ABSTRACT
Senescence is a natural anticancer defense program disabled in tumor cells. We discovered that deregulated CDK4 (cyclin dependant kinase 4) and CDK6 activities contribute to senescence bypass during tumorigenesis and that their inhibition restores the senescence response in tumor cells. CDK4 and CDK6 phosphorylate RB1/RB, preventing its inhibitory interaction with the E2Fs, the cell cycle transcription factors. However, we also found that CDK4 interacts and phosphorylates the DNMT1 (DNA methyltransferase 1) protein protecting it from macroautophagy/autophagy-mediated protein degradation. This discovery highlights a new epigenetic component of CDK4-CDK6 signaling that could be exploited in cancer treatment.
Although cancer is a leading cause of disease and death, it is overall surprising that its frequency in humans is low and mostly confined to advanced age, especially given human's high cell number and longevity. It follows that efficient endogenous anticancer defenses must operate to avoid tumor initiation and progression. Senescence is one of those mechanisms that consists of a stable cell cycle arrest where cells stay metabolically active and can signal to the immune system to eventually be cleared. Markers for senescence are numerous but they are not strictly specific, so the process has to be identified by combining several of them. These markers include cell cycle arrest, DNA damage, expression/secretion of cytokines, mitochondrial dysfunction and increased autophagy. The latter also includes the classic induction of the senescence-associated GLB1/β-galactosidase. Recently, many reports of senescent cells occurring naturally in vivo highlight a role for senescent cells in normal embryonic development, wound healing and tumor suppression. In particular, the demonstrations of senescent cells in various types of benign tumors such as nevi or benign prostatic hyperplasia exemplify the bona fide role of senescence to counteract tumorigenesis. For cancer to avoid or bypass senescence and form a malignant tumor, it is typically thought that mutations would be necessary. Interestingly, some of the cancer treatments currently in use seem to successfully trigger the senescence program. Indeed, senescence was proposed to be the mechanism leading to the complete remission of acute promyelocytic leukemia after treatment with retinoic acid and arsenic. This senescent response is mediated by restored signaling of the tumor suppressor PML.
In the course of our research on the mechanisms involved in PML-induced senescence, we observed that normal cells do not enter senescence in response to PML if CDK4 or CDK6 are overexpressed. Amplification of CDK4 or its increased activity via inactivation of CDKN2A/p16, a CDK4-CDK6 inhibitor, is quite frequent in many cancers and could be means through which cells escape senescence. Thus, we decided to study what would be the effects, in cancer cells, of inactivating CDK4-CDK6 either by shRNA knockdown or by chemical inhibition using palbociclib or flavopiridol along with the expression of PML. We noticed that although neither PML nor inhibition of CDK4-CDK6 alone could achieve a lasting cell cycle arrest in tumor cells, the combinations successfully result in a more lasting arrest with detectable senescence markers including high levels of autophagic foci as detected using anti-LC3B and anti-SQSTM1/p62 antibodies. Even in xenografts of PC3 prostate cancer cells expressing a control vector or PML, a pulse of 5 d of treatment with palbociclib provides a significant and sometimes complete remission in tumor growth.
As CDK4-CDK6 phosphorylate RB1/RB to prevent it from interacting and inhibiting the cell cycle transcription factors E2Fs, we expected that inhibition of CDK4-CDK6 along with the expression of PML would result in a greater inhibition of E2F target genes. Yet, expression of PML and inhibition of CDK4-CDK6 each individually reduced expression of classic E2F targets without the expected additive effect when we combined the 2 actions. However, and surprisingly, when a transcriptome analysis was performed, a gene signature corresponding to DNA methylation inhibition was uncovered. The importance of this signature was confirmed when we demonstrated that a more stable arrest with senescence markers was also achieved when combining PML expression with a pretreatment with the DNA methylation inhibitor 5-aza-deoxy-cytidine.
Further investigations revealed that overexpression of CDK4, even in normal cells, results in higher DNMT1 protein levels although the mRNA expression is unchanged suggestive of a post-transcriptional regulation. Moreover, CDK4-CDK6 knockdown by shRNA or their inhibition by palbociclib triggers a reduction of DNMT1 protein levels that cannot be rescued by the proteasome inhibitor MG132, but is rescued when we inhibit autophagy with bafilomycin A1. In vitro phosphorylation of DNMT1 with purified CDK4-CCND/cyclin D suggested putative new sites of phosphorylation, 2 of which correspond to consensus CDK target sites.
Interestingly, affecting DNA methylation through the destabilization of DNMT1 can potentially affect the cells in a more long-term manner. Indeed, we found that a 6-d pretreatment with palbociclib, to inhibit CDK4-CDK6 and reduce DNMT1 protein, potentiates the growth inhibitory effect of camptothecin treatment, a chemotherapeutic agent causing DNA damage, resulting in a more stable arrest of PC3 cancer cells. This result indicates that cells keep a memory of the treatment with the CDK4-CDK6 inhibitor, which is consistent with an epigenetic effect.
The specific autophagy of DNMT1 and, in opposition, CDK4s ability to protect it, is a novel concept that could now be taken into account in cancer treatments. Many clinical trials combining palbociclib with other treatments are underway. It is possible that autophagy-dependent DNMT1 degradation can help to erase epigenetic changes in tumor cells making them more susceptible to other antitumor mechanisms such as cellular senescence or immune-mediated cell clearance. An epigenetic effect will make it possible to schedule the treatment first with the CDK4-CDK6 inhibitor and then with additional chemotherapeutic drugs, avoiding the toxicity that the combined action of the 2 drugs can have in normal cells. Many studies have reported changes in DNA methylation in cancer cells that are suspected to play important roles in the tumorigenesis process. This observation along with the realization that gene mutations cannot always be found as drivers in some tumors highlights the need to better understand and take into account DNA methylation in our anticancer efforts. Of note, increased expression of some tumor suppressor genes that have their promoter methylated in cancer was also observed in our experiments with palbociclib.
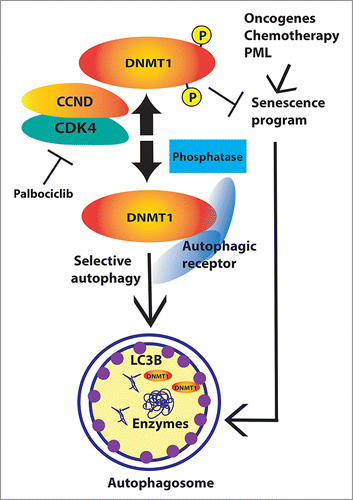
Our work adds DNMT1 as an additional target for autophagy-mediated protein degradation (). Further work is required to identify how DNMT1 is targeted for autophagy (via selective autophagy), and how CDK4-CDK6-dependent phosphorylation inhibits the process.
Figure 1. CDK4-CDK6 protect DNMT1 from autophagy. The effect of CDK4-CDK6 on DNMT1 levels can be inhibited by palbociclib or the autophagy inhibitor bafilomycin A1. The model predicts that a phosphatase can also target DNMT1 for autophagy and that an autophagy receptor able to link DNMT1 to the phagophore (the autophagosome precursor) would be involved. Inhibition of CDK4-CDK6 and targeting of DNMT1 for autophagy allow changes in epigenetic marks, which facilitate the induction of the senescence program in tumor cells. The senescence program itself involves an increase in the autophagy pathway generating a potential positive feedback mechanism to reinforce senescence.