
ABSTRACT
Proteins in eukaryotic cells are continually being degraded to amino acids either by the ubiquitin proteasome system (UPS) or by the autophagic-lysosomal pathway. The breakdown of proteins by these 2 degradative pathways involves totally different enzymes that function in distinct subcellular compartments. While most studies of the UPS have focused on the selective ubiquitination and breakdown of specific cell proteins, macroautophagy/autophagy is a more global nonselective process. Consequently, the UPS and autophagy were traditionally assumed to serve distinct physiological functions and to be regulated in quite different manners. However, recent findings indicate that protein breakdown by these 2 systems is coordinately regulated by important physiological stimuli. The activation of MTORC1 by nutrients and hormones rapidly suppresses proteolysis by both proteasomes and autophagy, which helps promote protein accumulation, whereas in nutrient-poor conditions, MTORC1 inactivation causes the simultaneous activation of these 2 degradative pathways to supply the deprived cells with a source of amino acids. Also this selective breakdown of key anabolic proteins by the UPS upon MTORC1 inhibition can help limit growth-related processes (e.g., cholesterol biosynthesis). Thus, the collaboration of these 2 degradative systems, together with the simultaneous control of protein translation by MTORC1, provide clear advantages to the organism in both growth and starvation conditions.
keywords:
The protein kinase MTOR is a major regulator of cell protein metabolism that determines whether a cell grows or atrophies.Citation1 This decision depends on the net balance between the overall rates of protein synthesis and degradation, both of which are precisely regulated. MTOR functions in 2 distinct complexes, MTORC1 and MTORC2, which have distinct regulatory roles. In response to adequate nutrients and growth factors, MTORC1 is activated and promotes overall protein synthesis by phosphorylating EIF4EBP and RPS6KB proteins, which stimulate translation of most mRNAs.Citation2 At the same time, MTORC1 also inhibits proteolysis by autophagy by phosphorylating and inactivating ULK/Atg1 kinases ().Citation3 Conversely, upon nutrient deprivation, MTORC1 is inactivated, which reduces protein synthesis to conserve amino acids and stimulates autophagy, which provides the starved cells with amino acids for the synthesis of essential proteins or for energy production. Because this generation of amino acids is important for cell survival in nutrient-poor environments, there has been extensive interest in this rapid activation of autophagy. However, in mammalian cells under normal conditions more than 2 thirds of the protein breakdown occurs by the UPS,Citation4-6 which thus continually provides a large fraction of the amino acids in intracellular pools. Therefore, we tested the possibility that overall proteolysis by the UPS also increases when MTOR is inhibited in order to provide the cells with an additional supply of amino acids.Citation6
Figure 1. Summary of the multiple actions of MTORC1 that synergize to promote protein accumulation when nutrient supply and growth factors are high. Recent findings demonstrate the coordinate suppression of overall protein degradation by the ubiquitin-proteasome system as well as autophagy, which occur simultaneously with the enhancement of protein synthesis. Conversely, nutrient deprivation or MTOR inhibitors cause rapid increases in ubiquitination and overall proteolysis by these 2 systems as protein synthesis decreases.
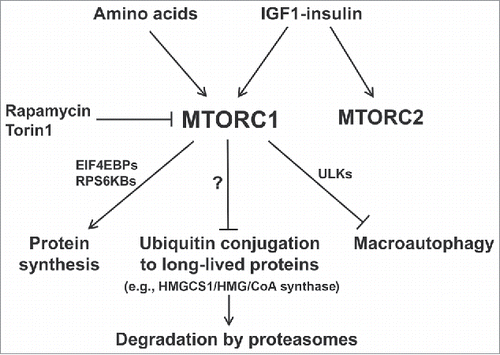
The UPS not only degrades newly synthesized short-lived cell proteins, which are mainly regulatory or misfolded proteins, but also catalyzes the slower breakdown of long-lived cell proteins, which comprise the great majority of cell constituents.Citation6 These general classes of proteins can be differentially labeled by exposing cells to pulses of radioactive amino acids of different durations. When degradation of short-lived proteins was measured in a “pulse-chase protocol” involving a 20 min pulse of 3H-phenyalanine, this process was not affected by the inhibitors of MTOR, rapamycin or Torin 1.Citation6 In contrast, in all cell lines tested (e.g., HEK293, C2C12, and MEFs), the breakdown of the long-lived proteins (which were selectively labeled for 20 h with 3H-phenyalanine and then chased for 2 h), increased rapidly upon MTOR inhibition.Citation6 Using selective inhibitors of proteasomes (i.e., bortezomib) or lysosomal acidification (i.e., concanamycin A) to measure substrate flux through each proteolytic pathway, we found that MTOR inhibition rapidly enhances the degradation of long-lived cell proteins by both proteasomes and lysosomes.Citation6 Accordingly, in MEF cells lacking the key autophagy gene Atg5, proteolysis by lysosomes does not increase upon MTOR inhibition, but proteasomal degradation does.Citation6 Thus, these 2 catabolic responses are simultaneous, but signaled independently.
This increase in degradation by the UPS seems to be due to a general stimulation of ubiquitination. Treatment with Torin 1 or rapamycin raises the cellular content of ubiquitinated proteins within 30 min. Furthermore, blocking protein ubiquitination prevents the enhancement of proteasomal proteolysis, but with little effect on the increase in lysosomal degradation.Citation6 Also, no change in proteasome function is detected after addition of rapamycin or Torin1.Citation6 Thus, the increase in ubiquitination upon MTOR inhibition appears responsible for the enhanced proteasomal degradation, while the increase in lysosomal proteolysis is largely unaffected by blocking ubiquitination and thus is not due to ubiquitin-mediated substrate targeting to autophagic vacuoles or to proteolysis by the endocytic (ESCRT) pathway.
As noted above, MTORC1 and MTORC2 activities are controlled by distinct mechanisms, have different substrates, and serve different physiological roles.Citation1 MTORC1 activity is regulated by the availability of amino acids, but MTORC2 is not. To determine if Torin 1 and rapamycin enhance proteasomal proteolysis by inactivating MTORC1 or MTORC2, we used MEFs that lack MTORC2 activity, and found that inhibition of MTORC1 alone increases degradation by the UPS. Furthermore, in nutrient-deprived cells that lack MTORC1 activity, addition of insulin to activate MTORC2 alone does not affect this process.Citation6 Thus, the enhancement of proteasomal degradation, like the activation of autophagy, is signaled primarily by MTORC1 inactivation. It is noteworthy that upon MTORC1 inhibition, the 2- to 3-fold increase in lysosomal proteolysis is a much more dramatic response than the small relative increase (∼30%) in substrate flux through the UPS. However, since normally the amount of protein breakdown by the UPS is about 3-fold greater in fed cells, upon nutrient restriction these 2 proteolytic systems actually make similar contributions in providing amino acids.
Upon nutrient deprivation, the enhanced protein breakdown by the UPS could in principle provide amino acids by accelerated degradation of proteins generally or through a selective increase in the breakdown of certain cell constituents. The lack of any change in the ubiquitination and degradation of short-lived proteins upon MTOR inhibition indicates some selectivity.Citation6 To learn whether some specific proteins undergo preferential ubiquitination and degradation, we isolated the ubiquitinated proteins from control and Torin 1-treated cells and analyzed their abundance by mass spectrometry. We identified several growth-related proteins that were ubiquitinated at clearly increased rates and showed that they were degraded faster upon Torin 1 treatment, including HMGCS1 (HMG-CoA synthase), the initial enzyme in the pathway for synthesis of cholesterol and isoprenoids. The half-life of this enzyme is >10 h in normal conditions, but falls to ∼2 h after Torin 1 addition.Citation6 This accelerated degradation of HMGCS1 seems to represent a new mechanism for rapid inhibition of the biosynthesis of cholesterol and isoprenoids upon nutrient or insulin deficiency. Thus, the activation of the UPS upon MTOR inhibition selectively decreases certain cell proteins, apparently ones important for cell growth. As many more proteins are likely to be regulated similarly by MTOR, the stabilization of key anabolic proteins is likely to be an additional general mechanism by which MTORC1 promotes growth.
Despite our appreciable efforts, it remains unclear as to what are the biochemical mechanisms by which MTORC1 kinase suppresses the ubiquitination and degradation of these proteins, or even if there exists a single mechanism regulating ubiquitination of various long-lived proteins. This rapid enhancement of degradation by the UPS upon MTOR inhibition does not require any new protein synthesis. Also, this process is not signaled by either the EIF4EBP or RPS6KB proteins, which mediate simultaneous changes in protein synthesis, or by ULKs, which trigger the activation of autophagy. Interestingly, the turnover of certain key proteins is regulated in an opposite manner to the bulk of cellular proteins. For example, DEPTOR, a negative regulator of MTOR signaling is ubiquitinated by SCF (BTRC/βTrCP) and degraded when MTOR activity is high.Citation7-9 Possibly the dephosphorylation of many cellular proteins upon MTOR inhibition enhances their susceptibility to ubiquitination and degradation, and such a mechanism has been reported for the regulator for growth factor signaling GRB10.Citation10,11 However, among the 4 proteins we found that show enhanced degradation upon MTOR inhibition, only SUPT6H is reported to be a putative substrate of MTOR,Citation10 and the increase in overall proteasomal degradation does not require CUL/cullin-based ubiquitin ligases,Citation6 which typically ubiquitinate phosphorylated proteins. Other possible mechanisms could be that phosphorylation by MTORC1 of certain ubiquitin ligases might reduce their activity, or, alternatively, phosphorylation of certain deubiquitinating enzymes could activate them and reduce ubiquitination generally.
This simultaneous activation of protein breakdown by the UPS and autophagy, and the degradation of critical growth-related enzymes upon MTORC1 inhibition would all appear to be highly advantageous to the organism during starvation and can help explain the rapid loss of cell proteins during fasting or the net accumulation on refeeding. Surprisingly, a recent study actually reported the opposite conclusion: MTORC1 inhibition reduces proteasomal degradation by suppressing the expression of new proteasomes.Citation12 However, as we demonstrated elsewhere,Citation6,13 the methodology used in that study for measuring protein degradation and therefore their conclusions on proteolysis are questionable and potentially misleading. In fact, that study did not detect any protein breakdown for many hours after synthesis (when proteolysis is most rapid), nor the well-established enhancement of autophagy upon MTORC1 inhibition.Citation12 Moreover, this delayed reduction in the transcription of proteasomal components after MTOR inhibition is unlikely to reduce proteasome content or protein degradation within 5–16 h as proposed by Zhang et al. because proteasomes have long half-lives (40 to 200 h) and are abundant cell constituents.Citation14 In fact, we did not observe any significant decrease in the content of several proteasome subunits after 24 h of rapamycin treatment.Citation6 The great majority of proteasomes in mammalian cells are inactiveCitation15 and rates of degradation are determined largely by rates of ubiquitination and not by rates of proteasome production. Therefore, changes in proteasome gene expression are unlikely to alter overall proteolysis rates upon changes in nutrient availability or during diurnal cycles as suggested recently.Citation12 When nutrients and growth factors are abundant for prolonged periods and MTORC1 activity is high, expression of new proteasomes and ribosomes may serve to enable cells to undergo cell division.
Because MTOR inhibition can enhance overall proteolysis via both the UPS and autophagy within 30 min, it presumably regulates these processes continuously, but especially with diurnal changes in nutrient intake (meals) and insulin levels (). Coordinate activation of protein breakdown by the UPS and autophagy by FOXO transcription factors have been shown previously to be important in the profound loss of cell protein in atrophying skeletal muscles.Citation5,16 This marked loss of muscle mass is a characteristic cellular response in specific muscles upon denervation or disuse and occurs systemically in muscles upon fasting, cancer cachexia, and other chronic illnesses.Citation17 Unlike the rapid changes induced by MTORC1, the profound loss of muscle protein seen in these catabolic states is a transcriptional adaptive response involving induction by FOXO transcription factors of a set of atrophy-related genes (“atrogenes”), including several specific ubiquitin ligases and many autophagy genes.Citation17 This FOXO-mediated stimulation of protein breakdown requires hours or days to cause prolonged activation of these 2 degradative systems. Thus, MTOR inhibition and FOXO activation appear to have complementary roles upon starvation in vivo or in disease, and probably function sequentially to activate overall proteolysis and to enable the nutrient-deficient organism to mobilize essential amino acids from cell proteins and even from extracellular proteinsCitation18 for gluconeogenesis, energy production, or for synthesis of essential proteins. Conversely, when nutrients and growth factors (e.g., insulin and IGF1) are in ample supply, MTORC1 is activated, FOXOs are inactivated, and the coordinate suppression of both proteolytic systems synergizes with the enhancement of translation to promote the net accumulation of cell proteins (i.e., more cell proteins are synthesized, and they are more stable).
The coordinated functioning of the UPS and autophagy may also help cells eliminate misfolded, potentially toxic proteins, which accumulate and form intracellular aggregates in many of the major neurodegenerative diseases (e.g., Huntington and Parkinson diseases), as well as during aging. In cellular and animal models of these diseases, inhibition of either the proteasome or autophagy can lead to the buildup of such aggregated proteins and the death of neurons.Citation19-22 Rapamycin was shown by Rubinsztein and colleagues in cell and animal models to stimulate the clearance of protein aggregates and to protect neurons against the cytotoxicity caused by such aggregation-prone proteins.Citation23 Beneficial effects of rapamycin have also been reported in animal models of a wide range of neurodegenerative diseases, including Parkinson, Alzheimer, and Huntington diseases, as well as amyotrophic lateral sclerosis.Citation24 In addition, rapamycin administration was reported to extend life span in lower organisms and even mice,Citation25 as was the genetic downregulation of MTOR, probably through decreasing MTORC1.Citation26,27 Therefore, MTOR inhibitors hold the potential to combat these aging-associated proteotoxic diseases in humans. The clearance of misfolded proteins by rapamycin has been attributed solely to the activation of autophagy. However, our findings, raise the possibility that activation of the UPS may also contribute to these protective effects by promoting the clearance of the potentially toxic proteins prior to aggregate formation. Thus, the coordinate enhancement of these 2 degradative processes could synergize to reduce the accumulation of misfolded proteins following rapamycin treatment or nutrient restriction. While the coordinate regulation of the UPS and autophagy by MTOR seems to have evolved to promote maximal growth when nutrients are abundant and to provide amino acids in poor environments, activating these catabolic processes pharmacologically also offers the possibility of more efficient protein quality control.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants to A.L.G. from the National Institute of Health (5R01AR055255 and 5R01GM051923), Muscular Dystrophy Association of America, and Target ALS.
References
- Chantranupong L, Wolfson RL, Sabatini DM. Nutrient-Sensing Mechanisms across Evolution. Cell 2015; 161:67-83; PMID:25815986; http://dx.doi.org/10.1016/j.cell.2015.02.041
- Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012; 485:109-13; PMID:22552098; http://dx.doi.org/10.1038/nature11083
- Feng Y, Yao Z, Klionsky DJ. How to control self-digestion: transcriptional, post-transcriptional, and post-translational regulation of autophagy. Trends Cell Biol 2015; 25:354-63; PMID:25759175; http://dx.doi.org/10.1016/j.tcb.2015.02.002
- Rock KL, Gramm C, Rothstein L, Clark K, Stein R, Dick L, Hwang D, Goldberg AL. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 1994; 78:761-71; PMID:8087844; http://dx.doi.org/10.1016/S0092-8674(94)90462-6
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472-83; PMID:18054316; http://dx.doi.org/10.1016/j.cmet.2007.11.004
- Zhao J, Zhai B, Gygi SP, Goldberg AL. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc Natl Acad Sci U S A 2015; 112:15790-7; PMID:26669439; http://dx.doi.org/10.1073/pnas.1521919112
- Duan S, Skaar JR, Kuchay S, Toschi A, Kanarek N, Ben-Neriah Y, Pagano M. mTOR generates an auto-amplification loop by triggering the betaTrCP- and CK1alpha-dependent degradation of DEPTOR. Mol Cell 2011; 44:317-24; PMID:22017877; http://dx.doi.org/10.1016/j.molcel.2011.09.005
- Gao D, Inuzuka H, Tan MK, Fukushima H, Locasale JW, Liu P, Wan L, Zhai B, Chin YR, Shaik S, et al. mTOR drives its own activation via SCF(betaTrCP)-dependent degradation of the mTOR inhibitor DEPTOR. Mol Cell 2011; 44:290-303; PMID:22017875; http://dx.doi.org/10.1016/j.molcel.2011.08.030
- Zhao Y, Xiong X, Sun Y. DEPTOR, an mTOR inhibitor, is a physiological substrate of SCF(betaTrCP) E3 ubiquitin ligase and regulates survival and autophagy. Mol Cell 2011; 44:304-16; PMID:22017876; http://dx.doi.org/10.1016/j.molcel.2011.08.029
- Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011; 332:1317-22; PMID:21659604; http://dx.doi.org/10.1126/science.1199498
- Yu Y, Yoon SO, Poulogiannis G, Yang Q, Ma XM, Villen J, Kubica N, Hoffman GR, Cantley LC, Gygi SP, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011; 332:1322-6; PMID:21659605; http://dx.doi.org/10.1126/science.1199484
- Zhang Y, Nicholatos J, Dreier JR, Ricoult SJ, Widenmaier SB, Hotamisligil GS, Kwiatkowski DJ, Manning BD. Coordinated regulation of protein synthesis and degradation by mTORC1. Nature 2014; 513:440-3; PMID:25043031; http://dx.doi.org/10.1038/nature13492
- Zhao J, Garcia GA, Goldberg AL. Control of proteasomal proteolysis by mTOR. Nature In Press.
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature 2011; 473:337-42; PMID:21593866; http://dx.doi.org/10.1038/nature10098
- Asano S, Fukuda Y, Beck F, Aufderheide A, Forster F, Danev R, Baumeister W. Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science 2015; 347:439-42; PMID:25613890; http://dx.doi.org/10.1126/science.1261197
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 2007; 6:458-71; PMID:18054315; http://dx.doi.org/10.1016/j.cmet.2007.11.001
- Cohen S, Nathan JA, Goldberg AL. Muscle wasting in disease: molecular mechanisms and promising therapies. Nat Rev Drug Discov 2015; 14:58-74; PMID:25549588; http://dx.doi.org/10.1038/nrd4467
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, Thompson CB. The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 2015; 162:259-70; PMID:26144316; http://dx.doi.org/10.1016/j.cell.2015.06.017
- Bedford L, Hay D, Devoy A, Paine S, Powe DG, Seth R, Gray T, Topham I, Fone K, Rezvani N, et al. Depletion of 26S proteasomes in mouse brain neurons causes neurodegeneration and Lewy-like inclusions resembling human pale bodies. J Neurosci 2008; 28:8189-98; PMID:18701681; http://dx.doi.org/10.1523/JNEUROSCI.2218-08.2008
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/10.1038/nature04724
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; http://dx.doi.org/10.1038/nature04723
- Rubinsztein DC. The roles of intracellular protein degradation pathways in neurodegeneration. Nature 2006; 443:780-6; PMID:17051204; http://dx.doi.org/10.1038/nature05291
- Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr Top Dev Biol 2006; 76:89-101; PMID:17118264; http://dx.doi.org/10.1016/S0070-2153(06)76003-3
- Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 2011; 12:437-52; PMID:21772323; http://dx.doi.org/10.1038/nrn3068
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460:392-5; PMID:19587680
- Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012; 335:1638-43; PMID:22461615; http://dx.doi.org/10.1126/science.1215135
- Wu JJ, Liu J, Chen EB, Wang JJ, Cao L, Narayan N, Fergusson MM, Rovira II, Allen M, Springer DA, et al. Increased Mammalian Lifespan and a Segmental and Tissue-Specific Slowing of Aging after Genetic Reduction of mTOR Expression. Cell Rep 2013; 4:913-20; PMID:23994476; http://dx.doi.org/10.1016/j.celrep.2013.07.030