
ABSTRACT
Lysosomes, the cell's recycling center, undergo nutrient-sensitive adaptive changes in function and biogenesis, i.e., lysosomal adaptation. We recently discovered that lysosomes also mediate the cell's “survival” response (i.e., autophagy) to oxidative stress through the activation of TFEB (transcription factor EB), a master regulator of lysosome biogenesis and autophagy. MCOLN1/TRPML1, the principal Ca2+ release channel on the lysosomal membrane, serves as the redox sensor in this process. Increasing reactive oxygen species (ROS) levels, either endogenously by mitochondrial damage or exogenously, directly activates MCOLN1 to induce lysosomal Ca2+ release, triggering PPP3/calcineurin-dependent TFEB nuclear translocation to enhance autophagy. Hence, ROS may induce autophagy by activating the MCOLN1-lysosome Ca2+-TFEB pathway, facilitating the removal of damaged mitochondria and excess ROS. Our findings have revealed a lysosomal signaling mechanism for cells to respond to oxidative bursts and adapt to oxidative stress.
One of the most important players in the cell's adaptive machinery is MTOR (mechanistic target of rapamycin [serine/threonine kinase]), a nutrient-sensitive kinase that is inhibited during nutrient starvation. TFEB (transcription factor EB), a master regulator of lysosome biogenesis and autophagy, is negatively regulated by MTOR, playing an essential role in starvation-induced autophagy. Dephosphorylation of TFEB caused by MTOR inhibition leads to the translocation of TFEB from the cytosol to the nucleus (i.e., TFEB activation). Once in the nucleus, TFEB induces the expression of a unique set of genes that are involved in lysosome function, e.g., lysosomal acidification, biogenesis, and degradation. In addition, TFEB activation also promotes the expression of genes required for multiple steps of autophagy, including autophagy induction and autophagosome biogenesis. Hence, TFEB promotes autophagy by increasing the biogenesis and function of both lysosomes and phagophores (the precursors to autophagosomes), facilitating the degradation of autophagic substrates. In this paradigm, as an interface for MTOR complex assembly and MTOR-TFEB interaction, lysosomes transduce the information about nutrient availability from the cytosolic sensor proteins to transcriptional factors via a lysosome-to-nucleus mechanism.
Oxidative stress occurs under both patho-physiological conditions and aging when oxidant (e.g., reactive oxygen species, ROS) production overwhelms the detoxification and antioxidant mechanisms in the cell, and is a common challenge for cell survival. Chronic oxidative stress may permanently damage cellular proteins and other constituents, hence acting as a “death” signal. A mild increase in ROS levels, however, activates the cell's “survival” mechanisms, such as autophagy, to remove oxidized biomaterials, damaged organelles, and excessive ROS. As with starvation-induced adaptive responses, it is reasonable to expect that ROS-induced autophagy also requires dual roles of lysosomes in both signal transduction and lysosomal degradation. However, whether and how ROS signal lysosomes to trigger the upregulation of lysosomes is not known.
In our recent whole-endolysosome patch-clamp studies, we found that the MCOLN1/TRPML1 (mucolipin 1), the major Ca2+ release channel in the lysosome, is activated by various oxidants including chloramine T and H2O2. Consistently, ROS production by mitochondrial decouplers such as carbonyl cyanide m-chlorophenylhydrazone (CCCP) also increases lysosomal MCOLN1 currents. In contrast, pretreatment with reducing or antioxidant reagents effectively prevents the channels from being activated by exogenous oxidants or mitochondrial ROS. The stimulatory effect of oxidants on MCOLN1 is likely to be direct, as the activation is quick in both whole-endolysosome and cell-free excised patches. This activation is also specific, as oxidants fail to activate other endo-lysosomal Ca2+ channels (i.e. MCOLN2/TRPML2, MCOLN3/TRPML3, and TPCN2/TPC2). Consistently, oxidants evoke endogenous MCOLN1-like currents in the lysosomes of wild-type, but not MCOLN1 knockout, fibroblasts.
MCOLN1-mediated lysosomal Ca2+ release activates PPP3/calcineurin-dependent dephosphorylation of TFEB, which is otherwise kept in the inactive, phosphorylated form by the kinase activity of lysosome-localized MTOR. Indeed, both CCCP and H2O2 treatments induce robust translocation of TFEB to the nucleus, and the translocation is prevented by a ROS scavenger (N-acetyl-cysteine, NAC), an intracellular Ca2+ chelator (BAPTA-AM), or synthetic inhibitors of MCOLN1 (ML-SIs). Furthermore, TFEB nuclear translocation is blocked by PPP3/calcineurin inhibitors. Conversely, artificial activation of MCOLN1 by synthetic agonists (ML-SAs) is sufficient to activate TFEB in a Ca2+-dependent but redox-independent (i.e., NAC-insensitive) manner. Control experiments demonstrate that the CCCP or ML-SAs fail to induce TFEB nuclear translocation in MCOLN1 knockout human fibroblasts. Hence, MCOLN1 is specifically required for ROS-induced TFEB activation.
MCOLN1-mediated lysosomal Ca2+ signaling plays a unique role in ROS-induced autophagy. MCOLN1 and lysosomal Ca2+ are not required for TFEB activation induced by MTOR inhibition. Torin-1, a potent MTOR inhibitor, induces strong TFEB signals in the nucleus within 1–2 h in MCOLN1 knockout cells or wild-type cells in the presence of ML-SIs or BAPTA-AM. Similarly, starvation still robustly induces TFEB activation when MCOLN1 is genetically inactivated or pharmacologically inhibited. However, BAPTA-AM treatment effectively blocks starvation-induced TFEB activation. Hence, starvation may induce lysosomal and intracellular Ca2+ release via MCOLN1-dependent and -independent mechanisms. Although ROS levels are increased during starvation, NAC does not block TFEB nuclear translocation. Hence, MCOLN1 and lysosomal Ca2+ have a specific role in lysosome adaptation to ROS elevation.
TFEB is a transcriptional regulator of autophagosome and lysosome biogenesis. Hence, as a consequence of ROS- and MCOLN1-induced TFEB activation, the biogenesis of autophagosomes and lysosomes are enhanced. Consistent with this prediction, ROS-induced accumulation of LC3 puncta is largely diminished by NAC or ML-SIs. In addition, LC3-II levels are increased by ROS in wild-type, but not MCOLN1 knockout, cells. Finally, the expression levels of LAMP1 (a lysosome marker as readout of lysosome biogenesis) are enhanced by a short-term treatment with CCCP, and the enhancement is reduced by ML-SIs. Hence, ROS activation of MCOLN1 may be essential for ROS regulation of autophagy.
Enhanced autophagy promotes the removal of damaged mitochondria and excessive ROS. Because damaged mitochondria lead to ROS elevation, autophagy (more specifically, mitophagy) may mediate a negative-feedback loop to prevent oxidative stress (see ). When the MCOLN1-TFEB pathway is compromised, cells exhibit mitochondrial fragmentation and elevated ROS levels. Hence, whereas lysosomes are focal sensing sites for nutritional and oxidative stress, preemptive activation of the MCOLN1-TFEB pathway may alleviate symptoms in patients with lysosomal storage diseases and neurodegenerative diseases, in both of which oxidative stress occurs.
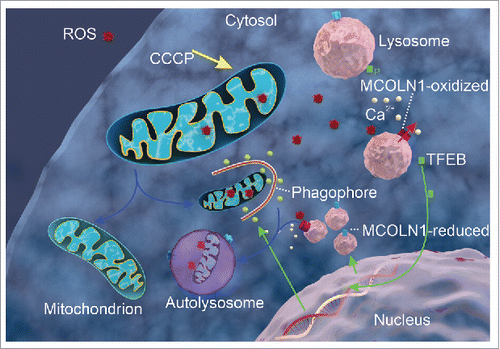
Figure 1. ROS activate MCOLN1 and lysosome calcium release to promote autophagy. A working model to illustrate the roles of MCOLN1 and lysosomal Ca2+ in ROS-induced TFEB activation and autophagy. An increase in mitochondrial ROS, e.g., by CCCP, may activate lysosomal MCOLN1 channels, inducing Ca2+ release from the lysosome lumen to mediate PPP3/calcineurin-dependent dephosphorylation of TFEB. Dephosphorylated TFEB enters the nucleus, promoting the mRNA expression of genes involved in autophagy induction, autophagosome and lysosome biogenesis, and lysosomal degradation. Subsequently, autophagy is enhanced to facilitate the clearance of damaged mitochondria and removal of excessive ROS.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.