
ABSTRACT
Autophagy is critical for maintaining cellular homeostasis. Organs such as the eye and brain are immunologically privileged. Here, we demonstrate that autophagy is essential for maintaining ocular immune privilege. Deletion of multiple autophagy genes in macrophages leads to an inflammation-mediated eye disease called uveitis that can cause blindness. Loss of autophagy activates inflammasome-mediated IL1B secretion that increases disease severity. Inhibition of caspase activity by gene deletion or pharmacological means completely reverses the disease phenotype. Of interest, experimental uveitis was also increased in a model of Crohn disease, a systemic autoimmune disease in which patients often develop uveitis, offering a potential mechanistic link between macrophage autophagy and systemic disease. These findings directly implicate the homeostatic process of autophagy in blinding eye disease and identify novel pathways for therapeutic intervention in uveitis.
Introduction
Autophagy is a process by which proteins, organelles, and cellular pathogens are isolated and transported for degradation within lysosomes.Citation1 There are 3 distinct forms of autophagy: microautophagy, chaperone-mediated autophagy, and macroautophagy (hereafter referred to as autophagy). During autophagy, targets in the cytoplasm such as damaged organelles are isolated and enclosed within a double-membrane compartment termed the phagophore. The phagophore expands and matures to form the autophagosome. Maturation of the autophagosome includes conversion of the cytosolic protein LC3-I into the lipidated membrane-associated LC3-II, following its recruitment to the phagophore membrane. The outer membrane of the autophagosome subsequently fuses with the membrane of a lysosome, forming the autolysosome. Targeted materials are then degraded within the autolysosome.Citation1 The autophagic cascade has been reviewed recently and involves numerous proteins in the initiation, elongation, and maturation phases of the process.Citation2 In addition to its role in cellular homeostasis, autophagy can regulate both innate and adaptive immunity by modulating signaling mediators and platforms such as the inflammasome.Citation3,4 Conversely, cellular and soluble factors in the immune system regulate the magnitude of autophagic flux.
The eye is an immune-privileged organ. Both innate and adaptive immune responses are actively dampened within the ocular micromilieu in order to protect the delicate ocular tissues and the visual axis from immune-mediated damage and inflammation that may obstruct the passage of light to the retina. Immune privilege is not simply a function of antigenic sequestration and immune evasion, as antigens introduced into the eye can be recognized by the immune system.Citation5 Macrophages are key antigen presenting cells (APCs) that under normal circumstances process intraocular antigens and induce a deviant immune response highlighted by induction of regulatory cells that lead to tolerance. The molecular mechanisms by which macrophages contribute to immune privilege in the eye are not completely understood, but they are critically important in preventing autoimmune diseases of the eye.Citation5 Loss of ocular immune privilege is associated with uveitis, a term used for a set of diseases characterized by intraocular inflammation of uveal tissues. Uveitis accounts for 10% of all legal blindness in the United States and can be classified according to the parts of the eye involved (anterior, intermediate, posterior, or panuveitis), or by etiology (infectious or noninfectious).Citation6 Noninfectious uveitis, which accounts for the majority of uveitis and blindness in the industrialized world, is thought to be a result of unbridled immune responses to either selfantigens in the eye (autoimmunity) or an innate immune reaction to tissue damage. Of interest, a subset of patients with diverse systemic autoimmune diseases such as lupus and Crohn disease develop uveitis, but the molecular mechanisms that link eye disease with systemic immunity in these disorders are unclear. Citation7 Current therapeutic approaches are not based on the molecular mechanisms that drive disease but on globally inhibiting inflammation.Citation8 Understanding the pathophysiology of immune-mediated uveitis might open novel therapeutic vistas for targeted approaches that are more efficacious than current treatment paradigms with fewer off-target effects.
In these studies, we investigate the role of autophagy in the pathobiology of uveitis. Using multiple models of uveitis, we demonstrate that inhibiting autophagy within macrophages leads to activation of the inflammasome and an IL1B-mediated severe uveitis disease phenotype, thus implicating autophagy proteins in this devastating disease. These studies also demonstrate that macrophage-mediated autophagy and the inflammasome are the shared molecular link that may drive uveitis in patients with systemic autoimmune disease.
Results
Selective deletion of Atg5 in macrophages abrogates autophagy and leads to increased expression of inflammatory cytokines
Germline deletion of Atg5 (autophagy-related 5) results in lethality soon after birth due to developmental defects.Citation9 To create a model in which Atg5 was specifically deleted in macrophages, we bred mice in which exon 3 of Atg5 was flanked by loxP sites (Atg5flox/flox), to mice expressing the Cre recombinase transgene at the Lyz2 (lysozyme 2) locus to yield mice in which Atg5 was conditionally ablated from circulating macrophages (Mφs) (Atg5 CKO) (Fig. S1A).Citation10 The genotype and efficacy of Cre-mediated gene deletion was confirmed by PCR (Fig. S1B), and gene expression analysis confirmed efficient reduction of Atg5 mRNA in peritoneal Mφs from Atg5 CKO mice compared to Atg5 floxed controls (Atgfl/fl) (). Electron micrographs of Atg5 CKO Mφs revealed a lack of intracellular double-membrane vacuoles compared to Atg5 floxed control Mφs (). Western blot analysis of Atg5 CKO Mφs stimulated with LPS and cotreated with bafilomycin A1 (BAF) to inhibit autophagosome degradation revealed an increased conversion of the cytosolic form of MAP1LC3A/B (microtubule-associated protein 1 light chain 3 α/β (LC3A/B-I or LC3-I henceforth in the text) to the phosphatidylethanolamine-conjugated form (LC3-II), associated with phagophore and autophagosome membranes after activation of autophagy. This conversion was absent in Mφs from Atg5 CKO mice. Additionally, Atg5 CKO Mφs showed an accumulation of SQSTM1 (sequestosome 1) compared to Mφs from floxed control mice, even at baseline conditions, indicating impaired autophagic flux (). Together, these data indicate that macrophages from Atg5 CKO mice have impaired autophagy.
Figure 1. Effects of Atg5 ablation on double-membrane vesicle formation, cytokine gene expression, and protein secretion. (A) Atg5 gene expression in Atg5 CKO Mφs (n = 3 mice) vs. floxed controls (n = 3 mice) as measured by qPCR. Student t test ****, P < 0.0001. (B) Representative electron micrographs of a Atg5fl/fl control or Atg5 CKO peritoneal Mφ. (C) Representative western blot from Mφ cell lysates treated with LPS alone or LPS and bafilomycin A1 for 18 h in culture. (D) Representative qPCR gene expression analyses of Mφ polarization markers (M1 markers Il1B (n = 4 vs n = 4 mice), Il6 (n = 4 vs n = 10 mice), Nos2 (n = 4 vs n = 4 mice), Ccl2 (n = 2 vs n = 6 mice), and Ptgs2 (n = 2 vs n = 6 mice) and M2 markers Il10 (n = 4 vs n = 10 mice) and Arg1 (arginase, liver; n = 2 vs n = 6 mice) from baseline or LPS-treated Atg5CKO Mφs and controls. One-way ANOVA with the Tukey post-hoc test **, P < 0.01; ***, P < 0.001. (E) IL6 (n = 7 vs n = 6) and (F) IL1B (n = 7 vs n = 6) secretion by Atg5fl/fl control or Atg5 CKO conditional knockout peritoneal Mφ at baseline or following 24 h of LPS stimulation in culture (n = 2 vs n = 2). One-way ANOVA with the Tukey post-hoc test **, P < 0.01; ***, P < 0.001. (G) TLR4 surface levels from Atg5 CKO (n = 12 mice) or control (n = 11 mice) peritoneal Mφs following INFG priming or INFG priming plus LPS stimulation as measured by FACS analysis. Ns, not significant. All data are represented as mean ± SEM.
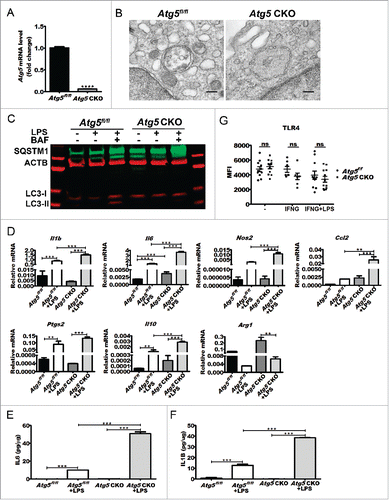
Upon LPS stimulation for 6 h in culture, Atg5 CKO Mφs exhibited significantly increased expression of markers of classically activated macrophages, such as Il6 (interleukin 6), Il1b (interleukin 1β), Nos2 (nitric oxide synthase 2, inducible), Ccl2 (chemokine [C-C motif] ligand 2), and Ptgs2 (prostaglandin-endoperoxide synthase 2) (). Examination of secreted cytokines at 24 h after LPS stimulation in culture revealed significantly increased IL6 and IL1B secretion by Atg5 CKO Mφs versus controls, though no significant difference was observed at baseline (). Of note, there was no difference in the systemic immune cell composition of Atg5 CKO mice compared to littermate Atg5 floxed controls, and Atg5 CKO Mφs were able to induce antigen-specific (peptide and protein) T-lymphocyte proliferation at levels comparable to littermate controls (data not shown). Additionally, differences in LPS responsiveness were not related to differences in TLR4 receptor frequency ().
Autophagy in macrophages regulates the severity of uveitis
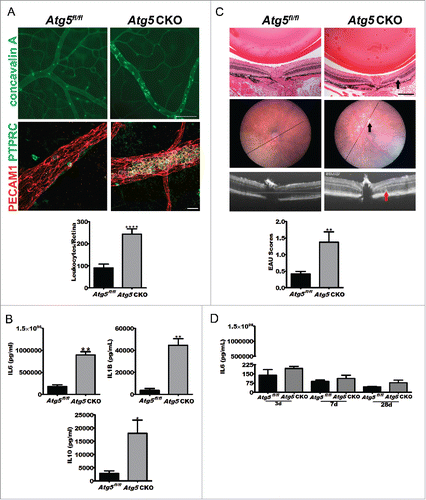
Two models for studying uveitis in mice and rats include endotoxin-induced uveitis (EIU) and experimental autoimmune uveitis (EAU). In the EIU model, ocular inflammation is induced by systemic injection of bacterial lipopolysaccharide (LPS), while induction of uveitis by EAU results from immunization with retinal antigen such as RBP3/IRBP (retinol binding protein 3) peptide (RBP31-20).Citation11,12 Given that macrophages are critical for maintaining ocular immune privilege, we examined the susceptibility of Atg5 CKO mice to LPS-induced uveitis. Atg5 CKO mice and littermate controls were treated intraperitoneally (i.p.) with LPS from E. coli (10 mg/kg), and the severity of uveitis was assessed 24 h later by quantifying the number of adherent leukocytes in the retinal vasculature. This was accomplished by imaging and counting the number of leukocytes in retinal whole mounts from mice perfused with FITC-conjugated concavalin A to identify both leukocytes and retinal vessels () or by immunohistochemical staining of whole retinas (). All EIU quantifications herein were obtained using concavalin A perfused retinas. At baseline, eyes of Atg5 CKO mice did not show any evidence of altered leukocyte stasis or adherence compared to littermate controls (Fig. S2A). Upon induction of EIU, eyes of Atg5 CKO mice developed severe uveitis, characterized by a 2.5-fold increase in the number of adherent leukocytes in the retinal vessels (). Serum collected 6 h post-EIU induction from Atg5 CKO mice also demonstrated a significant increase in IL6, IL1B and IL10 compared to littermate controls (). We next induced organ-specific inflammation in Atg5 CKO and littermate control mice via EAU by inoculation with RBP31-20. Uveitis severity in EAU is assessed by scoring histology of eye sections based on inflammatory cell infiltration and retinal damage. Twenty-eight d post-EAU induction, Atg5 CKO mice displayed EAU scores approximately 2.5-fold higher than littermate floxed controls (), similar to the differences observed using the LPS-based EIU model, demonstrating that macrophage deletion of Atg5 contributes to uveitis severity in 2 models of experimental uveitis. Animals injected with vehicle in place of RBP31-20 showed no signs of inflammation (Fig. S2B). No differences in serum IL6 levels were observed in the tissue-specific EAU model (), and the levels of these cytokines were more than 100-fold lower than those noted in mice with EIU. Both IL1B and IL10 levels were below the limit of detection by ELISA. These results highlight that impaired macrophage autophagy leads to increased severity of uveitis, both in the presence or absence of systemic inflammation.
Figure 2. Atg5 CKO mice develop severe uveitis and undergo a cytokine storm. (A) Representative concavalin A lectin perfused (top; scale bar: 100 µm) or PECAM1 (platelet/endothelial cell adhesion molecule 1) and PTPRC (protein tyrosine phosphatase, receptor type, C) costained (bottom; scale bar: 20 µm) retinal vasculature whole mount images from Atg5fl/fl control (n = 10) or Atg5 CKO (n = 10) conditional knockout mice 24 h postuveitis initiation by 10 mg/kg bacterial LPS and quantification of retinal leukostasis (graph). Student t test ****, P < 0.0001. (B) IL6, IL1B, and IL10 serum levels in Atg5 CKO (n = 3 mice) and control mice (n = 3) 6 h postLPS treatment. Student t test *, P < 0.05; **, P < 0.01. (C) Representative histology from Atg5fl/fl control (n = 19) or Atg5 CKO (n = 17) conditional knockout mice treated with 200 µg RBP31-20 to induce EAU (top; scale bar: 200 µm) with corresponding fundus (top) and OCT imaging (middle), and EAU grading (bottom). The black line in the eye fundus photo indicates the area used for the OCT image below. Arrows in Atg5 CKO images indicate an area of intra-retinal abnormality that was observed across histology, fundus imaging and OCT imaging. Mann-Whitney U-test *, P < 0.05 (D) IL6 serum levels 3, 7 or 28 d postRBP31-20 inoculation to initiate EAU in Atg5 CKO (n = 5) or floxed control (n = 4) mice. All data are represented as mean ± SEM.
It is possible that the increased severity of uveitis seen in Atg5 CKO mice is related to a deficiency in Atg5 but may not be related to the overall process of autophagy.Citation13-16 To rule out this possibility, we examined how macrophage-specific deletion of genes involved in the initiation of autophagy (Rb1cc1 and Atg14), and in autophagosome formation (Atg16l1, Atg7 and Atg3) affected the severity of uveitis using the LPS-based EIU model. As shown in , macrophage-specific deletion of each of these genes significantly increased the severity of uveitis compared to floxed littermate controls in the EIU model, while no evidence of uveitis could be detected in vehicle-treated mice (Fig. S3).
Figure 3. Loss of autophagy promotes uveitis. Representative images of concavalin A lectin- perfused retinal flatmounts from Rb1cc1 CKO (n = 17 vs. n = 10), Atg14 CKO (n = 8 vs. n = 16), Atg16l1 CKO (n = 7 vs. n = 14), Atg7 CKO (n = 9 vs n = 7), and Atg3 CKO (n = 17 vs n = 8) mice and littermate controls with corresponding quantification of adhered leukocytes per retina and serum IL1B levels for each genotype. Scale bar: 100 µm. All data are represented as mean ± SEM. Student t test or Mann-Whitney U-test *, P < 0.05; **, P < 0.01; ***, P < 0.001.
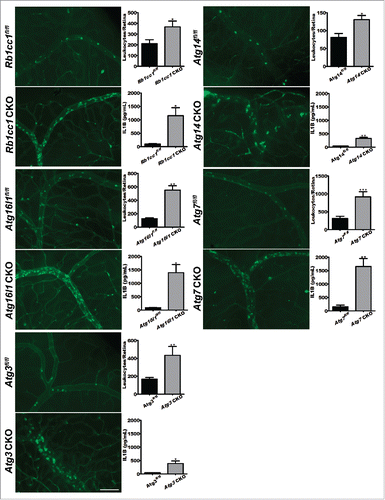
In addition, macrophages from these mice lacking various autophagy genes also showed a significant increase in serum IL1B compared to littermate controls (). Therefore, multiple genes essential for autophagy play key roles in controlling the severity of experimental uveitis, confirming that the homeostatic process of autophagy regulates intraocular inflammation.
Uveitis severity is increased in mice expressing the human ATG16L1T300A polymorphism
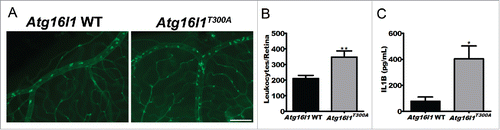
An important question is whether our discovery that autophagy regulates immune-mediated uveitis has relevance to human disease. To do this, we tested whether known human mutations in essential autophagy genes might predispose to uveitis in our animal model. It has been previously reported that a risk allele in humans, specifically a coding polymorphism (Thr300Ala or T300A), in ATG16L1, a gene orthologous to murine Atg16l1, increases the risk for an inflammatory bowel condition called Crohn disease.Citation17,18 Of interest, a subset of patients with Crohn disease can develop vision loss from immune-mediated uveitis, but the molecular basis of this association is unclear.Citation7 We wanted to test whether mice carrying the human T300A knock-in variant of Atg16l1 were more susceptible to uveitis. While noninjected mice show no evidence of spontaneous uveitis at 6 to 8 wk of age (Fig. S3), mice with the T300A polymorphism have a significant increase in the severity of uveitis in the context of the LPS-induced EIU model compared to their littermate controls (), as well as increased levels of IL1B serum levels (). Though further study is warranted, these findings suggest macrophage-mediated autophagy as a potential molecular link between systemic disease and uveitis in Crohn disease.
Figure 4. Assessment of uveitis in a murine model of Crohn disease. (A) Representative images of concavalin A lectin perfused retinal whole mount images from mice with genetic knock-in of the Crohn-associated variant ATG16L1T300A (n = 18) versus Atg16l1 WT controls (n = 14). Scale bar: 100 µm. (B) Corresponding quantification of retinal leukostasis. (C) Serum IL1B levels from Atg16l1T300A (n = 3) vs. Atg16l1 WT controls (n = 3). All data are represented as mean ± SEM. Student t test *, P < 0.05; **, P < 0.01.
Loss of autophagy induces uveitis by activation of the inflammasome
The results above demonstrate a role for autophagy genes and the overall autophagy pathway in regulating ocular inflammation, immune privilege, and secretion of proinflammatory cytokines. A leading potential mechanism to explain these observations is the antagonistic relationship between autophagy and the inflammasome. Inflammasome activation results in cleavage of IL1B and IL18 into their active forms.Citation3,4 We therefore compared the activity of the inflammasome in vitro by examining the secretion of IL1B after activation of autophagy by a combination of LPS, to stimulate autophagy, and ATP, to promote the release of IL1. Consistent with prior work with Atg16l1-deficient macrophagesCitation18, Atg5 CKO Mφs demonstrated significantly increased secreted IL1B after stimulation with LPS and ATP compared to littermate floxed control macrophages (). Floxed control macrophages demonstrated an increase in IL1B secretion after LPS stimulation; however, the amplitude of this response was significantly higher in Atg5 CKO Mφs (Student t test, *, P = 0.0242). Inhibition of general caspase-activation with Z-VAD or of more specific CASP1 and CASP4-inhibition with Z-YVAD resulted in a significant decrease in IL1B in both control and Atg5 CKO Mφs (), suggesting that activation of the inflammasome was involved in this phenotype. In order to directly assess the role of inflammasome activation and IL1B in the pathogenesis and severity of uveitis, we treated Atg5 CKO mice i.p. with an IL1 receptor antagonist (Anakinra™; 10 mg/kg) or vehicle prior to LPS treatment. Neutralization of IL1 signaling completely reversed the increased severity of uveitis in Atg5 CKO mice (). These experiments demonstrate that secreted IL1B is responsible for uveitis seen in Atg5 CKO mice.
Figure 5. Blocking IL1B reverses severe uveitis. (A) The level of IL1B secreted by Atg5 CKO and floxed control peritoneal macrophages in response to LPS treatment can be reduced in both knockouts and controls by treatment with the pancaspase inhibitor, Z-VAD-FMK or CASP1and CASP4 inhibitor Z -YVAD-FMK. Two-way ANOVA (confidence level=95%) with the Bonferroni post-hoc test ****, P < 0.0001. (B) Pharmacological IL1B inhibition in Atg5 CKO mice using the IL1R (interleukin 1 receptor) antagonist Anakinra™. Representative retinal whole mounts from FITC-concavalin A lectin-perfused vehicle (n = 11) and Anakinra™ (n = 12) treated mice and quantification of adhered leukocytes per retina. Scale bar: 100 µm. Student t test **, P < 0.01 (C) Images of concavalin A lectin-perfused retinal wholemounts from control (Atg5fl/fl) (n = 6), Atg5 CKO (n = 15), casp1−/− casp4−/− knockout (n = 7) or Atg5 CKO casp1 casp4 triple-KO mice (n = 8) and quantification of adhered leukocytes in the retinal vasculature and IL1B serum levels from each group. (D). Representative histology from Atg5fl/fl control (n = 10), or Atg5 CKO (n = 14), casp1−/− casp4−/− double knockout (n = 14), and Atg5 CKO casp1−/− casp4−/− triple knockout mice (n = 14) treated with 200 ug RBP31-20 to induce EAU (top; scale bar: 200 µm) with corresponding fundus and OCT imaging, and EAU grading. Arrows indicate clusters of inflammatory cells in the vitreous and the circled areas highlight a region of retinal abnormality observed across imaging modalities. Kruskal-Wallis test with the Dunn post-hoc test *, P < 0.05; **, P < 0.01; ***, P < 0.001. All data are represented as mean ± SEM.
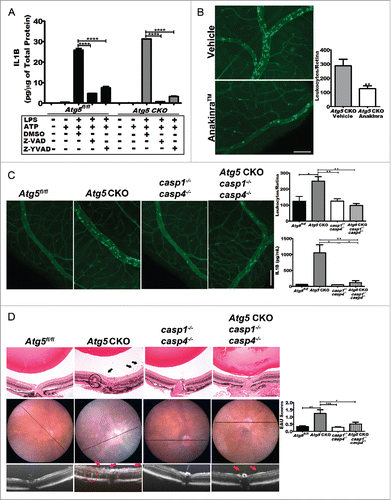
In order to confirm the role of canonical and noncanonical inflammasome-mediated caspase activation and subsequent IL1B secretion in the uveitis phenotype, we generated knockout mice that were deficient in Atg5 in macrophages and also had germline deletion of Casp1 and Casp4 (Atg5 CKO casp1−/− casp4−/−), and compared them to littermate Atg5 CKO mice. Deletion of Casp1 and Casp4 in Atg5 CKO mice (Atg5 CKO casp1−/− casp4−/−) reduced the severity of uveitis back to levels observed after inhibition of IL1 signaling and similar to the phenotype observed in Atg5 floxed control (Atg5fl/fl) or casp1 and casp4 knockout mice (casp1−/− casp4−/−) (). Vehicle treated controls showed no difference between any of the various genotypes and no evidence of uveitis (Fig. S3). Serum levels of IL1B were significantly reduced in Atg5 CKO casp1−/− casp4−/− knockout mice 24 h after LPS treatment compared to Atg5fl/fl littermates, and comparable to those seen in both casp1−/− casp4−/− mice and Atg5fl/fl control mice (). Furthermore, in response to LPS stimulation, macrophages from Atg5 CKO casp1−/− casp4−/− mice secreted significantly less IL1B than macrophages from Atg5 CKO mice (Fig. S4A). We next examined the contribution of caspase activation in the tissue-specific EAU model. Again, deletion of Casp1 and Casp4 ameliorated disease severity in Atg5 CKO mice, with no significant differences noted between Atg5 CKO casp1−/− casp4−/−, casp1−/− casp4−/− or Atg5fl/fl control mice (). There was no significant inflammation noted in vehicle-treated animals (Fig. S4). These experiments demonstrate that autophagy-deficient macrophages cause severe immune-mediated uveitis via activation of the inflammasome leading to caspase-mediated secretion of IL1B.
Discussion
Autophagy as a homeostatic mechanism may have evolved in order to efficiently eliminate intracellular pathogens. The process is evolutionarily conserved and plays a critical role in immune-mediated disease and the pathophysiology of cancers.Citation1,19 The eye is an immune-privileged organ where cellular and soluble factors normally contribute to both immune evasion and suppression leading to tolerance, a phenotype critical for maintaining the visual axis. Macrophages play a key role in this process.Citation5 In autoimmune or immune-mediated diseases of the eye, dysregulated immunity can trigger severe inflammation and uveitis, which left untreated, can lead to blindness. The molecular mechanisms that control ocular inflammation and thereby limit uveitis are not fully understood. Here, we have shown that autophagy genes in macrophages regulate ocular inflammation and prevent uveitis. By showing the involvement of many independent essential autophagy genes in this process, we feel it likely that the overall process of autophagy, rather than nonautophagic functions of autophagy proteins is important to regulate ocular inflammation. The mechanism by which impaired autophagy causes uveitis is through activation of the macrophage inflammasome. This process is dependent on caspase-mediated IL1B secretion and can be completely reversed by pharmacological neutralization or genetic deletion of either IL1B or Casp1 and Casp4.
Treatment of severe uveitis currently involves nonspecific systemic inhibition of immune responses that can have significant off-target effects and complications.Citation8 The findings reported here may provide a novel therapeutic avenue in the treatment of this blinding disease.
Materials and methods
Animals
All animal experiments were conducted in accordance with Washington University in St. Louis School of Medicine Animal Care and Use guidelines and after approval by the Animal Studies Committee. Mice in which exon 3 of the Atg5 gene was flanked by loxP sites (Atg5fl/fl) were kindly provided by Dr. Noboru Mizushima (Tokyo University, Japan) and have been previously characterized.Citation20 To obtain Mφ-specific Atg5 conditional knockout mice, we bred Atg5fl/fl mice to mice carrying the Lyz2-Cre recombinase transgene (The Jackson Laboratory, 004781), yielding mice in which Atg5 is conditionally ablated from circulating Mφs (Atg5 CKO). This breeding approach (Atg5fl/fl Lyz2-Cre+/−) X (Atg5fl/fl Lyz2-Cre−/−) produced litters in which roughly half of the mice were Cre-negative, or floxed controls, (Atg5fl/fl) and half were Cre-positive, or conditional knockout (Atg5 CKO). Other autophagy genes including Atg16l1, Atg14, Atg7, Rb1cc1, and Atg3 were conditionally deleted in macrophages using a similar protocol and have been previously characterized.Citation10,14,18,21-24 Casp1 knockout mice were originally created in the 129 background strain, which is naturally deficient in Casp4/Casp1. Due to the close genomic proximity of casp1 and casp4, the genes did not segregate upon backcrossing into the C57Bl/6 background. Thus, Casp1 knockout mice are also Casp4-deficientCitation25 and are designated within this paper as casp1−/− casp4−/−. casp1−/− casp4−/− mice were crossed with Atg5fl/fl Lyz2-Cre mice to create the Atg5 CKO casp1−/− casp4−/− mice used within this work.
Genotyping
For genotyping, mouse toe DNA was extracted using the DNeasy® Blood and Tissue Kit (Qiagen; 69506). PCR was performed to distinguish between Atg5WT and Atg5flox alleles using Atg5WT-specific (forward 5′-GAATATGAAGGCACACCCCTGAAATG-3′) and Atg5flox-specific (forward 5′-ACAACGTCGAGCACAGCTGCGCAAGG-3′) primers, combined with an Atg5 common reverse primer (forward 5′-GTACTGCATAATGGTTTAACTCTTGC-3′). The resulting Atg5WT fragment is 310 base pairs (bp), while the resulting Atg5flox product is 650 bp. The presence of Cre recombinase was determined using the following primers: forward 5′-CCCAAGAAGAAGAGGAAGGTGTCC-3′ and reverse 5′-CCCAGAAATGCCAGATTACG-3′. The resulting Cre product was 500 bp. For confirmation of excision of exon 3 of Atg5, a specific Atg5Δ primer was used in combination with the common reverse primer (forward 5′-cagggaatggtgtctcccac-3′). The resulting Atg5 mutant product was 300 bp. A complete list of primers used for all mice in this study can be found in Table S1.
Isolation of primary peritoneal Mφs
Peritoneal Mφs were elicited by intraperitoneal (i.p.) injection of 4% thioglycollate (Sigma-Aldrich, T9032). Five d postinjection, Mφs were harvested via peritoneal lavage using 10 mL of sterile 1X Dulbecco's phosphate-buffered saline (Life Technologies, 14190-144), centrifuged, resuspended, and cultured in RPMI-1640 (Life Technologies, 22400154) supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin and 1% GlutaMAX™ (Life Technologies, 35050061) overnight to allow adherence. Mφs were then washed twice with RPMI-1640 to remove nonadherent cells.
For macrophage polarization studies, cells were treated with 100 ng/ml LPS (Sigma-Aldrich, L9641) for 6 h.
Transmission electron microscopy
For ultrastructural analysis, peritoneal Mφs from Atg5fl/fl control and Atg5 CKO mice were fixed in 2% paraformaldehyde, 2.5% glutaraldehyde (Polysciences Inc.; 07710) in 100 mM phosphate buffer, pH 7.2 for 1 h at room temperature. Samples were washed in phosphate buffer and postfixed in 1% osmium tetroxide (Polysciences Inc., 0223C-10) for 1 h. Samples were then rinsed extensively in distilled H20 prior to en bloc staining with 1% aqueous uranyl acetate (Ted Pella Inc., 19481) for 1 h. Following several rinses in distilled H20, samples were dehydrated in a graded series of ethanol and embedded in Eponate 12 resin (Ted Pella Inc., 18005). Sections of 95 nm were cut with a Leica Ultracut UCT ultramicrotome (Leica Microsystems Inc., Buffalo Grove, IL), stained with uranyl acetate and lead citrate, and viewed on a JEOL 1200 EX transmission electron microscope (JEOL USA Inc., Peabody, MA).
Real-time PCR and gene expression analysis
Total RNA was prepared from peritoneal Mφs using the RNeasy mini kit (Qiagen, 74014). cDNA was prepared using the High Capacity cDNA Reverse Transcription Kit (Life Technologies, 4368813) and PCR amplifications of cDNA were performed using Taqman probe-based gene expression assay (Life Technologies) according to the manufacturer's protocols. Primer and probe sets were as follows: Actb, Mm00607939_s1; Il6, Mm00446190_m1; Il10, Mm99999062_m1; Il1b, Mm01336189_m1; Nos2, Mm00440502_m1; Ccl2, Mm00441242_m1; Ptgs2, Mm00478374_m1; Arg1, Mm00475988_m1.
Western blot analysis
Primary peritoneal Mφs were lysed using protease inhibitor-containing RIPA buffer (Thermo-Fisher, 89901) for 30 min followed by centrifugation at 18,400 × g for 10 min, both at 4°C. Lysates (20 µg) were resolved using Mini-PROTEAN TGX gradient gels (Bio-Rad, 456-1094) and Tris/Glycine/SDS Buffer (Bio-Rad, 161-0732) and electro-transferred to nitrocellulose membranes, pore size 0.2 µm, (Bio-Rad,162-0097) according to the manufacturer's instructions. Membranes were blocked in 5% BSA (Sigma-Aldrich, A7030) in TBST (50mM Tris HCL, 150mM NaCl, 0.1% Tween-20) for 1 h at room temperature then incubated with antibodies against LC3 (Cell Signaling Technology, 4108), SQSTM1/p62 (Progen, GP62-C) or ACTB (actin, β; Sigma-Aldrich, 5316) in 5% BSA in TBST at 4°C overnight. Blots were then washed and incubated with secondary antibodies (goat anti-rabbit IRDye® 680LT, donkey anti-guinea pig IRDye® 800CW and goat anti-mouse IRDye® 680LT, Licor, 926-68021, 926-32411 and 962-68020, respectively). Proteins were detected using the corresponding wavelengths on an Odyssey Infrared Imaging System (Licor, Lincoln, NE).
Flow cytometry
Peritoneal macrophages (1 × 106 per mouse) were primed with 10 ng/mL INFG (R&D Systems, 485-MI-100) for 16 h in culture followed by 24 h stimulation with 10 ng/mL LPS. Cells were stained for TLR4 (clone SA15-21; BioLegend, 145402) at a concentration of 1:200 and FACS data was acquired using a FACSCalibur or a FACSCanto flow cytometer (BD Biosciences, San Jose, CA) and analyzed using FlowJo software (Tree Star, Ashland, OR).
ELISA
The following assays were used according to the manufacturer's recommendations: IL6 (R&D Systems, M600B), IL1B (R&D Systems, MLB00C), and IL10 (R&D Systems, M1000B).
For cultured macrophages, cells pooled from 4 mice for each genotype were treated with 100 ng/mL LPS for 24 h. Supernatants were assayed for cytokine levels and cells were pelleted, lysed, and quantified by BCA assay (Thermo/Pierce, 23225) to enable normalization of cytokine levels to total cell protein.
For inflammasome inhibition studies, cells pooled from 4 mice for each genotype were treated for 1 h with 40 µM Z-VAD-FMK or Z-YVAD-FMK (R&D Systems, FMK001 and FMK005). 100 ng/mL LPS was added for 2 h followed by 2.5 mM ATP (Sigma-Aldrich, A6559) for 1 h. Supernatants were collected and assayed by ELISA (R&D Systems, MLB00C) according to the manufacturer's instructions, while cells were harvested and lysed to normalize for total protein content per sample as above.
For serum cytokine analysis, whole blood was collected via submandibular puncture into microtainer tubes (BD Biosciences, 365967) and separated by centrifugation at 6500g for 8 min. The resulting serum layer was decanted into a fresh tube and analyzed by the assays above.
Endotoxin-induced uveitis
Mice (6- to 12-wk old) were injected intraperitoneally with a bolus of 10 mg/kg of Escherichia coli LPS (Sigma-Aldrich, L9641). After 24 h, mice were anesthetized with 86.9 mg/kg ketamine and 13.4 mg/kg xylazine and injected intravenously via the femoral vein with 100 µl of 5 mg/mL FITC-concanavalin A lectin (Vector Labs, FL-1001) to label both blood vessels and leukocytes. After allowing 5 min for circulation, the mice were perfused with 7 to 10 mL of PBS by intracardiac perfusion to remove all nonadhered cells from the vasculature. Eyes were then enucleated, fixed in 4% paraformaldehyde, and retinas were carefully isolated and flatmounted onto glass slides using ProLong Gold with DAPI (Life Technologies, P36931). Leukocytes adhered within the retinal vasculature were imaged by a masked observer at room temperature on an Olympus BX51 microscope equipped with a SPOT RT slider digital camera using a UPlan Apo 20x/0.70 objective (N.A. = 0.17). Luekocytes were manually quantified using ImageJ.
For IL1 receptor antagonist studies, Atg5 CKO littermate mice were randomly divided for treatment with 10 mg Anakinra™ (Kineret®; Sobi, Inc., Waltham, MA) or vehicle twice a day, one day prior to uveitis initiation with LPS, and twice a day again on the day of LPS administration. Mice were sacrificed 24 h post-LPS and processed as above.
Immunofluorescence
EIU was initiated by LPS injection as described above. After 24 h, mice were perfused with 7 to 10 mL of PBS through the left ventricle to remove nonadhered leukoyctes. Following enucleation and 1 h fixation in 4% paraformaldehyde, retinas were dissected from the eye and incubated in 5% normal goat serum plus 1% Triton X-100 (Sigma-Aldrich, T8532) in PBS for 1 h to block nonspecific protein binding. Primary antibodies against PECAM1 (Millipore, MAB1398Z) and PTPRC (BD Bioscience, 550539) were each diluted 1:500 in blocking buffer applied to tissues for 48 h at 4°C. After washing with PBS, Alexa Fluor 594 donkey anti-Armenian hamster (Jackson Immuno, 127-585-160) was used to identify PECAM1 and Alexa Fluor 488 goat anti-rat (Molecular Probes/Thermo, A11006) was used to identify myeloid cells. Stained retinas were then flat mounted onto glass slides and coverslipped using ProLong Gold, as described above, and imaged using an Olympus FV1000 confocal system utilizing Fluoview software with a BX61-WI microscope equipped with a UPLFLN 40x Oil emersion objective lens (N.A. 1.3) and 3x digital zoom.
Experimental autoimmune uveitis
Atg5 CKO and littermate floxed control mice 6 to 8 wk of age were injected with an emulsion of 200 µg of human RBP3 fragment (RBP31-20; Anaspec, AS62297) in 200 µl of complete Freund Adjuvant (1:1 v/v) (BD Difco, 263810) supplemented with 2.5 mg/mL Mycobacterium tuberculosis strain H37RA (BD Difco, 231141). The dose was dispersed subcutaneously in equal volumes into each thigh and the base of the tail. Mice also received 0.3 µg pertussis toxin from Bordetella pertussis (Calbiochem/Millipore, 80501-120) I.P. in 100 µl PBS. Eyes were monitored using a Micron III fundus camera equipped with image-guided 830nm OCT (Phoenix Research Laboratories, Pleasanton, CA) under Ketamine/Xylazine anesthetization. Upon sacrifice, enucleated eyes were fixed in 4% glutaraldehyde in PBS for 1 h followed by overnight fixation in 4% paraformaldehyde. Samples were then processed for paraffin embedding, sectioned at a thickness of 4 µm, and stained with hematoxylin and eosin. Histology was observed from a minimum of 5 sections per eye, which bisected the optic nerve, and were scored based on previously published grading standards by a blinded observer.Citation6 Due to differences in variance between groups, statistical analysis was performed using the Mann-Whitney U-test or Kruskal-Wallis One-way ANOVA.
Statistics
Statistical analysis, unless otherwise noted, was determined via the 2-tailed Student t test, or One-way ANOVA at the 95% confidence level using GraphPad Prism Software (GraphPad Software). In instances where the variance between groups was significantly different, Mann-Whitney U-test (single comparisons) or Kruskal-Wallis with the Dunn post-hoc test (multiple comparisons) was used as indicated in the corresponding figure legend. Results are presented as mean ± SEM. Statistical significance was defined at P< 0.05.
Abbreviations
| Atg | = | autophagy related |
| BAF | = | bafilomycin A1 |
| Casp | = | caspase |
| Ccl2 | = | chemokine [C-C motif] ligand 2 |
| EAU | = | experimental autoimmune uveitis |
| EIU | = | endotoxin induced uveitis |
| Il | = | interleukin |
| RBP3 | = | retinol binding protein 3 |
| LC3 | = | microtubule-associated protein 1 light chain 3 α/β (MAP1LC3A/B) |
| LPS | = | lipopolysaccharide from E. coli |
| Lyz2 | = | lysozyme 2 |
| Mφs | = | macrophages |
| Nos2 | = | nitric oxide synthase 2, inducible |
| Rb1cc1 | = | RB1 inducible coiled-coil 1 |
| Ptgs2 | = | prostaglandin-endoperoxide synthase 2 |
| SQSTM1 | = | sequestosome 1 |
Disclosure of potential conflicts of interest
There were no potential conflicts of interest that needed to be disclosed.
KAUP_A_1207857_Supplementary_material.zip
Download Zip (36.8 MB)Acknowledgments
The authors would like to acknowledge the insights and constructive input of Gregory Wu (Washington University in St. Louis).
Funding
This work was supported by NIH Grant R01EY019287 (RSA), NIH Vision Core Grant P30EY02687, Carl Marshall Reeves and Mildred Almen Reeves Foundation Inc. Award (RSA), Research to Prevent Blindness Inc. Career Development Award and Physician Scientist Award (RSA), Jeffrey Fort Innovation Fund (RSA), NIH grants U19AI109725 and R01AI084887 (HWV), NIH Grant DK097845 (RX), and a Research to Prevent Blindness Inc. Unrestricted Grant to Washington University. Experimental support was also provided by the Speed Congenics Facility of the Rheumatic Diseases Core Center and the National Institute of Arthritis and Musculoskeletal and Skin Diseases (P30AR048335).
Related Research Data
References
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature 2011; 469:323-35; PMID:21248839; http://dx.doi.org/10.1038/nature09782
- Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. N Engl J Med 2013; 368:651-62; PMID:23406030; http://dx.doi.org/10.1056/NEJMra1205406
- Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012; 13:255-63; PMID:22286270; http://dx.doi.org/10.1038/ni.2215
- Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 2008; 456:264-8; PMID:18849965; http://dx.doi.org/10.1038/nature07383
- Niederkorn JY. See no evil, hear no evil, do no evil: the lessons of immune privilege. Nat Immunol 2006; 7:354-9; PMID:16550198; http://dx.doi.org/10.1038/ni1328
- Caspi RR. A look at autoimmunity and inflammation in the eye. J Clin Invest 2010; 120:3073-83; PMID:20811163; http://dx.doi.org/10.1172/JCI42440
- Mintz R, Feller ER, Bahr RL, Shah SA. Ocular manifestations of inflammatory bowel disease. Inflamm Bowel Dis 2004; 10:135-9; PMID:15168814; http://dx.doi.org/10.1097/00054725-200403000-00012
- Pasadhika S, Rosenbaum JT. Update on the use of systemic biologic agents in the treatment of noninfectious uveitis. Biologics 2014; 8:67-81; PMID:24600203
- Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 2004; 432:1032-6; PMID:15525940; http://dx.doi.org/10.1038/nature03029
- Choi J, Park S, Biering SB, Selleck E, Liu CY, Zhang X, Fujita N, Saitoh T, Akira S, Yoshimori T, et al. The parasitophorous vacuole membrane of Toxoplasma gondii is targeted for disruption by ubiquitin-like conjugation systems of autophagy. Immunity 2014; 40:924-35; PMID:24931121; http://dx.doi.org/10.1016/j.immuni.2014.05.006
- Li Q, Peng B, Whitcup SM, Jang SU, Chan CC. Endotoxin induced uveitis in the mouse: susceptibility and genetic control. Exp Eye Res 1995; 61:629-32; PMID:8654505; http://dx.doi.org/10.1016/S0014-4835(05)80056-9
- Kubota S, Kurihara T, Mochimaru H, Satofuka S, Noda K, Ozawa Y, Oike Y, Ishida S, Tsubota K. Prevention of ocular inflammation in endotoxin-induced uveitis with resveratrol by inhibiting oxidative damage and nuclear factor-kappaB activation. Invest Ophthalmol Vis Sci 2009; 50:3512-9; PMID:19279313; http://dx.doi.org/10.1167/iovs.08-2666
- Bestebroer J, V'Kovski P, Mauthe M, Reggiori F. Hidden behind autophagy: the unconventional roles of ATG proteins. Traffic 2013; 14:1029-41; PMID:23837619; http://dx.doi.org/10.1111/tra.12091
- DeSelm CJ, Miller BC, Zou W, Beatty WL, van Meel E, Takahata Y, Klumperman J, Tooze SA, Teitelbaum SL, Virgin HW. Autophagy proteins regulate the secretory component of osteoclastic bone resorption. Dev Cell 2011; 21:966-74; PMID:22055344; http://dx.doi.org/10.1016/j.devcel.2011.08.016
- Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 2008; 4:458-69; PMID:18996346; http://dx.doi.org/10.1016/j.chom.2008.10.003
- Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, Shrestha B, Diamond MS, Dani A, Sosnovtsev SV, et al. Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 2012; 11:397-409; PMID:22520467; http://dx.doi.org/10.1016/j.chom.2012.03.002
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008; 456:259-63; PMID:18849966; http://dx.doi.org/10.1038/nature07416
- Lassen KG, Kuballa P, Conway KL, Patel KK, Becker CE, Peloquin JM, Villablanca EJ, Norman JM, Liu TC, Heath RJ, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci U S A 2014; 111:7741-6; PMID:24821797; http://dx.doi.org/10.1073/pnas.1407001111
- Green DR, Levine B. To be or not to be? How selective autophagy and cell death govern cell fate. Cell 2014; 157:65-75; PMID:24679527; http://dx.doi.org/10.1016/j.cell.2014.02.049
- Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441:885-9; PMID:16625204; http://dx.doi.org/10.1038/nature04724
- Starr T, Child R, Wehrly TD, Hansen B, Hwang S, Lopez-Otin C, Virgin HW, Celli J. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe 2012; 11:33-45; PMID:22264511; http://dx.doi.org/10.1016/j.chom.2011.12.002
- Komatsu M, Wang QJ, Holstein GR, Friedrich VL, Jr., Iwata J, Kominami E, Chait BT, Tanaka K, Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci U S A 2007; 104:14489-94; PMID:17726112; http://dx.doi.org/10.1073/pnas.0701311104
- Gan B, Peng X, Nagy T, Alcaraz A, Gu H, Guan JL. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol 2006; 175:121-33; PMID:17015619; http://dx.doi.org/10.1083/jcb.200604129
- Cann GM, Guignabert C, Ying L, Deshpande N, Bekker JM, Wang L, Zhou B, Rabinovitch M. Developmental expression of LC3alpha and beta: absence of fibronectin or autophagy phenotype in LC3beta knockout mice. Dev Dyn 2008; 237:187-95; PMID:18069693; http://dx.doi.org/10.1002/dvdy.21392
- Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011; 479:117-21; PMID:22002608; http://dx.doi.org/10.1038/nature10558