
ABSTRACT
Accumulating evidence suggests that mitogen-activated protein kinases (MAPKs) regulate macroautophagy/autophagy. However, the involvement of dual-specificity protein phosphatases (DUSPs), endogenous inhibitors for MAPKs, in autophagy remains to be determined. Here we report that DUSP1/MKP-1, the founding member of the DUSP family, plays a critical role in regulating autophagy. Specifically, we demonstrate that DUSP1 knockdown by shRNA in human ovarian cancer CAOV3 cells and knockout in murine embryonic fibroblasts, increases both basal and rapamycin-increased autophagic flux. Overexpression of DUSP1 had the opposite effect. Importantly, knockout of Dusp1 promoted phosphorylation of ULK1 at Ser555, and BECN1/Beclin 1 at Ser15, and the association of PIK3C3/VPS34, ATG14, BECN1 and MAPK, leading to the activation of the autophagosome-initiating class III phosphatidylinositol 3-kinase (PtdIns3K) complex. Furthermore, knockdown and pharmacological inhibitor studies indicated that DUSP1-mediated suppression of autophagy reflected inactivation of the MAPK1-MAPK3 members of the MAPK family. Knockdown of DUSP1 sensitized CAOV3 cells to rapamycin-induced antigrowth activity. Moreover, CAOV3-CR cells, a line that had acquired cisplatin resistance, exhibited an elevated DUSP1 level and were refractory to rapamycin-induced autophagy and cytostatic effects. Knockdown of DUSP1 in CAOV3-CR cells restored sensitivity to rapamycin. Collectively, this work identifies a previously unrecognized role for DUSP1 in regulating autophagy and suggests that suppression of DUSP1 may enhance the therapeutic activity of rapamycin.
Introduction
Autophagy is a highly conserved process by which cytoplasmic materials are encapsulated in double-membrane compartments, phagophores, that mature into autophagosomes, allowing the subsequent degradation of the cargo following fusion of the latter with lysosomes.Citation1 Autophagy is a constitutive process and plays a critical role in the degradation of long-lived proteins and aged/dysfunctional organelles. However, autophagy is also induced by nutrient and energy deficiency, and as a response to cellular damage induced by a variety of exogenous stressors.Citation1,2 As such, constitutive and inducible autophagy has critical roles in cellular housekeeping and maintenance of homeostasis. Although generally viewed as being a prosurvival process, there are numerous cases in which autophagy appears to directly mediate or contributes to cell death.Citation3,4 Extensive literature documents the prosurvival functions of autophagy, and the induction of autophagy in tumors during chemotherapy often provides a survival advantage,Citation5,6 but there are cases in which autophagy mediates the cytotoxic effects of stressors and therapeutics, and promoting autophagy causes cancer cell death or sensitizes cancer cells to other chemotherapeutics.Citation3,4 In addition, autophagy is increasingly recognized as a critical player in cancer development, but its role in cancer is complex. For example, mutations resulting in the loss or silencing of Becn1 or Atg5 are pro-tumorigenic, and can promote chromosome rearrangements and aneuploidy.Citation7-9 Similarly, autophagy occurring in cancer-associated stromal cells can promote the survival and growth of neighboring tumors.Citation10,11
Mammalian MAPKs mainly consist of MAPK8/JNK1, MAPK9/JNK2, MAPK10/JNK3, MAPK11/p38β, MAPK12/p38γ, MAPK13/p38δ, MAPK14/p38α, MAPK1/ERK2, and MAPK3/ERK1 (hereafter referred to primarily as MAPK/JNK, MAPK/p38 and MAPK/ERK, respectively),Citation12,13 each of which are activated by diverse stimuli. In response to stimuli, MAPKs are activated through the reversible phosphorylation of both threonine and tyrosine residues of the TXY motif in the catalytic domain by upstream dual-specificity kinases. These upstream kinases, named MAP kinase kinases (MKKs/MEKs), include MAP2K1/MKK1, MAP2K2/MKK2, MAP2K3/MKK3, MAP2K4/MKK4, MAP2K6/MKK6 and MAP2K7/MKK7, which are in turn activated by MAPK kinase kinases.Citation14-16 Activated MAPKs phosphorylate a number of substrates and subsequently modulate many signaling pathways and processes including autophagy.Citation15,17-19
Since phosphorylation is required for the activation of MAPKs, dephosphorylation by members of the DUSP (dual-specificity protein phosphatase) family plays a critical role in controlling MAPK signaling. The DUSP family contains 11 members, including DUSP1, DUSP2, DUSP4, DUSP5, DUSP6, DUSP7, DUSP8, DUSP9, DUSP10, DUSP16 and STYXL1.Citation20 DUSP1 is the founding member of the DUSP family and was originally identified as a growth factor and stress inducible gene.Citation21-24 DUSP1 is a dual-specificity protein phosphatase that dephosphorylates both the threonine and tyrosine residues on members of all 3 major MAPK subfamilies—MAPK/JNK, MAPK/p38, and MAPK/ERK.Citation25-27 DUSP1 is involved in the regulation of the cell cycle and apoptosis.Citation28-33 Importantly, DUSP1 is overexpressed in several cancers, including ovarian cancer.Citation34-36 DUSP1 inhibits the induction of cell death by several apoptotic stimuli.Citation31,33,Citation37,38 Furthermore, studies with lung and ovarian cancer cells demonstrate a clear correlation between increased DUSP1 expression and acquired chemoresistance.Citation33,38 Furthermore, studies have shown that DUSP1 can protect cells from chemotherapy-induced apoptosis.Citation37 However, it is not known whether DUSP1 plays a role in autophagy.
In this study we investigated the effects of DUSP1 knockdown and overexpression on basal and rapamycin-induced autophagy in 3 different cellular models. In all 3 models both basal and inducible autophagic activities were inversely related to DUSP1 level. The effects of DUSP1 were primarily due to its inactivation of MAPK/ERK, which positively regulated autophagy. We also evaluated the therapeutic use of rapamycin to treat human ovarian cancer cells. Rapamycin significantly reduced proliferation via a mechanism that was dependent upon the induction of autophagy. However, sensitivity to rapamycin was significantly compromised in variant ovarian cancer cells that were unable to mount an autophagic response due to the upregulated expression of DUSP1.
Results
Knockdown or knockout of DUSP1 leads to an increase in MAP1LC3-II/LC3-II levels whereas its overexpression has an opposite effect
MAP1LC3/LC3 is a cytosolic protein that is incorporated into the membranes of phagophores. Prior to its incorporation, LC3-I is converted into LC3-II by a post-translational modification in which phosphatidylethanolamine is covalently attached. Cellular LC3-II content is commonly used as a marker of autophagy and as an index of autophagosome formation.Citation2 CAOV3 ovarian cancer cells exhibited basal expression of LC3-II (). Knockdown of DUSP1 in CAOV3 cells increased LC3-II levels (). In order to determine if the observed result was cell-line specific, we analyzed LC3-II levels in 2 additional model systems. Like DUSP1 knockdown CAOV3 cells, LC3-II levels were greater in dusp1−/− mouse embryonic fibroblasts (MEFs) derived from Dusp1 knockout mice, relative to MEFs derived from wild-type mice (). Conversely, DUSP1-overexpressing human ovarian cancer TOV112D cells exhibited lower LC3-II levels than empty vector control cells ().
Figure 1. Correlations between DUSP1, LC3-II and SQSTM1 levels. Western blot analyses of DUSP1, LC3-I, LC3-II, ACTB or SQSTM1 (upper panel) and quantification of LC3-II (lower panel) in (A and D) CAOV3 cells stably transfected with DUSP1 shRNA (sh-DUSP1) or nontarget control shRNA (Nontarget), (B, E and G) Dusp1+/+ and dusp1−/− MEFs, and (C and F) TOV112D cells stably transfected with DUSP1 cDNA or empty vector. Cells (D-F) were left untreated or treated with 5 µM rapamycin (Rap) for 20 h prior to being harvested for western blot analyses. Cells (G) were left untreated or treated with 20 μM cisplatin for 24 h prior to being harvested for western blot analysis. Data represent mean±SD of 3 independent experiments. *, P < 0.01 and **, P < 0.005, statistically significant.
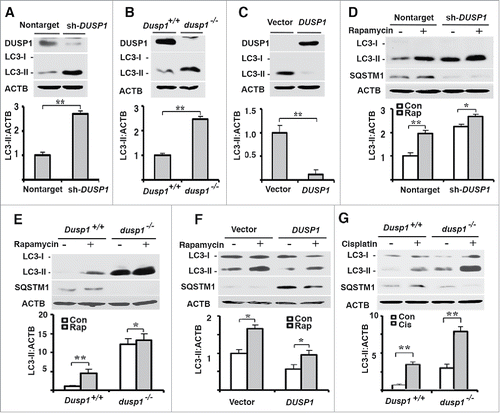
To further confirm the role of DUSP1 in regulating LC3-II levels, we treated nontarget shRNA control and DUSP1 knockdown CAOV3 cells with rapamycin, a well-characterized inducer of autophagy.Citation2,39 shows that both basal and rapamycin-induced LC3-II levels were higher in DUSP1 knockdown cells. Furthermore, SQSTM1/p62 levels were decreased in DUSP1 knockdown cells (). This is significant because SQSTM1 is involved in autophagic degradation of protein aggregates and damaged mitochondria, and reductions in SQSTM1, coupled with increased LC3-II levels, are indicative of functional autophagy.Citation2 Similar results were obtained in dusp1−/− MEFs (), and opposite results were obtained in DUSP1-overexpressing TOV112D cells (). Importantly, an inverse relationship between functional autophagy and DUSP1 level was also observed in Dusp1+/+ and dusp1−/− MEFs following treatment with cisplatin (), another inducer of autophagy.Citation40 Taken together, these data suggest that DUSP1 may negatively regulate autophagy.
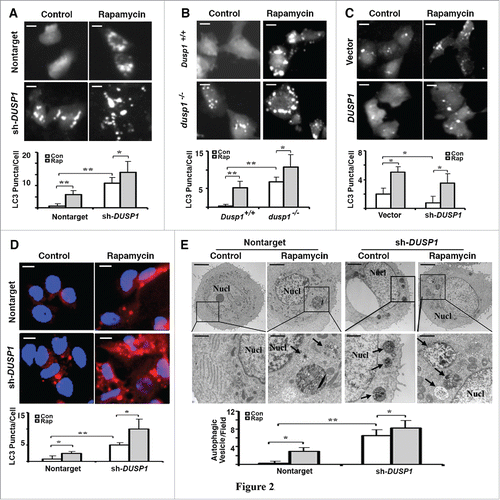
To further define the role of DUSP1 in regulating autophagy, we used multiple approaches to monitor autophagosome development/accumulation. The first approach employed fluorescence microscopy to assess the incorporation of stably expressed GFP-LC3 into fluorescent punctate structures (). Fluorescent GFP-LC3 puncta were relatively absent in untreated nontarget shRNA CAOV3 cells () and wild-type Dusp1+/+ MEFs (), but apparent in rapamycin-treated cells. In contrast, fluorescent GFP-LC3 puncta were easily observed in untreated DUSP1 knockdown CAOV3 () and dusp1−/− MEF () cells, and increased in number following rapamycin treatment (). This indicates that knockdown of DUSP1 increases autophagosome accumulation. Conversely, GFP-LC3 fluorescence was primarily punctate in nontreated vector control TOV112D cells, but mostly diffuse and nonpunctate in DUSP1-overexpressing TOV112D cells (). Although rapamycin treatment enhanced the conversion of GFP-LC3 into fluorescent puncta in DUSP1-overexpressing TOV112D cells, there was still considerable diffuse cytoplasmic fluorescence (). In addition, we used antibodies to LC3 and indirect immunofluorescence to monitor endogenous LC3 incorporation into puncta in CAOV3 cells stably expressing nontarget control shRNA or DUSP1 shRNA (). Untreated nontarget shRNA CAOV3 cells exhibited few LC3 puncta, which increased after treatment with rapamycin (). In contrast, endogenous LC3 puncta were easily observed in DUSP1 knockdown cells in the absence of rapamycin treatment, and markedly increased following rapamycin treatment (). Finally, we used transmission electron microscopy to determine if organelles consistent with autophagosomes could be identified, and if their abundance correlated with our proxy (i.e., LC3-II, GFP-LC3 puncta) measurements of autophagy. Vesicles with double membranes, a structural feature unique to autophagosomes, were easily observed and abundant in both nontreated and rapamycin-treated DUSP1 knockdown CAOV3 cells (). In contrast, autophagic vesicles were markedly fewer in nontarget shRNA-expressing CAOV3 cells (). Taken together, these data strongly suggest that DUSP1 negatively regulates both constitutive and inducible autophagy.
Figure 2. Inverse relationship between DUSP1 expression and autophagic vesicle formation. (A-C) Representative fluorescence images (upper panels) and quantification of GFP-LC3 puncta (lower panels) in (A) CAOV3 cells with DUSP1 shRNA (sh-DUSP1) or nontarget shRNA, (B) Dusp1+/+ and dusp1−/− MEFs and (C) TOV112D cells stably transfected with DUSP1 cDNA or empty vector. Cells were left untreated or treated with 5 µM rapamycin for 20 h prior to image capture. Bright puncta denote autophagosomal structures. (D) Representative indirect immunofluorescent images of endogenous LC3. DUSP1 shRNA or nontarget shRNA CAOV3 cells were left untreated or treated with 5µM rapamycin for 20 h prior to being processed for immunofluorescent detection of LC3. Red puncta denote autophagic vesicle structures (upper panel) and quantification of LC3 puncta (lower panel). Data represent mean ± SD of 3 independent experiments, in each of which 20 or more cells were counted. (E) Representative electron micrographs (upper panel) and quantification of autophagic vesicles (lower panel). DUSP1 shRNA or nontarget shRNA CAOV3 cells were left untreated or treated with rapamycin (5 µM, 20 h). Arrows denote autophagic vesicles. Nucl, nucleus. Scale bars: (A-D) 10 μm; (E) 1.7 µm (upper panels), 600 nm (lower panels). *, P < 0.01 and **, P < 0.005, statistically significant.
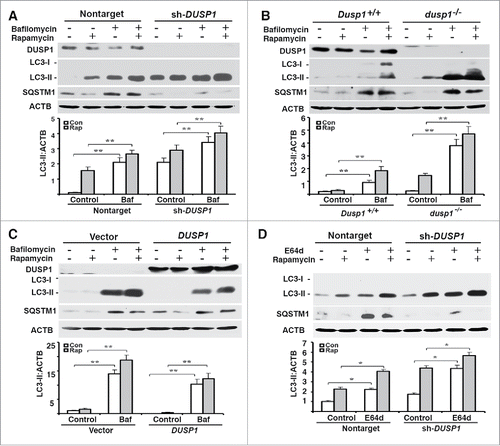
The cellular LC3-II level reflects not only the rate of autophagosome formation, but also the efficiencies of autophagosome fusion with lysosomes, and subsequent lysosomal degradation of LC3-II.Citation2 In order to determine if the observed inverse relationship between LC3-II and DUSP1 levels reflected effects on autophagosome maturation, we treated cells with bafilomycin A1 (BafA1), which inhibits both autophagosome-lysosome fusion and lysosome-mediated degradation of fused autophagosomes and their cargo.Citation41 If LC3-II levels are similar in BafA1-treated and nontreated cells, such results are generally interpreted as a block in autophagosome maturation/degradation and an inhibition of autophagic flux. Conversely, normal autophagic flux is assumed if BafA1 treatment increases LC3-II levels. Furthermore, if BafA1-treatment enhances the accumulation of LC3-II above what is observed following some treatment/manipulation, it is assumed that the increased LC3-II accumulation occurring as a consequence of the treatment/manipulation reflects increased autophagosome formation. Treatment of CAOV3 (), MEF () and TOV112D () cells with BafA1 increased both basal and rapamycin-induced LC3-II levels, as well as basal SQSTM1 levels. These effects of BafA1 were independent of DUSP1 level (). However, greater accumulations of LC3-II always occurred in DUSP1-deficient cells (). Similar results were obtained in CAOV3 cells treated with the lysosomal protease inhibitor E64d (). Collectively, these data suggest that autophagosome formation and flux are increased in DUSP1-deficient cells.
Figure 3. Effects of DUSP1 on autophagic flux. (A-D) Western blot analyses of DUSP1, LC3, SQSTM1 and ACTB (upper panel) and quantification of LC3 (lower panel). (A and D) DUSP1 shRNA (sh-DUSP1) or nontarget shRNA CAOV3 cells. (B) Dusp1+/+ and dusp1−/− MEFs. (C) Stable DUSP1 overexpression and empty vector control TOV112D cells. Cells were left untreated, or treated with 5 µM rapamycin and/or 10 nM bafilomycin A1 (A-C) or 10 μg/ml E64d (D) for 20 h before being harvested for western blot analyses. Data represent mean±SD of 3 independent experiments. *, P < 0.01 and **, P < 0.005, statistically significant.
DUSP1 inhibits MAPK/ERK-mediated autophagy
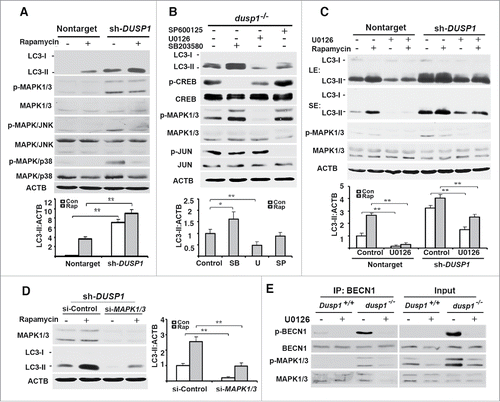
Given that phospho-MAPKs are the only known substrates of DUSP1,Citation35,42 it seemed logical that the effects of DUSP1 deficiency had to be related to the activities of one or more of these MAPKs. shows that DUSP1-deficient CAOV3 cells, relative to cells transfected with nontarget shRNA, exhibited higher basal levels of phosphorylated (activated) MAPKs. Interestingly, treatment of DUSP1-deficient CAOV3 cells with rapamycin decreased basal phosphorylated MAPK/JNK and MAPK/p38, without having an obvious effect on MAPK/ERK phosphorylation (). To address whether MAPK/ERK plays a role in DUSP1 modulation of autophagy, dusp1−/− MEFs were left untreated, or treated with the MAPK/p38 inhibitor SB203580, the MAPK/JNK inhibitor SP600125, or the MAP2K inhibitor U0126 for 24 h. Each agent suppressed the phosphorylation of its respective kinase target (). Whereas SP600125 had marginal effects on basal LC3-II levels, the MAPK/p38 and MAPK/ERK inhibitors markedly enhanced and reduced, respectively, basal LC3-II levels (). Furthermore, the MAPK/ERK inhibitor U0126 reduced both basal and rapamycin-induced LC3-II levels in both nontarget and DUSP1 shRNA knockdown CAOV3 cells (). Comparable results were obtained if we knocked down MAPK/ERK by siRNA (). Specifically, knocking down MAPK/ERK expression reduced both basal and rapamycin-induced LC3-II levels. Importantly, the phosphorylation of BECN1 at Ser15, a post-translational modification critical to BECN1's role in initiating autophagy,Citation43 was significantly increased in dusp1−/− MEFs, as compared to Dusp1+/+ MEFs (). Moreover, Dusp1 knockout enhanced the association of BECN1 and phosphorylated MAPK/ERK, and the inhibition of MAP2K activity by U0126 significantly decreased the association of p-BECN1 (Ser15) and phosphorylated active MAPK/ERK (). Collectively, these data suggest that MAPK/ERK activity plays a positive role in autophagy and that DUSP1 targets MAPK/ERK to inhibit MAPK/ERK-mediated autophagy.
Figure 4. Effects of DUSP1 on MAPK activities and role of MAPKs in autophagy. (A) Western blot analyses of LC3, MAPK/p38, p-MAPK/p38, MAPK/ERK, p-MAPK/ERK, MAPK/JNK, and p-MAPK/JNK in control nontarget shRNA (nontarget) and DUSP1 shRNA (sh-DUSP1) CAOV3 cells (upper panel) and quantification of LC3 (lower panel). Cells were left untreated or treated with 5 µM rapamycin for 20 h prior to being harvested. (B) Western blot analyses of LC3, CREB, p-CREB, MAPK/ERK, p-MAPK/ERK, JUN and p-JUN in dusp1−/− MEFs treated with MAPK inhibitors (upper panel) and quantification of LC3 (lower panel). Cells were left untreated or treated with 10 µM SP600125 (SP), 10 µM U0126 (U) or 5 µM SB203580 (SB) for 24 h prior to being harvested for western blot analyses. (C) Western blot analyses of LC3, MAPK/ERK, and p- MAPK/ERK in nontarget and sh-DUSP1 CAOV3 cells treated with rapamycin (5 µM, 20 h), in the presence or absence of 10 µM U0126 (upper panel; LE, long exposure; SE, short exposure) and quantification of LC3 (lower panel). (D) Western blot analyses of MAPK/ERK and LC3 in sh-DUSP1 CAOV3 cells transfected with control siRNA (si-Control) or MAPK1-MAPK3 siRNA (si-MAPK1/3) (left panel) and quantification of LC3 (right panel). Cells were left untreated or treated with 5 μM rapamycin for 20 h prior to being harvested. (E) Dusp1+/+ and dusp1−/− MEFs were left untreated or treated with U0126 and then harvested for immunoprecipitation (IP) with antibody to BECN1. Immunoprecipitated proteins were analyzed by western blot with anti-BECN1, BECN1 phosphorylated at Ser15 (p-BECN1), phosphorylated MAPK/ERK and total MAPK/ERK. Whole cell lysates were used as input control. Data in (A-D) represent mean ± SD of 3 independent experiments. *, P < 0.01 and **, P < 0.005, statistically significant.
Dusp1 knockout promotes autophagy through ULK-mediated formation/activation of an ATG14-BECN1-PIK3C3 complex
To understand the mechanism by which Dusp1 knockout or knockdown promotes autophagy, we first examined if MTOR (mechanistic target of rapamycin [serine/threonine kinase]), a well-characterized suppressor of the initiation of autophagy,Citation44 is altered in dusp1−/− MEFs. RPS6KB/p70S6K is a downstream substrate of MTOR kinase, and its phosphorylation at Thr389 by MTOR is commonly used as a surrogate index of MTOR activity. As shown in , phospho-RPS6KB (at Thr389) was detected in both Dusp1+/+ and dusp1−/− cells, and phosphorylation at Thr389 was inhibited by treatment with rapamycin, an inhibitor of MTOR activity. We detected in dusp1−/− cells, but not Dusp1+/+ cells, increased phosphorylation of ULK1 at Ser555 (), which is a pro-autophagic activating post-translational modification.Citation45 Levels of phosphorylated (p)-BECN1 (at Ser15), a site phosphorylated by ULK1,Citation43 were also increased in dusp1−/− cells (). Knockdown of Mapk1-Mapk3 by their siRNAs in dusp1−/− MEFs significantly decreased basal and rapamycin-induced p-ULK1 (Ser555) and p-BECN1 (Ser15) levels (). These data suggest that the activation of BECN1 by phosphorylation at Ser15 in part depends on MAPK/ERK activity.
Figure 5. Effects of DUSP1 on RPS6KB, ULK1, and BECN1 phosphorylation. (A) Western blot analyses of the levels of total and phosphorylated RPS6KB (at Thr389) in Dusp1+/+ and dusp1−/− MEFs. Cells were left untreated or treated with 5 μM rapamycin for 20 h prior to being harvested. (B) Western blot analyses of the levels of p-ULK1 (at Ser555), ULK1, p-BECN1 (at Ser15) and BECN1 (upper panel), and quantification of p-ULK1 (Ser555) (middle panel) and p-BECN1 (Ser15) (lower panel). Dusp1+/+ and dusp1−/− MEFs were left untreated or treated with 5 μM rapamycin for 20 h prior to being harvested. (C) Western blot analyses of the levels of p-ULK1, ULK1, p-BECN1, BECN1 and MAPK1-MAPK3 in dusp1−/− MEFs (upper panel), and quantification of p-ULK1 (middle panel) and p-BECN1 (lower panel). Cells transfected with control siRNA (si-Control) or siRNA against Mapk1/3 (si-Mapk1/3) were left untreated or treated with 5 μM rapamycin for 20 h prior to being harvested. Data represent mean ± SD of 3 independent experiments. *, P < 0.01 and **, P < 0.005, statistically significant.
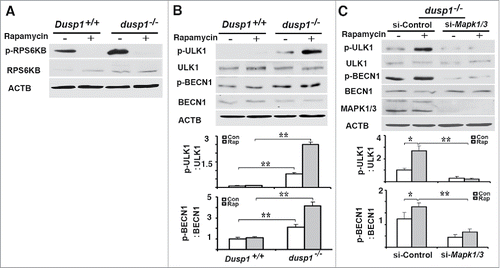
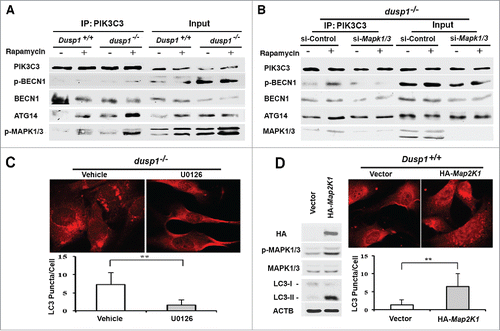
Increased levels of p-ULK1 (Ser555), and p-BECN1 (Ser15) in dusp1−/− cells are conducive to the formation and activation of PIK3C3 complexes needed for initiation of autophagosome formation.Citation43,45,Citation46 Coprecipitation analyses indicated that antibodies to PIK3C3 pulled down increased amounts of ATG14 and p-BECN1 (Ser15) in dusp1−/− cells, relative to Dusp1+/+ cells (). These increases were observed under basal conditions, as well as after rapamycin treatment. Furthermore, activated MAPK1/ERK2 and MAPK3/ERK1 coprecipitated with the PIK3C3-BECN1-ATG14 complexes (). Importantly, knockdown of Mapk1-Mapk3 in dusp1−/− MEFs decreased PIK3C3-associated p-BECN1 (S15) and ATG14 complexes (). In addition, we found that U0126 inhibits Dusp1 knockout-induced LC3 punctate staining (), and that overexpression of a constitutively active Map2k1 itself, an upstream activator of MAPK/ERK, is sufficient to increase the accumulation of LC3 punctate structures (). Thus, these data strongly suggest that MAPK/ERK promotes autophagy by affecting the formation of PIK3C3-BECN1-ATG14 complexes.
Figure 6. Effects of DUSP1 and MAPK/ERK on the association of PIK3C3, BECN1, ATG14, and p-MAPK/ERK. (A) Dusp1+/+ and dusp1−/− MEFs were left untreated or treated with 5 μM rapamycin for 20 h. Cell lysates were prepared, subjected to immunoprecipitation with antibody to PIK3C3, and then followed by western blot analyses using antibodies against BECN1, p-BECN1 (Ser15), ATG14 and p-MAPK1/ERK2-MAPK3/ERK1. Total cell lysates were used to assess input. (B) Dusp1−/− MEFs were transfected with control siRNA (si-Control) or siRNA against Mapk1-Mapk3 (si-Mapk1/3), and treatments as well as IP-western blot were performed as in (A). (C) Dusp1−/− MEFs were treated with 10 µM U0126 for 24 h prior to being processed for immunofluorescent detection of LC3. Red fluorescent puncta denote autophagic vesicle structures (upper panel) and were quantified (lower panel). (D) Dusp1+/+ MEFs transfected with pMCL-HA-Map2k1-R4F or control vector for 24 h were used for western blot analyses of HA, p-MAPK/ERK, MAPK/ERK, LC3 and ACTB (left panel) and immunofluorescent detection of LC3 (upper right panel). The lower right panel depicts quantification of red fluorescent puncta. The data in (C and D) represent means ± SD of analyses of minimally 20 cells. **, P < 0.005, statistically significant.
Cisplatin-resistant ovarian cancer cells upregulate DUSP1 expression and are resistant to rapamycin
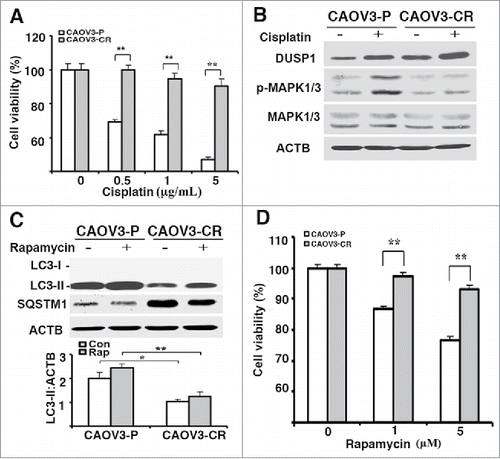
DUSP1 is often upregulated in cancer cells as they acquire resistance to chemotherapeutics such as cisplatin.Citation32,47 We established a cisplatin-resistant CAOV3 cell line (i.e., CAOV3-CR) by selection with increasing cisplatin doses over a 6-month period (). Compared to parental CAOV3-P cells, CAOV3-CR cells expressed higher levels of both basal and cisplatin-induced DUSP1, and lower levels of cisplatin-induced phosphorylated MAPK/ERK (). Importantly, CAOV3-CR cells expressed lower basal and rapamycin-induced accumulations of LC3-II and higher SQSTM1 levels than CAOV3-P cells (). These data are consistent with the results obtained from TOV112D cells () in which overexpression of DUSP1 suppressed both basal and inducible autophagy. In addition, we asked if the antiproliferative activities of rapamycin are dependent on the induction of autophagy. shows that CAOV3-P cells were more sensitive than CAOV3-CR cells to rapamycin, as scored in an MTT assay (), as well as in a cell counting assay (data not shown).
Figure 7. Cisplatin-resistant cells express high levels of DUSP1, low levels of activated MAPK/ERK and LC3-II, and decreased sensitivity to rapamycin. (A) CAOV3-P and CAOV3-CR cells were treated with different amounts of cisplatin for 48 h prior to being analyzed by MTT assays. (B) Western blot analyses of DUSP1 and MAPK/ERK in CAOV3-P and CAOV3-CR cells. Cells were left untreated or treated with 10 µM cisplatin for 20 h before being harvested. (C) Cisplatin-resistant cells have a reduced capacity for autophagy. CAOV3-P and CAOV3-CR cells were left untreated or treated with 5 µM rapamycin for 20 h prior to being harvested for western blot analyses of LC3 and SQSTM1 (upper panel) and quantification of LC3 (lower panel). (D) Cisplatin-resistant cells are less sensitive to rapamycin. CAOV3-P and CAOV3-CR cells were treated with the indicated doses of rapamycin for 48 h prior to MTT assays. Data represent mean ± SD of 3 independent experiments. *, P < 0.01 and **, P < 0.005, statistically significant.
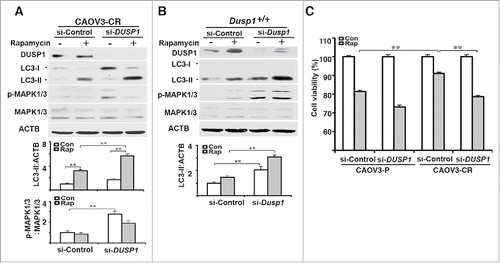
To further define the role of DUSP1 in modulating the sensitivity of CAOV3 cells to rapamycin we transfected CAOV3-CR and CAOV3-P cells with nontarget or DUSP1 siRNAs. shows that knockdown of DUSP1 in CAOV3-CR cells facilitated MAPK/ERK activation and greatly enhanced rapamycin-mediated accumulation of LC3-II. A similar result was obtained with MEFs transfected with Dusp1 siRNA (). MTT assays indicated the DUSP1 knockdown enhanced the effects of rapamycin in CAOV3-P cells, and sensitized previously resistant CAOV3-CR cells to rapamycin ().
Figure 8. DUSP1-mediated suppression of autophagy is associated with cisplatin resistance in human ovarian cancer cells. (A) Effect of DUSP1 knockdown on LC3 level in cisplatin-resistant ovarian cancer cells. CAOV3-CR cells were transfected with control nontarget siRNA (si-Control) or siRNA against DUSP1 (si-DUSP1). Three days later the cells were either left alone or treated with 5 µM rapamycin for 20 h prior to being harvested for western blot analyses of DUSP1, MAPK/ERK, and LC3 (upper panel), quantifications of LC3 (middle panel) and p-MAPK/ERK (lower panel). (B) Effect of DUSP1 knockdown on LC3 level in MEFs. Dusp1+/+ MEFs were transfected with siRNA against Dusp1 (si-Dusp1) or control nontarget siRNA (si-Control) and treated with rapamycin as in (A). DUSP1, MAPK/ERK, and LC3 were determined by western blot analysis (upper panel) and LC3-II was quantified (lower panel). (C) Effect of DUSP1 on rapamycin sensitivity. CAOV3-P and CAOV3-CR cells were transfected with DUSP1 siRNA or control nontarget siRNA as in (A), and then left alone or treated with 5 μM rapamycin for 48 h prior to harvesting for MTT assays. Data represent mean ± SD of 3 independent experiments. **, P < 0.005, statistically significant.
Discussion
Previous studies have implicated MAPK/ERK, MAPK/JNK and MAPK/p38 as being involved in, or a modifier of autophagy.Citation17-19 However, it was not known if their endogenous negative regulator, DUSP1, also plays a role in autophagy. In this study, we used multiple parameters to monitor autophagy and showed in 3 different cell model systems that knockdown or knockout of DUSP1 increased constitutive/basal and rapamycin-inducible autophagy; whereas overexpression of DUSP1 had opposite effects. Our data strongly suggest that DUSP1 negatively regulates autophagy.
Because knockdown or knockout of DUSP1 enhanced both inducible and constitutive autophagy, and MAPKs are the only known substrates for DUSP1,Citation35,42 these findings suggest that DUSP1 must act on MAPKs to inhibit autophagy. The role of individual MAPKs in autophagy is variable, and possibly cell-context dependent. Knockdown and inhibitor studies suggest that MAPK/ERK is necessary for the induction of autophagy in some cell models,Citation19,48 but that elevated, persistent activity can suppress autophagosome maturation.Citation49 MAPK/JNK appears to be a positive modulator of autophagy via its ability to phosphorylate BCL2 and BCL2L1/BCL-XL, thus disrupting BCL2-BECN1 binding and facilitating BECN1 interaction with the PIK3C3 complex, which is required for initiation of phagophore formation.Citation50,51 As for MAPK/p38, reports have appeared indicating that it is both an activatorCitation17,18,Citation52 and inhibitorCitation53,54 of autophagy. In this study, we showed that DUSP1 knockdown increased the level of phosphorylated/activated MAPK/ERKs, MAPK/JNKs and MAPK/p38. Of the 3 MAPKs, only inhibition of MAPK/ERK by the MAP2K inhibitor U0126 strongly suppressed autophagy in DUSP1 knockdown cells, a result that was confirmed by knockdown with siRNAs against MAPK1/ERK2-MAPK3/ERK1. In addition, we found that rapamycin-induced autophagy was abolished by U0126 or MAPK/ERK knockdown by siRNAs. Thus, we conclude that the enhancement of both basal and inducible autophagy by downregulating DUSP1 reflects, at least in part, a MAPK/ERK-dependent process.
The increased basal and rapamycin-induced autophagic activity in dusp1−/− cells, relative to Dusp1+/+ cells, correlated with increased content of the PIK3C3-BECN1-ATG14 complex. This complex is responsible for the initiation of phagophore formation. Furthermore, the BECN1 associated with the complex was phosphorylated at a site that enhances PIK3C3 kinase activity. Hence, the elevated autophagic activity observed in dusp1−/− cells reflects, at least in part, a greater capacity for initiating phagophore formation. Interestingly, coprecipitation studies indicated that phosphorylated/active MAPK/ERKs were a component of the PIK3C3-BECN1-ATG14 complex. We currently do not know the role MAPK/ERKs play in this complex, which is under investigation.
ULK1 activity plays a critical role in the generation/activation of the PIK3C3 complex needed to initiate phagophore formation. The activity of ULK1 is negatively regulated by MTOR, and inhibition of MTOR activates ULK, leading to autophagy induction.Citation43 In the current study, dusp1−/− cells exhibited elevated levels of phosphorylated ULK1 (at Ser555) and BECN1 (at Ser15). In essence, ULK1 phosphorylates BECN1 at Ser15 and promotes PIK3C3-BECN1-ATG14 complex formation and activation in dusp1−/− cells. Given that enhanced autophagy occurred in dusp1−/− cells, which could be suppressed by pharmacological inhibition or knockdown of MAPK/ERKs, it seems that ULK1 activation in dusp1−/− cells is related to elevated MAPK/ERK activity.
An interesting outcome of the MAPK inhibitor studies was that treatment with the MAPK/p38 inhibitor SB203580 markedly enhanced the basal accumulation of LC3-II in DUSP1-deficient cells. This result is consistent with the effects of the MAPK/p38 inhibitor SB202190 in colon cancer cells,Citation54,55 and suggests that MAPK/p38-mediated signaling may suppress basal autophagy. Since both MAPK/ERK and MAPK/p38 activities are increased in DUSP1-deficient CAOV3 cells (), the observed increase in basal autophagy most likely represents an aggregate of the 2 activities in which MAPK/ERK signaling predominates. Notably, the situation is less complicated in the case of rapamycin-induced autophagy in DUSP1-deficient cells since active MAPK/p38 levels were reduced in rapamycin-treated cells.
MTOR is both an inhibitor of autophagy and an activator of processes necessary for protein synthesis and cell division. As such, inhibitors of MTOR such as rapamycin are used as inducers of autophagy or as cancer therapeutics. Indeed, rapamycin and its analogs have been used as a single modality, and in combinational protocols, in the treatment of many forms of cancer, including ovarian cancer.Citation56,57 In our studies rapamycin suppressed the proliferation of ovarian CAOV3 cells in a concentration-dependent fashion. Knockdown of DUSP1 induced autophagy and markedly enhanced the efficacy of rapamycin's cytostatic effects. Thus, rapamycin-mediated induction of autophagy appears to contribute to the therapeutic properties of rapamycin in CAOV3 cells. We do not know the mechanism by which autophagy contributes to rapamycin's effects on cell viability. However, several studies have implicated autophagy as having a role in mediating the antiproliferative properties of some therapeutics.Citation5,40
Previous studies showed that DUSP1 can be induced by cisplatin and that its induction contributes to cisplatin resistance.Citation33,37,Citation38 Consistent with these findings, we observed that basal levels of DUSP1 were higher and MAPK/ERK activities were lower in cisplatin-resistant CAOV3-CR cells compared to the parental cells from which they were derived. Furthermore, basal and rapamycin-induced autophagy were markedly reduced in CAOV3-CR cells, as indicated by reduced LC3-II and elevated SQSTM1 levels. Because knockdown of DUSP1 in CAOV3-CR cells increased basal and rapamycin-induced LC3-II levels, it appears that DUSP1 plays a negative role in regulating autophagy in ovarian cancer cells that have developed acquired cisplatin resistance. Then, the question becomes what advantage does reducing autophagy provide CAOV3-CR cells. We speculate that the reduced capacity for autophagy in the CAOV3-CR cell line may be a secondary consequence (albeit a negative one) of the prosurvival mechanisms that were preferentially selected during continual exposure to cisplatin. For example, cisplatin causes the activation of MAPK/ERK, MAPK/JNK and MAPK/p38 in OV433 and 2008 ovarian cancer cells.Citation40,58 Apoptosis occurring in 2008 ovarian cancer cells following cisplatin treatment can be suppressed by cotreatment with MAPK/JNK and MAPK/p38 inhibitors.Citation58 Interestingly, the anti-apoptotic activities of BCL2 are reduced/eliminated by its phosphorylation at Ser87 and Thr56 by MAPK/p38,Citation59 and at Ser70 by MAPK/JNK.Citation60 Given that DUSP1 dephosphorylates and inactivates MAPK/ERK, MAPK/JNK and MAPK/p38, its overexpression would help preserve the anti-apoptotic activities of BCL2, but inadvertently suppress autophagy in CAOV3-CR cells due to an inhibition of MAPK/ERK activity.
We have also shown that knockdown of DUSP1 markedly enhanced the sensitivity of CAOV3-CR cells to the antigrowth activities of rapamycin. The antigrowth properties of rapamycin can be attributable to not only its effects on protein translation, but also its ability to activate MAP3K5/ASK1- MAPK/JNK-mediated apoptosis in cells with mutant TP53/p53.Citation61 The parental line from which CAOV3-CR cells were derived has a mutated TP53 that results in chain termination at codon 136.Citation62 Presumably, knockdown of DUSP1 in the CAOC3-CR cell line would facilitate activation of the MAP3K5 pathway. Irrespective of the basis for why DUSP1 knockdown sensitized CAOV3-CR cells to cisplatin, our finding is significant since ovarian tumors commonly develop resistance to cisplatin, and rapamycin analogs are increasingly being used as anticancer agents for ovarian cancer.Citation56,57 Suppression of DUSP1 activity or expression may be a means to potentiate the therapeutic activity of rapamycin analogs in cancer clinical trials.
In summary, this study makes the following novel observations: 1) DUSP1 negatively regulates autophagy mediated by MAPK/ERKs; 2) the antigrowth activity of rapamycin on ovarian cancer cells is autophagy dependent; and 3) autophagy is suppressed by DUSP1 in ovarian cancer cells that have developed acquired cisplatin resistance. Recognition that DUSP1 inhibits autophagy is fundamental to our understanding of the mechanisms involved in the regulation of autophagy.
Materials and methods
Reagents
Cisplatin (P4394), bafilomycin A1 (B1793) and anti-ACTB antibody (AC-74) were purchased from Sigma. Rapamycin (13346) was purchased from Cayman. Rabbit polyclonal DUSP1 antibody (SC-370) was purchased from Santa Cruz Biotechnology. Rabbit antibodies against SQSTM1 (5114), LC3 (2775), ATG5 (2630), and total and phosphorylated MAPK/ERK (9102 and 9104), MAPK/p38 (9212 and 9211), MAPK/JNK (9252 and 9251), CREB (9197 and 9198), JUN (9165 and 9164), RPS6KB (9202 and 9205), ULK1 (8054), p-ULK1 at Ser555 (5869), BECN1 (3738), p-BECN1 at Ser15 (13825), and PIK3C3/VPS34 (4263) were purchased from Cell Signaling Technology. Antibody against ATG14 (PD016) was purchased from MBL. Mouse antibody against SQSTM1 (ab56416) was purchased from Abcam. E64d (324890) was purchased from EMD Millipore. The constitutively active mutant Map2k1 plasmid (pMCL-HA-Map2k1-R4F) was kindly provided by Dr. Raymond Mattingly (Wayne State University), as described previously.Citation63
Cell lines, culture conditions and treatment
The human ovarian cancer cell lines TOV112D and CAOV3 were described previously.Citation64 Cell lines were maintained in MCDB105/M199 (Sigma, M2154 and M6395) or DMEM (Gibco, 12800-017), supplemented with 10% fetal bovine serum (Sigma, F2442) and antibiotics at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air. Dusp1 knockout MEFs (dusp1−/− MEFs) and their matched wild-type MEFs (Dusp1+/+ MEFs) were described previously.Citation37
Construction of DUSP1-expressing vectors and generation of DUSP1-overexpression and DUSP1-knockdown cell lines
A full-length DUSP1 cDNA was amplified from pIRES2-EGFP as describedCitation65 using a GC-rich PCR system (Roche Applied Science, 12140306001) and the following primers: 5′-GGAATTCATGGTCATGGAAGTGGGC-3′ and 5′-CGGGATCCTCAGCAGCTGGGAGAGG-3′. The PCR conditions were as follows: 95°C for 3 min; 10 cycles at 95°C for 30 sec, 62°C for 30 sec, and 72°C for 135 sec, and then 25 cycles at 95°C for 30 sec, 62°C for 30 sec, and 72°C for 215 sec. The amplified fragment was isolated from a 1% agarose gel, digested with BamHI and EcoRI and subcloned into pNTAP (Stratagene, 240101) to generate pNTAP-DUSP1. To generate DUSP1 stable expression cells, TOV112D cells were transfected with either pNTAP or pNTAP-DUSP1 using Lipofectamine 2000 reagent (Invitrogen, 11668027). Transfected cells were selected with 800 μg/mL G418 (Life Technologies, 10131027) for 4 wk, and individual clones were isolated as described elsewhere.Citation30 To generate cell lines whose DUSP1 was stably knocked down, CAOV3 cells were transfected with DUSP1 shRNA (SHGLY) or nontarget shRNA control vector (SHC002) from Sigma and selected by growth in medium containing 0.15 µg/ml puromycin. Western blot analyses were used to identify clones that either had markedly elevated or reduced levels of DUSP1.
Generation of cisplatin-resistant CAOV3 cells
Parental CAOV3 cells (CAOV3-P) were gradually exposed to increased concentrations of cisplatin starting from 0.1 to 0.8 μg/ml for over 6 mo to select cisplatin-resistant cells (CAOV3-CR), as described previously.Citation47
Electron microscopy
Cells were fixed with 0.1 M phosphate buffer (pH 7.2) containing 2.5% gluteraldehyde at room temperature for 2 h and then post-fixed with 1% osmium tetroxide for 90 min. The fixed cell pellets were dehydrated in graded ethanol and then embedded in 100% eponate resin (TED PELLA, INC, 18010). Tissue blocks were sectioned to a thickness of 80 nm and placed on 200 mesh rhodium/copper grids (Ernest F Fullam, Inc., 62130). The grids were counterstained with saturated uranyl acetate and lead citrate. The sections were then examined under a Zeiss EM 900 electron microscope (Carl Zeiss AG). Cells with double-membrane vacuoles and an intact nucleus were recorded as autophagic cells.
Knockdown of DUSP1, MAPK1/ERK2-MAPK3/ERK1 and Mapk1-Mapk3 expression by short interfering RNA (siRNA)
On-TARGETplus SMARTpool siRNAs for DUSP1 (L-003484-02), Dusp1 (M-040753-01), MAPK1/ERK2-MAPK3/ERK1 (L-003555-00/L-003592-00), Mapk1/Erk2-Mapk3/Erk1 (M-040126-01/M-040613-01) and corresponding control nontarget siRNAs (D-001810-10) were purchased from Dharmacon Research. The transfection was performed as suggested by the manufacturer's instructions with slight modifications, as described previously.Citation40 Briefly, cells were plated at 6 × 105 cells per well in 6-well plates. The next day, cells were transfected with DUSP1 siRNA, MAPK1-MAPK3 siRNA, Mapk1-Mapk3 siRNA, or nontarget control siRNA using Lipofectamine 2000. After 3 d, transfected cells were left untreated or treated with 5 μM rapamycin for 20 h, and then harvested for subsequent western blot analysis.
MTT assays
The MTT assay was performed in 96-well plates as previously described.Citation40 Typically, there were 4 wells per treatment group.
Detection of exogenous GFP-LC3 by direct fluorescence and endogenous LC3 by indirect immunofluorescence
Cells were transfected with a GFP-LC3 expression vector for 24 h and then selected with G418 for 3 wk. Cells stably expressing GFP-LC3 were obtained and live cell fluorescence was recorded under an inverted fluorescence microscope (Olympus IX70). To detect endogenous LC3 by indirect immunofluorescence, cells grown on chamber slides were fixed with 3.5% formaldehyde, followed by treatment with 0.2% Triton X-100 (Sigma, T8787) in phosphate-buffered saline (Life Technologies, 70-013-032) for 10 min. After incubation with 1% BSA (Sigma, A2153) for 30 min, cells were incubated with a primary rabbit antibody against LC3 at 4°C overnight. The slides were then incubated with Texas red-conjugated anti-rabbit secondary antibodies (ThermoFisher, T-2767) for 30 min. The resulting cells were then incubated with DAPI. The slides were mounted with a coverslip and sealed with nail polish. Fluorescence was observed under a Zeiss LSM780 confocal microscope. We scored 20 or more cells per experiment, in each of 3 independent experiments, for the quantification of fluorescent LC3 puncta.
Western blot analysis
Whole cell lysates were prepared as described previouslyCitation65 and protein concentration was determined using the Protein Assay Kit (Bio-Rad, 500-0006). Cell lysates (50 µg) were electrophoresed through 12% or 15% denaturing polyacrylamide gels and transferred to a PVDF membrane (Millipore, IPVH00010). The blots were probed or reprobed with primary antibodies, and bound antibody was detected using anti-rabbit or mouse HRP- or fluorescence-linked secondary antibodies (ThermorFisher, 35568 and Cell Signaling, 7076), and Enhanced Chemiluminescence (ECL) ECLplus Reagent (Pierce, 80196) or the Odyssey infrared imaging system (LI-COR Biosciences) according to the manufacturer's protocol.
Statistical analysis
Statistical analyses were performed using Student t test. The data are presented as the mean ± SD, and p value < 0.05 was considered significant.
Abbreviations
| BafA1 | = | bafilomycin A1 |
| BECN1 | = | Beclin 1, autophagy related |
| CREB | = | cAMP response element binding protein |
| DUSP1/MKP-1 | = | dual-specificity protein phosphatase 1 |
| MAPK | = | mitogen-activated protein kinase |
| MAP1LC3/LC3 | = | microtubule-associated protein 1 light chain 3 |
| MAP2K/MKK/MEK | = | mitogen-activated protein kinase kinase |
| MEFs | = | mouse embryonic fibroblasts |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| MTT | = | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PIK3C3 | = | phosphoinositide-3-kinase, class 3 |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| SQSTM1/p62 | = | sequestosome 1 |
| shRNA | = | short hairpin RNA |
| siRNA | = | small interfering RNA |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. James Hatfield for assistance with the electron microscopy and Dr. Yusen Liu for helpful discussion.
Funding
This work was supported, in part, by National Institutes of Health Grant 1R21CA178111 through the NCI.
References
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol 2007; 8:931-7; PMID:17712358; http://dx.doi.org/10.1038/nrm2245
- Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008; 4:151-75; PMID:18188003; http://dx.doi.org/10.4161/auto.5338
- Scarlatti F, Maffei R, Beau I, Codogno P, Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ 2008; 15:1318-29; PMID:18421301; http://dx.doi.org/10.1038/cdd.2008.51
- Denton D, Nicolson S, Kumar S. Cell death by autophagy: facts and apparent artefacts. Cell Death Differ 2012; 19:87-95; PMID:22052193; http://dx.doi.org/10.1038/cdd.2011.146
- Sui X, Chen R, Wang Z, Huang Z, Kong N, Zhang M, Han W, Lou F, Yang J, Shang Q, et al. Autophagy and chemotherapy resistance: a promising therapeutic target for cancer treatment. Cell Death Dis 2013; 4:e838; PMID:24113172; http://dx.doi.org/10.1038/cddis.2013.350
- Aredia F, Ortiz LMG, Giansanti V, Scovassi AI. Autophagy and cancer. Cells 2012; 1:520-34; PMID:24710488; http://dx.doi.org/10.3390/cells1030520
- Mathew R, Kongara S, Beaudoin B, Karp CM, Bray K, Degenhardt K, Chen G, Jin S, White E. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Dev 2007; 21:1367-81; PMID:17510285; http://dx.doi.org/10.1101/gad.1545107
- Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of P62. Cell 2009; 137:1062-75; PMID:19524509; http://dx.doi.org/10.1016/j.cell.2009.03.048
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci U S A 2003; 100:15077-82; PMID:14657337; http://dx.doi.org/10.1073/pnas.2436255100
- Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S, Martinez-Cantarin MP, Capozza F, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 2010; 9:3515-33; PMID:20855962; http://dx.doi.org/10.4161/cc.9.17.12928
- Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, Tanowitz HB, Casimiro MC, Wang C, Pestell RG, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle 2010; 9:3534-51; PMID:20864819; http://dx.doi.org/10.4161/cc.9.17.12908
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell 2000; 103:239-52; PMID:11057897; http://dx.doi.org/10.1016/S0092-8674(00)00116-1
- Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002; 298:1911-2; PMID:12471242; http://dx.doi.org/10.1126/science.1072682
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22:153-83; PMID:11294822
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature 2001; 410:37-40; PMID:11242034; http://dx.doi.org/10.1038/35065000
- Kennedy NJ, Davis RJ. Role of JNK in tumor development. Cell Cycle 2003; 2:199-201; PMID:12734425
- Moruno-Manchon JF, Perez-Jimenez E, Knecht E. Glucose induces autophagy under starvation conditions by a p38 MAPK-dependent pathway. Biochem J 2013; 449:497-506; PMID:23116132; http://dx.doi.org/10.1042/BJ20121122
- Matsuzawa T, Kim BH, Shenoy AR, Kamitani S, Miyake M, Macmicking JD. IFN-gamma elicits macrophage autophagy via the p38 MAPK signaling pathway. J Immunol 2012; 189:813-8; PMID:22675202; http://dx.doi.org/10.4049/jimmunol.1102041
- Pattingre S, Bauvy C, Codogno P. Amino acids interfere with the ERK1/2-dependent control of macroautophagy by controlling the activation of Raf-1 in human colon cancer HT-29 cells. The J Biol Chem 2003; 278:16667-74; PMID:12609989; http://dx.doi.org/10.1074/jbc.M210998200
- Dickinson RJ, Keyse SM. Diverse physiological functions for dual-specificity MAP kinase phosphatases. J Cell Sci 2006; 119:4607-15; PMID:17093265; http://dx.doi.org/10.1242/jcs.03266
- Lau LF, Nathans D. Identification of a set of genes expressed during the G0/G1 transition of cultured mouse cells. EMBO J 1985; 4:3145-51; PMID:3841511
- Charles CH, Abler AS, Lau LF. cDNA sequence of a growth factor-inducible immediate early gene and characterization of its encoded protein. Oncogene 1992; 7:187-90; PMID:1741163
- Keyse SM, Emslie EA. Oxidative stress and heat shock induce a human gene encoding a protein-tyrosine phosphatase. Nature 1992; 359:644-7; PMID:1406996; http://dx.doi.org/10.1038/359644a0
- Liu Y, Gorospe M, Yang C, Holbrook NJ. Role of mitogen-activated protein kinase phosphatase during the cellular response to genotoxic stress. Inhibition of c-Jun N-terminal kinase activity and AP-1-dependent gene activation. J Biol Chem 1995; 270:8377-80; PMID:7721728; http://dx.doi.org/10.1074/jbc.270.15.8377
- Sun H, Charles CH, Lau LF, Tonks NK. MKP-1 (3CH134), an immediate early gene product, is a dual specificity phosphatase that dephosphorylates MAP kinase in vivo. Cell 1993; 75:487-93; PMID:8221888; http://dx.doi.org/10.1016/0092-8674(93)90383-2
- Noguchi T, Metz R, Chen L, Mattei MG, Carrasco D, Bravo R. Structure, mapping, and expression of erp, a growth factor-inducible gene encoding a nontransmembrane protein tyrosine phosphatase, and effect of ERP on cell growth. Mol Cell Biol 1993; 13:5195-205; PMID:8355678; http://dx.doi.org/10.1128/MCB.13.9.5195
- Franklin CC, Kraft AS. Conditional expression of the mitogen-activated protein kinase (MAPK) phosphatase MKP-1 preferentially inhibits p38 MAPK and stress-activated protein kinase in U937 cells. J Biol Chem 1997; 272:16917-23; PMID:9202001; http://dx.doi.org/10.1074/jbc.272.27.16917
- Brondello JM, McKenzie FR, Sun H, Tonks NK, Pouyssegur J. Constitutive MAP kinase phosphatase (MKP-1) expression blocks G1 specific gene transcription and S-phase entry in fibroblasts. Oncogene 1995; 10:1895-904; PMID:7761091
- Wu GS. The functional interactions between the p53 and MAPK signaling pathways. Cancer Bio Ther 2004; 3:156-61; PMID:14764989; http://dx.doi.org/10.4161/cbt.3.2.614
- Li M, Zhou JY, Ge Y, Matherly LH, Wu GS. The phosphatase MKP1 is a transcriptional target of p53 involved in cell cycle regulation. J Biol Chem 2003; 278:41059-68; PMID:12890671; http://dx.doi.org/10.1074/jbc.M307149200
- Franklin CC, Srikanth S, Kraft AS. Conditional expression of mitogen-activated protein kinase phosphatase-1, MKP-1, is cytoprotective against UV-induced apoptosis. Proc Natl Acad Sci USA 1998; 95:3014-9; PMID:9501207; http://dx.doi.org/10.1073/pnas.95.6.3014
- Sanchez-Perez I, Martinez-Gomariz M, Williams D, Keyse SM, Perona R. CL100/MKP-1 JNK activation and apoptosis in response to cisplatin. Oncogene 2000; 19:5142-52; PMID:11064451; http://dx.doi.org/10.1038/sj.onc.1203887
- Chattopadhyay S, Machado-Pinilla R, Manguan-Garcia C, Belda-Iniesta C, Moratilla C, Cejas P, Fresno-Vara JA, de Castro-Carpeno J, Casado E, Nistal M, et al. MKP1/CL100 controls tumor growth and sensitivity to cisplatin in non-small-cell lung cancer. Oncogene 2006; 25:3335-45; PMID:16462770; http://dx.doi.org/10.1038/sj.onc.1209364
- Wang HY, Cheng Z, Malbon CC. Overexpression of mitogen-activated protein kinase phosphatases MKP1, MKP2 in human breast cancer. Cancer Lett 2003; 191:229-37; PMID:12618338; http://dx.doi.org/10.1016/S0304-3835(02)00612-2
- Wu GS. Role of mitogen-activated protein kinase phosphatases (MKPs) in cancer. Cancer Metastasis Rev 2007; 26:579-85; PMID:17717636; http://dx.doi.org/10.1007/s10555-007-9079-6
- Rojo F, Gonzalez-Navarrete I, Bragado R, Dalmases A, Menendez S, Cortes-Sempere M, Suarez C, Oliva C, Servitja S, Rodriguez-Fanjul V, et al. Mitogen-activated protein kinase phosphatase-1 in human breast cancer independently predicts prognosis and is repressed by doxorubicin. Clin Cancer Res 2009; 15:3530-9; PMID:19417026; http://dx.doi.org/10.1158/1078-0432.CCR-08-2070
- Wang Z, Xu J, Zhou JY, Liu Y, Wu GS. Mitogen-activated protein kinase phosphatase-1 is required for cisplatin resistance. Cancer Res 2006; 66:8870-7; PMID:16951204; http://dx.doi.org/10.1158/0008-5472.CAN-06-1280
- Wang J, Zhou JY, Wu GS. ERK-dependent MKP-1-mediated cisplatin resistance in human ovarian cancer cells. Cancer Res 2007; 67:11933-41; PMID:18089824; http://dx.doi.org/10.1158/0008-5472.CAN-07-5185
- Raught B, Gingras AC, Sonenberg N. The target of rapamycin (TOR) proteins. Proc Natl Acad Sci U S A 2001; 98:7037-44; PMID:11416184; http://dx.doi.org/10.1073/pnas.121145898
- Wang J, Wu GS. Role of autophagy in cisplatin resistance in ovarian cancer cells. J Biol Chem 2014; 289:17163-73; PMID:24794870; http://dx.doi.org/10.1074/jbc.M114.558288
- Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct Funct 1998; 23:33-42; PMID:9639028; http://dx.doi.org/10.1247/csf.23.33
- Keyse SM. Dual-specificity MAP kinase phosphatases (MKPs) and cancer. Cancer Metastasis Rev 2008; 27:253-61; PMID:18330678; http://dx.doi.org/10.1007/s10555-008-9123-1
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; PMID:23685627
- Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32:2-11; PMID:22025673; http://dx.doi.org/10.1128/MCB.06159-11
- Tian W, Li W, Chen Y, Yan Z, Huang X, Zhuang H, Zhong W, Chen Y, Wu W, Lin C, et al. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett 2015; 589:1847-54; PMID:25980607; http://dx.doi.org/10.1016/j.febslet.2015.05.020
- Fogel AI, Dlouhy BJ, Wang C, Ryu SW, Neutzner A, Hasson SA, Sideris DP, Abeliovich H, Youle RJ. Role of membrane association and Atg14-dependent phosphorylation in beclin-1-mediated autophagy. Mol Cell Biol 2013; 33:3675-88; PMID:23878393; http://dx.doi.org/10.1128/MCB.00079-13
- Wang J, Zhou JY, Zhang L, Wu GS. Involvement of MKP-1 and Bcl-2 in acquired cisplatin resistance in ovarian cancer cells. Cell Cycle 2009; 8:3191-8; PMID:19755862; http://dx.doi.org/10.4161/cc.8.19.9751
- Wang J, Whiteman MW, Lian H, Wang G, Singh A, Huang D, Denmark T. A non-canonical MEK/ERK signaling pathway regulates autophagy via regulating Beclin 1. J Biol Chem 2009; 284:21412-24; PMID:19520853; http://dx.doi.org/10.1074/jbc.M109.026013
- Corcelle E, Nebout M, Bekri S, Gauthier N, Hofman P, Poujeol P, Fenichel P, Mograbi B. Disruption of autophagy at the maturation step by the carcinogen lindane is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Cancer Res 2006; 66:6861-70; PMID:16818664; http://dx.doi.org/10.1158/0008-5472.CAN-05-3557
- Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 2008; 30:678-88; PMID:18570871; http://dx.doi.org/10.1016/j.molcel.2008.06.001
- Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011; 18:571-80; PMID:21311563; http://dx.doi.org/10.1038/cdd.2010.191
- Paillas S, Causse A, Marzi L, de Medina P, Poirot M, Denis V, Vezzio-Vie N, Espert L, Arzouk H, Coquelle A, et al. MAPK14/p38alpha confers irinotecan resistance to TP53-defective cells by inducing survival autophagy. Autophagy 2012; 8:1098-112; PMID:22647487; http://dx.doi.org/10.4161/auto.20268
- Keil E, Hocker R, Schuster M, Essmann F, Ueffing N, Hoffman B, Liebermann DA, Pfeffer K, Schultze-Osthoff K, Schmitz I. Phosphorylation of Atg5 by the Gadd45beta-MEKK4-p38 pathway inhibits autophagy. Cell Death Differ 2013; 20:321-32; PMID:23059785; http://dx.doi.org/10.1038/cdd.2012.129
- Comes F, Matrone A, Lastella P, Nico B, Susca FC, Bagnulo R, Ingravallo G, Modica S, Lo Sasso G, Moschetta A, et al. A novel cell type-specific role of p38alpha in the control of autophagy and cell death in colorectal cancer cells. Cell Death Differ 2007; 14:693-702; PMID:17159917; http://dx.doi.org/10.1038/sj.cdd.4402076
- Corcelle E, Djerbi N, Mari M, Nebout M, Fiorini C, Fenichel P, Hofman P, Poujeol P, Mograbi B.. Control of the autophagy maturation step by the MAPK ERK and p38: lessons from environmental carcinogens. Autophagy 2007; 3:57-9; PMID:17102581; http://dx.doi.org/10.4161/auto.3424
- Schlosshauer PW, Li W, Lin KT, Chan JL, Wang LH. Rapamycin by itself and additively in combination with carboplatin inhibits the growth of ovarian cancer cells. Gynecol Oncol 2009; 114:516-22; PMID:19576622; http://dx.doi.org/10.1016/j.ygyno.2009.06.002
- Mabuchi S, Hisamatsu T, Kimura T. Targeting mTOR signaling pathway in ovarian cancer. Curr Med Chem 2011; 18:2960-8; PMID:21651485; http://dx.doi.org/10.2174/092986711796150450
- Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y, Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38 MAPK pathways in response to cisplatin leads to Fas ligand induction and cell death in ovarian carcinoma cells. J Biol Chem 2003; 278:19245-56; PMID:12637505; http://dx.doi.org/10.1074/jbc.M208134200
- De Chiara G, Marcocci ME, Torcia M, Lucibello M, Rosini P, Bonini P, Higashimoto Y, Damonte G, Armirotti A, Amodei S, et al. Bcl-2 Phosphorylation by p38 MAPK: identification of target sites and biologic consequences. J Biol Chem 2006; 281:21353-61; PMID:16714293; http://dx.doi.org/10.1074/jbc.M511052200
- Srivastava RK, Mi QS, Hardwick JM, Longo DL. Deletion of the loop region of Bcl-2 completely blocks paclitaxel-induced apoptosis. Proc Natl Acad Sci U S A 1999; 96:3775-80; PMID:10097113; http://dx.doi.org/10.1073/pnas.96.7.3775
- Huang S, Shu L, Dilling MB, Easton J, Harwood FC, Ichijo H, Houghton PJ. Sustained activation of the JNK cascade and rapamycin-induced apoptosis are suppressed by p53/p21(Cip1). Mol Cell 2003; 11:1491-501; PMID:12820963; http://dx.doi.org/10.1016/S1097-2765(03)00180-1
- Yaginuma Y, Westphal H. Abnormal structure and expression of the p53 gene in human ovarian carcinoma cell lines. Cancer Res 1992; 52:4196-9; PMID:1638534
- Mansour SJ, Matten WT, Hermann AS, Candia JM, Rong S, Fukasawa K, Wande Woude GF, Ahn NG. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 1994; 265:966-70; PMID:8052857; http://dx.doi.org/10.1126/science.8052857
- Kwong J, Lee JY, Wong KK, Zhou X, Wong DT, Lo KW, Welch WR, Berkowitz RS, Mok SC. Candidate tumor-suppressor gene DLEC1 is frequently downregulated by promoter hypermethylation and histone hypoacetylation in human epithelial ovarian cancer. Neoplasia 2006; 8:268-78; PMID:16756719; http://dx.doi.org/10.1593/neo.05502
- Zhou JY, Liu Y, Wu GS. The role of mitogen-activated protein kinase phosphatase-1 in oxidative damage-induced cell death. Cancer Res 2006; 66:4888-94; PMID:16651445; http://dx.doi.org/10.1158/0008-5472.CAN-05-4229