein synthesis inhibitor) in comparison with cells treated with CHX alone (). These results demonstrate that nickel could promote SQSTM1 protein degradation. Thus, we anticipated that nickel exposure would upregulate SQSTM1 protein, mainly at the mRNA level, although it promoted SQSTM1 protein degradation mediated by autophagy. To test this notion, Beas-2B cells were exposed to nickel to determine the effects of nickel on SQSTM1 mRNA expression. The results from RT-PCR showed that nickel exposure resulted in a remarkable increase in SQSTM1 mRNA in both time- and dose-dependent manners (). Further, we found that nickel treatment only showed a slight effect on exogenous GFP-SQSTM1 mRNA expression, whereas it remarkably upregulated endogenous SQSTM1 mRNA under same experimental conditions in Beas-2B(GFP-SQSTM1) transfectants (). These results indicate that nickel is likely to upregulate SQSTM1 expression at the transcriptional level.
Figure 7. Nickel induced SQSTM1 transcription in Beas-2B cells. (A) 2×105 stable Beas-2B(GFP) and Beas-2B(GFP-SQSTM1) transfectants, as indicated, were seeded into each well of 6-well plates. The cells were exposed to 1.0 mM NiCl2 for the indicated time points. The cells were extracted with SDS-sample buffer and western blot was carried out. ACTB was used as protein loading control. (B) Beas-2B cells were treated with CHX (50 μg/ml) together with or without NiCl2 for the indicated times. The cell extracts were subjected to analysis of SQSTM1 protein degradation rate by western Blotting. (C, E and F) Beas-2B cells were exposed to 1.0 mM NiCl2 for different time points, as indicated (C), or treated with the indicated doses of NiCl2 for 12 h (E). The stable Beas-2B(GFP-SQSTM1) cells were exposed to 1.0 mM of NiCl2 for the indicated time periods (F). The cells collected from (C-F) were extracted with Trizol reagent for total RNA isolation and RT-PCR was performed to determine SQSTM1 or GFP-SQSTM1 expression with their specific primers. ACTB was used as an internal control. (D) Real-time PCR was carried out to determine the SQSTM1 mRNA expression using cDNA samples collected from Beas-2B cells exposed to 1.0 mM NiCl2 for 24 h obtained in (C). The symbol (*) indicates a significant increase as compared with the medium control (p < 0.05).
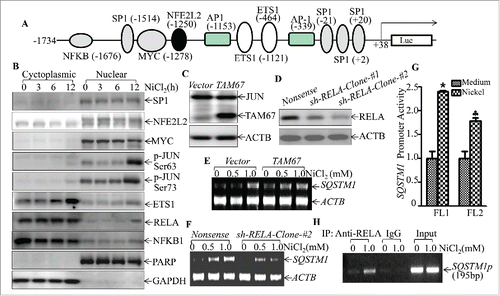
To evaluate this possibility, bioinformatics software was used to analyze the potential transcription factor binding sites in the SQSTM1 promoter region. The results showed that there were multiple potential transcription factor binding sites in the SQSTM1 promoter region, including those for JUN/AP-1, MYC/c-MYC, SP1, NFE2L2/NRF2, NFKB, and ETS1 (). We then determined the effects of nickel on the nuclear translocation of these potential transcription factors using a nuclear/cytosol fractionation assay. The results indicated that nickel exposure led to the marked translocation of JUN/c-JUN and NFKB, whereas it did not show any observable effect on other transcription factors, including MYC, SP1 and NFE2L2 in Beas-2B cells (). The lack of involvement of NFE2L2 in nickel-induced SQSTM1 was also supported by the results of testing the expression of NFE2L2-regulated genes HMOX1/HO-1 and NQO1. As shown (Fig. S7), mRNA induction of HMOX1 and NQO1 was observed in 24–36 h, but not within 12 h in nickel-exposed Beas-2B cells. Given that SQSTM1 protein upregulation by nickel exposure could be observed as early as 1 h following nickel exposure, excluding the possibility of NFE2L2 regulation of nickel-induced SQSTM1 expression. Although we noted that nickel upregulated ETS1 expression, the most of upregulated ETS1 protein was located in cytoplasm, suggesting that upregulated ETS1 protein might not act as a transcription factor in the nucleus. To identify the role of JUN and NFKB in nickel-induced SQSTM1 upregulation, dominant negative mutant JUN (TAM67) and RELA shRNA were stably transfected into Beas-2B cells. As shown in , the inhibition of SQSTM1 mRNA expression was only observed in Beas-2B cells stably knocked down for RELA by using RELA shRNA (), whereas the introduction of TAM67 had no observable effect on SQSTM1 mRNA expression ().
Figure 8. RELA was a key transcription factor mediating SQSTM1 transcription following nickel exposure. (A) Potential transcription factor binding sites in the SQSTM1 promoter region (-1734∼+38) were analyzed using the TRANSFAC 8.3 engine online. (B) 2 × 105 Beas-2B cells were seeded into each well of 6-well plates. The cells were exposed to 1.0 mM NiCl2 for the times indicated. The cell extracts were used to isolate cytoplasmic and nuclear fractions according to the protocol of the nuclear/cytosol fractionation kit. The isolated protein fractions were subjected to western blot. GAPDH and PARP were used as control for cytosol or nuclear protein loading, respectively. (C) Beas-2B(Vector) and Beas-2B(TAM67) cells were extracted with SDS-sample buffer and the cell extracts were analyzed by western blot with a specific anti-JUND antibody. (D) Stable Beas-2B(shRELA) and Beas-2B(Nonsense) cells were extracted with SDS-sample buffer and the cell extracts were analyzed by western blot with specific anti-RELA antibody. ACTB was used as a control for protein loading. (E and F) 2 × 105 Beas-2B(TAM67) (E) or Beas-2B(shRELA) (F) cells and their corresponding vector transfectants were seeded into each well of 6-well plates. After the cell density reached 80∼90%, the cells were exposed to NiCl2 for 12 h and the cells were then extracted with Trizol reagent for total RNA isolation. SQSTM1 was amplified with specific primers by RT-PCR. ACTB was used as an internal control. (G) 1×104 Besa-2B cells transfected with SQSTM1 full-length promoter-driven luciferase reporter (FL1) or NFKB-binding site-deleted SQSTM1 promoter-driven luciferase reporter (FL2) were seeded into each well of 96-well plates. After being cultured overnight, the cells were treated with 1.0 mM NiCl2 for 24 h. The luciferase activity was then determined and the results are presented as SQSTM1 promoter activity, relative to medium control. Each bar indicates the mean and SD of triplicate assay wells. The symbol (*) indicates a significant increase in comparison to the medium control (p < 0.05). The symbol (♣) indicates a dramatic decrease as compared to the Beas-2B cells transfected with FL1 (p < 0.05). (H) 1 × 106 Beas-2B cells were seeded into 100-mm dishes. After the cell density reached 80∼90%, the cells were exposed to 1.0 mM NiCl2 for 12 h. A ChIP assay was performed with anti-RELA antibody to determine RELA binding to the SQSTM1 promoter as described in “Materials and Methods.”
To provide direct evidence showing whether or not RELA regulated SQSTM1 transcription, we transfected either a full-length SQSTM1 promoter-driven luciferase reporter or short length SQSTM1 promoter-driven luciferase reporter lacking NFKB binding sites into Beas-2B cells (Fig. S8). As shown in , the promoter transcription activation was inhibited in the promoter with deletion of NFKB binding sites in comparison to the cells transfected with the full-length SQSTM1 promoter reporter. Moreover, a ChIP assay was performed to determine whether RELA could directly bind to the SQSTM1 promoter region that contains tentative RELA binding sites. The results showed that nickel exposure significantly increased RELA protein binding to the tentative DNA fragment (), revealing that RELA did bind to the SQSTM1 promoter region in nickel-exposed cells. Taken together, our results conclusively demonstrate that NFKB RELA activation plays an important role in nickel-induced SQSTM1 transcription and protein expression in Beas-2B cells.
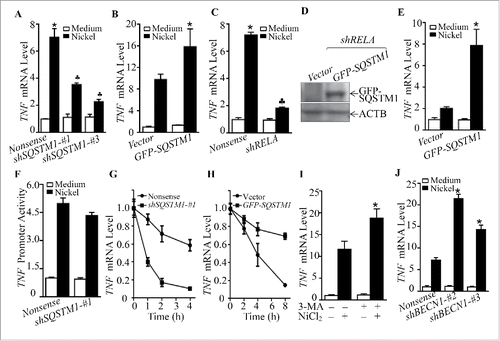
SQSTM1 upregulation contributes to an increase in inflammatory TNF/TNFα by nickel
Chronic lung inflammation is a potent force for driving development of lung cancers.Citation35 As shown above, our results demonstrate that SQSTM1 is important for nickel-induced transformation of Beas-2B cells. These results prompted us to determine the potential relationship between SQSTM1 upregulation and lung inflammatory responses upon nickel exposure. The results showed that knockdown of SQSTM1 expression reduced TNF mRNA level following nickel exposure (), whereas ectopic expression of GFP-SQSTM1 remarkably upregulated TNF levels induced by nickel exposure (). Consistent with RELA regulation of SQSTM1, knockdown of RELA also attenuated the TNF mRNA level following nickel exposure, whereas introduction of GFP-SQSTM1 could restore the TNF mRNA level upon nickel exposure in RELA knockdown cells (). To elucidate the molecular mechanism underlying SQSTM1 upregulation of TNF expression, the TNF promoter-driven transcription activity and TNF mRNA stability were evaluated in Beas-2B(shSQSTM1) transfectants in comparison to Beas-2B(Nonsense) transfectants following nickel exposure. As shown in , knockdown of SQSTM1 only showed a slight effect on nickel-induced TNF promoter transcriptional activity (), whereas it markedly reduced nickel-induced TNF mRNA stability (). Further, ectopic expression of GFP-SQSTM1 significantly increased nickel-induced TNF mRNA stability (). These results reveal that SQSTM1 mediates TNF mRNA abundance mainly through enhancing its mRNA stability. The results from determination of TNF protein in cell cultured medium indicated that SQSTM1-mediated stabilization of TNF mRNA following nickel exposure resulted in increased TNF release into the culture medium (Fig. S9). Moreover, inhibition of nickel-induced autophagy by 3-MA or knockdown of BECN1 could promote TNF mRNA abundance by nickel exposure (). Collectively, these results clearly demonstrate that RELA-mediated SQSTM1 upregulation plays an essential role in the increased TNF abundance upon nickel exposure, whereas autophagy exerts an inhibitory effect on TNF level following nickel exposure.
Figure 9. SQSTM1 upregulation contributed to inflammatory TNF induction by nickel. (A-C and J) 2 × 105 Beas-2B transfectants, as indicated, were seeded into each well of 6-well plates. After the cell density reached 80∼90%, the cells were exposed to 1.0 mM NiCl2 for 24 h. Cells were extracted with Trizol reagent for total RNA isolation. Real-time PCR was carried out to monitor the TNF mRNA. (D and E) GFP-SQSTM1 or its control vector was transfected into Beas-2B(shRELA) cells. Cell extracts were subjected to western Blot for determination of protein expression as indicated (D). 2×105 transfectants were seeded into each well of 6-well plates. The cells were then exposed to 1.0 mM NiCl2 for 24 h, and nickel-induced TNF mRNA was evaluated by real-time PCR (E). (F) 8 × 103 Beas-2B (Nonsense/TNF-Luc) cells and Beas-2B (shSQSTM1-#1/TNF-Luc) cells were seeded into each well of a 96-well plate. After being cultured at 37°C overnight, the cells were exposed to 1.0 mM nickel for 24 h. The luciferase activity was measured. (G and H) TNF mRNA degradation rate in Beas-2B cell transfectants was determined following nickel treatment in the presence of actinomycin D (RNA synthesis inhibitor, 10 μM) for the indicated time. (I) Beas-2B cells were pretreated with 5.0 mM 3-MA for 30 min and then exposed to 1.0 mM NiCl2 for 24 h. The cells were extracted with Trizol reagent for total RNA isolation and then subjected to real-time PCR. The symbol (*) indicates a significant increase (p < 0.05), while the symbol (♣) indicates a significant decrease (p < 0.05).
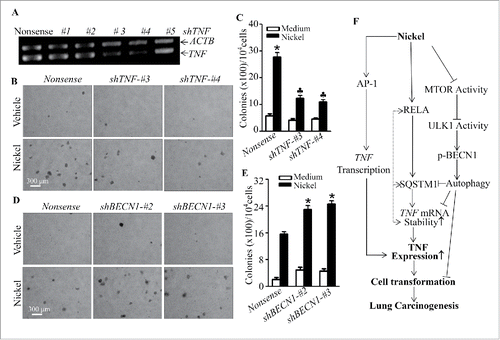
TNF induction mediates cell malignant transformation of human bronchial epithelial cells following nickel exposure.
Our previous studies demonstrate that TNF induction is critical for B[a]PDE-induced cell transformation in vitro in mouse epidermal Cl41 cells and human lung epithelial Beas-2B cells.Citation36,37 Our above results also indicate that SQSTM1 is important for nickel-induced cell malignant transformation and TNF levels in Beas-2B cells. Thus, we determined the role of TNF induction in malignant transformation of human lung bronchial epithelial cells upon nickel exposure. The results showed that the knockdown of TNF in Beas-2B or BEP2D cells, by stable transfection of TNF-specific shRNA-#3 and shRNA-#4, as shown in and Fig. S10A, dramatically inhibits transformation of either Beas-2B cells () or BEP2D cells (Fig. S10B and S10C) following nickel-repeated exposure in comparison to their nonsense transfectants, revealing that TNF induction plays an important role in nickel-induced transformation of human bronchial epithelial cells. Consistently, the stable knockdown of BECN1 expression in Beas-2B cells also significantly increased nickel-induced cell transformation of Beas-2B (), further demonstrating that TNF induction is important for nickel-induced malignant transformation of human bronchial epithelial cells.
Figure 10. TNF induction mediated cell transformation of Beas-2B cells due to nickel exposure. (A) Specific TNF shRNAs were stably transfected into Beas-2B cells. The cells were extracted with Trizol reagent for total RNA isolation. TNF was amplified with its specific primers by RT-PCR. ACTB was used as an internal control. (B and C) Beas-2B(Nonsense), Beas-2B(shTNF-#3), and Beas-2B(shTNF-#4) cells were repeatedly treated with 0.5 mM NiCl2 for 6 months, then 1×104 cells were subjected to a soft agar assay as described in “Materials and Methods.” The cell colonies were counted by microscopy. Each bar indicates the mean and SD from triplicate assays. The symbol (*) indicates a significant increase as compared to that of the medium control (p < 0.05), while the symbol (♣) indicates a dramatic decrease compared with Beas-2B(Nonsense) (p < 0.05). (D and E) Beas-2B(Nonsense), Beas-2B(shBECN1-#2), and Beas-2B(shBECN1-#3) cells were repeatedly treated with 0.5 mM NiCl2 for 4 months. 1 × 104 treated cells were subjected to a soft agar assay. The symbol (*) indicates a significant increase in comparison to Beas-2B(Nonsense) (p < 0.05). (F) A novel molecular mechanisms underlying SQSTM1 induction and its role in malignant transformation of human bronchial epithelial cells following nickel exposure. The symbol (*) indicates a significant increase (p < 0.05), while the symbol (♣) indicates a significant decrease (p < 0.05).
Discussion
Chronic exposure to the inorganic nickel compounds occurs not only in environmental air pollution and occupational workers, but also in tobacco smokers.Citation9 The major medical problem caused by nickel exposure is chronic lung inflammation.Citation38 Since nickel compounds do not show high affinity for DNA,Citation11 it is accepted that non-genotoxic mechanisms are responsible for their carcinogenic activity. Chronic lung inflammation caused by nickel exposure is most frequently associated with the development of lung cancer.Citation38-40 The link between nickel-caused chronic inflammation to lung carcinogenesis was observed as early as 30 y ago,Citation41 and has been verified in animal studies.Citation39,40 However, the molecular mechanisms by which nickel-initiated lung inflammation leads to lung cancer development largely remain unknown. Here, we discover a novel RELA-SQSTM1 axis that enhances TNF mRNA stability and expression, which promotes cell transformation following nickel exposure. Concurrently, nickel exposure activates the MTOR-ULK1-BECN1 autophagic cascade, subsequently causing SQSTM1 protein degradation, which exhibits an inhibitory effect on cell transformation upon nickel exposure. Our findings not only identify SQSTM1 as a new target for nickel initiation of lung tumorigenic effects, they also provide a novel scenario linking SQSTM1 and autophagy to the regulation of lung inflammation and malignant cell transformation upon environmental nickel exposure, as shown in .
Although SQSTM1 has been reported to be overexpressed in cancer tissues,Citation25,42 SQSTM1 induction and its contribution to environmental carcinogens has never been explored. We found that nickel exposure was able to upregulate SQSTM1 protein level in Beas-2B, BEP2D and NHBEC cells. This upregulation was also observed in the lung tissues of mice exposed to nickel. Consistent with these findings observed in nickel exposure in an in vitro cell culture model and in an in vivo mouse model, the results obtained from some human lung cancer tissues also displayed the SQSTM1 upregulation in comparison to those in the paired adjacent normal lung tissues. Of greatest importance is that our results demonstrate that SQSTM1 upregulation is important for nickel-induced malignant transformation of human bronchial epithelial cells (), cancer marker CCND1 (cyclin D1) and CCNE1 (cyclin E1) expression (Fig. S11A), and cell migration (Fig. S11B and S11C). Thus, our studies not only show, for the first time, that SQSTM1 is induced following nickel exposure, but also demonstrate the contribution of SQSTM1 upregulation to nickel's lung carcinogenic effects.
SQSTM1 protein expression is regulated at multiple levels, including protein degradation and mRNA transcription.Citation43,44 Autophagy is an important cell biological function that could help cell in degrading unnecessary or dysfunctional cellular components through the actions of lysosomes.Citation29 SQSTM1 is a receptor and substrate of autophagy, and autophagy defects lead to accumulation of SQSTM1 protein.Citation20 In the current studies, we evaluated the effects of nickel on cell autophagy and unexpectedly found that nickel activated autophagy rather than inhibited cell autophagy in Beas-2B cells. Remarkably induction of LC3A-II and LC3B-II, and GFP-LC3B puncta was observed in Beas-2B cells exposed to nickel, whereas the pretreatment of cells with the autophagy inhibitor 3-MA impaired nickel-induced formation of GFP-LC3B puncta in Beas-2B cells under the same experimental conditions (Fig. S4). We also found that nickel exposure induced autophagy of Beas-2B cells via inhibition of MTOR, which in turn activated the ULK1-BECN1 autophagic cascade. Our results showed that ectopic expression of activated-PI3K or knockdown of ULK1 or BECN1 expression dramatically inhibited nickel-induced autophagy, accompanied by the accumulation of GFP-SQSTM1 protein expression, revealing that autophagy induced by nickel exposure promoted SQSTM1 protein degradation. It is of great interest to note that nickel exposure inhibits endogenous SQSTM1 protein expression, which is different from previous findings that arsenite upregulates SQSTM1 expression through inhibiting cell autophagy.Citation45 Moreover, we found that ectopic expression of activated-PI3K or knockdown of ULK1 or BECN1, dramatically inhibited nickel-induced autophagy and elevated nickel-induced SQSTM1 protein expression, revealing that nickel-induced autophagy negatively regulates SQSTM1 protein expression following nickel exposure.
We found that the final outcome of nickel-upregulated SQSTM1 protein expression resulted from mRNA induction, which overcame SQSTM1 protein degradation mediated by autophagy due to nickel exposure. The exploration of the mechanisms underlying nickel regulation of SQSTM1 mRNA showed that nickel upregulated SQSTM1 mRNA due to SQSTM1 promoter transcriptional activation, as demonstrated using a SQSTM1 promoter-driven luciferase reporter that had been cloned from Beas-2B genomic DNA, as shown in Fig. S8. By analyzing the SQSTM1 promoter, we observed multiple transcription binding sites, including NFE2L2, SP1, ETS1, JUN, MYC and NFKB. NFE2L2 has been reported as enhancing tumor growth in some cancer tissues by upregulating SQSTM1 transcription,Citation46 whereas SP1 has been reported as regulating SQSTM1 promoter activity following H2O2 treatment in HEK293 cells.Citation47 The results from our studies showed that nickel exposure did not induce expression and nuclear translocation of either NFE2L2 or SP1. The SQSTM1 overexpression in breast tumors and regulation by prostate-derived ETS1 factor in breast cancer cells has been reported.Citation42 The results from our studies indicated that nickel exposure only induced a slightly nuclear translocation, although ETS1 expression was inducible in nickel-treated Beas-2B cells, suggesting that ETS1 might not be a major transcriptional factor mediating SQSTM1 transcription. A potential MYC binding site has also been observed in the SQSTM1 promoter, however, nickel failed to induce MYC nuclear translocation in Beas-2B cells. Our previous studies demonstrate that nickel exposure is able to activate JUN and NFKB in Beas-2B cells and the results from the current studies consistently indicated that nuclear translocation of both transcription factors was observable in Beas-2B cells exposed to nickel. We further found that blockage of JUN activation by ectopic expression of a dominant negative JUN mutant (TAM67)Citation48 did not show any observable effect on SQSTM1 mRNA induction in Beas-2B cells following nickel exposure, whereas knockdown of RELA expression dramatically attenuated SQSTM1 mRNA induction under the same experimental conditions, demonstrating that RELA plays an important role for SQSTM1 transcriptional induction following nickel exposure. The results from ChIP assay showed that nickel-activated RELA is able to bind to the SQSTM1 promoter sequence. Therefore our results, for the first time, identify RELA acting as a key transcription factor binding to the SQSTM1 promoter and initiating SQSTM1 transcription. This notion is consistently supported by the findings obtained from in vivo studies showing that RELA phosphorylation at Ser536 is markedly increased in mouse lung tissues following nickel inhalation (Fig. S12).
Growing evidence indicates that a chronic inflammatory microenvironment in the lung is a major driving force for the development of lung cancers.Citation15,49,50 At least 10 cohort studies have shown that chronic obstructive pulmonary disease is an independent predictor of lung cancer risk.Citation51,52 Following a short period of silica exposure, type-2 pneumocytes and bronchiolar epithelial cells become hyperplastic. The subsequent appearance of adenomas is followed by the appearance of adenocarcinomas and then squamous cell carcinomas. The progression from inflammatory granuloma to lung carcinoma in this model clearly points to the important role of inflammation in human lung carcinogenesis.Citation53 The strong association between airway inflammation and susceptibility to lung cancer was confirmed by epidemiological and clinical studies.Citation54,55 The chronic inflammation microenvironment is predominated by macrophages,Citation49,56 which together with other leukocytes, may generate high levels of reactive oxygen and nitrogen species, and eventually may cause DNA damage and the activation of signaling pathways that lead to cancer-associated gene expression.Citation9,15 In addition, the cells in the inflammatory site may release TNF, which plays a regulatory role in growth and the inhibition of apoptosis.Citation57,58
The link between nickel-caused chronic inflammation to lung carcinogenesis was observed as early as 30 y agoCitation41 and has been verified in animal studies.Citation39,40 Our recent studies have shown that nickel exposure induces the activation of the key inflammatory transcription factor NFKB, which in turn leads to the induction of the key inflammatory mediator PTGS2/COX-2 expression, subsequently protecting nickel-treated HBECs from apoptosis.Citation59 Our published studies also show that nickel exposure leads to JUN activation, NFAT activation and TNF expression in Beas-2B cells.Citation60 The results from the current studies revealed that SQSTM1 expression played an important role in TNF induction by nickel exposure mainly through upregulating TNF mRNA stability, which further promoted nickel-induced cell transformation. Moreover, SQSTM1 protein increase by inhibition of autophagy with either 3-MA or knockdown of BECN1 also enhanced the TNF mRNA level following nickel exposure. Our studies further demonstrate that TNF induction is essential for nickel-induced malignant transformation of human bronchial epithelial cells (), increased expression of cell growth markers CCND1 and CCNE1 (Fig. S11D), and promotion of cell migration (Fig. S11E and S11F). This notion was supported by finding that the increased TNF mRNA level in BECN1 knockdown cells also promoted nickel-induced Beas-2B cell transformation. It has been reported that NFKB acts as a key SQSTM1 downstream target that is responsible for TNF expression.Citation61 Our previous studies also indicate that TNF could be a feedback player in the activation of NFKB.Citation62 Given our previous studies demonstrating that JUN is important for nickel-induced TNF transcription, we anticipate that the TNF induction mediated by both JUN-dependent transcription and RELA-SQSTM1-dependent mRNA stability will promote and maintain the sustained inflammatory microenvironment as schematically depicted in .
In the current studies, we determined the potential molecular mechanisms underlying SQSTM1-mediated stabilization of TNF mRNA. As shown in Figure S13, knockdown of SQSTM1 did not affect expression of NCL (nucleolin) and ELAVL1/HUR, both of which have been reported to regulate their targeted mRNA stability.Citation63,64 Since MAPK14/p38 and MAPK1/ERK2-MAPK3/ERK1 activation has also been reported to be involved in the modulation of TNF mRNA stability,Citation65 we also evaluated the effect of knockdown of SQSTM1 expression on activation of MAPK14 and MAPK1/3. The results did show that knockdown of SQSTM1 led to an inhibition of MAPK14 and MAPK1/3 phosphorylation as compared to these observed in Beas-2B(Nonsense) transfectants, suggesting their possible involvement in SQSTM1-mediated regulation of TNF mRNA stability. The studies toward this direction as well as elucidation of how MAPK14 and MAPK1/3 activation lead to TNF mRNA stabilization is a current ongoing project in our group.
In summary, our current studies, for the first time, discover SQSTM1 upregulation by the environmental lung carcinogen nickel both in vitro and in vivo via a RELA-dependent pathway, and demonstrate the essential role of this SQSTM1 upregulation in nickel-induced malignant transformation of human bronchial epithelial cells. We have also identified a novel signaling cascade underlying SQSTM1s incentive modulation of TNF upregulation following nickel exposure. The results from these current studies clearly demonstrate the formation of inflammatory positive feedback loops by NFKB RELA, SQSTM1 and TNF, as well as providing evidence that SQSTM1-autophagy are essential regulators of the entire nickel-induced tumorigenic network. The current studies facilitate our understanding of the molecular mechanism(s) underlying SQSTM1 upregulation and its role in lung cancer development due to nickel exposure. Such novel information will spur the efficacious development of preventive and therapeutic approaches that specifically target SQSTM1.
Materials and methods
Chemical reagents
NiCl2 (451193) and Ni(OH)2 (283622) were purchased from Sigma Aldrich Corporation. The autophagy inhibitor 3-MA (189490) was bought from Calbiochem. PolyJetTM DNA In Vitro Transfection Reagent (SL100468) was purchased from SignaGen Laboratories. TRIzol reagent (15596026) and SuperScriptTM First-Strand Synthesis system (18080051) were bought from Invitrogen Corporation. The dual luciferase assay kit (E1960) was purchased from Promega Corporation. The nuclear/cytosol fractionation kit (K266–100) was obtained from Biovision Incorporated.
Plasmids
The shRNA plasmids specifically targeting SQSTM1 (human, RHS3979-201739507), BECN1 (human, RHS3979-201763046), and RELA (human, RHS3979-201746261) were purchased from Open Biosystems, Inc. Another set of shRNA plasmids that target human TNF (TRCN0000003757) was bought from Sigma Aldrich Corporation. GFP-LC3B and its control vector were a kind gift from Dr. Gang Chen (University of Kentucky, Lexington, KY, USA),Citation66 and the GFP-SQSTM1 expression construct was from Dr. Sung Ouk Kim (University of Western Ontario, London, Ontario, Canada).Citation67 Dominant active mutant PI3K expression plasmid, PI3K-DA was from Dr. Lewis T. Williams (University of California, San Francisco, CA, USA).Citation68 The shRNA targeting ULK1 expression construct was obtained from Addgene (27633, Reuben Shaw Lab).Citation69 The plasmid of TNF promoter-driven luciferase reporter, dominant negative JUN plasmid (TAM67), was used as described in our previous studies.Citation48,70
SQSTM1 promoter (full length, from −1734 to +38)-driven luciferase reporter was constructed using genomic DNA purified from Beas-2B cells based on the NCBI database. Nested PCR was carried out. The forward outside primer sequence was (F1): 5′-CCT ATT ACG ACA GCG GTC ATG G-3′ and the reverse primer was (R1): 5′-AGC TGG CGG AAA ACG GG-3′. The PCR product was next used for subsequent PCR using the nested forward primer (F2): 5′-GGA AGA TCT CTG ACT CAC TGC CGG CCA GAC-3′ containing a BglII restriction site and the reverse primer (R2): 5′-CCC AAG CTT TGT AGC GAA CGC GGA GGC-3′ containing a HindIII restriction site. To construct a NFKB binding site deletion promoter (short, from −1656 to +38) the nested forward primer of (F3): 5′-GGA AGA TCT GTT TAC CTC CGG GAG GCC TCG-3′ was used. The PCR product was digested and cloned into the pGL3-Basic vector (Promega, E1751) and verified by DNA sequencing.
Cell culture and transfection
Beas-2B cells were cultured at 37°C with 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; 11995065) supplemented with 10% fetal bovine serum (FBS; 26140079), 1% penicillin/streptomycin (15140163), 2 mM L-glutamine (25030164), all from Life Technologies.Citation48 NHBECs were cultured as described in a previous study.Citation71 BEP2D cells were cultured as described in a previous study.Citation28 Cell transfection was performed using PolyJetTM DNA In Vitro Transfection Reagent, according to the manufacturer's instruction. Twenty-four h after transfection, the transfected cells including Beas-2B(shSQSTM1), Beas-2B(shBECN1), Beas-2B(shTNF), Beas-2B(shRELA), and Beas-2B(shULK1) were subjected to puromycin (0.3 µg/ml) (Alexis, BML-A260-0050) for stable selection, while Beas-2B(GFP-SQSTM1) and its control cell lines were selected for over 3 weeks in 10% FBS DMEM containing G418 (1000 μg/ml; Invitrogen, 10131027). The stable transfectants were cultured in the selective drug-free medium for at least 2 passages before being used for each experiment.
RT-PCR
Cells were exposed to nickel for the indicated time points, and then 5.0 μg total RNA was used for first-strand cDNA synthesis with oligodT (20) primer by SuperScriptTM First-Strand Synthesis system (Invitrogen, 11904018).Citation72 The SQSTM1 and TNF were monitored by PCR. The results were imaged with Α Innotech SP image system (Α Innotech Corporation, San Leandro, CA, USA). Three pairs of oligonucleotides (Forward: 5′- GAG AGT GTG GCA GCT GCC CT-3′, Reverse: 5′-GGC AGC TTC CTT CAG CCC TG-3′; Forward: 5′-GTG ATC GGC CCC CAG AGG GA-3′ Reverse: 5′-ACT GGA GCT GCC CCT CAG CTT-3′and Forward: 5′- CGC CGA CCA CTA CCA GCA GAA-3′, Reverse: 5′-CAC ACG GTG GGC GGT GGT CC-3′) were used as the specific primers to amplify human SQSTM1 (420 base pairs [bp]), human TNF (148 bp) and GFP-SQSTM1 (550 bp). ACTB/β-Actin (268 bp) (Forward: 5′-CTC CAT CCT GGC CTC GCT GT-3′, Reverse: 5′-GCT GTC ACC TTC ACC GTT CC-3′) was used as a loading control.
Quantitative RT-PCR
Real-time PCR was conducted following the protocol for Fast SYBR Green Master Mix kit (Applied Biosystems, 4385614) in the 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using the same cDNAs that were used for RT-PCR.
Western blot
The antibodies specific against JUN (9165S), JUND (5000S), P-JUN Ser63 (2361S), P-JUN Ser73 (9164S), AKT (9272S), p-AKT Ser473 (9271S), RELA (8242S), PIK3CA (4255S), PARP (9542S), GAPDH (5174S), NFE2L2 (12721S), MTOR Pathway Antibody Sampler Kit (9964S), Autophagy Antibody Sampler Kit (4445S), RPS6KB1 Substrates Antibody Sampler Kit (2903S) and ULK1 Antibody Sampler Kit (8359T) were purchased from Cell Signaling Technology. Antibodies to GFP (sc-390394), ETS1 (sc-55581), SP1 (sc-14027) and MYC (sc-788) were bought from Santa Cruz Biotechnology. Antibodies that are specific against NFKB1 (ab32360) and SQSTM1 (ab155686) were bought from Abcam. Antibodies against ACTB (A1978) and TUBA/α-tubulin (T6199) were bought from Sigma Aldrich Corporation. Western blotting was performed as described in our previous publication.Citation73,74
Animal experiments and lung tissue sample preparation
Male C57BL/6J mice were obtained from Taconic Farms (Germantown, NY) and housed, as described in our previous publication.Citation75 All procedures involving animals were conducted in compliance with guidelines for ethical animal research and approved by the New York University School of Medicine Animal Care and Use Committee. Nickel nanoparticles were generated by electricarc discharge (Palas GmbH) between 2 opposing high-purity rods (99.995% purity, ESPI, Ashland, OR) in an ultra pure argon chamber. One mg/m3 of Nano-Ni(OH)2 exposure was performed as described previously.Citation24 The mice were sacrificed at different periods, as indicated, and the lung tissues were extracted with SDS-sample buffer (10 mM Tris-HCl, pH 7.4, 1% SDS [Thermo Fisher Scientific Inc., BP16650], 1 mM Na3VO4, containing 2 × complete protease inhibitor [Roche, 04693116001]) and kept on ice for 2 h. The extracts were then sonicated and denatured by heating at 100°C for 5 min, and quantified with a Dc protein assay kit (Bio-Rad, 5000116). Equal aliquots of mouse lung tissue extracts were subjected to protein gel blot for protein analysis.
The clinical specimens used in the current study were lung squamous cell carcinomas as shown Table S1, which were obtained from the First Affiliated Hospital of Wenzhou Medical University (Wenzhou, Zhejiang, China) with appropriate informed consent from the patients and a supportive grant obtained from the Medical Ethics Committee of Wenzhou Medical University. Adjacent normal lung tissue specimens were taken from a standard distance (3 cm) from the margin of resected neoplastic tissues of patients with tumors who endured surgical lung ablation. Specimens were examined by pathologists,Citation37 and then frozen at −80°C until shipment on dry ice. The protein extracts were prepared using the same protocol for mouse lung tissues, and then subjected to western blot for determination of SQSTM1 expression.
Luciferase reporter assay
SQSTM1 promoter driven-luciferase or TNF promoter-driven luciferase reporter plasmids were transiently transfected into cells. The transfectants were seeded into each well of 96-well plates (8×103 cells per well) and subjected to the various treatments, as described in our previous study.Citation76 Luciferase activities were determined with the Dual-Luciferase Reporter Assay System using a luminometer as described previously.Citation77
Fluorescence microscopy
Beas-2B cell transfectants were cultured on cover slides in 10% FBS DMEM medium for 48 h. The cells were exposed to 1.0 mM nickel for the indicated time and fixed with 4% paraformaldehyde (Sigma Aldrich Corporation, 158127) in PBS (135 mM NaCl, 4.7 mM KCl, 10 mM Na2HPO4, 2.0 mM NaH2PO4, pH 7.4) at room temperature for 15 min, and then stained with 0.1 mg/ml DAPI (Sigma Aldrich Corporation, 9542) for 1 min. The slides were washed 3 times with PBS and mounted with antifade reagent (Molecular Probes, P36930). All the cell images were captured using an inverted Leica fluorescence microscope (Wetzlar, Germany). For quantification of autophagic cells, GFP-LC3B puncta were determined by counting at least 30 cells per slide.Citation22
Chromatin immunoprecipitation (ChIP) assay
ChIP was performed as described in our previous publication.Citation78 Briefly, Beas-2B cells were treated with 1.0 mM nickel for 12 h. Then genomic DNA and proteins were cross-linked with 1% formaldehyde. The complex was sonicated to generate 200- to 500-bp chromatin DNA fragments. The chromatin was then subjected to immunoprecipitation using antibodies specific to RELA. After immunoprecipitation, the protein-DNA cross-links were used to extract DNA and then subjected to PCR analysis. The following pair of primers: 5′-CCT ATT ACG ACA GCG GTC ATG-3′ and 5′-ACA CCC GGC TCT GGC CCT TC-3′ was used to amplify 195-bp fragments which contains RELA binding sites in the SQSTM1 promoter. The PCR products were separated on 2% agarose gels and the images were captured under UV light with the Α Innotech SP image system.
Cell transformation
Cell transformation was performed as described in our studies previously.Citation79 For the first 2 mo of nickel exposure, Beas-2B cells or BEP2D cells were exposed to 0.5 mM nickel for 24 h following which nickel-containing medium was replaced with fresh 10% FBS DMEM and cultured for 48 h. We continued to treat the nickel-exposed cells with 0.5 mM nickel for 12 h at which point the nickel-containing medium was replaced with fresh 10% FBS DMEM and cultured for 60 h. The cultures were split and subjected to another round of treatment. The described nickel exposure was repeated twice a wk for 4–6 mo when the anchorage-independent growth capability of the treated cells was subjected to soft agar assay.
Soft agar assay
The anchorage-independent growth ability of the nickel-treated Beas-2B cells was evaluated in soft agar, as described in our previous study.Citation80 Briefly, 3 ml of 0.5% agar (Becton, Dickinson and Company, 214010) in basal modified Eagle's medium supplemented with 10% FBS was layered onto each well of 6-well tissue culture plates. Cells (1×104 cells) suspended in 1 ml of normal medium (Sigma-Aldrich Corporation, B9638) were mixed with 2 ml of 0.5% agar in basal modified Eagle's medium supplemented with 10% FBS; 1 ml of mixture was added into each well on top of the 0.5% agar layer. Plates were incubated at 37°C in 5% CO2 for 3 wk, and the colonies were scored and presented as colonies/104 cells.
Statistical methods
Statistical analysis was performed using Prism 5.0 Software (Graph-pad software, San Diego, CA, USA). Student t test was employed to determine the significance of differences between various groups. The differences were considered significant at p < 0.05.
Abbreviations
| Act D | = | actinomycin D |
| ATG | = | autophagy-related |
| CHX | = | cycloheximide |
| FBS | = | fetal bovine serum |
| LC3 | = | microtubule associated protein 1 light chain 3 |
| NHBEC | = | normal human bronchial epithelial cell |
| NSCLC | = | non-small cell lung carcinoma |
| PI3K | = | phosphoinositide 3-kinase |
| SCLC | = | small cell lung carcinoma |
| shRNA | = | short hairpin RNA |
| SQSTM1/p62 | = | sequestosome 1 |
| 3-MA | = | 3-methyladenine |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary files
Download MS Word (2.2 MB)Acknowledgments
We thank Dr. Gang Chen (University of Kentucky, Lexington, KY, USA) and Dr. Sung Ouk Kim (University of Western Ontario, London, Ontario, Canada) for providing GFP-LC3B construct and GFP-SQSTM1 construct, respectively. We thank Dr. Lewis T. Williamst (University of California, San Francisco, CA, USA) for sharing with us the PI3K-DA expression plasmid.
Funding
This work was partially supported by the grants of NIH/NCI CA112557, CA177665 and CA165980, and NIH/NIEHS ES000260; the Natural Science Foundation of China (NSFC81229002, NSFC81372946) and Key Project of Science and Technology Innovation Team of Zhejiang Province (2013TD10).
References
- American Cancer Society. Cancer Facts and Figures 2014. Atlanta: American Cancer Soceity. 2014.
- Pass HMJ, Johnson D. Lung Cancer: Principles and Practice. Lippincott Williams & Wilkins, Philadelphia (pubs) 2000:453-517.
- Yesner R. Heterogeneity of so-called neuroendocrine lung tumors. Exp Mol Pathol 2001; 70:179-82; PMID:11417996; http://dx.doi.org/10.1006/exmp.2001.2373
- Dong XY, Lu YJ, Tong T, Wang YJ, Guo SP, Bai JF, Han NJ, Cheng SJ. Molecular cytogenetic alterations in the early stage at human bronchial epithelial cell carcinogenesis. J Cell Biochem Suppl 1997; 28–29:74-80; PMID:9589351; http://dx.doi.org/10.1002/(SICI)1097-4644(1997)28/29+%3c74::AID-JCB8%3e3.0.CO;2-T
- She J, Yang P, Hong Q, Bai C. Lung cancer in China: challenges and interventions. Chest 2013; 143:1117-26; PMID:23546484; http://dx.doi.org/10.1378/chest.11-2948
- The 2004 United States Surgeon General's Report: The Health Consequences of Smoking. N S W Public Health Bull 2004; 15:107; PMID:15543245
- Schottenfeld D, Beebe-Dimmer JL, Buffler PA, Omenn GS. Current perspective on the global and United States cancer burden attributable to lifestyle and environmental risk factors. Annu Rev Public Health 2013; 34:97-117; PMID:23514316; http://dx.doi.org/10.1146/annurev-publhealth-031912-114350
- Kasprzak KS, Sunderman FW, Jr., Salnikow K. Nickel carcinogenesis. Mutat Res 2003; 533:67-97; PMID:14643413; http://dx.doi.org/10.1016/j.mrfmmm.2003.08.021
- Lu H, Shi X, Costa M, Huang C. Carcinogenic effect of nickel compounds. Mol Cell Biochem 2005; 279:45-67; PMID:16283514; http://dx.doi.org/10.1007/s11010-005-8215-2
- Seilkop SK, Oller AR. Respiratory cancer risks associated with low-level nickel exposure: an integrated assessment based on animal, epidemiological, and mechanistic data. Regul Toxicol Pharmacol 2003; 37:173-90; PMID:12726752; http://dx.doi.org/10.1016/S0273-2300(02)00029-6
- Salnikow K, Costa M. Epigenetic mechanisms of nickel carcinogenesis. J Environ Pathol Toxicol Oncol 2000; 19:307-18; PMID:10983897
- Salnikow K, Zhitkovich A. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem Res Toxicol 2008; 21:28-44; PMID:17970581; http://dx.doi.org/10.1021/tx700198a
- Horie A, Haratake J, Tanaka I, Kodama Y, Tsuchiya K. Electron microscopical findings with special reference to cancer in rats caused by inhalation of nickel oxide. Biol Trace Elem Res 1985; 7:223-39; PMID:24259158; http://dx.doi.org/10.1007/BF02989248
- Yao H, Rahman I. Current concepts on the role of inflammation in COPD and lung cancer. Curr Opin Pharmacol 2009; 9:375-83; PMID:19615942; http://dx.doi.org/10.1016/j.coph.2009.06.009
- Lu H, Ouyang W, Huang C. Inflammation, a key event in cancer development. Mol Cancer Res 2006; 4:221-33; PMID:16603636; http://dx.doi.org/10.1158/1541-7786.MCR-05-0261
- Witz IP. Yin-yang activities and vicious cycles in the tumor microenvironment. Cancer Res 2008; 68:9-13; PMID:18172289; http://dx.doi.org/10.1158/0008-5472.CAN-07-2917
- Duran A, Serrano M, Leitges M, Flores JM, Picard S, Brown JP, Moscat J, Diaz-Meco MT. The atypical PKC-interacting protein p62 is an important mediator of RANK-activated osteoclastogenesis. Dev Cell 2004; 6:303-9; PMID:14960283; http://dx.doi.org/10.1016/S1534-5807(03)00403-9
- Rodriguez A, Duran A, Selloum M, Champy MF, Diez-Guerra FJ, Flores JM, Serrano M, Auwerx J, Diaz-Meco MT, Moscat J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab 2006; 3:211-22; PMID:16517408; http://dx.doi.org/10.1016/j.cmet.2006.01.011
- Sugimoto R, Warabi E, Katayanagi S, Sakai S, Uwayama J, Yanagawa T, Watanabe A, Harada H, Kitamura K, Noguchi N, et al. Enhanced neointimal hyperplasia and carotid artery remodelling in sequestosome 1 deficient mice. J Cell Mol Med 2010; 14:1546-54; PMID:19780870; http://dx.doi.org/10.1111/j.1582-4934.2009.00914.x
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell 2009; 137:1062-75; PMID:19524509; http://dx.doi.org/10.1016/j.cell.2009.03.048
- Wei H, Wang C, Croce CM, Guan JL. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev 2014; 28:1204-16; PMID:24888590; http://dx.doi.org/10.1101/gad.237354.113
- Ren F, Shu G, Liu G, Liu D, Zhou J, Yuan L. Knockdown of p62/sequestosome 1 attenuates autophagy and inhibits colorectal cancer cell growth. Mol Cell Biochem 2013; 385:95-102; PMID:24065390; http://dx.doi.org/10.1007/s11010-013-1818-0
- Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, Watanabe M, Sakurada A, Endo C, Uruno A, et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci 2012; 103:760-6; PMID:22320446; http://dx.doi.org/10.1111/j.1349-7006.2012.02216.x
- Gillespie PA, Kang GS, Elder A, Gelein R, Chen L, Moreira AL, Koberstein J, Tchou-Wong KM, Gordon T, Chen LC. Pulmonary response after exposure to inhaled nickel hydroxide nanoparticles: short and long-term studies in mice. Nanotoxicology 2010; 4:106-19; PMID:20730025; http://dx.doi.org/10.3109/17435390903470101
- Puissant A, Fenouille N, Auberger P. When autophagy meets cancer through p62/SQSTM1. Am J Cancer Res 2012; 2:397-413; PMID:22860231
- Sunderman FW Jr, Morgan LG, Andersen A, Ashley D, Forouhar FA. Histopathology of sinonasal and lung cancers in nickel refinery workers. Ann Clin Lab Sci 1989; 19(1):44-50; PMID:2537054
- Zhang C, Elkahloun AG, Robertson M, Gills JJ, Tsurutani J, Shih JH, Fukuoka J, Hollander MC, Harris CC, Travis WD, et al. Loss of cytoplasmic CDK1 predicts poor survival in human lung cancer and confers chemotherapeutic resistance. PLoS One 2011; 6:e23849; PMID:21887332; http://dx.doi.org/10.1371/journal.pone.0023849
- Zhou H, Calaf GM, Hei TK. Malignant transformation of human bronchial epithelial cells with the tobacco-specific nitrosamine, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Int J Cancer 2003; 106:821-6; PMID:12918058; http://dx.doi.org/10.1002/ijc.11319
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 2000; 290:1717-21; PMID:11099404; http://dx.doi.org/10.1126/science.290.5497.1717
- Rabinowitz JD, White E. Autophagy and metabolism. Science 2010; 330:1344-8; PMID:21127245; http://dx.doi.org/10.1126/science.1193497
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8(4):445-544; PMID:22966490; http://dx.doi.org/10.4161/auto.19496
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010; 22:124-31; PMID:20034776; http://dx.doi.org/10.1016/j.ceb.2009.11.014
- Blagosklonny MV. Hypoxia, MTOR and autophagy: converging on senescence or quiescence. Autophagy 2012; 9:260-2; PMID:23192222; http://dx.doi.org/10.4161/auto.22783
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and MTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; http://dx.doi.org/10.1038/ncb2152
- Baniyash M, Sade-Feldman M, Kanterman J. Chronic inflammation and cancer: suppressing the suppressors. Cancer Immunol Immunother 2013; 63:11-20; PMID:23990173; http://dx.doi.org/10.1007/s00262-013-1468-9
- Ouyang W, Hu Y, Li J, Ding M, Lu Y, Zhang D, Yan Y, Song L, Qu Q, Desai D, et al. Direct evidence for the critical role of NFAT3 in benzo[a]pyrene diol-epoxide-induced cell transformation through mediation of inflammatory cytokine TNF induction in mouse epidermal Cl41 cells. Carcinogenesis 2007; 28:2218-26; PMID:17522069; http://dx.doi.org/10.1093/carcin/bgm115
- Huang H, Pan X, Jin H, Li Y, Zhang L, Yang C, Liu P, Liu Y, Chen L, Li J, et al. PHLPP2 Downregulation Contributes to Lung Carcinogenesis Following B[a]P/B[a]PDE Exposure. Clin Cancer Res 2015; 21:3783-93; PMID:25977341; http://dx.doi.org/10.1158/1078-0432.CCR-14-2829
- Chromium, nickel and welding. IARC Monogr Eval Carcinog Risks Hum. 1990; 49:1-648.
- Trubnikov GV, Fomin AA. [Blood plasma iron, copper, manganese, aluminum, nickel and chromium content in inflammatory diseases of the lungs and bronchial asthma]. Sov Med 1968; 31:137-8; PMID:5741583
- Kasprzak KS, Marchow L, Breborowicz J. Pathological reactions in rat lungs following intratracheal injection of nickel subsulfide and 3,4-benzpyrene. Res Commun Chem Pathol Pharmacol 1973; 6:237-45; PMID:4741096
- Trubnikov GV. [Level of microelements in the sputum of patients with inflammatory and tumorous processes of the lungs]. Ter Arkh 1975; 47:132-7; PMID:1188646
- Thompson HG, Harris JW, Wold BJ, Lin F, Brody JP. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene 2003; 22:2322-33; PMID:12700667; http://dx.doi.org/10.1038/sj.onc.1206325
- Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor NFE2L2 through inactivation of Keap1. Nat Cell Biol 2010; 12:213-23; PMID:20173742
- Vadlamudi RK, Shin J. Genomic structure and promoter analysis of the p62 gene encoding a non-proteasomal multiubiquitin chain binding protein. FEBS Lett 1998; 435:138-42; PMID:9762895; http://dx.doi.org/10.1016/S0014-5793(98)01021-7
- Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, Zhang DD. Arsenic inhibits autophagic flux, activating the NFE2L2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol 2013; 33:2436-46; PMID:23589329; http://dx.doi.org/10.1128/MCB.01748-12
- Fujita K, Maeda D, Xiao Q, Srinivasula SM. NFE2L2-mediated induction of p62 controls Toll-like receptor-4-driven aggresome-like induced structure formation and autophagic degradation. Proc Natl Acad Sci U S A 2011; 108:1427-32; PMID:21220332; http://dx.doi.org/10.1073/pnas.1014156108
- Du Y, Wooten MC, Wooten MW. Oxidative damage to the promoter region of SQSTM1/p62 is common to neurodegenerative disease. Neurobiol Dis 2009; 35:302-10; PMID:19481605; http://dx.doi.org/10.1016/j.nbd.2009.05.015
- Ding J, Huang Y, Ning B, Gong W, Li J, Wang H, Chen CY, Huang C. TNF-α induction by nickel compounds is specific through ERKs/AP-1-dependent pathway in human bronchial epithelial cells. Curr Cancer Drug Targets 2009; 9:81-90; PMID:19200052; http://dx.doi.org/10.2174/156800909787313995
- Coussens LM, Werb Z. Inflammation and cancer. Nature 2002; 420:860-7; PMID:12490959; http://dx.doi.org/10.1038/nature01322
- Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet 2001; 357:539-45; PMID:11229684; http://dx.doi.org/10.1016/S0140-6736(00)04046-0
- Wistuba II, Gazdar AF. Lung cancer preneoplasia. Annu Rev Pathol 2006; 1:331-48; PMID:18039118; http://dx.doi.org/10.1146/annurev.pathol.1.110304.100103
- Xu H, Verbeken E, Vanhooren HM, Nemery B, Hoet PH. Pulmonary toxicity of polyvinyl chloride particles after a single intratracheal instillation in rats. Time course and comparison with silica. Toxicol Appl Pharmacol 2004; 194:111-21; PMID:14736492; http://dx.doi.org/10.1016/j.taap.2003.09.018
- Nelson SM, Hajivassiliou CA, Haddock G, Cameron AD, Robertson L, Olver RE, Hume R. Rescue of the hypoplastic lung by prenatal cyclical strain. Am J Respir Crit Care Med 2005; 171:1395-402; PMID:15778486; http://dx.doi.org/10.1164/rccm.200409-1284OC
- Wu AH, Fontham ET, Reynolds P, Greenberg RS, Buffler P, Liff J, Boyd P, Henderson BE, Correa P. Previous lung disease and risk of lung cancer among lifetime nonsmoking women in the United States. Am J Epidemiol 1995; 141:1023-32; PMID:7771438
- Seril DN, Liao J, Yang GY, Yang CS. Oxidative stress and ulcerative colitis-associated carcinogenesis: studies in humans and animal models. Carcinogenesis 2003; 24:353-62; PMID:12663492; http://dx.doi.org/10.1093/carcin/24.3.353
- Nathan C. Points of control in inflammation. Nature 2002; 420:846-52; PMID:12490957; http://dx.doi.org/10.1038/nature01320
- Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer 2004; 4:71-8; PMID:14708027; http://dx.doi.org/10.1038/nrc1256
- Hudson JD, Shoaibi MA, Maestro R, Carnero A, Hannon GJ, Beach DH. A proinflammatory cytokine inhibits p53 tumor suppressor activity. J Exp Med 1999; 190:1375-82; PMID:10562313; http://dx.doi.org/10.1084/jem.190.10.1375
- Ding J, Zhang X, Li J, Song L, Ouyang W, Zhang D, Xue C, Costa M, Meléndez JA, Huang C. Nickel compounds render anti-apoptotic effect to human bronchial epithelial Beas-2B cells by induction of cyclooxygenase-2 through an IKKbeta/RELA-dependent and IKKalpha- and p50-independent pathway. J Biol Chem 2006; 281:39022-32; PMID:16982623; http://dx.doi.org/10.1074/jbc.M604798200
- Cai T, Li X, Ding J, Luo W, Li J, Huang C. A cross-talk between NFAT and NF-kappaB pathways is crucial for nickel-induced COX-2 expression in Beas-2B cells. Curr Cancer Drug Targets 2011; 11:548-59; PMID:21486220; http://dx.doi.org/10.2174/1568009117-95656001
- Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell 2008; 13:343-54; PMID:18394557; http://dx.doi.org/10.1016/j.ccr.2008.02.001
- Kojima K, Kitaoka Y, Munemasa Y, Hirano A, Sase K, Takagi H. Axonal protection by modulation of p62 expression in TNF-induced optic nerve degeneration. Neurosci Lett 2014; 581:37-41; PMID:25150927; http://dx.doi.org/10.1016/j.neulet.2014.08.021
- Viiri J, Amadio M, Marchesi N, Hyttinen JM, Kivinen N, Sironen R, Rilla K, Akhtar S, Provenzani A, D'Agostino VG, et al. Autophagy activation clears ELAVL1/HuR-mediated accumulation of SQSTM1/p62 during proteasomal inhibition in human retinal pigment epithelial cells. PLoS One 2013; 8:e69563; PMID:23922739; http://dx.doi.org/10.1371/journal.pone.0069563
- Zhang D, Liang Y, Xie Q, Gao G, Wei J, Huang H, Li J, Gao J, Huang C. A novel post-translational modification of nucleolin, SUMOylation at Lys-294, mediates arsenite-induced cell death by regulating gadd45alpha mRNA stability. J Biol Chem 2014; 290:4784-800; http://dx.doi.org/10.1074/jbc.M114.598219
- Deleault KM, Skinner SJ, Brooks SA. Tristetraprolin regulates TNF TNF-α mRNA stability via a proteasome dependent mechanism involving the combined action of the ERK and p38 pathways. Mol Immunol 2008; 45:13-24; PMID:17606294; http://dx.doi.org/10.1016/j.molimm.2007.05.017
- Chen G, Ke Z, Xu M, Liao M, Wang X, Qi Y, Zhang T, Frank JA, Bower KA, Shi X, et al. Autophagy is a protective response to ethanol neurotoxicity. Autophagy 2012; 8:1577-89; PMID:22874567; http://dx.doi.org/10.4161/auto.21376
- Park S, Ha SD, Coleman M, Meshkibaf S, Kim SO. p62/SQSTM1 enhances NOD2-mediated signaling and cytokine production through stabilizing NOD2 oligomerization. PLoS One 2013; 8:e57138; PMID:23437331; http://dx.doi.org/10.1371/journal.pone.0057138
- Hu Q, Klippel A, Muslin AJ, Fantl WJ, Williams LT. Ras-dependent induction of cellular responses by constitutively active phosphatidylinositol-3 kinase. Science 1995; 268:100-2; PMID:7701328; http://dx.doi.org/10.1126/science.7701328
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011; 331:456-61; PMID:21205641; http://dx.doi.org/10.1126/science.1196371
- Zhang D, Li J, Gao J, Huang C. JUN/AP-1 pathway-mediated cyclin D1 expression participates in low dose arsenite-induced transformation in mouse epidermal JB6 Cl41 cells. Toxicol Appl Pharmacol 2009; 235:18-24; PMID:19059425; http://dx.doi.org/10.1016/j.taap.2008.11.002
- Zuo Z, Cai T, Li J, Zhang D, Yu Y, Huang C. Hexavalent chromium Cr(VI) up-regulates COX-2 expression through an NFkappaB/JUN/AP-1-dependent pathway. Environ Health Perspect 2012; 120:547-53; PMID:22472290; http://dx.doi.org/10.1289/ehp.1104179
- Che X, Liu J, Huang H, Mi X, Xia Q, Li J, Zhang D, Ke Q, Gao J, Huang C. p27 suppresses cyclooxygenase-2 expression by inhibiting p38beta and p38delta-mediated CREB phosphorylation upon arsenite exposure. Biochim Biophys Acta 2013; 1833:2083-91; PMID:23639288; http://dx.doi.org/10.1016/j.bbamcr.2013.04.012
- Zhu J, Zhang J, Huang H, Li J, Yu Y, Jin H, Li Y, Deng X, Gao J, Zhao Q, et al. Crucial Role of JUN Phosphorylation at Ser63/73 Mediated by PHLPP Protein Degradation in the Cheliensisin A Inhibition of Cell Transformation. Cancer Prev Res (Phila) 2014; 7:1270-81; PMID:25281487; http://dx.doi.org/10.1158/1940-6207.CAPR-14-0233
- Huang H, Ma L, Li J, Yu Y, Zhang D, Wei J, Jin H, Xu D, Gao J, Huang C. NF-kappaB1 inhibits c-Myc protein degradation through suppression of FBW7 expression. Oncotarget 2014; 5:493-505; PMID:24457827; http://dx.doi.org/10.18632/oncotarget.1643
- Kang GS, Gillespie PA, Gunnison A, Rengifo H, Koberstein J, Chen LC. Comparative pulmonary toxicity of inhaled nickel nanoparticles; role of deposited dose and solubility. Inhal Toxicol 2011; 23:95-103; http://dx.doi.org/10.3109/08958378.2010.543440
- Yan Y, Li J, Ouyang W, Ma Q, Hu Y, Zhang D, Ding J, Qu Q, Subbaramaiah K, Huang C. NFAT3 is specifically required for TNF-α-induced cyclooxygenase-2 (COX-2) expression and transformation of Cl41 cells. J Cell Sci 2006; 119:2985-94; PMID:16803872; http://dx.doi.org/10.1242/jcs.03014
- Gao G, Chen L, Li J, Zhang D, Fang Y, Huang H, Chen X, Huang C. Isorhapontigenin (ISO) inhibited cell transformation by inducing G0/G1 phase arrest via increasing MKP-1 mRNA Stability. Oncotarget 2014; 5:2664-77; PMID:24797581; http://dx.doi.org/10.18632/oncotarget.1872
- Fang Y, Yu Y, Hou Q, Zheng X, Zhang M, Zhang D, Li J, Wu XR, Huang C. The Chinese herb isolate isorhapontigenin induces apoptosis in human cancer cells by down-regulating overexpression of antiapoptotic protein XIAP. J Biol Chem 2012; 287:35234-43; PMID:22896709; http://dx.doi.org/10.1074/jbc.M112.389494
- Zhang D, Li J, Costa M, Gao J, Huang C. JNK1 mediates degradation HIF-1alpha by a VHL-independent mechanism that involves the chaperones Hsp90/Hsp70. Cancer Res 2010; 70:813-23; PMID:20068160; http://dx.doi.org/10.1158/0008-5472.CAN-09-0448
- Zhang J, Gao G, Chen L, Li J, Deng X, Zhao QS, Huang C. Hydrogen peroxide/ATR-Chk2 activation mediates p53 protein stabilization and anti-cancer activity of cheliensisin A in human cancer cells. Oncotarget 2014; 5:841-52; PMID:24553354; http://dx.doi.org/10.18632/oncotarget.1780