
ABSTRACT
AR (androgen receptor) signaling is crucial for the development and maintenance of the prostate as well as the initiation and progression of prostate cancer. Despite the AR's central role in prostate cancer progression, it is still unclear which AR-mediated processes drive the disease. Here, we identified 4 core autophagy genes: ATG4B, ATG4D, ULK1, and ULK2, in addition to the transcription factor TFEB, a master regulator of lysosomal biogenesis and function, as transcriptional targets of AR in prostate cancer. These findings were significant in light of our recent observation that androgens promoted prostate cancer cell growth in part through the induction of autophagy. Expression of these 5 genes was essential for maximal androgen-mediated autophagy and cell proliferation. In addition, expression of each of these 5 genes alone or in combination was sufficient to increase prostate cancer cell growth independent of AR activity. Further, bioinformatic analysis demonstrated that the expression of these genes correlated with disease progression in 3 separate clinical cohorts. Collectively, these findings demonstrate a functional role for increased autophagy in prostate cancer progression, provide a mechanism for how autophagy is augmented, and highlight the potential of targeting this process for the treatment of advanced prostate cancer.
Introduction
AR (androgen receptor) is a hormone-regulated member of the nuclear receptor (NR) family of transcription factors that plays a crucial role in both the development and maintenance of the prostate gland and is an overall regulator of male secondary sexual traits and reproductive function.Citation1 Upon binding of androgens such as dihydrotestosterone (DHT), AR translocates to the nucleus where it dimerizes, interacts with regulatory regions of the DNA, and recruits coregulators and transcriptional machinery.Citation2 Beyond its basic physiological functions, AR is also a central factor in the development and progression of prostate cancer.Citation3
If prostate cancer is diagnosed early, it can often be treated successfully with surgery and/or radiation alone.Citation4 However, many patients progress to the advanced stages of the disease. As such, prostate cancer remains the second leading cause of cancer-related deaths in American men.Citation5 Because AR is a primary driver of prostate cancer growth and metastasis, patients with advanced prostate cancer are treated with systemic hormone therapy to prevent the spread of the disease.Citation6 Unfortunately, despite continuous androgen ablation therapy, patients inevitably develop resistance to this therapy. This relapsed stage of the disease, termed castration-resistant prostate cancer (CRPC) and for which there is currently no cure, interestingly is still driven largely by AR signaling.Citation6 In this context, the transcriptional targets of AR potentiate the progression of prostate cancer by altering cellular processes that ultimately allow the cell to rapidly proliferate, migrate and withstand adverse conditions. Despite AR's key role in prostate cancer, it is still poorly understood which specific AR-mediated processes drive the disease progression. This knowledge gap has precluded the development of therapies with new mechanisms of action.
Macroautophagy (hereafter referred to as autophagy) is a process in which nonessential cellular components are degraded into amino acids, lipids, nucleotides, and carbohydrates that can then be reallocated within the cell. Targeted components are first encircled by a growing double membrane known as a phagophore, which upon maturation and closure is known as an autophagosome. The autophagosome then fuses with a lysosome creating a structure known as an autolysosome that allows for the recycling of the degraded components.Citation7,8 Autophagy has long been implicated in cancer where it appears to have contrasting roles depending on context such as disease stage.Citation9 For example, while autophagy is at first generally considered a tumor suppressor, more recent evidence indicates an oncogenic role in advanced cancers.Citation10-17 Such changes can then help a cancer cell survive harsh conditions such as low oxygen, decreased nutrient supply, or chemotherapy.Citation18 Additionally, the enhanced recycling afforded by autophagy may, over longer periods, even facilitate anabolic tumor processes. Although many cancers exhibit increased autophagy, it is not often clear how cancer cells stimulate this process.
We have previously shown using a variety of approaches that androgens increase both autophagy and autophagic flux in prostate cancer cells in part by increasing reactive oxygen species (ROS).Citation19 Moreover, molecular or pharmacological inhibitors of autophagy significantly impair androgen-mediated prostate cancer cell growth. However, this androgen-mediated increase in ROS could only account for part of the increased autophagy we observed in response to androgens. Given AR's known role as a transcription factor, we hypothesized that AR could also be directing the expression of core autophagy components. These genes would therefore be required for induction of autophagy and subsequent androgen-mediated prostate cancer growth. We further hypothesized that these genes would represent a subset of AR-regulated genes that contribute to disease progression and hence be potential new therapeutic targets.
Results
Androgens increase the transcription of several core autophagy genes
We previously demonstrated that androgens increased autophagy using a combination of assays including immunoblot, immunofluorescence microscopy and transmission electron microscopy.Citation19 While AR was found to augment autophagy in part through increasing intracellular ROS levels, we suspected that the androgen-mediated induction of autophagy was mediated through additional molecular mechanisms. Since AR primarily functions as a transcription factor, we hypothesized that AR could be controlling the transcription of several components of the core autophagy machinery, thereby further increasing functional autophagy. Accordingly, androgen treatment of 2 different androgen-sensitive prostate cancer cell models, LNCaP and VCaP, resulted in consistently increased mRNA () and protein ( and Table S1) levels of 4 core autophagy genes: ULK1, ULK2, ATG4B, and ATG4D. The protein products of ULK1 and ULK2 (unc-51 like autophagy activating kinase 1 and 2), homologs of the yeast autophagy related (Atg) 1, are crucial kinases at the initiation of autophagy.Citation20,21 ATG4B and ATG4D are both homologs of the yeast ATG4 and make the initial cleavage of MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3 β) family members, a marker of autophagy and an important component of phagophore elongation and closure.Citation20,22 To assess whether AR could also regulate these genes in patients, we leveraged a previously published, curated AR gene signature of identified AR target genes (genes that were increased in response to androgens and modulated by AR antagonists).Citation23 Using an informatics approach, we determined that this AR gene signature positively correlated with increased mRNA transcript levels of ATG4B, ATG4D, ULK1, and ULK2 in a well-known clinical prostate cancer cohortCitation24 (), suggesting AR may also regulate the expression of these core autophagy genes in patients.
Figure 1. Androgens increase the expression of a subset of core autophagy genes in prostate cancer. (A) LNCaP and VCaP cells were treated with vehicle (ethanol) or 2 different concentrations (100 pM or 10 nM) of the synthetic androgen R1881 for 24 or 72 h. Cells were then harvested and assayed for mRNA levels using a curated qPCR-based array of core autophagy genes and normalized to RPLP0 mRNA levels. Duplicate samples for each condition are shown. (B) Validation (qPCR) in biological triplicate of results shown in (A) confirming the androgen induction of ATG4B, ATG4D, ULK1 and ULK2. *, significant (P < 0.05) changes from vehicle. (C) Four genes (ATG4B, ATG4D, ULK1 and ULK2) were confirmed with western blot. Cells were treated with increasing concentrations of R1881 (LNCaP: 0, 0.1, 1 and 10 nM shown; VCaP: 0, 0.1 and 10 nM shown). A summary of these data with the full dose response (0, 0.1, 1 and 10 nM) and statistical analyses of the experimental repeats is presented in Table S1. (D) A gene signature of the 4 autophagy genes (ATG4B, ATG4D, ULK1, ULK2) correlated significantly with a previously described AR gene signatureCitation23 in prostate cancer patients from the Taylor et alCitation24 clinical cohort. Similar results were obtained using additional AR activity signatures across multiple clinical cohorts.
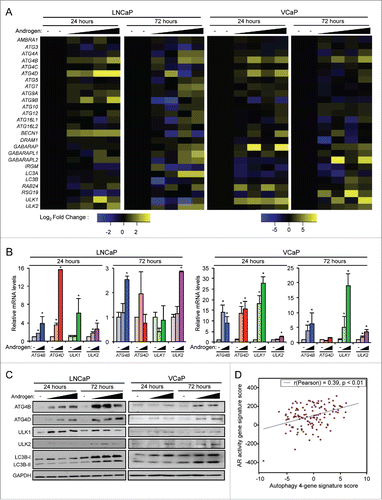
Significant increases in ATG4B and ULK2 mRNA levels were observed as early as 1 h post-androgen treatment, whereas ATG4D and ULK1 transcript levels were significantly elevated by 6 h after androgen exposure (). These rapid inductions indicated AR may increase the transcription of these genes. To test this, we first treated prostate cancer cells for just 8 h with androgens in the presence or absence of actinomycin D, an inhibitor of transcription, and assessed the expression of FKBP5/FKBP51 (a known transcriptional target of AR), ATG4B, ATG4D, ULK1 and ULK2. Similar to the results with FKBP5, actinomycin D blocked the androgen-mediated expression of ATG4B, ATG4D, ULK1 and ULK2 (). To determine whether these genes could be primary or secondary AR targets, we next treated prostate cancer cells for 16 h with androgens in the presence or absence of the translational inhibitor cycloheximide and assessed the expression of ATG4B, ATG4D, ULK1 and ULK2. Similar to FKBP5, a gene that is at least in part directly regulated by AR, but in contrast to a previously described secondary AR target gene, CXCR4, cycloheximide did not fully block androgen-mediated ATG4B, ATG4D, or ULK1 expression ().Citation25 In addition, cycloheximide had no effect on androgen-mediated ULK2 expression (). These cycloheximide studies further suggested that AR may directly regulate ULK2 transcription and possibly could regulate ATG4B, ATG4D and ULK1 expression through a combination of direct and/or indirect mechanisms.
Figure 2. ATG4B, ATG4D, ULK1, and ULK2 are transcriptional targets of AR. (A) LNCaP cells were treated with vehicle (ethanol) or androgen (100 pM R1881) for the indicated times before RNA was collected and subjected to qPCR. Data are normalized to RPLP0 and expressed as mean fold induction ± SE. *, significant (P < 0.05) changes from vehicle. (B) LNCaP cells were treated for 8 h with vehicle or androgen ± 1 μg/ml actinomycin D. FKBP5 is a known transcriptional target of AR.Citation25 Data are normalized to RPLP0 and expressed as mean fold induction + SE. *, significant (P < 0.05) changes from vehicle. (C) LNCaP cells were treated for 16 h with vehicle or androgen (100 pM R1881) ± 1 μg/μl cycloheximide. FKBP5 is a direct transcriptional target of AR, CXCR4 is an indirect transcriptional target of AR.Citation25 Data are normalized to RPLP0 and expressed as mean fold induction + SE. *, significant (P < 0.05) changes from vehicle. (D) ChIP-Seq tracks of LNCaP cells treated with vehicle or DHT for 2 h. AR binding sites in the intronic regions of ULK1 and ULK2 are highlighted. Similar data for VCaP and C4–2B cells are presented in Fig. S1. (E) Various enhancer luciferase reporter constructs including those containing the potential AR binding sites identified in (D) were transfected into LNCaP cells and treated overnight with an androgen (R1881) dose response (0, 0.1, 1, and 10 nM). After treatment, cells were harvested and assayed for luciferase activity. Luciferase values were normalized to the β-galactosidase control. Data are the mean relative light units (RLUs) + SEM for one representative experiment conducted in triplicate (n = 3). *, significant (P < 0.05) changes from vehicle-treated cells.
Mining of existing chromatin-immunoprecipation sequencing (ChIP-Seq) data sets from several prostate cancer cell models indicated that AR directly bound to intronic regions of ULK1 and ULK2 in the presence of DHT ( and Fig. S1).Citation26,27 Despite the observed induction of ATG4B and ATG4D (), interestingly, no AR binding sites were located within the ATG4B or ATG4D genes or within 50 kb upstream or downstream of either gene (data not shown). However, this finding was not surprising given that many NR response elements are often located far away (sometimes even on different chromosomes) from the genes they regulate.Citation28,29 To confirm which, if any, of the identified AR bindings sites were functional androgen receptor elements and thus primary AR targets, we cloned out the genomic regions surrounding the identified binding sites and tested their ability to confer androgen responsiveness to an enhancerless luciferase reporter gene. Interestingly, while the single AR binding site identified within the ULK1 gene was not androgen-responsive, it did display remarkably high basal expression (note Y-axis scales, compare with negative/empty vector control; ). This suggests that either a) ULK1 may not be a primary AR target or b) perhaps some other, more complicated mechanism of regulation is occurring here that confounds the reporter gene assays (e.g., additional chromatin remodeling, etc). When we tested the 3 identified AR binding sites located within the ULK2 gene, we determined that the third binding site (ARBS 3) conferred androgen responsiveness, indicating that ULK2 is a direct AR target. Thus, taken together these data suggest that all 4 genes are transcriptional targets of AR and that, at a minimum, ULK2 is a primary AR target gene.
ATG4B, ATG4D, ULK1 and ULK2 are necessary for maximal androgen-mediated autophagy and cell proliferation and also correlate with poor patient prognosis
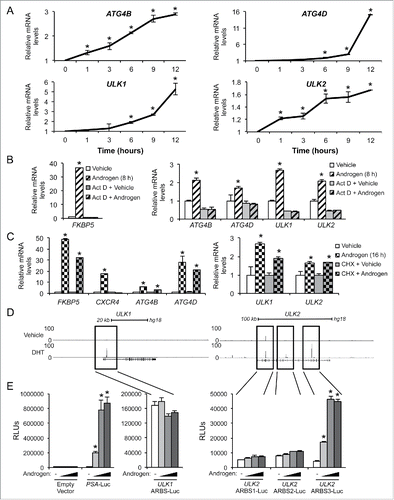
To assess whether the increases in these androgen-mediated genes functionally affected autophagy, we quantified the conversion of LC3B-I to LC3B-II, a canonical marker of autophagy.Citation30 As previously shown using immunoblot analysis,Citation19 androgen treatment increased conversion to LC3B-II ( and Fig. S2). Two different siRNAs were then used to target 4 genes: ATG4B, ATG4D, ULK1 and ULK2 ( and Figs. S2 and S3). In each case, knockdown of the 4 genes resulted in decreased androgen-mediated LC3B-II conversion ( and Figs. S2 and S3). To determine whether these androgen-mediated effects increased autophagic flux, we performed a similar experiment to that described in but this time treated cells in the presence or absence of chloroquine, an inhibitor of lysosomal function and autophagic flux. Here, treatment with androgens in the presence of chloroquine lead to a further increase in LC3B-II levels ( and Table S2), confirming our previous observation that androgens promote autophagic flux.Citation19 We next simultaneously knocked down ATG4B and ATG4D or ULK1 and ULK2 expression to account for any potential compensation and determined that these genes were indeed required for maximal androgen-mediated autophagic flux ( and Table S2). When expressed within the cell, the GFP-LC3B fusion protein in the pre-lipidated, LC3B-I form appears throughout the cytoplasm as a diffuse GFP signal. However, once the GFP-LC3B has had the phosphatidylethanolamine (PE) moiety attached, it will locate to the growing phagophore and be visible as GFP punctate shapes.Citation30 Using previously described hormone-sensitive LNCaP cells stably expressing GFP-LC3B,Citation19 we transfected cells with siRNAs targeting scramble control alone or with siRNAs jointly targeting ATG4B and ATG4D or ULK1 and ULK2 to again account for any additional feedback mechanisms. We followed the siRNA transfections with treatment ± androgens and then assessed GFP-LC3B puncta formation. Similar to what we have previously reported,Citation19 we observed an increase in the number of GFP-positive puncta per cell following androgen treatment in comparison to vehicle control (). In addition, siRNA-mediated knockdown of ATG4B and ATG4D or ULK1 and ULK2 resulted in a significantly reduced number of GFP-positive puncta per cell, further indicating that ATG4B, ATG4D, ULK1, and ULK2 are necessary for maximal androgen-mediated increases in autophagy. Importantly, knockdown of these 4 core autophagy genes resulted in decreased androgen-mediated prostate cancer growth ( and Fig. S4). Interestingly, the androgen-mediated increase in cell numbers appeared to be due to increased proliferation rather than changes in cell survival, effects that were decreased following ATG4 or ULK knockdown (). Conversely, overexpression of each of these 4 autophagy genes, alone or in combination, was sufficient to increase prostate cancer cell growth independent of AR activity ( and Fig. S5). When we compared the protein levels, hormone-sensitive cell lines, LNCaP and CWR22, expressed lower levels of ATG4B (LNCaP), ATG4D (CWR22), ULK1 (LNCaP), ULK2 (LNCaP and CWR22) and LC3B-II (LNCaP and CWR22) than their more aggressive CRPC derivatives, C4–2 and 22Rv1 (). Moreover, increased expression of any of these 4 genes in clinical samples correlated with poor patient prognosis ().
Figure 3. ATG4B, ATG4D, ULK1, and ULK2 are necessary for maximal androgen-mediated autophagy and proliferation. (A) LNCaP cells were transfected with siRNAs and then treated with vehicle (ethanol) or androgen (R1881) for 72 h before cells were harvested and cell lysates were subjected to immunoblot analysis. Densitometry results for these data with statistical analyses from experimental repeats are presented in Fig. S2. (B) LNCaP cells were transfected with siRNAs as in (A) except this time siRNAs were pooled to maximize knockdowns. Cells were then treated with vehicle (ethanol) or androgen (R1881) ± 20 μM chloroquine for 72 h and then subjected to immunoblot analysis as in (A). Densitometry results for these data with statistical analyses from experimental repeats are presented in Table S2. (C) LNCaP cells stably expressing eGFP-LC3B were transfected with siRNAs and then treated with vehicle (ethanol) or androgen (R1881) for 72 h before cells were fixed and stained with DAPI. Experiments were performed in triplicate and 10 random images from each treatment group were taken. One representative image of each is shown. Quantification of eGFP-LC3B puncta/nuclei is shown in the graph on the right. *, significant (P < 0.05) changes from vehicle-treated cells. #, significant (P < 0.05) changes from siControl-transfected cells. (D) LNCaP cells were transfected with siControl or siRNAs directed against ATG4B and ATG4D or ULK1 and ULK2. Transfected cells were then treated with vehicle or androgen (R1881) for 7 d. Cells were then lysed, and the relative number of cells was quantified using a fluorescent DNA-binding dye. Results are expressed as mean relative fold induction + SE. *, significant (P < 0.05) changes from siControl + vehicle. (E) LNCaP cells were transfected as in (D) and treated with vehicle, androgen (R1881) or paclitaxel (positive control) for 72 h. Cells were then subjected to PI staining and subsequent FACS analysis to assess sub G0/G1 DNA levels. #, significant (P < 0.05) changes from siControl-transfected cells. No significant androgen-mediated effects were observed. (F) LNCaP cells were transfected and treated as in (E). Cells were then subjected to PI staining and FACS analysis to assess S phase DNA levels. *, significant (P < 0.05) changes from vehicle-treated cells. #, significant (P < 0.05) changes from siControl-transfected cells. (G) Untreated LNCaP cells were transfected with mock or the indicated mammalian expression vectors for 3 d. Cells were then lysed and subjected to western blot analysis to confirm overexpression (Fig. S5) or the relative number of cells was quantified as in (E). *, significant (P < 0.05) changes from empty vector (Control). (H) Hormone-sensitive LNCaP and CWR22 cells, and their respective CRPC derivatives, C4–2 and 22Rv1, were probed for basal ATG4B, ATG4D, ULK1, ULK2 and LC3B protein levels. (I) The AR-regulated autophagy gene signature of ATG4B, ATG4D, ULK1 and ULK2 was subjected to Kaplan-Meier analysis using clinical data from The Cancer Genome Atlas (TCGA).Citation63
AR increases TFEB expression and potentiates its activity
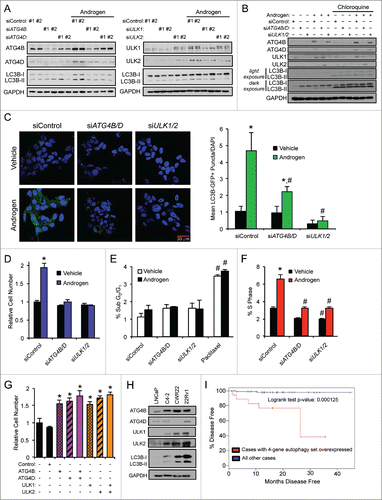
The above 4 identified core autophagy genes (ATG4B, ATG4D, ULK1, ULK2) all have defined roles in the early steps of autophagy.Citation31 Given our previous data demonstrating that androgens not only promote the initial steps of autophagy, but also autophagic flux,Citation19 we could not rule out the possibility that AR may also regulate the later steps of the process. In this regard, it was of interest to us that AR had recently been shown to exogenously interact with TFEB (transcription factor EB), a master regulator of lysosomal function and biogenesis, in HEK 293 cells.Citation32 While we were not able to detect an endogenous interaction in prostate cancer cell models, we did observe an increase in TFEB mRNA and protein levels in the hormone-sensitive LNCaP cells in response to androgens ( and Fig. S6). Furthermore, siRNA-mediated knockdown of AR in CRPC C4–2 cells, a CRPC derivative of LNCaP cells, significantly reduced TFEB protein levels (). To mechanistically understand how AR was regulating TFEB expression, we analyzed ChIP-Seq data from multiple prostate cancer cell models. This allowed us to determine that AR was strongly recruited to intron 1 of TFEB as well as an enhancer region ∼12 kb upstream of the gene, indicating that TFEB is a direct transcriptional target of AR ( and Fig. S7). Accordingly, similar to the primary AR transcriptional target FKBP5 but in contrast to the secondary AR target CXCR4,Citation25 cycloheximide treatment did not abolish androgen-mediated TFEB expression (), further suggesting that TFEB may at least in part be directly regulated by AR. To confirm this, we cloned out the genomic regions surrounding the 2 AR binding sites identified in and determined that they were sufficient to confer androgen responsiveness to an enhancerless luciferase reporter gene construct, indicating that TFEB is a direct AR target gene (). Interestingly, similar to what we observed for the other AR-regulated autophagy genes, hormone-sensitive cell lines, LNCaP and CWR22, expressed lower protein levels of TFEB than their more aggressive CRPC derivatives, C4–2 and 22Rv1 (). In support of this finding, expression of TFEB alone tracked with metastasis and trended with increased Gleason score (Fig. S8).Citation24
Figure 4. AR stimulates TFEB expression and activity. (A) LNCaP cells treated with vehicle (ethanol) or androgen (100 pM or 10 nM R1881) for 24 or 72 h and then subjected to qPCR analysis. (B) LNCaP cells treated for 24 h with an androgen dose response (0, 0.1, 1 or 10 nM R1881) and then subjected to immunoblot analysis. Left, representative images. Right, densitometry (n = 3). *, significant (P < 0.05) changes from vehicle. (C) C4–2, a castration-resistant prostate cancer (CRPC) cell line-derivative of LNCaPs, were transfected with siRNAs targeting AR or control, lysed and probed for TFEB. (D) ChIP-Seq tracks of LNCaP cells treated with vehicle or DHT 2 h. AR binding sites in the intronic region and direct upstream region of TFEB are highlighted. Similar data for VCaP and C4–2B cells are presented in Fig. S7. (E) LNCaP cells were treated with vehicle or androgen (R1881) ± 1 μg/μl cycloheximide and subjected to qPCR analysis as in . FKBP5 is a known direct transcriptional target of AR, while CXCR4 is a known indirect AR transcriptional target.Citation25 Data are normalized to RPLP0 and expressed as mean fold induction ± SE. *, significant (P < 0.05) changes from vehicle. (F) Various enhancer luciferase reporter constructs including those containing the potential AR binding sites identified in (D) were transfected into LNCaP cells and treated overnight with an androgen (R1881) dose response (0, 0.1, 1, and 10 nM). After treatment, cells were harvested and assayed for luciferase activity. Luciferase values were normalized to β-galactosidase control. Data are the mean relative light units (RLUs) + SEM for one representative experiment conducted in triplicate (n = 3). *, significant (P < 0.05) changes from vehicle-treated cells. (G) Hormone-sensitive LNCaP and CWR22 cells, and their respective CRPC derivatives, C4–2 and 22Rv1, were probed for basal TFEB protein levels. (H) LNCaP cells were transfected with a KLK3/PSA-luciferase reporter plasmid or a 4X-CLEAR (TFEB DNA binding sequence)-luciferase reporter plasmid in conjunction with a CMV-β-galactosidase control plasmid. After transfection, cells were treated with vehicle or increasing concentrations of R1881 for 24 h. Cells were harvested and assayed for luciferase activity. All luciferase values were normalized to β-galactosidase transfection controls. Data are expressed as normalized relative light units (RLUs) ± SE. *, significant (P < 0.05) changes from vehicle. (I) VCaP cells were treated as indicated and subjected to qPCR analysis to quantitate the mRNAs levels of 6 established transcriptional targets of TFEB34,69: ATP6AP1, LAMP1, PPARGC1A, GLB1, MCOLN1, and SQSTM1.
We next assessed the functional activity of this androgen-mediated increase in TFEB expression in prostate cancer cells. To do so, we first took advantage of a luciferase reporter plasmid that harbored 4 upstream TFEB binding sites in tandem (4X-coordinated lysosomal expression and regulation sequence [CLEAR]).Citation32 Similar to a well-characterized AR target enhancer of KLK3, the gene that codes for prostate-specific antigen (PSA),Citation33 androgens increased 4X-CLEAR-mediated luciferase activity in a dose-dependent manner (). We then checked whether androgen treatment could increase the expression of endogenous TFEB target genes, which have been well curated.Citation34 Androgen treatment led to an increase in the mRNA levels of all 6 TFEB target genes (ATP6AP1, LAMP1A, PPARGC1A, GLB1, MCOLN1, and SQSTM1) that were checked (). Of note and as previously described, several of these TFEB-regulated genes (e.g., SQSTM1) have well known functions in autophagy.Citation35 Taken together, our data indicate that AR regulates the expression and transcriptional activity of TFEB in prostate cancer cells.
TFEB is required for maximal androgen-mediated flux through autophagy
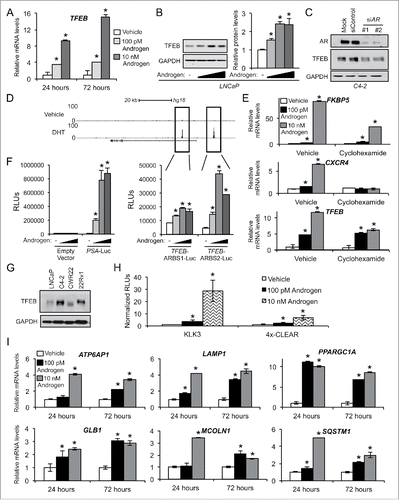
To test the functional significance of the androgen-mediated TFEB induction, we knocked down TFEB expression using 2 different siRNAs and then treated prostate cancer cells ± androgens. Knockdown of TFEB resulted in decreased androgen-mediated LC3B-II accumulation in the hormone-sensitive cell line LNCaP and decreased basal LC3B-II accumulation in the CRPC cell line, C4–2 (). These findings are in agreement with earlier reports demonstrating that TFEB regulates multiple stages of autophagy.Citation35 Since TFEB has also been reported to regulate several autophagy genes,Citation35 we also tested whether TFEB was required for the androgen-mediated expression of ATG4B, ATG4D, ULK1 or ULK2. While TFEB was required for LC3B conversion, it did not appear necessary for the expression of ATG4B, ATG4D, ULK1 or ULK2 at the protein () or mRNA level () with the lone exception of ATG4D mRNA expression (). However, as can be seen in , depletion of TFEB did not appear to translate into significant changes in ATG4D protein levels. These results are consistent with previous work that suggested a role for TFEB in the regulation of ATG4D mRNA levels but did not detect regulation of ATG4D at the protein level.Citation35 That seminal study,Citation35 similar to our data presented here, also failed to detect any regulation of ATG4B, ULK1 or ULK2 expression by TFEB at either the mRNA or protein level.
Figure 5. TFEB is required for maximal androgen-mediated autophagic flux, cell proliferation and lysosomal biogenesis. (A) LNCaP or CRPC C4–2 cells were transfected with siRNAs and then treated with vehicle (ethanol) or androgen (R1881) for 72 h before cells were lysed and subjected to immunoblot analysis. (B) LNCaP cells were transfected and treated as in (A) and then subjected to qPCR analysis. *, significant (P < 0.05) changes from vehicle-treated cells. #, significant (P < 0.05) changes from siControl-transfected cells. (C) LNCaP cells transiently expressing a mCherry-GFP-LC3B construct were transfected with siRNAs and then treated with vehicle or androgen (R1881) for 72 h before cells were fixed, nuclei stained with DAPI (blue) and imaged. Experiments were performed in triplicate and 10 random images from each treatment group were taken. One representative image of each is shown. Quantification of puncta/cell is shown in the graph on the right. *, significant (P < 0.05) changes from vehicle-treated cells. #, significant (P < 0.05) changes from siControl-transfected cells. (D) LNCaP cells were transfected with siRNAs targeting scramble control or TFEB. 24 h post-transfection, cells were treated ± androgen and cultured for 72 h, fixed and then stained with LysoTracker Red (red) and DAPI (blue). Total lysosomal area (red) per image was taken as a fraction of nuclear area (blue) using an ImageJ script. *, significant (P < 0.05) changes from vehicle-treated cells. #, significant (P < 0.05) changes from siControl-transfected cells. (E) LNCaP cells were transfected with siRNAs directed against scramble control or TFEB and then treated with vehicle or androgen (R1881) for 7 d. Relative cell numbers were then quantified as in . Results are expressed as mean relative fold induction + SE. *, significant (P < 0.05) changes. (F) LNCaP cells were transfected as in (E) and treated with vehicle, androgen (R1881) or paclitaxel (positive control) for 72 h. Cells were then subjected to PI staining and subsequent FACS analysis to assess sub G0/G1 DNA levels. #, significant (P < 0.05) changes from siControl-transfected cells. No significant androgen-mediated effects were observed. (G) LNCaP cells were transfected and treated as in (F). Cells were then subjected to PI staining and FACS analysis to assess S phase DNA levels. *, significant (P < 0.05) changes from vehicle-treated cells. #, significant (P < 0.05) changes from siControl-transfected cells. (H) Untreated LNCaP cells were transfected with mock or the indicated mammalian expression vectors for 3 d. Cells were then lysed and subjected to western blot analysis to confirm overexpression (Figs. S5 and S10) or the relative number of cells was quantified as in (E). *, significant (P < 0.05) changes from empty vector (Control).
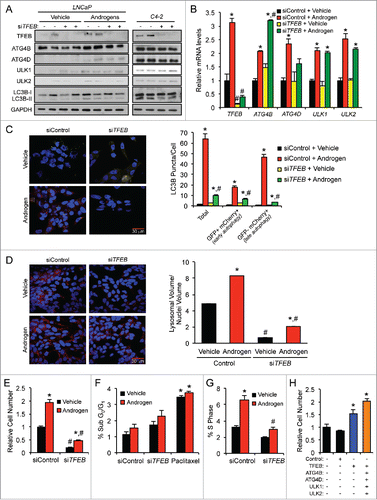
To study the flux through autophagy, a mCherry-GFP-LC3B plasmid was used.Citation36,37 This approach was used instead of the lysosomal block (e.g., ) because, as previously reported,Citation38 inhibition of lysosomal function leads to a decrease in total TFEB levels (Fig. S9), complicating data interpretation. Thus, we chose to examine LC3B subcellular relocalization and GFP quenching as a measure for autophagic flux when altering TFEB levels. When this LC3B-II fusion protein is associated with the phagophore or autophagosome, both the mCherry and GFP will be active resulting in a merged yellow signal. Once fused with the lysosome, the GFP signal will be lost due to the presence of the acidic environment, resulting in only a detectable mCherry-LC3B-II signal. Similar to what we have previously reported,Citation19 androgen treatment resulted in increased LC3B-II overall punctate signal that was largely due to the increased mCherry-LC3B-II levels (). Knockdown of TFEB resulted in decreased total LC3B-II levels as well as mCherry-LC3B-II levels. These data indicate that the AR transcriptional target TFEB is required for androgen-mediated autophagic flux.
Since TFEB is a master regulator of lysosomal biogenesis and lysosomes are required for the completion of autophagy, we hypothesized that TFEB might also regulate autophagic flux by modulating the amount of lysosomes per cell. To test this, we again silenced TFEB using siRNAs in the presence or absence of androgens and assessed the total lysosomal area per cell by fixing and costaining cells with LysoTracker Red and 4′,6-diamidino-2-phenylindole (DAPI). We then quantitated the lysosomal area per cell in an unbiased manner using a customized ImageJ script that allowed us to quantify in a high-throughput manner the total lysosomal area (LysoTracker) normalized to the total area of nuclei (DAPI) for multiple captured fields. As expected, androgens dramatically increased the amount of lysosomes per cell (). Additionally, we observed a robust decrease in both the basal and androgen-mediated lysosomal area when TFEB was knocked down, indicating that the androgen-mediated increase in lysosomal area was mediated through TFEB.
As with the silencing of ATG4B, ATG4D, ULK1 and ULK2 ( and Fig. S4) as well as ATG7,Citation19 knockdown of TFEB significantly decreased androgen-mediated growth (). This reduction in cell numbers again appeared to be due to changes in proliferation rather than rates of cell survival/apoptosis (). Importantly, overexpression of TFEB alone or in combination with the other AR-regulated autophagy genes was sufficient to increase cell proliferation independent of AR activity ( and Fig. S10).
Androgen-induced core autophagy genes and TFEB clinically correlate with AR activity and metastasis
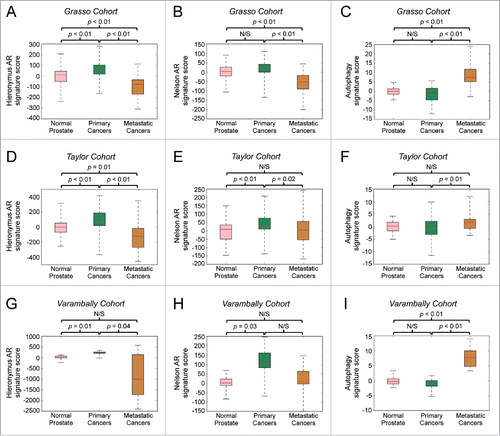
To understand the potential translational significance of our findings, we analyzed existing clinical data sets to determine whether expression of the identified genes of interest 1) correlated with AR activity in cohorts of primary and metastatic cancer and 2) tracked with disease progression. Using the same curated AR gene signature used in ,Citation24 we first determined that general AR activity positively correlated with a 5-gene signature consisting of ATG4B, ATG4D, ULK1, ULK2, and TFEB in primary and metastatic prostate cancer cohortsCitation24 (). We next assessed the expression of the above-mentioned (Hieronymus) AR signatureCitation23 and, to minimize bias/dependence on a single defined AR signature, a second AR activity gene signature (Nelson AR signature)Citation39 in samples from normal prostates, primary prostate cancers and metastatic prostate cancers in 3 separate clinical cohorts.Citation24,40,41 As expected, expression of the 2 curated, general AR gene signatures was increased and positively correlated with progression from normal prostate to primary prostate cancer in all 3 different clinical cohorts ().Citation23,24,39-41 However, both of these AR gene signatures were significantly decreased in the same clinical cohorts in metastatic cancers compared with primary prostate cancers (). Given that it is now known that AR remains a central driver of metastatic prostate cancer,Citation6,24,42 these data confirm that AR is reprogrammed during disease progression. As such, either new AR target genes or a subset of existing AR target genes drives advanced disease progression. This would be different from the majority of classically defined AR-regulated genes, most of which likely are left over from development and/or basic physiology. In this regard, it was of note that when we then tracked the expression of the specific AR-regulated autophagy 5 gene signature (ATG4B, ATG4D, ULK1, ULK2, TFEB), we discovered that while it did not correlate with cancer initiation, it was consistently elevated in metastatic cancers in all 3 clinical cohorts examined, suggesting this subset of genes contributes to clinical disease progression ().
Table 1. Correlation of AR activity with an autophagy gene signature in patients. The expression of a previously annotated AR activity gene signatureCitation23 was correlated with an autophagy gene signature consisting of ATG4B, ATG4D, ULK1, ULK2 and TFEB in primary cancers and metastatic cancers from the Taylor et al. 201024 prostate cancer clinical data set.
Figure 6. An autophagy gene signature positively correlates with the transition to prostate cancer metastasis, whereas general AR activity gene signatures positively correlate with cancer initiation but not metastatic progression. Bioinformatic analysis of 3 separate prostate cancer clinical cohorts (A-C: Grasso et al., 201240; D-F: Taylor et al., 201024; G-I: Varambally et al., 200541). The expression of 2 previously described AR activity gene signatures (Hieronymus et al., 200623: A, D, G and Nelson et al., 200239: B, E, H) or an autophagy gene signature consisting of ATG4B, ATG4D, ULK1, ULK2 and TFEB were compared between samples from normal prostates, primary cancers and metastatic cancers in each cohort.
Discussion
Our previous study highlighted the importance of AR-mediated autophagy in prostate cancer.Citation19 Because of the sustained induction of autophagy by androgens, we suspected that AR could modulate the process via multiple mechanisms of action. Here, we identified 4 core autophagy genes: ULK1, ULK2, ATG4B, and ATG4D, and a transcription factor, TFEB, that is a master regulator of lysosomal biogenesis and function, all of which are transcriptionally regulated by AR in prostate cancer. Knockdown of each of these genes resulted in a decrease in androgen-mediated autophagy and decreased prostate cancer cell proliferation (, and Fig. S4), indicating that autophagy can augment prostate tumor growth. In support of this, upregulation of these 5 genes, which correlated with AR activity in patient samples (), also tracked with progression to the advanced stages of the disease in multiple clinical cohorts ( and Fig. S8). Given that this autophagy gene signature correlated with cancer progression and not cancer initiation, these findings are consistent with previous reports suggesting contrasting roles for this process in different disease stages.Citation10,12,14-17,43 Further supporting what we have reported here, others have found that high ULK1 protein levels in primary prostate cancers positively correlated with increased biochemical reoccurrence (rising KLK3/PSA).Citation44 One of the challenges moving forward will be determining when autophagy is being stimulated and therefore should be targeted.
The lack of reliable, high-throughput clinical biomarkers of autophagic activity has been a major limitation in the cancer field.Citation45 To that end, our results here demonstrate that a commonly used biomarker of autophagic flux, SQSTM1/p62 protein turnover,Citation30 should be used with extreme caution and may not be a reliable marker of autophagy in prostate cancer. This is because SQSTM1 is regulated by AR in a TFEB-mediated manner (). Hence, SQSTM1 turnover may not be a reliable marker of autophagy in prostate cancer because changes in SQSTM1 protein levels will likely be significantly confounded by effects on SQSTM1 gene expression. Alternatively, our elucidation of key genes upregulated in prostate cancer, suggest these 5 genes may have value as a surrogate marker of autophagic activity in a disease-specific context.
Targeting AR in metastatic CRPC remains a challenge in the clinic. Therefore, we rationalize that targeting the pathways downstream of AR could be an alternative and viable pharmacological option for men suffering from this aggressive disease stage. In this regard, an inhibitor of a gene or protein that is specifically upregulated in prostate cancer would be desirable. Several ATG4 inhibitors are currently under development including an ATG4B antagonist that had promising effects in blocking autophagy and inhibiting osteosarcoma tumor formation in vivo.Citation46 Whether these antagonists will be beneficial for prostate cancer patients remains unknown.Citation46 Additionally, a small molecule inhibitor of ULK1 has recently been synthesized, but its efficacy in vivo in preclinical animal models of cancer has not yet been tested.Citation47
In agreement with what others have reported, our data support the hypothesis that autophagy is promoting not just cell survival but proliferation as well.Citation48-53 Our previous data implicated a role for altered lipid metabolism in this process.Citation19 However, it is entirely possible and perhaps likely that additional alterations contribute to net anabolic tumor processes. In this regard, several studies have demonstrated that an autophagy-mediated breakdown of proteins supplies cells with amino acids such as glutamine that can fuel cancer growth.Citation11,48,54,55 Whether such metabolic changes in lipid, amino acid and potentially sugar metabolism occur in prostate cancer is an area we are actively investigating.
In summary, we have outlined the mechanism by which AR upregulates the transcription of autophagy genes required for maximal cancer cell growth. This work significantly adds to the knowledge of prostate cancer, linking for the first time a role for AR-mediated autophagy in metastatic lesions. This work also further confirms that despite AR's known role in metastatic CRPCs,Citation6,24,42 many classically defined AR target genes likely do not contribute to prostate cancer progression. Accordingly, general AR activity gene signatures, while elevated during cancer initiation, were actually decreased during the transition to advanced, metastatic prostate cancer (). These findings contrasted with what was observed for the 5-gene autophagy signature that was not altered during cancer initiation but was significantly upregulated in metastatic prostate cancers (). Taken together, the data suggest these autophagy genes may represent a subset of AR-regulated target genes driving disease progression. Importantly, this work also highlights the potential for the rational development of improved therapeutic agents targeting autophagy, and perhaps more specifically distinct steps of autophagy, for the treatment of advanced prostate cancer.
Materials and methods
Cell culture and reagents
LNCaP, 22Rv1, PC-3, and HEK293 cell lines were obtained from American Type Culture Collection (CRL-1740, CRL-2505, CRL-1435, CRL-1573). C4–2 and CWR22 cell lines were from Donald McDonnell (Duke University School of Medicine). Cells were maintained and validated as previously described.Citation56-59 In brief, cell lines are validated biannually by genotyping and mycoplasma-free confirmation through the use of a PCR-based assay. The synthetic androgen methyltrienolone (R1881) was purchased from PerkinElmer. Actinomycin D (A1410), cycloheximide (C7698) and anti-GAPDH (glyceraldehyde 3-phosphate dehydrogenase) antibody (G8795) were obtained from Sigma. Anti-ULK2 (ab97695), and anti-ATG4D (ab137621) antibodies were purchased from Abcam. Anti-ULK1 (4773), anti-ATG4B (13507), anti-LC3B (2775), and anti-TFEB (4240) antibodies were purchased from Cell Signaling Technology. Agarose A/G beads (sc-2003) and anti-AR antibody (sc-816) were from Santa Cruz Biotechnology. Enhanced GFP-LC3B and mCherry-GFP-LC3B constructs have been previously described.Citation19 pCMV-Myc-hATG4B and pCMV-Myc-hATG4D were from Sino Biological Inc. (HG20407-NM and HG15537-NM). pcDNA3.1 is from ThermoFisher Scientific. pcDNA3.1-hULK1 was a gift from Mondira Kundu (St. Jude Children's Research Hospital). pRK5-myc-hULK2 (Addgene plasmid 31966) was a gift from Do-Hyung Kim (University of Minnesota).Citation60 pcDNA3.1-TFEB was a gift from Marco Sardiello (Baylor College of Medicine). Empty vector, ATG4B, ATG4D, ULK1, ULK2 and TFEB mammalian expression vectors were all driven by a CMV promoter.
RNA isolation, cDNA preparation, and quantitative RT-PCR (qPCR)
RNA isolation, cDNA preparation and qPCR were performed as previously described using RPLP0/36B4 as an internal control.Citation56 The sequences of the primers are listed in Table S3.
Immunoblot analysis
Immunoblotting was conducted as previously described.Citation19,61 Densitometry was performed using ImageJ software (National Institutes of Health) and samples were normalized to GAPDH (loading control).
Bioinformatic analyses of gene expression in clinical data sets
To analyze the relationship of core autophagy gene expression with patient prognosis we generated a Kaplan-Meier survival plot in March 2015 using data from The Cancer Genome Atlas (TCGA) available at www.cancergenome.nih.gov.Citation62,63 Prostate adenocarcinoma tumors with mRNA for ATG4B, ATG4D, ULK1 or ULK2 expression greater than 2-fold above the mean were compared with the remaining samples.
For the gene expression signature comparisons, gene expressions of human prostate cancer cohorts including Grasso et al., (GSE35988),Citation40 Taylor et al., (GSE21034),Citation24 and Varambally et al., (GSE3325),Citation41 were downloaded from GEO. Androgen-induced signatures (Hieronymus AR and Nelson AR) were generated from previously defined data.Citation23,39 For each of the signatures, an activity score for each sample in each cohort was generated as described previously.Citation24 Briefly, gene expressions of prostate cancer cohorts were converted z-scores with respect to normal samples. The activity score for each sample for a signature was evaluated by adding the z-scores of upregulated genes and subtracting the z-scores of downregulated genes. Differences between the distributions of activity scores between different subtypes were estimated using the t test (P < 0.05) as implemented in the Python statistical library SciPy. Correlation between pairs of gene signature activity scores were evaluated using the Pearson Correlation Coefficient as implemented in the Python statistical library SciPy.
Correlation of TFEB mRNA levels with clinical variables was done as previously describedCitation33 using the Taylor et al. data set.Citation24
Chromatin-immunoprecipitation-sequencing (ChIP-Seq)
ChIP-Seq analysis was performed as previously described.Citation26,27
Plasmid transfection and reporter gene assays
Unless otherwise noted, for all experiments cells were first steroid-starved for 72 h in phenol red-free medium containing 8% charcoal-stripped fetal bovine serum (Gemini Bio-Products, 100–119). Plasmids were then transfected using Lipofectamine 2000 (ThermoFisher Scientific, 11668019) according to the manufacturer's instructions. Cells were treated with hormones approximately 16 h before the assay. Luciferase and β-galactosidase (transfection normalization) activities were measured as previously described.Citation33 Each treatment was performed in triplicate and results are expressed as mean ± SE. Each experiment was repeated at least 3 times, with a representative experiment shown. The pGL4.26-ULK1 (ULK1-ARBS-Luc), pGL4.26-ULK2 (ULK2-ARBS1-Luc, ULK2-ARBS2-Luc, ULK2-ARBS3-Luc) and pGL4.26-TFEB (TFEB-ARBS1-Luc, TFEB-ARBS2-Luc) enhancer constructs were created by PCR amplifying sequences that encompassed the potential AR binding sites identified using ChIP-SeqCitation26,27 and shown in and and Figures S1 and S7. These fragments were then cloned into the pGL4.26 vector (Promega, E8441) using NheI and HindIII or XhoI restriction sites. All primers used for the creation of constructs are listed in Table S4. All sequences were confirmed using restriction digests and sequencing. The PSA-Luc and 4X-CLEAR reporter plasmids were previously described.Citation32,56
Small interfering RNA (siRNA) transfections
Stealth and Silencer Select siRNA (Life Technologies) transfections were performed as previously described,Citation33 with the exception that Silencer Select siRNAs were transfected at a final concentration of 10 nM. The sequences of the siRNAs are listed in Table S5.
Generation of stable cell lines
Cell lines stably expressing GFP-LC3B were previously described.Citation19,59
Fluorescence microscopy
Fluorescence microscopy and quantification were performed as previously described unless otherwise noted.Citation19,33,61,64
Cell proliferation assays
Cell proliferation assays were performed as previously described by measuring the cellular double-stranded DNA content using a Hoechst-based dye.Citation19,65
FACS analysis
The percentage of cells in different phases of the cell cycle was determined based on relative DNA content as assessed by flow cytometry analysis. LNCaP cells were transfected 24 h prior to treatment with androgens. After 72 h, cells were detached by incubating with 0.25% trypsin-EDTA, washed with phosphate-buffered saline (PBS; Corning, 21–040-CV) and fixed overnight in 70% ethanol at 4°C. Fixed cells were then centrifuged (100 × g, 5 min), washed 1x in PBS, resuspended in PBS containing RNase A and propidium iodide (PI) (50 μg/ml each; ThermoFisher, EN0531 and P1304MP) and analyzed on a Gallios Flow Cytometer (Beckman Coulter, Inc.). The percentages of sub-G1 population and cell cycle distribution were determined using the MULTICYCLE software program (Phoenix Flow Systems).
Co-immunoprecipiation assays
Co-immunoprecipiation assays were performed as previously described.Citation33,64
Lysosomal quantification
LNCaP cells were seeded on coverslips in charcoal-stripped media and allowed to adhere for 72 h before transfection and treatment. After the last treatment, 1 μl/ml of LysoTracker® Red (Life Technologies, L7528) was added to each well and allowed to incubate at 37°C for 30 min before cells were fixed, stained with DAPI and mounted onto slides for imaging. Each coverslip was repeated in triplicate and at least 10 randomly selected images per coverslip were taken. Images were analyzed using the open-source software ImageJ (Fiji package) via a customized script to enable automated high-throughput analysis. The script (macro) uses built-in functions provided by the ImageJ developer community. To begin, each image is split into the red, green and blue color channels, and then the red lysosomal (LysoTracker Red) and blue nuclear (DAPI) images are further analyzed to compute the lysosomal and nuclear areas, respectively. The background subtraction algorithm is first applied to separate the fluorescence signal from background noise,Citation66 and the lysosomal/nuclear regions are automatically segmented by applying the Renyi Entropy algorithm.Citation67 Next, each image is convolved with a median filter of 2-pixel radius to reduce any random pixel noise. In order to count nuclei, a hole-filling and erosion operations were executed sequentially followed by the watershed algorithmCitation68 to facilitate separation of any touching nuclei. Finally, individual/clusters of nuclei are counted using the Analyze Particles function of ImageJ, which also provides morphological features such as area, circularity and average and integrated intensity. The ratio of the lysosomal area to the nuclear area is also determined and results are exported to a text file, which can be further analyzed in any data processing software (e.g., Excel). The script is written to operate in the batch mode, which enables high-throughput analysis of multiple images in a given folder. To enable manual validation, the script also saves a copy of each analyzed image with segmented nuclei outlined, and an additional file with the numerical measurements of all images in the folder being analyzed.
Statistical analyses
Unless otherwise noted, 2-sample comparisons were performed using Student t tests. Multiple comparisons were performed using a one-way ANOVA and post hoc Dunnett's test with GraphPad Prism, Version 4 (GraphPad Software, Inc.). Statistically significant changes were determined at the P < 0.05 level.
Abbreviations
| AMBRA1 | = | autophagy and Beclin 1 regulator 1 |
| AR | = | androgen receptor |
| ATG | = | autophagy related |
| ATP6AP1 | = | ATPase H+ transporting accessory protein 1 |
| BECN1 | = | Beclin 1 |
| ChIP-Seq | = | chromatin-immunoprecipation sequencing |
| CLEAR | = | coordinated lysosomal expression and regulation |
| CRPC | = | castration-resistant prostate cancer |
| CXCR4 | = | C-X-C motif chemokine receptor 4 |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| DHT | = | dihydrotestosterone |
| DRAM1 | = | DNA damage regulated autophagy modulator 1 |
| FACS | = | fluorescence-activated cell sorting |
| FKBP5/FKBP51 | = | FK506 binding protein 5 |
| GABARAP | = | GABA type A receptor-associated protein |
| GABARAPL1/2 | = | GABA type A receptor associated protein like 1/2 |
| GAPDH | = | glyceraldehyde 3-phosphate dehydrogenase |
| GFP | = | green fluorescent protein |
| IRGM | = | immunity related GTPase M |
| LAMP1 | = | lysosomal associated membrane protein 1 |
| MAP1LC3B/LC3B | = | microtubule associated protein 1 light chain 3 β |
| NR | = | nuclear receptor |
| qPCR | = | quantitative reverse transcription polymerase chain reaction |
| R1881 | = | methyltrienolone |
| ROS | = | reactive oxygen species |
| SQSTM1 | = | sequestosome 1 |
| siRNA | = | small interfering RNA |
| TFEB | = | transcription factor EB |
| ULK | = | unc-51 like autophagy activating kinase |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplementary files
Download Zip (2.1 MB)Acknowledgments
We thank members of the Frigo laboratory for helpful discussions and editorial assistance during the preparation of the manuscript. We also thank Dr. Kathleen Gajewski and the University of Houston Biology & Biochemistry Imaging Core for help with microscopy.
Funding
This work was supported by a grant from the American Cancer Society (RSG-16–084–01 – TBE to D.E.F.), National Institutes of Health grants R01CA184208 and R03DK092434 (D.E.F.), R01AG033082 and R01NS100023 (A.R.L.) and funding from the Golfers Against Cancer (D.E.F.) and a University of Houston GEAR award (D.E.F.). This work was also partially supported by a CPRIT Proteomics and Metabolomics Core Facility Support Award RP120092 (C.C. and K.R.).
Related Research Data
References
- Koochekpour S. Androgen receptor signaling and mutations in prostate cancer. Asian J Androl 2010; 12:639-57; PMID:20711217; http://dx.doi.org/10.1038/aja.2010.89
- Heinlein CA, Chang C. Androgen receptor (AR) coregulators: an overview. Endocr Rev 2002; 23:175-200; PMID:11943742; http://dx.doi.org/10.1210/edrv.23.2.0460
- Knudsen KE, Scher HI. Starving the addiction: new opportunities for durable suppression of AR signaling in prostate cancer. Clin Cancer Res 2009; 15:4792-8; PMID:19638458; http://dx.doi.org/10.1158/1078-0432.CCR-08-2660
- Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev 2010; 24:1967-2000; PMID:20844012; http://dx.doi.org/10.1101/gad.1965810
- Society AC. Cancer Facts & Figures 2015. Atlanta: American Cancer Society, 2015.
- Augello MA, Den RB, Knudsen KE. AR function in promoting metastatic prostate cancer. Cancer Metastasis Rev 2014; 33:399-411; PMID:24425228; http://dx.doi.org/10.1007/s10555-013-9471-3
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; http://dx.doi.org/10.1146/annurev-genet-102808-114910
- Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, et al. Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 2010; 90:1383-435; PMID:20959619; http://dx.doi.org/10.1152/physrev.00030.2009
- Jin S, White E. Role of autophagy in cancer: management of metabolic stress. Autophagy 2007; 3:28-31; PMID:16969128; http://dx.doi.org/10.4161/auto.3269
- Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, et al. Autophagy in malignant transformation and cancer progression. EMBO J 2015; 34:856-80; PMID:25712477; http://dx.doi.org/10.15252/embj.201490784
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 2014; 4:914-27; PMID:24875857; http://dx.doi.org/10.1158/2159-8290.CD-14-0363
- Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nat Rev Cancer 2005; 5:726-34; PMID:16148885; http://dx.doi.org/10.1038/nrc1692
- Levy JM, Thompson JC, Griesinger AM, Amani V, Donson AM, Birks DK, Morgan MJ, Mirsky DM, Handler MH, Foreman NK, et al. Autophagy inhibition improves chemosensitivity in BRAF(V600E) brain tumors. Cancer Discov 2014; 4:773-80; PMID:24823863; http://dx.doi.org/10.1158/2159-8290.CD-14-0049
- Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat Rev Cancer 2007; 7:961-7; PMID:17972889; http://dx.doi.org/10.1038/nrc2254
- Thorburn A, Thamm DH, Gustafson DL. Autophagy and cancer therapy. Mol Pharmacol 2014; 85:830-8; PMID:24574520; http://dx.doi.org/10.1124/mol.114.091850
- White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res 2009; 15:5308-16; PMID:19706824; http://dx.doi.org/10.1158/1078-0432.CCR-07-5023
- Hippert MM, O'Toole PS, Thorburn A. Autophagy in cancer: good, bad, or both? Cancer Res 2006; 66:9349-51; PMID:17018585; http://dx.doi.org/10.1158/0008-5472.CAN-06-1597
- Amaravadi RK, Lippincott-Schwartz J, Yin XM, Weiss WA, Takebe N, Timmer W, DiPaola RS, Lotze MT, White E. Principles and current strategies for targeting autophagy for cancer treatment. Clin Cancer Res 2011; 17:654-66; PMID:21325294; http://dx.doi.org/10.1158/1078-0432.CCR-10-2634
- Shi Y, Han JJ, Tennakoon JB, Mehta FF, Merchant FA, Burns AR, Howe MK, McDonnell DP, Frigo DE. Androgens promote prostate cancer cell growth through induction of autophagy. Mol Endocrinol 2013; 27:280-95; PMID:23250485; http://dx.doi.org/10.1210/me.2012-1260
- Klionsky DJ, Codogno P, Cuervo AM, Deretic V, Elazar Z, Fueyo-Margareto J, Gewirtz DA, Kroemer G, Levine B, Mizushima N, et al. A comprehensive glossary of autophagy-related molecules and processes. Autophagy 2010; 6:438-48; PMID:20484971; http://dx.doi.org/10.4161/auto.6.4.12244
- Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15:741-50; PMID:23685627; http://dx.doi.org/10.1038/ncb2757
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 2008; 9:859-64; PMID:18704115; http://dx.doi.org/10.1038/embor.2008.163
- Hieronymus H, Lamb J, Ross KN, Peng XP, Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, et al. Gene expression signature-based chemical genomic prediction identifies a novel class of HSP90 pathway modulators. Cancer Cell 2006; 10:321-30; PMID:17010675; http://dx.doi.org/10.1016/j.ccr.2006.09.005
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010; 18:11-22; PMID:20579941; http://dx.doi.org/10.1016/j.ccr.2010.05.026
- Frigo DE, Sherk AB, Wittmann BM, Norris JD, Wang Q, Joseph JD, Toner AP, Brown M, McDonnell DP. Induction of Kruppel-like factor 5 expression by androgens results in increased CXCR4-dependent migration of prostate cancer cells in vitro. Mol Endocrinol 2009; 23:1385-96; PMID:19460858; http://dx.doi.org/10.1210/me.2009-0010
- Chng KR, Chang CW, Tan SK, Yang C, Hong SZ, Sng NY, Cheung E. A transcriptional repressor co-regulatory network governing androgen response in prostate cancers. Embo j 2012; 31:2810-23; PMID:22531786; http://dx.doi.org/10.1038/emboj.2012.112
- Tan PY, Chang CW, Chng KR, Wansa KD, Sung WK, Cheung E. Integration of regulatory networks by NKX3-1 promotes androgen-dependent prostate cancer survival. Mol Cell Biol 2012; 32:399-414; PMID:22083957; http://dx.doi.org/10.1128/MCB.05958-11
- Carroll JS, Liu XS, Brodsky AS, Li W, Meyer CA, Szary AJ, Eeckhoute J, Shao W, Hestermann EV, Geistlinger TR, et al. Chromosome-wide mapping of estrogen receptor binding reveals long-range regulation requiring the forkhead protein FoxA1. Cell 2005; 122:33-43; PMID:16009131; http://dx.doi.org/10.1016/j.cell.2005.05.008
- Liu MH, Cheung E. Estrogen receptor-mediated long-range chromatin interactions and transcription in breast cancer. Mol Cell Endocrinol 2014; 382:624-32; PMID:24071518; http://dx.doi.org/10.1016/j.mce.2013.09.019
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; http://dx.doi.org/10.1080/15548627.2015.1100356
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy 2011; 7:279-96; PMID:21189453; http://dx.doi.org/10.4161/auto.7.3.14487
- Cortes CJ, Miranda HC, Frankowski H, Batlevi Y, Young JE, Le A, Ivanov N, Sopher BL, Carromeu C, Muotri AR, et al. Polyglutamine-expanded androgen receptor interferes with TFEB to elicit autophagy defects in SBMA. Nat Neurosci 2014; 17:1180-9; PMID:25108912; http://dx.doi.org/10.1038/nn.3787
- Blessing AM, Ganesan S, Rajapakshe K, Ying Sung Y, Bollu LR, Shi Y, Cheung E, Coarfa C, Chang JT, McDonnell DP, et al. Identification of a novel coregulator, SH3YL1, that interacts with the androgen receptor N-terminus. Mol Endocrinol 2015; 29(10):1426-39: me20151079
- Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum Mol Genet 2011; 20:3852-66; PMID:21752829; http://dx.doi.org/10.1093/hmg/ddr306
- Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, et al. TFEB links autophagy to lysosomal biogenesis. Science 2011; 332:1429-33; PMID:21617040; http://dx.doi.org/10.1126/science.1204592
- Kimura S, Fujita N, Noda T, Yoshimori T. Monitoring autophagy in mammalian cultured cells through the dynamics of LC3. Methods Enzymol 2009; 452:1-12; PMID:19200872; http://dx.doi.org/10.1016/S0076-6879(08)03601-X
- Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 2007; 3:452-60; PMID:17534139; http://dx.doi.org/10.4161/auto.4451
- Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 2012; 5:ra42; PMID:22692423; http://dx.doi.org/10.1126/scisignal.2002790
- Nelson PS, Clegg N, Arnold H, Ferguson C, Bonham M, White J, Hood L, Lin B. The program of androgen-responsive genes in neoplastic prostate epithelium. Proc Natl Acad Sci U S A 2002; 99:11890-5; PMID:12185249; http://dx.doi.org/10.1073/pnas.182376299
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012; 487:239-43; PMID:22722839; http://dx.doi.org/10.1038/nature11125
- Varambally S, Yu J, Laxman B, Rhodes DR, Mehra R, Tomlins SA, Shah RB, Chandran U, Monzon FA, Becich MJ, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer Cell 2005; 8:393-406; PMID:16286247; http://dx.doi.org/10.1016/j.ccr.2005.10.001
- Augello MA, Burd CJ, Birbe R, McNair C, Ertel A, Magee MS, Frigo DE, Wilder-Romans K, Shilkrut M, Han S, et al. Convergence of oncogenic and hormone receptor pathways promotes metastatic phenotypes. J Clin Invest 2013; 123:493-508; PMID:23257359; http://dx.doi.org/10.1172/JCI64750
- Kimura T, Takabatake Y, Takahashi A, Isaka Y. Chloroquine in cancer therapy: a double-edged sword of autophagy. Cancer Res 2013; 73:3-7; PMID:23288916; http://dx.doi.org/10.1158/0008-5472.CAN-12-2464
- Liu B, Miyake H, Nishikawa M, Tei H, Fujisawa M. Expression Profile of Autophagy-related Markers in Localized Prostate Cancer: Correlation With Biochemical Recurrence After Radical Prostatectomy. Urology 2015; 85:1424-30; PMID:25881865; http://dx.doi.org/10.1016/j.urology.2015.03.006
- Martinet W, Schrijvers DM, Timmermans JP, Bult H, De Meyer GR. Immunohistochemical analysis of macroautophagy: recommendations and limitations. Autophagy 2013; 9:386-402; PMID:23242143; http://dx.doi.org/10.4161/auto.22968
- Akin D, Wang SK, Habibzadegah-Tari P, Law B, Ostrov D, Li M, Yin XM, Kim JS, Horenstein N, Dunn WA, Jr. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy 2014; 10:2021-35; PMID:25483883; http://dx.doi.org/10.4161/auto.32229
- Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell 2015; 59:285-97; PMID:26118643; http://dx.doi.org/10.1016/j.molcel.2015.05.031
- Guo JY, Karsli-Uzunbas G, Mathew R, Aisner SC, Kamphorst JJ, Strohecker AM, Chen G, Price S, Lu W, Teng X, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes Dev 2013; 27:1447-61; PMID:23824538; http://dx.doi.org/10.1101/gad.219642.113
- Rosenfeldt MT, O'Prey J, Morton JP, Nixon C, MacKay G, Mrowinska A, Au A, Rai TS, Zheng L, Ridgway R, et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013; 504:296-300; PMID:24305049; http://dx.doi.org/10.1038/nature12865
- Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011; 25:1510-27; PMID:21764854; http://dx.doi.org/10.1101/gad.2051011
- Xie X, Koh JY, Price S, White E, Mehnert JM. Atg7 Overcomes Senescence and Promotes Growth of BrafV600E-Driven Melanoma. Cancer Discov 2015; 5:410-23; PMID:25673642; http://dx.doi.org/10.1158/2159-8290.CD-14-1473
- Yang A, Rajeshkumar NV, Wang X, Yabuuchi S, Alexander BM, Chu GC, Von Hoff DD, Maitra A, Kimmelman AC. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discov 2014; 4:905-13; PMID:24875860; http://dx.doi.org/10.1158/2159-8290.CD-14-0362
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell'antonio G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev 2011; 25:717-29; PMID:21406549; http://dx.doi.org/10.1101/gad.2016111
- Strohecker AM, Guo JY, Karsli-Uzunbas G, Price SM, Chen GJ, Mathew R, McMahon M, White E. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov 2013; 3:1272-85; PMID:23965987; http://dx.doi.org/10.1158/2159-8290.CD-13-0397
- White E, Mehnert JM, Chan CS. Autophagy, Metabolism, and Cancer. Clin Cancer Res 2015; 21:5037-46; PMID:26567363; http://dx.doi.org/10.1158/1078-0432.CCR-15-0490
- Frigo DE, Howe MK, Wittmann BM, Brunner AM, Cushman I, Wang Q, Brown M, Means AR, McDonnell DP. CaM kinase kinase beta-mediated activation of the growth regulatory kinase AMPK is required for androgen-dependent migration of prostate cancer cells. Cancer Res 2011; 71:528-37; PMID:21098087; http://dx.doi.org/10.1158/0008-5472.CAN-10-2581
- Frigo DE, McDonnell DP. Differential effects of prostate cancer therapeutics on neuroendocrine transdifferentiation. Mol Cancer Ther 2008; 7:659-69; PMID:18347151; http://dx.doi.org/10.1158/1535-7163.MCT-07-0480
- Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, Hammond M, Patterson JR, Thompson SK, Kazmin D, et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res 2008; 68:7475-83; PMID:18794135; http://dx.doi.org/10.1158/0008-5472.CAN-08-1047
- Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, Zhang A, Xia X, Ilkayeva OR, Xin L, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1alpha-mediated metabolic switch. Oncogene 2014; 33:5251-61; PMID:24186207; http://dx.doi.org/10.1038/onc.2013.463
- Jung CH, Jun CB, Ro SH, Kim YM, Otto NM, Cao J, Kundu M, Kim DH. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell 2009; 20:1992-2003; PMID:19225151; http://dx.doi.org/10.1091/mbc.E08-12-1249
- Blessing A, Xu L, Gao G, Bollu LR, Ren J, Li H, Wu X, Su F, Huang WC, Hung MC, et al. Sodium/Glucose Co-transporter 1 expression increases in human diseased prostate. Cancer Science and Therapy 2012; 4:306-12.
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012; 2:401-4; PMID:22588877; http://dx.doi.org/10.1158/2159-8290.CD-12-0095
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013; 6:l1; PMID:23550210; http://dx.doi.org/10.1126/scisignal.2004088
- Bollu LR, Katreddy RR, Blessing AM, Pham N, Zheng B, Wu X, Weihua Z. Intracellular activation of EGFR by fatty acid synthase dependent palmitoylation. Oncotarget 2015; 6(33):34992-5003; PMID:26378037
- Gaur S, Wen Y, Song JH, Parikh NU, Mangala LS, Blessing AM, Ivan C, Wu SY, Varkaris A, Shi Y, et al. Chitosan nanoparticle-mediated delivery of MiRNA-34a decreases prostate tumor growth in the bone and its expression induces non-canonical autophagy. 2015; 6(30):29161-77
- Sternberg SR. Biomedical Image-Processing. Computer 1983; 16:22-34; http://dx.doi.org/10.1109/MC.1983.1654163
- Kapur JN, Sahoo PK, Wong AKC. A new method for gray-level picture thresholding using the entropy of the histogram. Computer Vision Graphics and Image Processing 1985; 29:273-85; http://dx.doi.org/10.1016/0734-189X(85)90125-2
- Vincent L, Soille P. Watersheds in digital spaces - an efficient algorithm based on immersion simulations. Ieee Transactions on Pattern Analysis and Machine Intelligence 1991; 13:583-98; http://dx.doi.org/10.1109/34.87344
- Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, et al. A gene network regulating lysosomal biogenesis and function. Science 2009; 325:473-7; PMID:19556463