
ABSTRACT
A growing number of studies have demonstrated that both macroautophagy/autophagy and apoptosis are important inner mechanisms of cell to maintain homeostasis and participate in the host response to pathogens. We have previously reported that a functional autophagy pathway is trigged by infection of classical swine fever virus (CSFV) and is required for viral replication and release in host cells. However, the interplay of autophagy and apoptosis in CSFV-infected cells has not been clarified. In the present study, we demonstrated that autophagy induction with rapamycin facilitates cellular proliferation after CSFV infection, and that autophagy inhibition by knockdown of essential autophagic proteins BECN1/Beclin 1 or MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) promotes apoptosis via fully activating both intrinsic and extrinsic mechanisms in CSFV-infected cells. We also found that RIG-I-like receptor (RLR) signaling was amplified in autophagy-deficient cells during CSFV infection, which was closely linked to the activation of the intrinsic apoptosis pathway. Moreover, we discovered that virus infection of autophagy-impaired cells results in an increase in copy number of mitochondrial DNA and in the production of reactive oxygen species (ROS), which plays a significant role in enhanced RLR signaling and the activated extrinsic apoptosis pathway in cultured cells. Collectively, these data indicate that CSFV-induced autophagy delays apoptosis by downregulating ROS-dependent RLR signaling and thus contributes to virus persistent infection in host cells.
Introduction
Classical swine fever virus (CSFV), an enveloped virus with a positive-stranded RNA genome, is classified as a Pestivirus within the Flaviviridae family.Citation1 CSFV is the causative agent of classical swine fever (CSF), a highly virulent disease of swines.Citation2,3 In vivo, CSFV infection mainly causes high fever, multiple hemorrhages, and leukopenia, leading to high morbidity and mortality, the severity of which might be due to host quality and the virulence of the viral strains.Citation2,3 Interestingly, in vitro, CSFV replication in cells suppresses type I IFN (interferon)-inducible antiviral activity and apoptosis by interfering with IFN production, thereby resulting in the persistent survival of CSFV in host cells.Citation4 Although there have been extensive studies on the pathogenesis of CSFV,Citation2,5-7 the underlying mechanisms of apoptosis inhibition in cells in response to infection with CSFV are only beginning to be elucidated.
Apoptosis (formerly referred to as programmed cell death type I) is regulated by CASPs/caspases, which are apoptosis-related cysteine peptidases.Citation8,9 Two main signals induce apoptosis, including the intrinsic and extrinsic pathways. The induction of the intrinsic pathway results in mitochondrial outer membrane permeabilization, therefore triggering CASP3/caspase-3 by activating CASP9/caspase-9.Citation8 The extrinsic pathway activates CASP3 via cleavage of CASP8/caspase-8 in a death receptor-mediated manner.Citation8 Apoptosis also could be considered a defense mechanism against virus replication by triggering cell death.Citation10
Autophagy is an intracellular bulk degradation pathway by which cytosolic constituents can be cleared and recycled in the cytosol for the maintenance of cellular homeostasis.Citation11 Dysfunctional autophagy is associated with the causation of human diseases such as cancer, aging, and neurodegenerative disorders.Citation12-14 In addition, recent studies demonstrate that an autophagic mechanism is required to combat viral infections via influencing the innate and adaptive immune defenses.Citation15-17 Interestingly, several viruses have evolved diverse strategies to subvert autophagy for their own replication.Citation18-21 We have shown that CSFV infection triggers the autophagy pathway in host cells, which is essential for viral replication.Citation22 Based on these findings, we postulate that CSFV-activated autophagy may be a potential mechanism used by viruses for establishing a persistent infection. Therefore, further work investigating the precise molecular mechanisms of the relationship between CSFV and autophagy is important for controlling viral infection.
Currently, several genetic connections have emerged between autophagy and apoptosis, such functional links mainly depend on the regulation of the same proteins in both autophagy and apoptosis.Citation23-26 To date, it is not yet known whether a crosstalk might exist between autophagic and apoptotic pathways in cells subjected to CSFV infection. However, in view of the known functions of autophagy and apoptosis in physiological homeostasis and in the viral infection process, it seems likely that apoptosis inhibition is related to CSFV-induced autophagy. Herein, our research focused on the role of CSFV-induced autophagy in apoptotic pathways through the regulation of autophagic activity, and reveals a novel regulatory mechanism by which autophagy limits apoptosis and contributes to CSFV infection in host cells.
Results
Autophagy promotes cell survival under CSFV infection
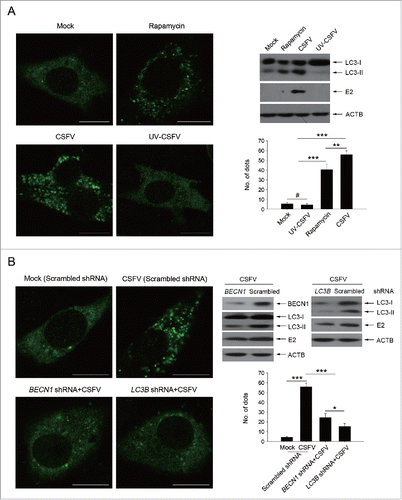
We have reported earlier that CSFV-infected cells displayed induction of autophagy.Citation22 In addition to the autophagic pathway, other forms of stress on cell survival may be induced in cells subjected to viral infection, such as cell death pathways. However, in vitro studies showed that CSFV infection does not induce a cytopathic effect (CPE) and therefore has no obvious effect on cell death.Citation27 Autophagy, a protective mechanism in cells, can prolong cell survival under different stresses. As CSFV infection cannot trigger a pronounced CPE and cell death, it is important and necessary to know more about the function of autophagy induced by CSFV on cell death. To this end, rapamycin, an inducer of autophagy, was used to promote the cellular autophagic pathway.Citation28,29 Meanwhile, short hairpin RNA (shRNA)-mediated knockdown treatments were performed to decrease the activity of autophagy in PK-15 and 3D4/2 cells. The pharmacological regulation of autophagy and RNA interference efficiency are shown in . Significant fluorescence signals of the mostly autophagy-specific protein LC3 were observed in both CSFV-infected and rapamycin-treated cells with puncta accumulation (). In contrast, the classical LC3 puncta accumulation was not observed in mock-infected and ultraviolet (UV)-CSFV infected cells (), indicating that replication-inhibited CSFV lost its capability of triggering the functional autophagic pathway. The corresponding number of LC3 puncta in cells subjected to different treatments were also counted (). Additionally, western blot analysis was used to measure the conversion of LC3 and the expression of CSFV envelope protein E2 (), and the results were consistent with the data from the detection of fluorescence signals. Meanwhile, PK-15 cells were transfected with scrambled, BECN1 or LC3B shRNAs, and then were mock infected or infected with CSFV. Followed by observation of fluorescence signals and protein level measurements. Our results showed that a significant inhibition of autophagy was observed in BECN1 or LC3B shRNA-transfected cells (). As expected, rapamycin treatment and autophagic protein knockdown could be used for regulating autophagy activity in our research.
Figure 1. Rapamycin and autophagy-specific shRNAs treatments alter autophagic activity in host cells. (A) PK-15 cells were treated with rapamycin (100 nM) or DMSO (Mock) for 48 h as control groups. Other groups were infected with CSFV or UV-CSFV for 48 h at an MOI of 1. Then cells were fixed and processed for fluorescence signal detection of LC3 accumulation with corresponding antibodies as described in Materials and Methods. The fluorescence signals were visualized with confocal immunofluorescence microscopy. In images, LC3 staining is shown in green. Scale bar: 10 μm. Average number of punctate LC3 in each cell were determined from at least 100 cells in each group. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. **, P < 0.01; ***, P < 0.001; #, P > 0.05. Meanwhile, at the end of the infection, some cell samples were analyzed by immunoblotting with specific antibodies as described in Materials and Methods. Similar results were obtained in 3 independent experiments. (B) PK-15 cells were transfected with shRNAs targeting BECN1 or LC3B or scramble shRNA for 48 h, followed by mock infection and CSFV infection for 48 h at an MOI of 1. Then the cells were treated and analyzed as in (A). The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; ***, P < 0.001. Meanwhile, after infection, some cell samples were analyzed by immunoblotting with specific antibodies as described in Materials and Methods. Similar results were obtained in 3 independent experiments.
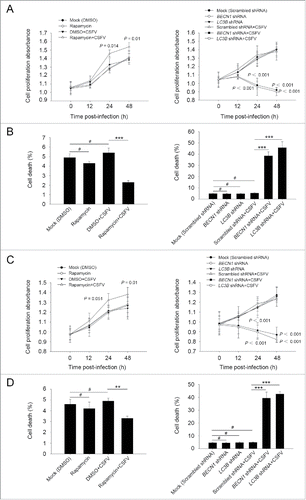
Furthermore, we chose to use rapamycin and specific shRNAs in examining whether regulation of autophagy activity alters CSFV-infected cell growth. As demonstrated in , the induction of autophagy with rapamycin showed an enhancement effect on cell viability during infection with CSFV, from 0 to 48 h post-infection, whereas there were no significant differences in cell viability between cells with single infection of CSFV and mock-infected cells (). In contrast, knockdown of autophagy with shRNAs targeting endogenous BECN1 and LC3B reduced cell viability in PK-15 and 3D4/2 cells during CSFV infection (). Notably, a decreased percentage of cell death was observed in the rapamycin-treated cells, and an increased percentage of cell death was obtained in autophagy-impaired cells under virus infection, compared with all necessary controls (), suggesting that CSFV subverts autophagy to promote cell proliferation or inhibit cell death.
Figure 2. Alteration of autophagic activity affects viability of CSFV-infected cells. (A and C) PK-15 (A) and 3D4/2 (C) cells were pretreated with rapamycin (100 nM) or DMSO for 1 h. Cells were then mock infected or infected with CSFV at an MOI of 1. After 1 h of virus absorption at 37°C, the cells were incubated in fresh medium containing rapamycin (100 nM ) or DMSO for 0, 12, 24 and 48 h. Cells only treated with rapamycin were considered as one control. Meanwhile, 2 cell lines were transfected with shRNAs targeting BECN 1 or LC3B or scramble shRNA for 48 h, followed by mock infection and CSFV infection for 0, 12, 24 and 48 h at the same MOI. Then the cell viability was determined by the CCK-8 assay. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. (B and D) PK-15 (B) and 3D4/2 (D) cells were pretreated and transfected as described in (A and C), followed by mock infection and CSFV infection for 48 h at an MOI of 1. Percentage of cell death was then detected using a live-dead cell staining solution and analyzed by flow cytometry. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. **, P < 0.01; ***, P < 0.001; #, P > 0.05.
CSFV infection induces apoptosis in autophagy knockdown cells
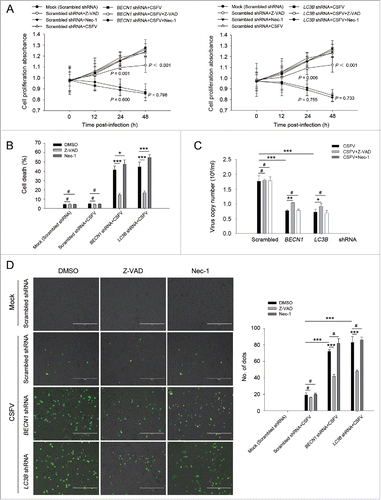
Increasing evidence suggests that in some instances autophagy and apoptosis are cross inhibitory.Citation30 Our results showed that CSFV infection promotes cell death in autophagy-impaired cells (). To further determine if the autophagic machinery influences apoptosis during CSFV infection, we detected cell viability and percentage of cell death in autophagy knockdown cells by using Z-VAD-FMK (Z-VAD) and necrostatin-1 (Nec-1), which are usually used as inhibitors for apoptosis and necrosis.Citation31-34 As shown in and , infected autophagy knockdown cells treated with Z-VAD exhibited an enhanced level of cell viability and a reduced percentage of cell death, whereas Nec-1 treatment had no obvious role in cell viability and percentage of cell death in autophagy knockdown cells infected with CSFV (). To further analyze the correlation between autophagy and apoptosis during the progression of CSFV infection, the TUNEL staining method was performed to detect apoptosis. Our results showed that knockdown of autophagy with BECN1 and LC3B shRNAs promoted apoptosis in PK-15 and 3D4/2 cells during CSFV infection compared with cells only infected with CSFV. Autophagy-impaired cells treated with Nec-1 exhibited a similar level of apoptotic “dots” compared with both BECN1 and LC3B shRNA-transfected cells during CSFV infection, whereas Z-VAD rescued autophagy knockdown cells from CSFV-induced apoptosis (). Notably, virus production was significantly enhanced after Z-VAD treatment in both autophagy-impaired PK-15 and 3D4/2 cells (). These results indicate that CSFV infection induces cell death in autophagy knockdown cells, and apoptosis is the principal pathway of cell death.
Figure 3. CSFV infection in autophagy knockdown cells induces apoptotic cell death in PK-15 cells. (A) PK-15 cells were transfected with the scrambled or BECN1 or LC3B shRNA for 48 h. Cells were then mock infected or infected with CSFV at an MOI of 1. After 1 h of CSFV absorption, the cells were further cultured in fresh medium in the absence or presence of Z-VAD (10 μM) or Nec-1 (10 μM) at the indicated time points. Then the cell viability was measured by the CCK-8 assay. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. (B) PK-15 cells were transfected and infected as described in (A). Percentage of cell death was then measured using a live-dead cell staining solution and assayed by flow cytometry. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P< 0.05; ***, P < 0.001; #, P > 0.05. (C) PK-15 cells transfected with the targeted shRNAs were incubated with CSFV at an MOI of 0.5, and were further cultured as described in (A). At 48 h after CSFV infection, the copy number of virus was detected by qRT-PCR as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. ***, P < 0.001; #, P > 0.05. (D) PK-15 cells were transfected and infected as described in (A). Apoptosis was then measured using a TUNEL Apoptosis Assay Kit as described in Materials and Methods. In the images, apoptotic cells are shown in green. Scale bar: 400 μm. Average number of apoptotic green “dots” was determined from at least 3 images. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. ***, P < 0.001; #, P > 0.05.
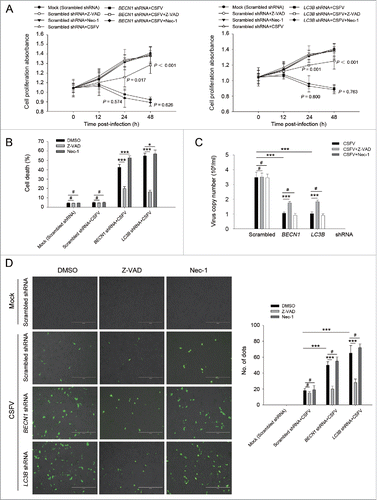
Figure 4. Autophagy knockdown promotes apoptosis in 3D4/2 cells under CSFV infection. (A) 3D4/2 cells were transfected and infected as described in the legend to . After 1 h of CSFV absorption, the cells were further cultured in fresh medium in the absence or presence of Z-VAD (10 μM) and Nec-1 (10 μM) at the indicated time points. Then the cell viability was measured by the CCK-8 assay. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. (B) 3D4/2 cells were treated as described in (A). Cell death was then detected using a live-dead cell staining solution and assayed by flow cytometry. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P< 0.05; ***, P < 0.001; #, P > 0.05. (C) 3D4/2 cells transfected with the autophagy-specific shRNAs were incubated with CSFV at an MOI of 0.5, and were further cultured as described in (A). At 48 h after CSFV infection, virus production was measured by qRT-PCR as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; #, P > 0.05. (D) 3D4/2 cells were treated as described in (A). Apoptosis was then measured using a TUNEL Apoptosis Assay Kit as described in Materials and Methods. In images, apoptotic cells are shown in green. Scale bar: 400 μm. Average number of apoptotic green “dots” were from at least 3 images. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. ***, P < 0.001; #, P > 0.05.
CSFV infection triggers the intrinsic and extrinsic apoptosis signaling pathways in autophagy knockdown cells
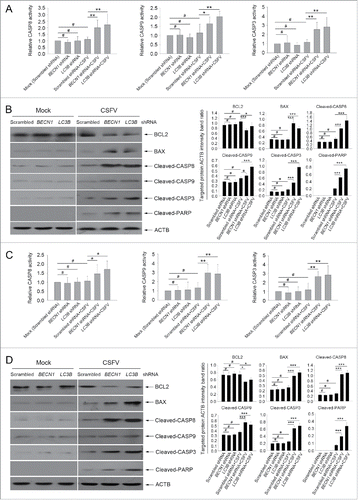
The apoptosis pathway is regulated by a series of action of CASPs.Citation8,9 To further define the relationship between autophagy and apoptosis in CSFV-infected cells, the activity of the CASP-mediated pathway was detected. As shown in , the activities of CASP3, CASP8, and CASP9 were promoted in autophagy-deficient cells during CSFV infection compared with the corresponding CASP activity in normal infected PK-15 and 3D4/2 cells (). Furthermore, our results showed that both cell lines transfected with BECN1 and LC3B shRNAs presented significantly increased levels of pro-apoptotic molecules including BAX, cleaved-CASP8, cleaved-CASP9, cleaved-CASP3, and cleaved-PARP proteins, and reduced the expression of the anti-apoptotic molecule BCL2/Bcl-2 compared with cells transfected with nontargeting (scrambled) shRNAs during CSFV infection (). These results indicate that CSFV-induced autophagy limits apoptosis by inhibiting both the intrinsic and extrinsic mechanisms.
Figure 5. Autophagy deficiency leads to the activation of apoptosis pathways in CSFV-infected cells. ((A)and C) PK-15 (A) and 3D4/2 (C) cells were transfected and infected as described in the legend to . At 48 h after infection, the activity of CASP3, CASP8, and CASP9 in cells was measured by the CASP activity assay as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; #, P > 0.05. ((B)and D) PK-15 (B) and 3D4/2 (D) cells were transfected and infected as described in the legend to . After 48 h, the expression of BCL2, BAX, cleaved-CASP3, cleaved-CASP8, cleaved-CASP9, cleaved-PARP, and ACTB (loading control) were analyzed by western blot with specific antibodies as described in Materials and Methods. The relative expression ratios of these proteins were analyzed by densitometric scanning. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; ***, P < 0.001; #, P > 0.05.
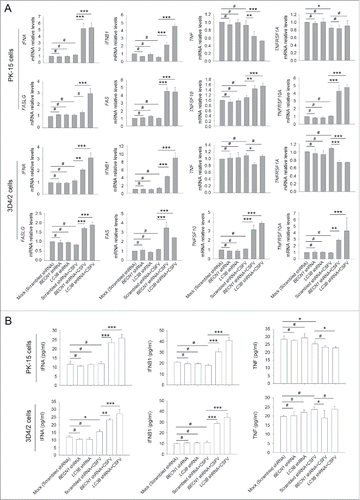
CSFV infection in autophagy knockdown cells promotes mRNA levels of type I IFN and IFN-stimulated genes (ISGs)
in vitro studies showed that inhibition of apoptosis in CSFV-infected cells is mainly attributed to suppression of type I IFN production.Citation4 Given that autophagy may suppress innate antiviral immunity in some viral infections, including vesicular stomatitis Indiana virus and hepatitis C virus,Citation35-37 and the role for autophagy in type I IFN production has not previously been reported in CSFV infection, we next asked whether knockdown of autophagy proteins following CSFV infection modulates type I IFN production; this was important to reveal the molecular mechanism by which CSFV inhibits IFN-mediated apoptosis in host cells. For this, the mRNA levels of IFNA/IFN-α, IFNB1/IFN-β, TNFSF10/TRAIL, TNFRSF10A/DR4, FASLG/TNFSF6/FASL, FAS/TNFRSF6, TNF/TNF-α, and TNFRSF1A/TNFR from CSFV-infected cells and noninfected cells, which were transfected with shBECN1, shLC3B, or scrambled shRNA (control), were examined to verify if deficiency of autophagy has an effect on the interferon signaling pathway. TNFSF10 and FAS, the well-known apoptosis-related ISGs, are involved in death receptor-mediated apoptotic processes in CSFV infection in vivo.Citation38 Our results showed a significant increase in mRNA expression of apoptosis-related genes in autophagy-impaired PK-15 and 3D4/2 cells infected with CSFV, at 48 h post-infection, compared with CSFV-infected (scrambled shRNA) cells (). However, a significant difference in expression of these genes in CSFV-infected (scrambled shRNA) cells compared with mock-infected (scrambled shRNA) cells was not observed (). Furthermore, similar results were also obtained in type I IFN production (). Importantly, TNF production was not affected by autophagy deficiency (), consistent with previous results that Nec-1 treatment had no obvious role in cell viability and percentage of cell death in autophagy knockdown cells infected with CSFV (). These findings indicate that CSFV-induced autophagy inhibits apoptosis by suppressing type I IFN production and the expression of apoptosis-related ISGs.
Figure 6. CSFV infection enhances the expression of IFN and ISGs in autophagy-deficient cells. (A) PK-15 and 3D4/2 cells were transfected and infected as described in the legend to . At 48 h after CSFV infection, the mRNA levels of IFNA, IFNB1, TNFSF10, TNFRSF10A, FASLG, FAS, TNF, and TNFRSF1A were detected by qRT-PCR as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P< 0.05; **, P < 0.01; ***, P < 0.001; #, P > 0.05. (B) PK-15 and 3D4/2 cells were transfected and infected as described in the legend to . After 48 h, the production of IFNA, IFNB1, and TNF was assessed by the ELISA assay. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; #, P > 0.05.
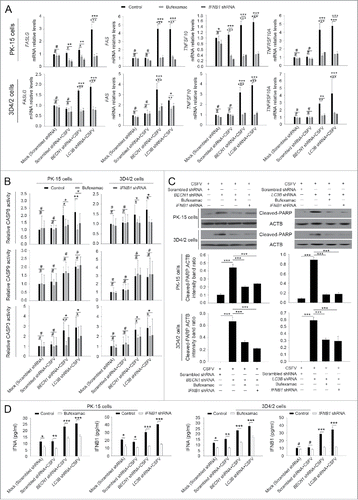
To further analyze the role of type I IFN in apoptosis, the inhibitor bufexamac targeting IFNA release and IFNB1 sequence-specific shRNA were used in this study.Citation39 As shown in , inhibition of IFNA or IFNB1 production significantly decreased mRNA levels of ISGs and therefore reduced the activity of CASP3 and CASP8, then the expression of cleaved-PARP was downregulated in autophagy knockdown PK-15 and 3D4/2 cells infected with CSFV, at 48 h post-infection, compared with CSFV-infected control (scrambled shRNA) cells and mock-infected control (scrambled shRNA) cells (). Additionally, the interference efficiency of bufexamac and IFNB1 shRNA was measured using an ELISA assay for type I IFN production detection (). These results further indicate that CSFV-induced autophagy suppresses type I IFN mediated extrinsic apoptosis.
Figure 7. Type I IFN production is required for the activation of the extrinsic apoptosis pathway. (A) PK-15 and 3D4/2 cells were transfected and infected as described in the legend to , which were considered as the control groups. For inhibition of IFNA and IFNB1 induction, cells were cultured with bufexamac (10 μM) after 1 h of virus absorption, or cotransfected with shRNA targeting IFNB1 prior to CSFV infection. At 48 h after infection, the mRNA levels of TNFSF10, TNFRSF10A, FASLG, and FAS were measured by qRT-PCR. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; #, P > 0.05. (B) PK-15 and 3D4/2 cells were treated as described in (A). After 48 h, the activity of CASP3, CASP8, and CASP9 in cultured cells was detected by the CASP activity assay as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; #, P > 0.05. (C) PK-15 and 3D4/2 cells were treated as described in (A). After 48 h, cell samples were analyzed by western blot with antibodies against PARP and ACTB (loading control). The relative expression ratios of the targeted proteins were analyzed by densitometric scanning. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. ***, P < 0.001. (D) PK-15 and 3D4/2 cells were treated as described in (A). After 48 h, IFNA and IFNB1 production and the inhibition efficiency of type I IFN induction were detected by the ELISA assay. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.001; #, P > 0.05.
Absence of autophagy in cells enhances RLR signaling upon CSFV infection
RNA viruses are recognized by the members of the RLRs in cells, such as DDX58/RIG-I (DEXD/H-box helicase 58) and IFIH1/MDA5 (interferon induced with helicase C domain 1).Citation40,41 Type I IFN production is closely related to the regulation of RLR signaling.Citation41 Given that the absence of autophagy leads to an increase in the production of type I IFN in host cells after infection with CSFV (), we reasoned that autophagy may limit type I IFN production through regulating the activation of RLR signaling. To test this hypothesis, we determined the levels of DDX58, IFIH1, and their adaptor protein MAVS in autophagy-impaired cells under CSFV infection by using western blot.Citation40 As shown in , knockdown of BECN1 and LC3 in CSFV-infected PK-15 cells leads to a significant increase in the expression of DDX58, IFIH1, and MAVS compared with cells transfected with nontargeting (scrambled) shRNAs (). Similar results were also observed in infected 3D4/2 cells (). The data thus far indicate that the absence of autophagy not only increases type I IFN levels but also enhances RLR signaling.
Figure 8. Absence of autophagy in CSFV-infected cells promotes the production of type I IFN by upregulating the RLR signaling. (A and B) PK-15 and 3D4/2 cells were transfected and infected as described in the legend to . At 48 h, the expression of DDX58, IFIH1, MAVS, and ACTB (loading control) were analyzed by western blot with specific antibodies as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P< 0.05; **, P < 0.01; ***, P < 0.001; #, P > 0.05. (C) PK-15 and 3D4/2 cells were cotransfected with the indicated shRNAs for 48 h, and were then infected with CSFV at an MOI of 1. At 48 h after infection, IFNA and IFNB1 production was detected by the ELISA assay. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; ***, P< 0.001; #, P > 0.05. (D) PK-15 cells were transfected with shRNAs targeting DDX58, IFIH1, or MAVS for 48 h, followed by CSFV infection at an MOI of 1. After 48 h, the expression of the targeted proteins was detected by western blot using anti-DDX58, anti-IFIH1, and anti-MAVS antibodies. Similar results were obtained in 3 independent experiments.
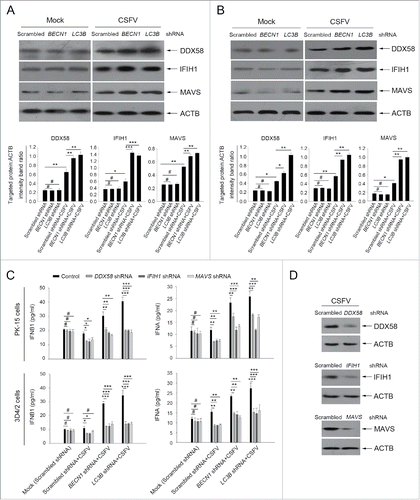
To support these data, we chose to perform gene silencing of DDX58, IFIH1, and MAVS; the silencing efficiency is shown in . Importantly, we further confirmed that inhibition of RLR signaling significantly reduced the production of type I IFN in autophagy-impaired cells during CSFV infection (). Thus, we suggest that CSFV-induced autophagy may limit type I IFN production by inhibiting RLR signaling.
CSFV infection induces ROS accumulation and leads to RLR signaling amplification in autophagy knockdown cells
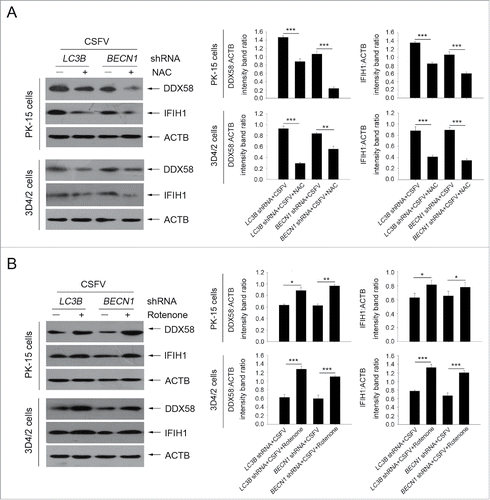
Previous studies have reported that RLR signaling amplification is related to ROS levels.Citation42 Next, we detected whether a decrease in ROS production affects RLR signaling in autophagy-defective cells during infection by CSFV. To this end, N-acetyl-L-cysteine (NAC) and rotenone were used in this study to alter ROS production; these compounds are usually considered as regulators for ROS release.Citation43,44 As shown in , an obvious increase in ROS levels were observed in autophagy knockdown PK-15 and 3D4/2 cells infected with CSFV, at 24 and 48 h post-infection, compared with CSFV-infected (scrambled shRNA) cells and mock-infected (scrambled shRNA) cells (). The regulation efficiency of NAC and rotenone were also measured by ROS detection (). Inhibition of ROS release with NAC reduced the levels of DDX58 and IFIH1 (). In contrast, ROS induction with rotenone increased the levels of DDX58 and IFIH1 (). Interestingly, autophagy defects also increased the copy number of mitochondrial DNA during CSFV infection, which is associated with the enhanced MAVS levels and ROS accumulation (). The data indicate that ROS accumulation is necessary for RLR signaling amplification in autophagy-impaired cells after infection with CSFV.
Figure 9. CSFV infection increases ROS levels in autophagy knockdown cells. (A) PK-15 and 3D4/2 cells were transfected with the scrambled, BECN1 or LC3B shRNAs for 48 h. After 1 h of mock infection or infection by CSFV at an MOI of 1, the cells were further cultured in fresh medium in the absence or presence of NAC (10 mM) or rotenone (1 μM) for 24 and 48 h. Levels of ROS in cultured cells were analyzed by MitoSOX staining as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P< 0.05, Dunnett's T3 (3) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01. (B) PK-15 and 3D4/2 cells were transfected and infected as described in the legend to . After 48 h, the copy number of mitochondrial DNA was assessed by qPCR as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. **, P < 0.01; ***, P < 0.001; #, P > 0.05.
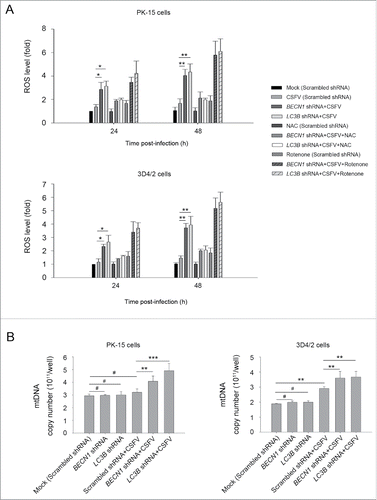
Figure 10. Alteration of ROS levels affects RLR signaling in cultured cells. (A) PK-15 and 3D4/2 cells were transfected with the BECN1 or LC3B shRNAs for 48 h. After 1 h of mock infection or infection by CSFV at an MOI of 1, the cells were further cultured in fresh medium in the absence or presence of NAC (10 mM) for 48 h. The expression of DDX58, IFIH1, and ACTB (loading control) were analyzed by western blot using specific antibodies. The relative expression ratios of these proteins were analyzed by densitometric scanning. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P< 0.05; **, P < 0.01; ***, P < 0.001. (B) PK-15 and 3D4/2 cells were transfected and infected as described in (A). After 1 h of CSFV infection at an MOI of 1, the cells were further cultured in fresh medium in the absence or presence of rotenone (1 μM) for 48 h. The expression of DDX58, IFIH1, and ACTB (loading control) were analyzed by western blot using specific antibodies. The relative expression ratios of these proteins were analyzed by densitometric scanning. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Enhanced ROS production is required for activating the intrinsic apoptosis pathway
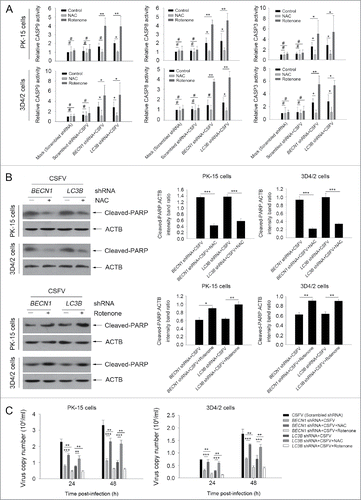
Our present results showed that the intrinsic apoptosis signaling pathway was triggered by CSFV in autophagy-impaired cells. To analyze the role of ROS in the activation of the intrinsic apoptosis pathway, we tested the activity of CASP3, CASP8 and CASP9, and the expression of cleaved-PARP with NAC or rotenone in cultured PK-15 cells. Our results showed that NAC treatment reduced both the activity of CASPs and the level of cleaved-PARP in autophagy-impaired PK-15 cells, at 48 h post-infection, compared with only CSFV-infected (BECN1 and LC3B shRNAs) cells (). We also found that rotenone treatment enhanced both the CASP activity and cleaved-PARP expression in autophagy-impaired PK-15 cells, at 48 h post-infection, compared with normal CSFV-infected (BECN1 and LC3B shRNAs) cells. Similar results were also observed in infected 3D4/2 cells (). Interestingly, NAC treatment increased virus production in both cell lines. In contrast, rotenone treatment reduced the generation of progeny virus in both cell lines, at 24 and 48 h post-infection, compared with normal CSFV-infected (BECN1 and LC3B shRNAs) (). These results further indicate that CSFV-induced autophagy suppresses apoptosis by downregulating ROS levels in host cells.
Figure 11. Regulation of ROS levels within autophagy-impaired cells triggers the extrinsic apoptosis pathway during CSFV infection. (A) PK-15 and 3D4/2 cells were treated and cultured as described in the legend to . The activities of CASP3, CASP8, and CASP9 in cell samples were assessed by the CASP activity assay as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P < 0.05, Dunnett's T3 (3) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; #, P > 0.05. (B) PK-15 and 3D4/2 cells were treated and cultured as described in the legend to . The levels of cleaved-PARP and ACTB (loading control) were detected by western blot using specific antibodies. The relative expression ratios of these proteins were assessed by densitometric scanning. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (C) PK-15 and 3D4/2 cells were transfected as described in the legend to . At 24 and 48 h after CSFV infection, virus copy number was detected by qRT-PCR as described in Materials and Methods. The data represent the mean ± SD of 3 independent experiments. One-way ANOVA test; test of homogeneity of variances, P > 0.05, LSD (L) was used for correction of post-hoc test. **, P < 0.01; ***, P < 0.001.
Discussion
Autophagy is a catabolic degradation pathway that is characterized by the formation of autophagosomes that engulf cytosolic constituents and fuse with lysosomes for degradation of their lumenal content.Citation11 Autophagy plays a key role in cytoprotective mechanisms of host defense and cell survival,Citation45-47 for instance, in Sindbis virus and tobacco mosaic virus infections.Citation48,49 Apoptosis is considered as a key pathway of host defense under viral infection, which may limit viral propagation.Citation10 We have seen earlier that a functional autophagy pathway induced by CSFV is required for enhancing virus replication and release in host cells.Citation22 However, in vitro studies have shown that CSFV infection cannot induce CPE and cell death.Citation27 Many studies have shown the importance of the interface between autophagy and apoptosis; for example, TP53/p53, a well-known inducer of apoptosis, also activates autophagy through a damage-regulated modulator of autophagy.Citation50 BECN1, an upstream molecule for autophgosome formation, also regulates apoptosis by interacting with both BCL2 and BCL2L1/BCL-XL.Citation51 Additionally, apoptosis may inhibit autophagy as CAPN/calpain cleaves ATG5 that triggers CASP-dependent cell death.Citation52,53 Clearly, the same proteins in some instances are confirmed as a link between autophagy and apoptosis that regulate both autophagic and apoptotic processes at the cellular level, and such processes can cross-inhibit each other under different environmental conditions. Based on the findings about the crosstalk between autophagy and apoptosis, an autophagic mechanism is likely involved in apoptosis inhibition in CSFV-infected cells. Here, we examined the role of CSFV-induced autophagy in apoptotic pathways and reveal a novel mechanism by which autophagy inhibits apoptosis and contributes to CSFV infection.
In this study, we used PK-15 and 3D4/2 cells as the model cell lines to extend our results, because they are targets for CSFV infection and are usually used to study CSFV infection in vitro.Citation5,7,54,55 In order to clarify the connections between the autophagic and apoptotic processes in CSFV-infected PK-15 and 3D4/2 cells, cell death was first detected when autophagy were either induced by a chemical inducer of autophagy or inhibited by knockdown of autophagy with gene-specific shRNA targeting LC3B or BECN1. As LC3 is widely used as a marker protein for assessing autophagosome formation,Citation56,57 and BECN1 is one of the upstream molecules for recruiting other autophagy proteins to initiate the autophagic pathway,Citation58,59 we chose to use BECN1 and LC3B shRNAs for examining whether knockdown of autophagy genes alters the cell death pathway in CSFV-infected cells. We found that induction of autophagy with rapamycin enhanced cell viability and reduced cell death in response to CSFV infection (). We also found that inhibition of autophagy with BECN1 and LC3 shRNAs increased the cell death rate of CSFV-infected cells (). Furthermore, the results of Z-VAD treatment showed that inhibition of the CASP activity with Z-VAD effectively suppressed cell death in autophagy-impaired cells infected with CSFV (). Further detection of CASP-mediated pathway activation confirmed that intrinsic and extrinsic apoptosis were both triggered by CSFV in autophagy knockdown cells (). These data indicate that CSFV subverts autophagy to promote cell proliferation and inhibit apoptosis in infected cells. In this study, we also found that UV-inactivated CSFV lost its capability of triggering the functional autophagic pathway (), suggesting that CSFV replication is the key prerequisite for autophagy induction and subsequent apoptosis inhibition.
Autophagy and apoptosis after viral infection are widely considered to be the essential mechanisms of host defense; however, autophagy is required for CSFV replication,Citation22 and CSFV infection could inhibit cell death and CPE.Citation35 Therefore, CSFV may subvert both 2 mechanisms of host defense for their own survival benefit. In this study, we provide the first strong evidence that CSFV-induced autophagy limits apoptosis, which is beneficial to virus persistent survival in host cells. Similar results were obtained with other virus infections, including influenza A virus, hepatitis C virus, and chikungunya virus, where autophagy protein deficiency induces apoptosis, suggesting that autophagy favors survival of infected cells for contributing to virus propagation.Citation60-62
Apoptosis is an important mechanism of host defense during virus infection.Citation10 Previous studies reported that in vitro infection with CSFV prevent cell death by interfering with IFN production, which is associated with the inhibition effect of the viral Npro protein on IRF3, a key inducer of type I IFN expression.Citation63,64 Previously, type I IFN was shown to regulate apoptosis by regulating FASLG-FAS signaling and the TNFSF10 pathway.Citation65-68 However, the role for autophagy in type I IFN production has not previously been reported in CSFV infection. It is possible that the autophagic machinery may inhibit apoptosis by suppressing type I IFN production. To this end, we have documented an increase in the mRNA level of IFNA, IFNB1, TNFSF10, TNFRSF10A, FASLG and FAS in CSFV-infected autophagy-impaired cells (), and we also found that downregulating production of type I IFN reduced the mRNA levels of TNFSF10, TNFRSF10A, FASLG and FAS in CSFV-infected autophagy-impaired cells and therefore led to a decrease in activity of the CASP8-mediated apoptosis pathway reflecting that CSFV-induced autophagy suppresses type I IFN production and then inhibits the expression of apoptosis-related ISGs (). These results were consistent with those of other studies, in which the lack of autophagy genes in certain cells can result in enhanced production of type I IFN.Citation42,61 Importantly, TNFSF10-TNFRSF10A and FASLG-FAS, the well-known apoptosis-related ISGs, are both involved in death receptor-mediated apoptosis pathways in CSFV infection in vivo.Citation38 Meanwhile, TNF-TNFRSF1A, another key member of death receptor-mediated apoptosis pathways, was also detected in this study; however, TNF production had no obvious changes in the same cultured cells (), consistent with the Nec-1 treatment data shown in and . These findings suggest that CSFV-induced autophagy inhibits cell death by suppressing type I IFN-mediated apoptosis.
In addition to the death receptor-mediated apoptotic pathway, BCL2 family members are also involved in apoptosis as an extrinsic pathway. BCL2 and BAX are 2 key factors that regulate apoptosis.Citation69 BCL2 is an antiapoptotic factor that is fully capable of promoting cell survival, whereas BAX is an antagonist of BCL2 that initiates apoptosis in cells.Citation69,70 Importantly, both BCL2-BAX-mediated apoptosis and receptor-mediated apoptotic pathways can result in the activation of CASP3, leading to apoptotic cell death.Citation8 It is worth noting that the expression of BCL2 and BAX changed in autophagy-impaired cells under CSFV infection; however, there was no significant difference between shBECN1- and shLC3B-treated cells; these changes were even more obvious in shLC3B-treated cells under CSFV infection. Thus, the effect of BECN1 on apoptosis regulation by interacting with both BCL2 and BCL2L1 is not the main mechanism in CSFV infection.
Type I IFN production is regulated by some receptor proteins, which directly recognize virus RNA in virus-infected cells, such as toll like receptors and RLRs.Citation71-73 In this study, we examined the effect of autophagy on RLR signaling during CSFV infection. We demonstrated that the expression of DDX58, IFIH1 and MAVS increased in autophagy knockdown cells (). We further found that inhibition of RLR signaling significantly reduced the mRNA level of type I IFN in autophagy-impaired cells (). Together, these findings suggest that CSFV-induced autophagy may limit type I IFN production by inhibiting RLR signaling. Importantly, our study provides a novel strategy by which autophagy limits apoptotic cell death, thus contributing to CSFV infection.
Prior study demonstrates that increased ROS production is required for RLR signaling enhancement.Citation42 Regarding the function of ROS, autophagy induction is usually described to be closely linked to oxidative stress. ROS production is also limited by autophagy, although virus infection and replication increase ROS production and maintain it at a certain level.Citation42 We chose to use the regulators NAC and rotenone in examining the function of ROS on enhanced RLR signaling in autophagy-impaired cells subjected to CSFV infection. Our results showed that RLR signaling was reduced or enhanced after NAC or rotenone treatment, respectively, in cultured cells (). Importantly, the higher ROS level and MAVS expression are related to the increased mitochondria in autophagy knockdown cells infected with CSFV (). These findings reflect that ROS accumulation is required for RLR signaling amplification in autophagy-impaired cells after infection with CSFV. Furthermore, our results showed that regulation of ROS level altered the activity of CASP and the level of cleaved-PARP, therefore affecting progeny virus production in cultured cells, indicating that CSFV-induced autophagy suppresses apoptosis by downregulating ROS levels in host cells ().
Collectively, our study points out that autophagy is a critical process for homeostasis maintenance in cells by clearing redundant components, especially damaged and excess mitochondria under CSFV infection and therefore regulating the levels of MAVS, which is a key adaptor in RLR signaling. It is worth noting that this connection results in the accumulation of excess mitochondria and increased level of mitochondrial-associated ROS in autophagy-impaired cells under CSFV infection, which plays a key role in RLR signaling enhancement. More importantly, the amplified RLR signaling induces the activation of the extrinsic apoptosis pathway by type I IFN regulation, which has not previously been reported. Our results suggest that ROS is a positive regulator of RLR; however, a critical unanswered question is, what are the possible targets of ROS, which could play a key role in RLR signaling enhancement? And the precise interaction targets have not yet been reported so far. It is conceivable that the possible targets are related to the mitochondria, because the location of RLR signaling amplification is associated with the mitochondria. We also speculate that there may be some key proteins involved in ROS function on RLR signaling activation or amplification. It will be important to identify the precise molecular mechanism by which ROS enhances the RLR antiviral signaling pathway.
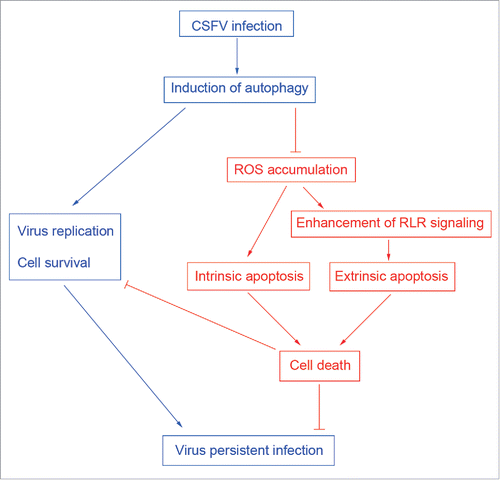
In conclusion, our in vitro study demonstrates for the first time that CSFV-induced autophagy not only induces virus replication but also inhibits the apoptosis pathway by limiting ROS-dependent RLR signaling, and therefore leads to virus persistent infection (). These findings suggest that CSFV-induced autophagy is a potential immune escape mechanism for virus persistent infection. However, the interaction of CSFV with the autophagy machinery still needs to be characterized in the future, which is important for further investigating the pathogenesis of CSFV and controlling viral infection.
Figure 12. Role of autophagy as a potential immune escape mechanism for CSFV persistent infection.
Materials and methods
Antibodies and chemicals
Anti-LC3B antibody (2775) and rapamycin (9904) were purchased from Cell Signaling Technology. Anti-DDX58/RIG-I antibody (IMG-6705A) was procured from Imgenex. NAC (V900429), rotenone (R8875), and anti-IFIH1/MDA5 antibody (SAB2101127) were obtained from Sigma-Aldrich. Anti-MAVS antibody (A300-782A) was procured from Bethyl. The following antibodies were purchased from Beyotime: ACTB (AA128), CASP8 (AC056), CASP9 (AC062), BCL2 (AB112-1), and PARP (AP102-1), as well as horseradish peroxidase (HRP)-labeled goat anti-rabbit (A0208) and anti-mouse (A0216) secondary antibodies, and Alexa Fluor 488-conjugated goat anti-rabbit secondary antibodies (A0423). Anti-BECN1 (NB110-87318) antibody was purchased from Novus Biologicals. Anti-CASP3 (BS7004) and anti-BAX (BS2538) antibodies were obtained from Bioworld Technology. Z-VAD (C1202) was purchased from Beyotime. Nec-1 (S8037) and Bufexamac (S3023) were obtained from Selleckchem. Mouse anti-CSFV E2 antibodies were prepared in our laboratory.
Cell culture
The swine kidney cell line PK-15 (ATCC, CCL-33) and porcine macrophage cell line 3D4/2 (ATCC, CRL-2845) were used in this study. PK-15 cells were grown in complete Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum and 1% antibiotics. 3D4/2 cells were maintained in complete RPMI 1640 medium (Thermo Fisher Scientific, 11875093) containing 10% FBS and 1% antibiotics. Then the cells were cultured at 37°C in a 5% CO2 incubator.
Viral infection
The CSFV strain (Shimen) was used in the study. The virus titers were determined by 50% tissue culture infectious doses assay on PK-15 and 3D4/2 cells. The multiplicity of infection (MOI) was confirmed according to the virus titer from the respective cell line. For comparison of viral replication in cells, virus infections were performed at an MOI of 0.5. For other experiments, viral stocks were prepared at an MOI of 1. PK-15 and 3D4/2 cells were infected at varying MOIs according to the requirements of different experiments. The mock was infected with phosphate-buffered saline (PBS; Thermo Fisher Scientific, 10010023). After 1 h, the inoculum was removed by aspiration. The cells were then washed with PBS and cultured in complete medium at 37°C for various times until harvesting. UV-CSFV was obtained by irradiating with UV light for 30 min at room temperature.
CCK-8 assay
Cell viability was determined using Cell Counting Kit-8 (CCK-8; Beyotime, C0038 ) according to the manufacturer's protocol. Briefly, approximately 1 × 104 cells (PK-15 or 3D4/2) per well were seeded in 96-well culture plates and cultured for 24 h at 37°C in a CO2 incubator. Two cell lines were treated according to experimental needs. Cells were then cultured with 100 μl of fresh medium supplemented with 10 μl of CCK-8 solution, and the plates were further incubated for 4 h at 37°C. Subsequently, the optical density was measured at 450 nm using a microplate reader (Bio-Rad, USA).
shRNA transfection
PK-15 and 3D4/2 cells grown to 60% confluence in 6-well cell culture plates were transfected with the targeted shRNA using X-tremeGENE HP DNA transfection reagent (Roche, 06366236001) according to the manufacturer's instructions. At 48 h after shRNA transfection, cells were infected with CSFV as described above. The protein knockdown was assessed by western blot analysis. The specific shRNAs for targeted genes and the scrambled shRNA were designed by and obtained from Cyagen. The shRNA specific sequences are described in .
Table 1. shRNA sequences of targeted genes.
Flow cytometry assay
For analysis of cell death, cultured cells were collected, washed with PBS and analyzed on a FACS Canto II flow cytometer (BD, USA) by using a Live-Dead Cell Staining Kit (BioVision, K501-100) according to the manufacturer's instruction. For detection of ROS levels, cells were stained with MitoSOX (Thermo Fisher Scientific, M36008) at 1 M for 30 min at 37°C, and then were washed and collected in PBS for FACS analysis.
CASP activity assay
The activation of CASP3, CASP8 and CASP9 in cells were measured using the CASP3/CPP32 Fluorometric Assay Kit (BioVision, K105-100), CASP8/FLICE Fluorometric Assay Kit (BioVision, K112-100), and CASP9 Fluorometric Assay Kit (BioVision, K118-100) following the manufacturer's instructions, respectively. The samples were assayed using a fluorescence microplate reader (Molecular Devices, USA).
Confocal immunofluorescence microscopy
Cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min, and then were treated with 0.2% Triton X-100 (Sigma-Aldrich, T8787) for 10 min. The cells were blocked in PBS containing 5% bovine serum albumin (BSA; Beyotime, ST023) for 30 min. Next, cells were incubated for 1 h in the presence of primary antibodies at 37°C, followed by 1 h of incubation in relevant secondary antibodies conjugated to Alexa Fluor 488. The fluorescence signals were visualized with a TCS SP2 confocal fluorescence microscope (Leica).
TUNEL staining
TUNEL staining was performed using a TUNEL Apoptosis Assay Kit (Beyotime, C1088). For measurement of apoptosis, samples were prepared according to the manufacturer's manual. Briefly, cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min, and then were treated with 0.1 % Triton X-100 for 5 min. Followed by incubation with TdT enzyme solution for 1 h at 37°C. TUNEL-positive cells were imaged with a EVOs fluorescence microscope (AMG). The cells with green fluorescence were considered as apoptotic cells.
Quantification of RNA
Real-time quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was used in the study. For measurement of targeted gene expression, total RNA was isolated using RNAiso Plus (TAKARA, 9108) according to the manufacturer's protocol. Quantitative RT-PCR was performed using the One Step SYBR® PrimeScript™ RT-PCR Kit II (TAKARA, RR086)on an iQ5 iCycler detection system (Bio-Rad, USA). Relative changes in mRNA levels of genes were assessed using the 2−ΔΔCT method and normalized to the housekeeping gene GAPDH. The primers used are indicated in .
Table 2. Primers used in this study.
For virus copy detection, viral RNA was extracted using MiniBEST Viral RNA/DNA Extraction Kit Ver.5.0 (TAKARA, 9766) and reverse-transcribed using PrimeScript®RT Master Mix (Perfect Real Time; TAKARA, RR036A) following the manufacturer's directions. The resulting cDNA was then amplified using SYBR® Premix Ex Taq™ (Tli RNaseH Plus; TAKARA, RR420B) and an iQ5 iCycler detection system (Bio-Rad, USA). The primers (CSFV1: CCTGAGGACCAAACACATGTTG, CSFV2: TGGTGGAAGTTGGTTGTGTCTG), targeting a region corresponding to the CSFV NS5B gene, were used in the study. The recombinant plasmid containing the CSFV NS5B gene was used to construct a reference curve for calculating the viral copy number.
ELISA
Levels of IFNA, IFNB1 and TNF in the supernatant were detected using ELISA kits (USCNK, SEA033Po, SEA222Po and SEA133Po) following the manufacturer's instructions. The samples were quantified using a microplate reader (Bio-Rad, USA).
Mitochondrial DNA quantification
Total DNA was isolated using MiniBEST Universal Genomic DNA Extraction Kit Ver.5.0 (TAKARA, 9765) following the manufacturer's direction, and then amplified using SYBR® Premix Ex Taq™ for quantification with an iQ5 iCycler detection system. The primers (F: AGACCGCAACCTGAACACAA, R: TCCGAATCCTGGTAAGATGAGA) were used in the study, which targeted to a region from the mitochondrial MT-CO1 gene. The recombinant plasmid with the MT-CO1 gene was used as a standard sample for calculating the copy number of the mitochondrial DNA in different samples.
Western blot analysis
After treatment, cells were washed with cold PBS and incubated on ice with RIPA lysis buffer (Beyotime, P0013B) supplemented with 1 mM PMSF (Beyotime, ST506) for 10 min. Cell lysates were then clarified by centrifugation at 14,500 g for 20 min at 4°C. The protein concentration was determined using a BCA protein assay kit (Beyotime, P0012). Equal amounts of protein samples were diluted in 5 × SDS-PAGE loading buffer and boiled for 5 min. Proteins (20 μg) were separated by SDS-PAGE and transferred onto polyvinylidene fluoride membranes (Beyotime, FFP30). After blocking with PBS containing 2% nonfat milk powder and 0.05% Tween 20 (Sigma-Aldrich, P2287) for 2 h at 25°C, the membrane was incubated with specific primary antibodies overnight at 4°C and then with the corresponding HRP-conjugated secondary antibodies at 37°C for 2 h at appropriate dilutions. The protein bands were visualized using an ECL Plus kit (Beyotime, P0018). Images of protein blots were obtained from a CanoScan LiDE 100 scanner (Canon, Japan).
Statistical analysis
The data are expressed as the mean ± standard deviation (SD) and were analyzed by one-way ANOVA using the SPSS software (version 17.0). A p value of < 0.05 was considered statistically significant.
Abbreviations
| BSA | = | bovine serum albumin |
| CASP/caspase | = | apoptosis-related cysteine peptidase |
| CPE | = | cytopathic effect |
| CSFV | = | classical swine fever virus |
| DDX58/RIG-I | = | DEXD/H-box helicase 58 |
| FBS | = | fetal bovine serum |
| HRP | = | horseradish peroxidase |
| IFIH1/MDA5 | = | interferon induced with helicase C domain 1 |
| IFN | = | interferon |
| ISGs | = | IFN-stimulated genes |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MOI | = | multiplicity of infection |
| PBS | = | phosphate-buffered saline |
| qRT-PCR | = | real-time quantitative reverse transcriptase polymerase chain reaction |
| RLRs | = | RIG-I-like receptors |
| ROS | = | reactive oxygen species |
| shRNA | = | short hairpin RNA |
| UV | = | ultraviolet |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. U1405216, 31172321 and 31472200), the Special Project for Science and Technology projects of Guangdong, China (Nos. 2015A050502044 and 2015B020230009), and the Special Fund for Agro-Scientific Research in the Public Interest (Nos. 201203056 and 201203082).
References
- Becher P, Avalos RR, Orlich M, Cedillo RS, König M, Schweizer M, Stalder H, Schirrmeier H, Thiel HJ. Genetic and antigenic characterization of novel pestivirus genotypes: implications for classification. Virology 2003; 311:96-104; PMID:12832207; http://dx.doi.org/10.1016/S0042-6822(03)00192-2
- Lohse L, Nielsen J, Uttenthal A. Early pathogenesis of classical swine fever virus (CSFV) strains in Danish pigs. Vet Microbiol 2012; 159:327-36; PMID:22608103; http://dx.doi.org/10.1016/j.vetmic.2012.04.026
- Kleiboeker SB. Swine fever: classical swine fever and African swine fever. Vet Clin North Am Food Anim Pract 2002; 18:431-51; PMID:12442576; http://dx.doi.org/10.1016/S0749-0720(02)00028-2
- Bensaude E, Turner JL, Wakeley PR, Sweetman DA, Pardieu C, Drew TW, Wileman T, Powell PP. Classical swine fever virus induces proinflammatory cytokines and tissue factor expression and inhibits apoptosis and interferon synthesis during the establishment of long-term infection of porcine vascular endothelial cells. J Gen Virol 2004; 85:1029-37; PMID:15039545; http://dx.doi.org/10.1099/vir.0.19637-0
- Knoetig SM, Summerfield A, Spagnuolo-Weaver M, McCullough KC. Immunopathogenesis of classical swine fever: role of monocytic cells. Immunology 1999; 97:359-66; PMID:10447754; http://dx.doi.org/10.1046/j.1365-2567.1999.00775.x
- Tautz N, Meyers G, Thiel HJ. Pathogenesis of mucosal disease, a deadly disease of cattle caused by a pestivirus. Clin Diagn Virol 1998; 10:121-7; PMID:9741637; http://dx.doi.org/10.1016/S0928-0197(98)00037-3
- Ressang AA. Studies on the pathogenesis of hog cholera. II. Virus distribution in tissue and the morphology of the immune response. Zentralbl Veterinarmed B 1973; 20:272-88; PMID:4751662; http://dx.doi.org/10.1111/j.1439-0450.1973.tb01127.x
- Kurokawa M, Kornbluth S. Caspases and kinases in a death grip. Cell 2009; 138:838-54; PMID:19737514; http://dx.doi.org/10.1016/j.cell.2009.08.021
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell 2004; 116:205-19; PMID:14744432; http://dx.doi.org/10.1016/S0092-8674(04)00046-7
- Everett H, McFadden G. Apoptosis: an innate immune response to virus infection. Trends Microbiol 1999; 7:160-5; PMID:10217831; http://dx.doi.org/10.1016/S0966-842X(99)01487-0
- Reggiori F. One. Membrane origin for autophagy. Curr Top Dev Biol 2006; 74:1-30; PMID:16860663; http://dx.doi.org/10.1016/S0070-2153(06)74001-7
- Todde V, Veenhuis M, van der Klei IJ. Autophagy: principles and significance in health and disease. Biochim Biophys Acta 2009; 1792:3-13; PMID:19022377; http://dx.doi.org/10.1016/j.bbadis.2008.10.016
- Alirezaei M, Kiosses WB, Flynn CT, Brady NR, Fox HS. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS One 2008; 3:e2906; PMID:18682838; http://dx.doi.org/10.1371/journal.pone.0002906
- Chen N, Karantza-Wadsworth V. Role and regulation of autophagy in cancer. Biochim Biophys Acta 2009; 1793:1516-23; PMID:19167434; http://dx.doi.org/10.1016/j.bbamcr.2008.12.013
- Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009; 5:527-49; PMID:19527881; http://dx.doi.org/10.1016/j.chom.2009.05.016
- Schmid D, Münz C. Innate and adaptive immunity through autophagy. Immunity 2007; 27:11-21; PMID:17663981; http://dx.doi.org/10.1016/j.immuni.2007.07.004
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat Rev Immunol 2007; 7:767-77; PMID:17767194; http://dx.doi.org/10.1038/nri2161
- Lee YR, Lei HY, Liu MT, Wang JR, Chen SH, Jiang-Shieh YF, Lin YS, Yeh TM, Liu CC, Liu HS. Autophagic machinery activated by dengue virus enhances virus replication. Virology 2008; 374:240-8; PMID:18353420; http://dx.doi.org/10.1016/j.virol.2008.02.016
- Wong J, Zhang J, Si X, Gao G, Mao I, McManus BM, Luo H. Autophagosome supports coxsackievirus B3 replication in host cells. J Virol 2008; 82:9143-53; PMID:18596087; http://dx.doi.org/10.1128/JVI.00641-08
- Zhou Z, Jiang X, Liu D, Fan Z, Hu X, Yan J, Wang M, Gao GF. Autophagy is involved in influenza A virus replication. Autophagy 2009; 5:321-8; PMID:19066474; http://dx.doi.org/10.4161/auto.5.3.7406
- Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A 2009; 106:14046-51; PMID:19666601; http://dx.doi.org/10.1073/pnas.0907344106
- Pei J, Zhao M, Ye Z, Gou H, Wang J, Yi L, Dong X, Liu W, Luo Y, Liao M, et al. Autophagy enhances the replication of classical swine fever virus in vitro. Autophagy 2014; 10:93-110; PMID:24262968; http://dx.doi.org/10.4161/auto.26843
- Zalckvar E, Yosef N, Reef S, Ber Y, Rubinstein AD, Mor I, Sharan R, Ruppin E, Kimchi A. A systems level strategy for analyzing the cell death network: implication in exploring the apoptosis/autophagy connection. Cell Death Differ 2010; 17:1244-53; PMID:20150916; http://dx.doi.org/10.1038/cdd.2010.7
- Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy 2008; 4:600-6; PMID:18497563; http://dx.doi.org/10.4161/auto.6260
- Boya P, Gonzalez-Polo RA, Casares N, Perfettini JL, Dessen P, Larochette N, Métivier D, Meley D, Souquere S, Yoshimori T, et al. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 2005; 25:1025-40; PMID:15657430; http://dx.doi.org/10.1128/MCB.25.3.1025-1040.2005
- Eisenberg-Lerner A, Bialik S, Simon HU, Kimchi A. Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ 2009; 16:966-75; PMID:19325568; http://dx.doi.org/10.1038/cdd.2009.33
- Summerfield A, Knötig SM, McCullough KC. Lymphocyte apoptosis during classical swine fever: implication of activation-induced cell death. J Virol 1998; 72:1853-61; PMID:9499036
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991; 253:905-9; PMID:1715094; http://dx.doi.org/10.1126/science.1715094
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC. Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and α-synuclein. J Biol Chem 2007; 282:5641-52; PMID:17182613; http://dx.doi.org/10.1074/jbc.M609532200
- Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis 2008; 13:1-9; PMID:17990121; http://dx.doi.org/10.1007/s10495-007-0154-9
- Khan SZ, Hand N, Zeichner SL. Apoptosis-induced activation of HIV-1 in latently infected cell lines. Retrovirology 2015; 12:42; PMID:25980942; http://dx.doi.org/10.1186/s12977-015-0169-1
- Chen Y, Peng GF, Han XZ, Wang W, Zhang GQ, Li X. Apoptosis prediction via inhibition of AKT signaling pathway by neogrifolin. Int J Clin Exp Pathol 2015; 8:1154-64; PMID:25973001
- Wu JR, Wang J, Zhou SK, Yang L, Yin JL, Cao JP, Cheng YB. Necrostatin-1 protection of dopaminergic neurons. Neural Regen Res 2015; 10:1120-4; PMID:26330837; http://dx.doi.org/10.4103/1673-5374.160108
- Chang YJ, Hsu SL, Liu YT, Lin YH, Lin MH, Huang SJ, Ho JA, Wu LC. Gallic acid induces necroptosis via TNF-α signaling pathway in activated hepatic stellate cells. PLoS One 2015; 10:e0120713; PMID:25816210; http://dx.doi.org/10.1371/journal.pone.0120713
- Ke PY, Chen SS. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J Clin Invest 2011; 121:37-56; PMID:21135505; http://dx.doi.org/10.1172/JCI41474
- Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, Ishii KJ, Kawai T, Akira S, Suzuki K, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A 2007; 104:14050-5; PMID:17709747; http://dx.doi.org/10.1073/pnas.0704014104
- Takeshita F, Kobiyama K, Miyawaki A, Jounai N, Okuda K. The non-canonical role of Atg family members as suppressors of innate antiviral immune signaling. Autophagy 2008; 4:67-9; PMID:17921696; http://dx.doi.org/10.4161/auto.5055
- Renson P, Blanchard Y, Le Dimna M, Felix H, Cariolet R, Jestin A, Le Potier MF. Acute induction of cell death-related IFN stimulated genes (ISG) differentiates highly from moderately virulent CSFV strains. Vet Res 2010; 41:7; PMID:19793538; http://dx.doi.org/10.1051/vetres/2009055
- Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon AM, Schlegl J, Abraham Y, Becher I, Bergamini G, et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat Biotechnol 2011; 29:255-65; PMID:21258344; http://dx.doi.org/10.1038/nbt.1759
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006; 441:101-5; PMID:16625202; http://dx.doi.org/10.1038/nature04734
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 2004; 5:730-7; PMID:15208624; http://dx.doi.org/10.1038/ni1087
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc Natl Acad Sci U S A 2009; 106:2770-5; PMID:19196953; http://dx.doi.org/10.1073/pnas.0807694106
- Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci 2003; 60:6-20; PMID:12613655; http://dx.doi.org/10.1007/s000180300001
- Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem 2003; 278:8516-25; PMID:12496265; http://dx.doi.org/10.1074/jbc.M210432200
- Crotzer VL, Blum JS. Autophagy and adaptive immunity. Immunology 2010; 131:9-17; PMID:20586810; http://dx.doi.org/10.1111/j.1365-2567.2010.03309.x
- Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science 2004; 306:990-5; PMID:15528435; http://dx.doi.org/10.1126/science.1099993
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov 2007; 6:304-12; PMID:17396135; http://dx.doi.org/10.1038/nrd2272
- Liu Y, Schiff M, Czymmek K, Tallóczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell 2005; 121:567-77; PMID:15907470; http://dx.doi.org/10.1016/j.cell.2005.03.007
- Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl-2-interacting protein. J Virol 1998; 72:8586-96; PMID:9765397
- Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006; 126:121-34; PMID:16839881; http://dx.doi.org/10.1016/j.cell.2006.05.034
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005; 122:927-39; PMID:16179260; http://dx.doi.org/10.1016/j.cell.2005.07.002
- Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, Simon HU. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol 2006; 8:1124-32; PMID:16998475; http://dx.doi.org/10.1038/ncb1482
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571-80; PMID:21311563; http://dx.doi.org/10.1038/cdd.2010.191
- Sun J, Jiang Y, Shi Z, Yan Y, Guo H, He F, Tu C. Proteomic alteration of PK-15 cells after infection by classical swine fever virus. J Proteome Res 2008; 7:5263-9; PMID:19367723; http://dx.doi.org/10.1021/pr800546m
- Grummer B, Fischer S, Depner K, Riebe R, Blome S, Greiser-Wilke I. Replication of classical swine fever virus strains and isolates in different porcine cell lines. Dtsch Tierarztl Wochenschr 2006; 113:138-42; PMID:16716048
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/10.4161/auto.5338
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy 2007; 3:181-206; PMID:17224625; http://dx.doi.org/10.4161/auto.3678
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672-6; PMID:10604474; http://dx.doi.org/10.1038/45257
- Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 2007; 17:839-49; PMID:17893711; http://dx.doi.org/10.1038/cr.2007.78
- Joubert PE, Werneke SW, de la Calle C, Guivel-Benhassine F, Giodini A, Peduto L, Levine B, Schwartz O, Lenschow DJ, Albert ML. Chikungunya virus-induced autophagy delays caspase-dependent cell death. J Exp Med 2012; 209:1029-47; PMID:22508836; http://dx.doi.org/10.1084/jem.20110996
- Shrivastava S, Raychoudhuri A, Steele R, Ray R, Ray RB. Knockdown of autophagy enhances the innate immune response in hepatitis C virus-infected hepatocytes. Hepatology 2011; 53:406-14; PMID:21274862; http://dx.doi.org/10.1002/hep.24073
- Gannagé M, Dormann D, Albrecht R, Dengjel J, Torossi T, Rämer PC, Lee M, Strowig T, Arrey F, Conenello G, et al. Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 2009; 6:367-80; http://dx.doi.org/10.1016/j.chom.2009.09.005
- Bauhofer O, Summerfield A, Sakoda Y, Tratschin JD, Hofmann MA, Ruggli N. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J Virol 2007; 81:3087-96; PMID:17215286; http://dx.doi.org/10.1128/JVI.02032-06
- Ruggli N, Bird BH, Liu L, Bauhofer O, Tratschin JD, Hofmann MA. N(pro) of classical swine fever virus is an antagonist of double-stranded RNA-mediated apoptosis and IFN-α/β induction. Virology 2005; 340:265-76; PMID:16043207; http://dx.doi.org/10.1016/j.virol.2005.06.033
- Chawla-Sarkar M, Leaman DW, Jacobs BS, Borden EC. IFN-β pretreatment sensitizes human melanoma cells to TRAIL/Apo2 ligand-induced apoptosis. J Immunol 2002; 169:847-55; PMID:12097388; http://dx.doi.org/10.4049/jimmunol.169.2.847
- Kaser A, Nagata S, Tilg H. Interferon α augments activation-induced T cell death by upregulation of Fas (CD95/APO-1) and Fas ligand expression. Cytokine 1999; 11:736-43; PMID:10525311; http://dx.doi.org/10.1006/cyto.1998.0484
- Shigeno M, Nakao K, Ichikawa T, Suzuki K, Kawakami A, Abiru S, Miyazoe S, Nakagawa Y, Ishikawa H, Hamasaki K, et al. Interferon-α sensitizes human hepatoma cells to TRAIL-induced apoptosis through DR5 upregulation and NF-kappa B inactivation. Oncogene 2003; 22:1653-62; PMID:12642868; http://dx.doi.org/10.1038/sj.onc.1206139
- Oshima K, Yanase N, Ibukiyama C, Yamashina A, Kayagaki N, Yagita H, Mizuguchi J. Involvement of TRAIL/TRAIL-R interaction in IFN-α-induced apoptosis of Daudi B lymphoma cells. Cytokine 2001; 14:193-201; PMID:11448118; http://dx.doi.org/10.1006/cyto.2001.0873
- Warzych E, Wrenzycki C, Peippo J, Lechniak D. Maturation medium supplements affect transcript level of apoptosis and cell survival related genes in bovine blastocysts produced in vitro. Mol Reprod Dev 2007; 74:280-9; PMID:16955406; http://dx.doi.org/10.1002/mrd.20610
- Flaws JA, Hirshfield AN, Hewitt JA, Babus JK, Furth PA. Effect of bcl-2 on the primordial follicle endowment in the mouse ovary. Biol Reprod 2001; 64:1153-9; PMID:11259262; http://dx.doi.org/10.1095/biolreprod64.4.1153
- Kato H, Takahasi K, Fujita T. RIG-I-like receptors: cytoplasmic sensors for non-self RNA. Immunol Rev 2011; 243:91-8; PMID:21884169; http://dx.doi.org/10.1111/j.1600-065X.2011.01052.x
- Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011; 34:637-50; PMID:21616434; http://dx.doi.org/10.1016/j.immuni.2011.05.006
- Honda K, Takaoka A, Taniguchi T. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 2006; 25:349-60; PMID:16979567; http://dx.doi.org/10.1016/j.immuni.2006.08.009