
ABSTRACT
The drug 2-hydroxypropyl-β-cyclodextrin (HPβCD) reduces lysosomal cholesterol accumulation in Niemann-Pick disease, type C (NPC) and has been advanced to human clinical trials. However, its mechanism of action for reducing cholesterol accumulation in NPC cells is uncertain and its molecular target is unknown. We found that methyl-β-cyclodextrin (MβCD), a potent analog of HPβCD, restored impaired macroautophagy/autophagy flux in Niemann-Pick disease, type C1 (NPC1) cells. This effect was mediated by a direct activation of AMP-activated protein kinase (AMPK), an upstream kinase in the autophagy pathway, through MβCD binding to its β-subunits. Knockdown of PRKAB1 or PRKAB2 (encoding the AMPK β1 or β2 subunit) expression and an AMPK inhibitor abolished MβCD-mediated reduction of cholesterol storage in NPC1 cells. The results demonstrate that AMPK is the molecular target of MβCD and its activation enhances autophagy flux, thereby mitigating cholesterol accumulation in NPC1 cells. The results identify AMPK as an attractive target for drug development to treat NPC.
Introduction
Niemann-Pick disease, type C (NPC) is a lysosomal storage disease caused by mutations in either NPC1 or NPC2. Deficiency in NPC1 or NPC2 protein results in endolysosomal accumulation of unesterified cholesterol.Citation1,Citation2 Clinical symptoms of NPC include hepatosplenomegaly, and progressive neurodegeneration and CNS dysfunction, including ataxia, seizures, motor impairment, and decline of intellectual function.Citation1,Citation2 There is no FDA-approved therapy for NPC, although miglustat, an enzyme inhibitor for the glycosphingolipid pathway, has been approved for NPC treatment in 45 countries.Citation3
β-cyclodextrins are cyclic oligosaccharides containing a heptameric glucose ring. They are commonly used in drug formulations as pharmaceutical excipients due to the capacity of their hydrophobic cores to form complexes with hydrophobic drugs, enhancing their water solubility. 2-hydroxypropyl-β-cyclodextrin (HPβCD) was found to delay disease onset, reduce accumulation of cholesterol and other lipids, and extend the life span in Npc1−/− mice.Citation4,Citation5 HPβCD has been used to treat NPC1-patients, resulting in partial alleviation of hepatosplenomegaly and central nervous system dysfunction,Citation6 and is currently being evaluated in a phase 3 clinical trial. However, the mechanism of action and molecular target for HPβCD in the reduction of cholesterol accumulation in NPC1 cells is poorly understood. Due to its cholesterol complexation capacity, it was initially assumed that HPβCD acted therapeutically through bulk removal of cellular cholesterol. More recent studies, however, have shown that the cyclodextrin enters cells through endocytosis,Citation7,Citation8 and at the concentrations achieved in vivo, acts by promoting redistribution of cholesterol within the cell.Citation9 HPβCD may also reduce cholesterol storage through stimulation of lysosomal exocytosis.Citation7,Citation8 The potency (EC50) of HPβCD in NPC1-patient fibroblast cells lines is in the range of 1–3 mM,Citation7,Citation10-Citation12 whereas the EC50 of methyl-β-cyclodextrin (MβCD), another more potent β-cyclodextrin derivative, is ∼20 μM for reducing cholesterol accumulation in NPC1 cells.Citation8,Citation13
In addition to lysosomal lipid accumulation, defective autophagy has also been implicated in the pathogenesis of lysosomal storage diseases including NPC1.Citation14 Autophagy is a conserved cellular process, essential for cellular homeostasis and implicated in the turnover of damaged proteins, lipids, carbohydrates, and organelles by the lysosomal degradation pathway.Citation15 Autophagy flux is a dynamic process involving the generation of autophagosomes, and their fusion with late endosomes to form amphisomes, which in turn fuse with lysosomes to form autolysosomes.Citation16,Citation17 Accumulation of autophagosomes was reported in various tissues and cells including Npc1-deficient mouse embryonic fibroblasts (MEFs),Citation12 brain and liver of the Npc1-deficient mice,Citation18-Citation21 NPC1 knockout human embryonic stem cell (hESC)-derived neurons,Citation22 NPC1 fibroblasts,Citation23 NPC1 induced pluripotent stem cells (iPSCs) and hepatocyte-like cells, neural progenitors, and neurons.Citation10,Citation11 Lysosomes play an important role in autophagy flux and impaired autophagy is observed in many other lysosomal storage diseases.Citation14 Autophagy malfunction is also implicated in most neurodegenerative diseases, such as Alzheimer disease,Citation24 Parkinson disease,Citation25 Huntington disease,Citation26 and amyotrophic lateral sclerosis,Citation27 which share a basic characteristic of aberrant misfolded proteins or peptide aggregations.Citation28
Here we report the identification of AMPK as a direct target of MβCD. Our results indicate that MβCD binds the β-subunits of AMPK, activating AMPK and the AMPK-dependent autophagy pathway. The ability of MβCD to reduce cholesterol accumulation in NPC1 cells was nearly abolished after knockdown of the PRKAB1 or PRKAB2 (encoding the AMPK β1 or β2 subunit) or treatment with an AMPK inhibitor. Conversely, AMPK activators mimicked the effect of MβCD, reducing cholesterol accumulation in NPC1 cells. Knockdown of PRKAB1 or PRKAB2 also recapitulated the lysosomal accumulation of cholesterol in wild-type (WT) cells. These findings identify AMPK as a novel target for drug development to treat NPC and lysosomal storage diseases and potentially may extend to treatment of other neurodegenerative disorders.
Results
β-cyclodextrin enters cells through the endocytic pathway
To determine how β-cyclodextrins penetrate the plasma membrane and enters cells, we labeled a per-methylated β-cyclodextrin with a BODIPY fluorophore (BODIPY-CD) and studied the kinetics of its cellular trafficking. We found that it entered cells rapidly reaching a plateau in 1 h (). The amount of BODIPY-CD inside cells correlated with the concentration of labeled cyclodextrin in the medium (Fig. S1A). The cells quickly eliminated BODIPY-CD after removing the labeled cyclodextrin from the medium, with the bulk of the intracellular fluorescence intensity eliminated after 2 h. The kinetic profiles of BODIPY-CD entering and exiting cells were similar in both WT and NPC1 fibroblasts as well as in the U2OS cells and neural stem cells (NSCs) differentiated from WT and NPC1 iPSCs (Fig. S1B). BODIPY-CD, similar to MβCD, reduced cholesterol accumulation in NPC1 fibroblasts (Fig. S1C), indicating that the pharmacological property is retained by fluorphore-labeled β-cyclodextrin.
Figure 1. Kinetics, cellular trafficking and distribution of BODIPY-CD. (A) Kinetics of BODIPY-CD entering and leaving WT and NPC1 fibroblasts. The images were acquired after incubation with 10 µM BODIPY-CD for the indicated times. (B) Cellular distribution of BODIPY-CD in U2OS cells localized with the RFP-tagged RAB5A (early endosome, EE), RAB7A (late endosome, LE), LAMP1 (lysosome, LY) and LC3B protein (LC3B vesicles, LC3/V). The fluorescence colocalization of BODIPY-CD and RFP were measured at the indicated times after BODIPY-CD was added to the cells. Only colocalized images are displayed and kinetic images are shown in Fig. S1D. Green triangles and *CD: BODIPY-CD. Scale bar: 10 µm.
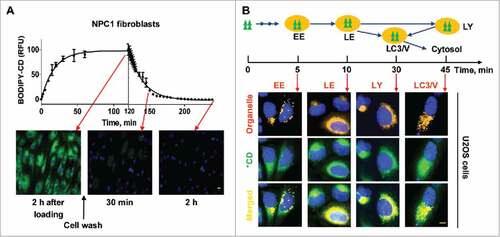
Although endocytosis of β-cyclodextrin has been demonstrated,Citation29 its intracellular trafficking itinerary remains unclear. To study the distribution of β-cyclodextrin inside cells, we used cells expressing red fluorescent protein (RFP)-labeled vesicles and organelles, and examined the colocalization of BODIPY-CD within these organelles (, Fig. S1D). We observed strong colocalization of BODIPY-CD with early endosomes, late endosomes, and lysosomes after 5-, 10- and 30-min incubation, respectively. BODIPY-CD also colocalized with MAP1LC3B/LC3B (microtubule associated protein 1 light chain 3B)-positive autophagic vesicles after 20–30 min incubation. Cytosolic BODIPY-CD, which was measured by cell membrane permeabilization, increased after a 20-min incubation, reaching a significantly higher level after a 30-min incubation (Fig. S1E). Together, the results demonstrate that BODIPY-CD enters cells rapidly via the endocytic pathway, then traffics to LC3B-positive autophagic vesicles and cytosol. BODIPY-CD also exits cell quickly, presumably through an exocytic pathway.
MβCD increases autophagosome formation and restores impaired autophagy flux in NPC1 cells
To further understand the pharmacodynamic activity of MβCD, we measured its time course effect on autophagy in WT and NPC1 fibroblasts, and in iPSC-derived NSCs and neurons. We first determined the effect of MβCD on autophagosome markers LC3B and SQSTM1/p62. LC3-II is the membrane-attached form of cytosolic LC3-I that has been conjugated with phosphatidylethanolamine and is present predominantly on phagophores and autophagosomes.Citation30,Citation31 The polyubiquitin-binding protein SQSTM1 is degraded by autophagy through direct interaction with LC3-II.Citation32 Basal levels of LC3B-II and SQSTM1 in the untreated NPC1 cells were higher than these in WT cells (). After 24-h treatment with MβCD, LC3B-II and SQSTM1 increased in NPC1 cells, whereas the LC3B-II was unchanged and SQSTM1 decreased in WT cells. LC3B-II and SQSTM1 levels increased in both WT and NPC1 cells upon treatment with bafilomycin A1, an inhibitor of the vacuolar-type H+-translocating ATPase that blocks autophagosome-lysosome fusion and decreases autophagy flux, indicating the impairment of autophagosome-lysosome fusion is not complete in NPC1 cells. Both proteins further increased after the treatment with MβCD plus bafilomycin A1, suggesting that MβCD stimulates autophagosome formation but does not block autophagy flux. We then performed a 6-d time course study to examine changes of LC3B-II and SQSTM1 levels after MβCD treatment. In NPC1 fibroblasts and NSCs, these markers gradually increased following MβCD treatment, peaking at 2–3 d and declining to levels even lower than basal expression at d 6. In NPC1 neurons the time course was accelerated, with LC3B-II and SQSTM1 levels in NPC1 peaking at d 1 and rapidly declining to sub-basal levels by d 3 (). By contrast, in WT cells we did not observe increased LC3B-II and SQSTM1 levels during the 6-d MβCD treatment in fibroblasts and NSCs or the 3-d treatment in neurons, though LC3B-I was increased and SQSTM1 was decreased slightly (Fig. S2A). Similar results were observed by immunofluorescence staining with anti-LC3B and SQSTM1 antibodies in fibroblasts and NSCs (, Fig. S2B), and in U2OS cells (Fig. S2C). Furthermore, we performed the time course of filipin staining and found the decrease of cholesterol accumulation in NPC1 cells correlated with the decreases in LC3B-II and SQSTM1 levels (). The restoration of autophagy flux correlated with the decrease of cellular cholesterol in NPC1 cells. Together, these results suggested biphasic effects of MβCD in NPC1 cells. LC3B-II and SQSTM1 levels initially are increased due to the stimulation of autophagosome formation and accumulation of increased autophagosomes caused by impaired autophagy flux, and then decreased, presumably as a result of MβCD-mediated restoration of autophagy flux and cholesterol reduction.
Figure 2. MβCD increases autophagosome formation and restores impaired autophagy flux in NPC1 cells. (A) Autophagy detection in both WT and NPC1 fibroblasts, NSCs and neurons. Cells were treated with the indicated compounds (100 µM MβCD, 100 nM bafilomycin A1 (baf. A1) or MβCD plus baf. A1) for 24 h, followed by western blot analysis. For this and all subsequent figures, a representative western blot is shown, and the bar graph represents the mean ± SEM of at least 3 replicates, unless otherwise noted. The intensity of LC3B-II and SQSTM1 were normalized with GAPDH. (B) Time-courses of LC3B and SQSTM1 levels in NPC1 cells after treatment with 100 µM MβCD. Cells were treated for the indicated times and analyzed by western blot. LC3B-II and SQSTM1 expression were normalized with GAPDH expression. (C) Quantification of LC3B and SQSTM1 immunofluorescence in WT and NPC1 fibroblasts and NSCs displayed in Fig. S2A. The normalized relative fluorescence intensities of LC3B and SQSTM1 dots are shown in bar graphs (mean ± SEM for at least 3 fields). (D) Time courses for effect of MβCD on reduction of cholesterol accumulation in NPC1 fibroblasts, NSCs and neurons. NPC1 cells were cultured in 96-well plates and treated with 100 µM MβCD for the indicated days, followed by the filipin staining assay. Data represent mean ± SEM of 3 replicates. For this and all subsequent figures, * p < 0.05, ** p < 0.01 and *** p < 0.001 compared to control, unless otherwise noted. (E) Images of mRFP-GFP-LC3B tandem sensor in NPC1 fibroblasts. Cells expressing this sensor were treated with the compounds for the indicated times and then stained with Hoechst 33342. GFP signal shows autophagosomes, whereas RFP signal indicates autophagosomes combined with autolysosomes. The number of GFP+ and RFP+ merged punctate structures, as well as the number of non-merged RFP+ punctate structures, were quantified for at least 10 cells. Scale bar: 10 µm. RFU, relative fluorescent units.
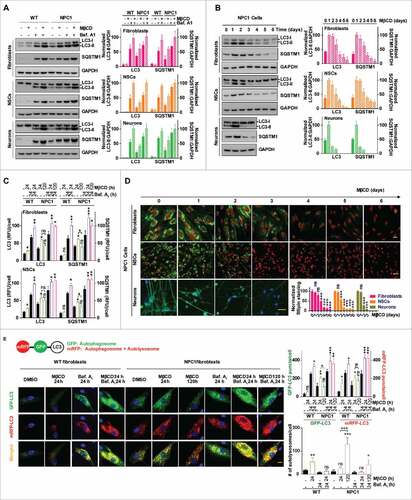
To further examine the effect of MβCD on autophagy flux, we employed a mRFP-GFP-LC3B tandem sensor to report the fusion of autophagosomes with lysosomes to form autolysosomes.Citation31,Citation32 This sensor consists of an acid-sensitive GFP mutant, which fluoresces at acidic pH, and an acid-insensitive RFP, which fluoresces in both acidic and neutral pH, permitting the progression from the autophagosome with a neutral pH to autolysosome with an acidic pH to be monitored. Consistent with previous results, we found that both RFP and GFP fluorescence signals were greater in NPC1 than in WT cells (p < 0.05, ), indicating accumulation of autophagosomes in NPC1 cells. After 24-h treatment with MβCD, both the RFP (p < 0.05) and GFP (p < 0.01) signals increased in NPC1 fibroblasts, indicating increased autophagosome formation without a corresponding increase in autolysosomes (p < 0.05). In WT cells, on the other hand, only the RFP fluorescence signal was increased (p < 0.05), indicating increased autolysosome formation through enhancing of the normal autophagy flux (p < 0.01). By contrast, after 120-h treatment in NPC1 cells, the GFP signal decreased (p < 0.05) and unmerged RFP puncta increased dramatically (p < 0.001). These findings were not accompanied by significant change of RFP signal (p>0.05), indicating an increase of autolysosomes (p < 0.001). Treatment of WT and NPC1 fibroblasts either with bafilomycin A1 or MβCD plus bafilomycin A1 greatly enhanced both GFP and RFP signals (p < 0.001), consistent with increased autophagosome formation (p < 0.05). Similar results were also obtained in U2OS cells (Fig. S2D). Together, these findings indicate that MβCD rapidly increased autophagosome formation and gradually restored impaired autophagy flux in NPC1 cells. In WT cells, MβCD increased autolysosomes and autophagy flux without the biphasic effects.
MβCD activates AMPK and the autophagy pathway
To explore the molecular target of MβCD for stimulation of autophagy, we profiled the MTOR (mechanistic target of rapamycin)-dependent autophagy pathway using a phosphorylation antibody array.Citation33 Among the altered phosphorylation events in response to MβCD, was Thr172 phosphorylation of the α-subunit of AMPK (PRKAA), an upstream component of the autophagy pathway. In addition, Ser2448 and Ser2481 phosphorylation in MTOR were reduced by MβCD, indicating inhibition of MTOR function (Fig. S3A). One of the functions of MTOR is to inhibit ULK1, a regulatory protein in the autophagy pathway. These results led us to hypothesize that MβCD increases autophagy through activation of AMPK. To determine the effect of MβCD on AMPK and the AMPK-MTOR autophagy pathway, we examined the protein phosphorylation status in this pathway. We found phosphorylation of Thr172 on PRKAA in fibroblasts, NSCs and neurons increased after a 30-min treatment with MβCD, peaking at 1.5 to 2 h, while the total PRKAA level was unchanged (, Fig. S3B). We next employed a time-resolved fluorescence energy transfer (TR-FRET) assay to quantitatively measure the Thr172 phosphorylation level to determine the potency of MβCD. The EC50 for AMPK activation by MβCD was 1–3 µM in NPC1 NSCs (), similar to the EC50 for reduction of cholesterol accumulation.
Figure 3. MβCD activates AMPK and the autophagy pathway. (A) Time courses of AMPK activation indicated by an increase in the phosphorylation of Thr172 on PRKAA in NPC1 cells. Indicated cells were treated with 100 µM MβCD for the indicated times, followed by western blot analysis with anti-PRKAA Thr172 antibodies. The ratio of the intensity of phosphorylated PRKAA at Thr172 and total PRKAA is shown. (B) Concentration-dependent activation of AMPK in NPC1 NSCs measured by a TR-FRET assay. The up panel shows the TR-FRET assay to detect the phosphorylation levels of Thr172 on PRKAA using an anti-PRKAA antibody for the nonphosphorylated (i.e., total) portion and an anti-phospho-Thr172 antibody. After cells in 384-well plates were treated with MβCD for 1.5 h, cells were lysed. The TR-FRET assay reagent mixture was added and TR-FRET signals were detected. (C) Correlation of AMPK activation with the activation of downstream proteins (TSC2, ULK1, RPTOR) and inhibition of MTOR in WT and NPC1 cells. Cells were treated with 100 µM MβCD for 1.5 h and lysed followed by western blotting. The ratio of the intensity of phosphorylated protein and total protein is shown.
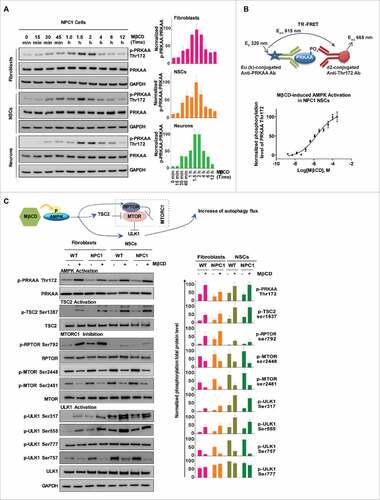
We further determined the effect of MβCD on downstream proteins of the AMPK-MTOR autophagy pathway (). We found an increase in phosphorylation of Ser792 of RPTOR/Raptor, a direct target of AMPK and essential for RPTOR-containing MTOR complex 1 (MTORC1) inhibition.Citation34 Phosphorylation levels of Ser79 of the AMPK-substrate ACACA/ACC (Fig. S3C), and Ser1387 of TSC2 also increased. The increase in RPTOR and TSC2 phosphorylation correlated with the dephosphorylation of Ser2448 and Ser2481 in MTOR, which leads to the inhibition of MTORC1 activity. ULK1 (unc-51 like autophagy activating kinase 1) is the mammalian ortholog of the yeast protein kinase Atg1 that plays a key role in the initial stage of autophagy activation. Ser317, Ser555 and Ser777 on ULK1 are directly phosphorylated by AMPK under conditions of energy stress that activate the autophagy pathway.Citation35 We found that MβCD increased phosphorylation levels of Ser317 and Ser555 but not Ser777. Active MTORC1 causes phosphorylation of Ser757 at ULK1, disrupting the interaction between ULK1 and AMPK.Citation35 We further found that MβCD decreased the phosphorylation of Ser757, which enabled ULK1 activation by AMPK. In addition, MβCD did not change the expression levels of STK11/LKB1 and CAMKK2, 2 upstream activators of AMPK (Fig. S3D). Although the ULK1 activity is suppressed under nutrition-rich conditions by MTORC1,Citation35 the pharmacological effect of MβCD on activation of AMPK-ULK1 seems to alter this activity. Previously, it had been shown that pharmacological activation of AMPK could prevent DNM1L/DRP1-mediated mitochondrial fission.Citation36 We found that MβCD, similar to the AMPK activator AICAR, could enhance DNM1L phosphorylation (Fig. S3E). Taken together, MβCD directly activates AMPK, which in turn activates RPTOR and TSC2 followed by inhibition of MTORC1. The inhibition of ULK1 by MTORC1 is then relieved, further stimulating the autophagy pathway.
MβCD binds to PRKAB1 and PRKAB2 with a higher affinity to PRKAB1
We next explored AMPK activation by investigating whether MβCD binds directly to one of its subunits. Glycogen, a glucose polysaccharide containing α-1,4 and 1,6 glycosidic bonds that functions as an energy reserve source, binds to PRKAB, inhibiting PRKAA kinase activity.Citation37 Therefore, we hypothesized that other glucose-containing polymers such as β-cyclodextrins, might also have the capacity to modulate AMPK activity. We employed a cellular thermal shift assay (CETSA) to determine the direct binding of a compound to its target protein.Citation38 This method uses a specific antibody to measure a compound's ability to protect its binding protein in cell lysates from heat-induced protein denaturation and aggregation. The aggregated proteins that precipitate out of the solution are not detected by western blot. Values of apparent aggregation temperature (Tagg) of PRKAB1 and PRKAB2 in cell lysates were measured in the absence or presence of MβCD at temperatures ranging from 37°C to 63°C (). MβCD increased the resistance of PRKAB1 and PRKAB2 in fibroblasts to aggregation caused by heating. The Tagg increased from 53°C to 58.8°C for PRKAB1 and from 52.8°C to 57.6°C for PRKAB2. In NSCs, the Tagg value increased from 53.2°C to 59.3°C for PRKAB1, the only subunit expressed in NSCs. The Tagg values were not significantly changed by DMSO (a solvent control). These results support direct binding of MβCD to PRKAB1 and PRKAB2.
Figure 4. MβCD binds to PRKAB1 and PRKAB2 with a higher affinity for PRKAB1. (A) Temperature melting curves of PRKAB1 and PRKAB2 in the presence or absence of 300 µM MβCD. The relative chemiluminescent intensity of each sample at different temperatures was used to generate temperature-dependent melting curves and the apparent aggregation temperature (Tagg) was calculated by nonlinear regression. (B) Apparent binding affinities of MβCD with PRKAB1 and PRKAB2 measured by CETSA. Cell lysates were treated with MβCD and heated to 53°C for 3 min. The supernatants obtained after centrifugation were analyzed by western blotting using anti-PRKAB1 or anti-PRKAB2 antibody. A representative blot was shown and data represent mean ± SEM of at least 3 replicates. (C) MβCD reduced NPC1 phenotypes in fibroblasts, NSCs and neurons. NPC1 cells were cultured in 96-well plates and treated with various concentrations of MβCD. After a 4-d (fibroblasts and NSCs) or 3-d (neurons) incubation filipin or LysoTracker Red staining was performed. Data represent mean ± SEM of 3 replicates. (D) A structural model of AMPK (PRKAA2B1G1, PDB code 4CFF) bound with MβCD (yellow), activator A769662 (brown), and inhibitor dorsomorphin (gray). The 3 subunits of AMPK are shown in green (PRKA2), magenta (PRKAB1), and cyan (PRKAG1). (E) Binding model of MβCD with PRKAB1. (F) Binding model of MβCD with PRKAB2. MβCD is shown in sticks in yellow (carbon atom). AMPK is shown in ribbons and key interacting residues are shown in sticks in green (PRKAB1) or cyan (PRKAB2). Residue L146 within a flexible loop, which is positioned in the MβCD hole, is shown in magenta.
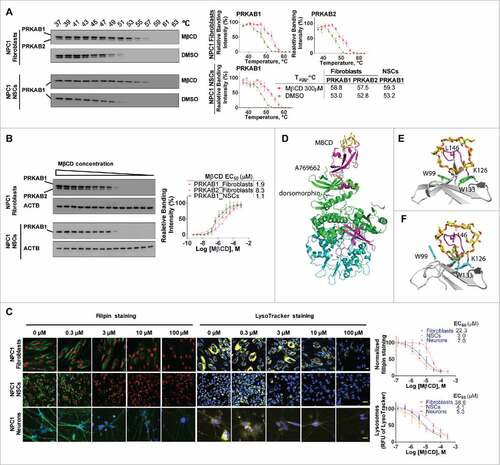
Further evidence of MβCD binding to PRKAB1 and PRKAB2 was obtained by measuring binding affinities using isothermal dose-response fingerprints (ITDRF), involving incubation of cell lysates with various concentrations of MβCD at 53°C (Tagg). At this temperature, the majority of PRKAB1 and PRKAB2 aggregate and precipitate in the absence of MβCD. We found that MβCD reduced aggregation of PRKAB1 and PRKAB2 in a concentration-dependent manner (). The binding affinities (EC50) of MβCD to PRKAB1 and PRKAB2 in NPC1 fibroblasts were 1.9 µM and 8.3 µM, respectively, and 1.1 µM for the PRKAB1 subunit in NSCs. By contrast, MβCD did not stabilize PRKAA (Fig. S4A). Taken together, these findings provide support for direct binding of MβCD to PRKAB1 and PRKAB2, with a 4-fold higher binding affinity for PRKAB1 as compared to PRKAB2.
MβCD was more effective in reducing cholesterol storage in NPC1 NSCs and neurons (EC50 = 3.0 µM in a filipin staining assay and 4.1 µM in a LysoTracker Red staining assay for NSCs, and 7.0 µM and 5.3 µM for neurons) than in NPC1 fibroblasts (EC50 = 22.3 µM in a filipin staining assay and 38.6 µM in a LysoTracker Red staining assay) ().Citation13 In addition, NSCs and neurons mainly express the PRKAA2/AMPK α2-subunit, whereas fibroblasts predominantly express the PRKAA1/AMPK α1-subunit (Fig. S4B and C). There were no significant differences in the subunit expression patterns between WT and NPC1 cells. These results suggest that differences in isoform expression patterns of AMPK subunits and the different affinities of MβCD toward PRKAB1 and PRKAB2 may account for the observed differences in EC50 values for MβCD-mediated cholesterol reduction across these 3 cell types.
To further determine the structural basis of the differences for efficacy of MβCD in different cell types, we modeled the binding interactions of MβCD with PRKAB1 and PRKAB2. The structural binding models were derived from the crystal structures of β-cyclodextrin bound with the human PRKAB2 and with a truncated rat PRKAB1,Citation37,Citation39,Citation40 and employed energy minimization and molecular dynamics simulations to refine the interactions and conformational changes of the protein upon MβCD binding. Initial analysis of the binding mode indicated that MβCD binds to PRKAB1 in a similar manner to that observed with PRKAB2 (). Key interactions including hydrogen bonding with residues Lys126 and Asn150, and aromatic stacking interactions with 2 Trp residues in PRKAB1 and PRKAB2, are generally conserved. However, structural differences between the 2 subunits with respect to interactions with MβCD were also observed, in particular the induced conformational changes of residue Trp100/Trp99 within PRKAB1 and PRKAB2 and a flexible binding loop, which is characterized by a hydrophobic interaction with L146. Moreover, the analysis indicates that the degree and localization of CD-substituents might promote or impair conformation shifts and therefore activation of each subunit, raising the possibility of developing isoform-specific activators.
Pharmacological effect of MβCD is blocked by an AMPK inhibitor and mimicked by AMPK activators
To examine the effect of the MβCD-AMPK interaction on cholesterol reduction and enhancement of autophagy flux, we used both an AMPK inhibitor and AMPK activators. Dorsomorphin, a known AMPK inhibitor, significantly increased lysosomal cholesterol accumulation in WT cells (), inducing a cellular phenotype of cholesterol storage similar to NPC1 cells. In the NPC1 cells, dorsomorphin further exacerbated cholesterol accumulation and also blunted MβCD's effect on cholesterol reduction (). In addition, dorsomorphin attenuated the ability of MβCD to increase LC3-II and SQSTM1 levels in NPC1 cells (). By contrast, AMPK activators RSVA405 and A769662 reduced cholesterol accumulation (p < 0.01, ) and activated autophagy by increasing LC3B-II and SQSTM1 levels in NPC1 fibroblasts (), mimicking the effect of MβCD. Together, the results indicate that MβCD acts through the AMPK-dependent autophagy pathway to regulate disposal of cholesterol in NPC1 cells.
Figure 5. Pharmacological effect of MβCD is blocked by an inhibitor and mimicked by activators of AMPK. (A, B) Filipin staining for unesterified cholesterol accumulation in WT and NPC1 fibroblasts treated with the AMPK inhibitor dorsomorphin (DM) in the presence and absence of 100 µM MβCD. (C) Western blotting for LC3B and SQSTM1 levels in NPC1 fibroblasts treated with the indicated compounds (2 µM DM, 100 µM MβCD, or 100 nM baf. A1) for 24 h. LC3B-II and SQSTM1 expression were normalized to GAPDH expression. (D) Filipin staining after the cells were treated with the indicated compounds (1.25 µM RSVA 405, 200 µM A769662 or 100 nM baf. A1) for 4 d. (E) Western blotting for LC3B and SQSTM1 levels in NPC1 fibroblasts treated with AMPK activators (1.25 µM RSVA 405 or 200 µM A769662) for 24 h. LC3-II and SQSTM1 expression were normalized to GAPDH expression. Scale bar: 10 µm.
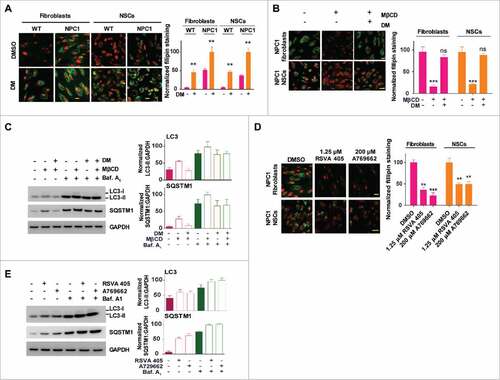
PRKAB1 or PRKAB2 is required for MβCD's effect on cholesterol reduction, autophagy activation and increase of autophagy flux
To further explore the requirement of PRKAB1 or PRKAB2 for the MβCD's mechanism of action, we performed experiments using cells in which PRKAB1 or PRKAB2 expression was silenced and then re-expressed (, Fig. S5A-C). Knockdown of PRKAB1 or PRKAB2 and pharmacological inhibition of AMPK by dorsomorphin did not have an effect on the cellular dynamics of BODIPY-CD (Fig. S5A). Knockdown of PRKAB1 or PRKAB2 either alone or together further increased the cholesterol accumulation in NPC1 fibroblasts (). Reduction in PRKAB1 or PRKAB2 expression, again either alone or in combination, markedly attenuated MβCD's effect on phosphorylation of Thr172 on PRKAA, as well as on reduction of cholesterol accumulation (, Fig. S5B). We then re-expressed PRKAB1 or PRKAB2 in the PRKAB1- or PRKAB2-silenced cells using activation vectors. The re-expression of PRKAB1 or PRKAB2 or both subunits in the double-knockdown cells restored the effect of MβCD on phosphorylation of Thr172 of PRKAA and cholesterol reduction (, Fig. S5B). We further found that the increases in LC3-II and SQSTM1 levels by MβCD treatment were abolished in PRKAB1 and PRKAB2 double-knockdown cells (Fig. S5C). Similarly, overexpression of PRKAB1 or PRKAB2 restored MβCD's effect on LC3 and SQSTM1 proteins (Fig. S5C). In order to determine whether the effect of MβCD on reduction of cholesterol in NPC1 cells was autophagy dependent, we performed ATG12 knockdown in NPC1 fibroblasts. ATG12 conjugates with ATG5 to form an ATG12–ATG5 complex that is essential for autophagy. Silencing of either ATG12 or ATG5 expression also impairs autophagy flux, similar to that induced by lysosomal cholesterol accumulation,Citation41 however as a result of defective autophagosome formation.Citation42 We found that ATG12 knockdown attenuated the effect of MβCD on reduction of accumulated cholesterol in NPC1 fibroblasts () as well as its effects on LC3B-II and SQSTM1 levels (). However, these effects were not completely abolished, likely due to the incomplete knockdown of the ATG12 protein. Together, our results provide strong evidence that the pharmacological effect of MβCD on reduction of cholesterol accumulation requires intact AMPK activity and is dependent on autophagy activation.
Figure 6. PRKAB1 or PRKAB2 is required for the effect of MβCD on cholesterol reduction, autophagy induction, and increase of autophagy flux. (A, B) Filipin staining of NPC1 fibroblasts treated with MβCD. (A) PRKAB1 or PRKAB2 expression was silenced by shRNA and the subunits re-expressed with transient transfection of either PRKAB1 or PRKAB2 activation vectors. (B) ATG12 expression was silenced by shRNA. All of the cells were treated with 100 µM MβCD or DMSO control for 4 d followed by the filipin staining assay. (C) NPC1 fibroblasts transfected with ATG12 shRNA or control shRNA were treated with 100 µM MβCD or DMSO control for 24 h, followed by western blot analysis with the indicated antibodies. LC3B-II, SQSTM1 and ATG12 expression were normalized to GAPDH expression. (D-G) MβCD effects on SNARE proteins interactions. (D) Immunoprecipitation and western blot analysis of 3 SNARE proteins (VAMP8, STX17 and SNAP29). WT fibroblasts with the PRKAB/AMPK β-subunit silenced or with AMPK β-subunit re-expression was treated with MβCD or DMSO for 24 h. Cells were lysed and directly immunoprecipitated with anti-VAMP8 antibody followed by western blot analysis with the indicated antibodies. (E) NPC1 and WT fibroblasts were treated with MβCD for the indicated times, followed by immunoprecipitation with anti-SNAP29 and western blot analysis with the indicated antibodies. (F) Immunofluorescence staining and colocalization of LC3 with VAMP8. Indicated WT fibroblasts, transiently transfected with TagRFP-LC3 lentiviral particles, were treated with 100 µM MβCD or DMSO for 24 h and stained with anti-VAMP8 antibody. The punctate structures of VAMP8 were colocalized with RFP-LC3 (yellow color in the merged images). Data represent mean ± SEM of 10 images. (G) NPC1 and WT fibroblasts, transiently transfected with TagRFP-LC3 lentiviral particles were treated with MβCD for the indicated times, followed by staining with anti-VAMP8 antibody. The colocalization of VAMP8 and RFP-LC3 puncta was analyzed as above. Abbreviations: PRKAB1 or PRKAB2 act., PRKAB1 or PRKAB2 activation vectors; Mock vec., mock vector; LY, lysosome; AP, autophagosome. Scale bar: 10 µm (yellow) and 1 µm (white).
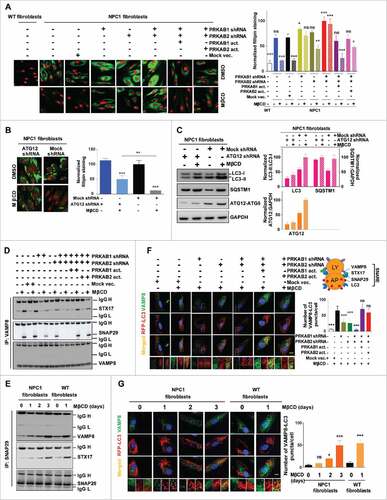
To further investigate the effect of MβCD on the late stage of autophagy flux (autophagosome-lysosome fusion), we examined the formation of soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes, a critical step in autophagosome-lysosome fusion, which is impaired in lysosomal storage disorders such as NPC1, multiple sulphatase deficiency and mucopolysaccharidosis type IIIA.Citation12,Citation43,Citation44 STX17 (syntaxin 17) resides in the outer membrane of autophagosomal vesicles and interacts with the endosomal-lysosomal VAMP8 and SNAP29 proteins, all of which are part of a SNARE complex during fusion.Citation43 We performed co-immunoprecipitation of STX17, VAMP8 and SNAP29 to determine whether MβCD enhances the process of autophagosome-lysosome fusion ( and , Fig. S6A). In WT fibroblasts, the expression levels of these 3 SNARE proteins were independent of PRKAB1 and PRKAB2 expression. STX17, VAMP8 and SNAP29 co-immunoprecipitated in the WT fibroblasts, and the interactions of these SNARE proteins were increased by MβCD, indicating enhanced autophagosome-lysosome fusion. MβCD-stimulated interactions of STX17, VAMP8 and SNAP29 were decreased after knockdown of either PRKAB1 or PRKAB2, or almost completely abrogated after knockdown of both PRKAB1 and PRKAB2. As expected, re-expression of PRKAB1 or PRKAB2 restored the MβCD-stimulated interactions of these SNARE proteins. In NPC1 fibroblasts, the interactions of these SNARE proteins were increased by MβCD, although the increases were not as significant as in WT cells (). We also used cells expressing a RFP-LC3B marker to examine colocalization of LC3B with STX17, VAMP8 and SNAP29, another indication of increased autophagosome-lysosome fusion ( and , Fig. S6B). MβCD-dependent colocalization of RFP-LC3B with STX17, VAMP8 and SNAP29 were partially or completely attenuated in the WT fibroblasts with reduced expression of PRKAB1 or PRKAB2. Colocalization of the SNARE proteins was restored by re-expression of either PRKAB1 or PRKAB2 (, Fig. S6B). In addition, the colocalization of RFP-LC3B with VAMP8 increased gradually in NPC1 cells during 3-d treatment with MβCD, whereas in WT cells a significant increase in colocalization was observed after 24-h treatment (). The results indicated that the effect of MβCD on SNARE complex formation relies on autophagy induction through its interaction with PRKAB1 and PRKAB2. The delayed interaction in NPC1 cells may be due to the lysosomal accumulation of cholesterol.
Discussion
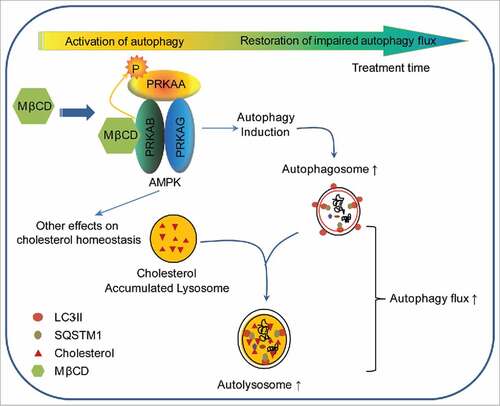
Our data provide important new insight into the mechanism of action by which MβCD alleviates cholesterol accumulation in NPC1 cells. We show that MβCD enters cells by endocytosis and binds to PRKAB1 and PRKAB2 leading to activation of AMPK and the AMPK-dependent autophagy pathway. In the NPC1 cells, the observed impairment in autophagy flux correlates with a reduced autophagosome-lysosome fusion, presumably due to lysosomal dysfunction as consequence of cholesterol accumulation.Citation14,Citation44 MβCD activation of AMPK promotes autophagy flux and gradually mobilizes accumulated cholesterol in lysosomes of NPC1 cells, restoring cholesterol homeostasis ().
Figure 7. Schematic diagram of the mechanism of action of MβCD in NPC1 cells. After entering NPC1 cells by endocytosis, MβCD activates AMPK by binding to PRKAB/β-subunit, enabling PRKAA/α-subunit to be phosphorylated and activated. The activated AMPK sequentially phosphorylates downstream proteins, inducing autophagy. As a consequence of autophagy induction, autophagosome formation increases and autophagy flux is enhanced, as evidenced by an increase in SNARE complexes and consequential autophagosome-lysosome fusion. Ultimately, the impaired autophagy flux within NPC1 cells is restored by MβCD treatment.
There are conflicting reports as to whether autophagy is increased or blocked in NPC1-patients cells,Citation19,Citation21 and Npc1 mutant animal models.Citation18 It remains unclear whether the observed elevations in LC3-II and SQSTM1 protein levels and increase in the number of autophagosomes represent a primary increase in autophagic activity or a secondary reduction in autophagy flux caused by impairment of autophagosome-lysosome fusion. Evidence for autophagy induction was based on an increase in LC3-II level in the cerebellar and liver extracts of NPC1-deficient mice,Citation18,Citation19,Citation21 and in NPC1-patient fibroblasts.Citation19,Citation21 Conversely, increases in LC3-II or SQSTM1 protein levels in NPC1-patient fibroblasts, iPSC-derived hepatocyte like cells and neural progenitor cells,Citation10,Citation11,Citation23 cerebellar or liver extracts of an NPC1-deficient mice model,Citation10,Citation12,Citation20,Citation41 and NPC1-deficient MEFs and CHO cellsCitation12 have been proposed to be secondary to impaired autophagy flux. Impaired autophagy flux may indicate reduced cellular capacity for lysosomal proteolysis, as suggested by an increase in autolysosomes,Citation15 and impaired SNARE assembly, while the cellular lysosomal proteolytic function might remain unaffected.Citation12,Citation44,Citation45 Others have proposed that NPC1 is characterized by both increased autophagy induction and impaired autophagy flux.Citation14,Citation22 It seems likely that differences in methods and experimental designs may account for the variable results. Our findings are consistent with the underlying impairment in autophagosome-lysosome fusion and autophagy flux in NPC1 cells. We also examined the impairment of the autophagy pathway in the patient cells of 4 other lysosomal storage diseases. We did not find significant impairment in the autophagy pathway nor cholesterol accumulation in patient cells derived from Batten, Farber, Tay-Sachs and Wolman diseases (Fig. S7). The negative results on impairment of the autophagy pathway may be explained by the examination of limited patient cell lines, and/or the fact that no cholesterol accumulation was found in these patients’ cells.
The effects of cyclodextrin on autophagy are also controversial. At high concentrations, HPβCD has been reported to inhibit autophagy,Citation11,Citation12,Citation45 whereas at lower concentrations autophagy was unaffected.Citation12 HPβCD increased autophagosome formation through an activation of TFEB (transcription factor EB) and an increase of LC3 expression,Citation46 along with amelioration of impaired autophagy flux.Citation10,Citation20,Citation23 After MβCD treatment we found dynamic changes in LC3-II and/or SQSTM1 protein levels in NPC1 cells, and increased LC3-I and decreased SQSTM1 levels in WT cells. The increased LC3-II and SQSTM1 levels in NPC1 cells in the early stage of MβCD treatment appears to be caused by the imbalance between enhanced autophagy induction and impaired autophagy flux, which was not observed in WT cells. With longer MβCD treatment (> 96 h), autophagy flux was restored, likely by promoting SNARE protein interactions. However, the SNARE protein interactions were enhanced upon 24-h treatment with MβCD, both in NPC1 and WT cells (before reduction of accumulated cholesterol in NPC1 cells). In NPC1 cells, MβCD enhanced SNARE protein interactions were weaker than these in WT cells, presumably due to the lysosomal accumulation of cholesterol.Citation44 The restoration of SNARE protein interactions by MβCD further led to reductions of accumulated cholesterol. Previous studies have shown that the impairment of autophagy in NPC1 can be restored by stimulating autophagy with rapamycin or CBZ,Citation11,Citation12 and the cholesterol accumulation phenotype also can be alleviated by rapamycin treatment.Citation47 The induction of autophagy can promote cholesterol efflux through a lysosomal pathway by enhanced autophagosome and lysosome fusion.Citation48 In addition, the basal autophagy level may affect the response time of MβCD-induced restoration of impaired autophagy and cholesterol reduction. Since neurons may have a higher basal autophagy level and more efficient autophagic turnover, they showed a faster response to MβCD treatment that is consistent with a previous report in which cholesterol is removed from NPC1 neurons after 24 h of HPβCD treatment.Citation49 Cyclodextrins may also have multiple effects on cholesterol homeostasis including the activation of AMPK-dependent autophagy, an increase in lysosomal exocytosis,Citation7 and by removing cholesterol from the plasma membrane.Citation50
The activation of AMPK could promote ABCA1-mediated cholesterol efflux to APOA1 (apolipoprotein A1), which is a major housekeeping mechanism for cellular cholesterol homeostasis.Citation51 Additionally, AMPK activation suppresses the activity of cholesterol synthesis post-translationally through phosphorylation of HMGCR (3-hydroxy-3-methylglutaryl-CoA reductase), the rate-limiting enzyme in cholesterol biosynthesis, and inhibition of sterol-regulatory element-binding proteins (SREBPs), transcription factors that are master regulators of cholesterol biosynthesis and lipoprotein cholesterol uptake.Citation52,Citation53 AMPK activation might also be an attractive drug development target for treatment of NPC. Computer modeling can be a useful tool to design a new generation of AMPK activators that have a more selective effect on restoration of impaired autophagy and reduction of accumulated cholesterol in NPC cells. Such AMPK activators will also need to have improved CNS penetration in order to treat neurological symptoms in NPC.
Impaired autophagy has been implicated in other neurodegenerative diseases. Importantly, aggregated proteins in post-mitotic neurons cannot be diluted by cell division, and therefore autophagy plays a fundamental role in mature neural cells maintaining protein quality-control processes and executing the cleavage of life-threatening protein aggregates.Citation54 Since abnormalities in autophagy flux have been implicated in the pathogenesis of neurodegenerative diseases, autophagy has been considered as an emerging therapeutic target.Citation28 Activation of autophagy ameliorates beta-amyloid peptides and MAPT/tau pathology in an Alzheimer disease mouse model,Citation55 enhances autophagic clearance of SNCA/α-synuclein oligomers in dopaminergic neurons,Citation56 alleviates behavioral motor abnormalities and neuropathology in Huntington disease models,Citation57 and promotes clearance of mutant SOD1 (superoxide dismutase 1) in models of amyotrophic lateral sclerosis.Citation58 Specifically, stimulation of autophagy through AMPK activation is protective and delays disease progression in a variety of neurodegenerative disorders.Citation28,Citation59-Citation61
HPβCD has advanced to late-stage clinical trials for treatment of NPC1 disease and has the potential to become the first FDA-approved drug for this disorder. Nonetheless, limitations of the drug are well recognized, including ototoxicity,Citation62 and a requirement for direct CNS delivery due to inability of the drug to efficiently cross the blood-brain barrier.Citation9,Citation63 Our findings provide a plausible mechanism of action of β-cyclodextrins for restoration of cholesterol homeostasis in NPC1 cells. Future drug discovery efforts focused on the development of CNS-penetrant small molecule activators of AMPK may offer effective and more tractable therapies for treatment of NPC disease.
Materials and methods
Compounds and antibodies
Three different methyl-β-cyclodextrins (MβCD) were purchased from MP Biomedicals (157320; lot number 9355K and M1322) and Sigma-Aldrich (C4555; lot number SLBH6893V). However, only the MβCD from MP Biomedicals (lot number 9355K) exhibited potent activity on reduction of lysosomal cholesterol accumulation and AMPK activation. BODIPY-labeled cyclodextrin (BODIPY-CD) was synthesized in house, and was used to monitor the distribution and trafficking in cells with excitation of 488 nm and emission of 525 nm. RSVA 405, A769662, dorsomorphin and bafilomycin A1 were purchased from Tocris Bioscience (5138, 3336, 3093 and 1334). AICAR and palmitate (PA) were purchased from Sigma-Aldrich (A9978 and P9767). All the antibodies are summarized in .
Table 1. Summary of the antibodies used in experiments.
Cell culture
Wild-type fibroblasts (Coriell Cell Repository, GM05659) and NPC1-patient fibroblasts (Coriell Cell Repository, GM03123; a heterozygote, 1 allele carries a missense mutation C > T at nucleotide 709 (709C > T) in exon 6 of the NPC1 gene, resulting in a substitution of a serine for a proline at codon 237; the second allele also carries a missense mutation T > C at nucleotide 3182 (3182T > C) in exon 21 which results in the substitution of a threonine for an isoleucine at codon 1061 in a transmembrane domain) were cultured in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, 12430047) with 10% fetal bovine serum (GE, SH30071) and 100 U/ml penicillin-streptomycin (Thermo Fisher Scientific, 15140-122,) in a 37°C, 5% CO2, and 75% humidity incubator. The cells were reprogrammed and the induced pluripotent stem cells both from WT and NPC1-patient cells were generated, followed by neural induction to neural stem cells (NSCs) and differentiation into neurons, as described previously.Citation13 Both the WT and NPC1 NSCs were cultured in StemPro NSC SFM (Life Technologies, A1050901) containing knockout Dulbecco's modified Eagle's medium-F12, StemPro neural supplement, 20 ng/ml bFGF, 20 ng/ml EGF and 1X GlutaMAX on Matrigel (Corning, 354263)-coated flasks. The medium for neuron differentiation was composed of Neurobasal medium (Life Technologies, 21103-049), B27 (Life Technologies, 17504-044), GlutaMAX (Life Technologies, 35050–061), 10 ng/ml BDNF (Life Technologies, PHC7074), 10 ng/ml GDNF (Life Technologies, PHC7074), 1 μM cAMP (Sigma, D0627) and 200 ng/ml of L-ascorbic acid (Sigma, A4403). Briefly, NSCs were seeded in the poly-L-ornithine and LAM/laminin dual pre-coated 96-well or 6-well plates (Corning, 354658 or 3546587) as indicated at 3000 cells/well (cell density of 5 × 104 cells/cm2). Cells were cultured for 4 wk in the neural differentiation medium and half of the medium was replaced twice a wk during continuous culturing. U2OS cells (ATCC, HTB-96) were cultured in McCoy's 5a Medium (Life Technologies, 16600-108) with 10% fetal bovine serum and 100 U/ml penicillin-streptomycin.
Cell transfection and generation of stable cell lines
RAB5A-RFP, RAB7A-RFP and LAMP1-RFP Bacmam 2.0 baculovirus (Thermo Fisher Scientific, C10587, C10589 and C10504) were used to transiently transfect U2OS cells and respectively label early endosomes, late endosomes and lysosomes with RFP in live cells per the manufacturer's instructions. TagRFP-LC3B lentiviral particles (EMD Millipore, 17–10143) were used to transfect fibroblasts at the indicated multiplicity of infection and label the autophagic vesicles including autophagosomes and autolysosomes. Tandem mRFP-GFP-LC3B BacMam 2.0 baculovirus (Thermo Fisher Scientific, P36239) was used for transient transduction of fibroblasts and for monitoring autophagosome formation and fusion with lysosomes into autolysosomes as previously described.Citation32
PRKAB1 and PRKAB2 shRNA Lentiviral Particles Gene Silencer system (Santa Cruz Biotechnology, sc-38925 and sc-38927) were used to generate stable cell lines with silencing of PRKAB1 or/and PRKAB2 expression per the manufacturer's instructions. Briefly, WT and NPC1 fibroblasts were seeded in 6-well plates (100,000 cells/well) with complete growth medium, and incubated overnight. The cells were ∼50% confluent on the day of infection. Medium was replaced with the medium containing polybrene (Santa Cruz Biotechnology, sc-134220) at a final concentration of 5 µg/ml. The lentiviral particles were handled per the manufacturer's instruction and added to the plates to achieve a multiplicity of infection of 2–10. The cells were incubated with lentiviral particles for 48 h, followed by a 1:3 split and cultured for another 48 h without lentiviral particles and polybrene. Stable clones expressing PRKAB1 and PRKAB2 shRNA were selected by growth in 8 µg/ml puromycin dihydrochloride (Santa Cruz Biotechnology, sc-108071). The resistant colonies were selected and expanded, followed by western blotting analysis of stable shRNA expression. PRKAB1 and PRKAB2 CRISPR Activation Plasmids (Santa Cruz Biotechnology, sc-402952-ACT and sc-403537-ACT) were used to transiently overcome PRKAB1 or PRKAB2 shRNA-induced gene silencing. Briefly, the selected clones with stable expression of PRKAB1 or/and PRKAB2 shRNA were seeded in 6-well plates (100,000 cells/well) in culture medium without antibiotics, and the cells were approximately 40–80% confluent after overnight incubation. Medium was replaced with antibiotic-free medium by adding UltraCruz transfection reagent (Santa Cruz Biotechnology, sc-395739) and plasmid DNA solution at optimized concentrations. The cells were incubated for 48 h, followed by medium change and cultured for another 24 h. Cells were examined by western blotting to evaluate PRKAB1 or PRKAB2 gene expression.
Live-cell imaging for study of kinetics and distribution of cyclodextrin in cells
The kinetics of BODIPY-CD was measured in WT and NPC1 fibroblasts, NSCs, and U2OS cells. After live staining of the nucleus (Life Technologies, R37605), the cells were cultured in black, clear-bottom, 96-well plates (Greiner Bio-One, 655097), and incubated with 10 µM BODIPY-CD for the indicated times. Cells with different BODIPY-CD incubation time were immediately imaged after 2 rinses with phosphate-buffered saline (PBS; Life Technologies, 14190-169). Cells after 120 min incubation with 10 µM BODIPY-CD incubation and cell rinses were imaged at 1 Hz for 120 min at 37°C to evaluate the kinetics of this labeled compound leaving cells. All the images were acquired on an INCell2200 automated fluorescence microscope (GE Healthcare Life Sciences) with a 40 × 0.75 High NA objective lens and FITC (BODIPY-CD) and DAPI (nuclei) filter sets. The intracellular fluorescence intensity of the FITC channel was analyzed using INCell Analyzer software.
U2OS cells transiently transfected with RAB5A-RFP, RAB7A-RFP and LAMP1-RFP BacMam 2.0 baculovirus. TagRFP-LC3B lentiviral particles were used to determine the cellular distribution of BODIPY-CD. The cells were incubated with 10 µM BODIPY-CD for the indicated times (5-60 min). After 2 rinses with PBS, cells were imaged on an INCell2200 imaging system with a 40 × 0.75 High NA objective lens and FITC (BODIPY-CD), CY3 (RFP-labeled organelles) and DAPI (nuclei) filter sets. The colocalization was analyzed by the merging of images of FITC and CY3 channels using the INCell Analyzer software.
Filipin staining
Filipin dye (Sigma-Aldrich, F9765) detects the unesterified cholesterol in cells. Cells were cultured at densities of 500 cells/well for fibroblasts and 1000 cells/well for NSCs in black, clear-bottom 96-well plates and treated with compounds for the indicated days. After rinsing twice with PBS, cells were fixed with 100 μl/well of 3.2% paraformaldehyde for 15 min followed by a cell rinse. The cells were stained with 100 μl/well of 50 ng/ml filipin (freshly dissolved in DMSO at 10 mg/ml and then diluted in PBS) at room temperature for 1 h. After rinsing with PBS, cell nuclei were stained with 16 µM red nuclear dye (AAT Bioquest, 17552) for 30 min at room temperature followed by a cell wash. The cells were then imaged using the INCell2200 imaging system with a 20X or 40X objective lens using DAPI and Cy5 filter sets. Image analysis was conducted using the INCell Analyzer software. With a multitarget analysis protocol, nuclei were segmented using the top-hat segmentation method with a minimum area set at 125 μm and a sensitivity set at 60. Filipin staining was identified as “organelles” by the analysis software and was segmented using the multi scale top-hat algorithm. Settings for filipin detection involved identification of filipin aggregates ranging in size from 10 to 125 μm (30 to 300 pixels) and a sensitivity setting of 50. Total filipin intensity was calculated from cells exceeding the user-defined threshold for whole-cell intensity.
LysoTracker Red staining
LysoTracker Red dye stains cellular acidic compartments and allows visualization of enlarged lysosomes at the proper dye concentration in NPC1-patient cells. Cell seeding and treatment were exactly same as for the filipin staining assay. After washing with PBS, cells were incubated with 100 μl/well 50 nM LysoTracker Red dye (Life Technologies, L-7528) at 37°C for 1 h. Cells were then fixed in 100 μl/well 3.2% paraformaldehyde solution containing 1 μg/ml Hoechst 33342 (Life Technologies, H1399) in PBS and incubated at room temperature for 30 min. After washing twice with PBS, cells were imaged in an INCell Analyzer 2200 using DAPI and DsRed filter sets. Images were analyzed with the multi-target analysis protocol as with the filipin staining assay.
Immunocytochemistry
Cells were grown and stained in black, clear-bottom 96-well plates. All the liquid handling steps including aspiration, dispensing and washing were conducted using an automated microtiter plate liquid handling system (GNF system, San Diego, CA, USA) for blocking, antibody incubation and nucleus staining at 50 µl volume and cell wash with 200 µl PBS. The cell staining buffer (Biolegend, 420201) was used to block and dilute antibodies. Briefly, 4% paraformaldehyde was dispensed into each well and cells were fixed for 30 min. The fixation solution was aspirated and 50 µl 0.1% Triton X-100 (Sigma, T8787) was dispensed for permeabilization at 15 min, followed by 1 h blocking. The cells were incubated overnight at 4°C with primary antibodies at the optimized concentration. After rinsing 3 times with PBS, secondary antibodies of correlated species were added. Cells were then stained with Hoechst 33342 nuclear dye for 30 min after rinsing with PBS 3 times and imaged in the INCell2200 imaging system with a 40X objective lens, and imaging detection was performed using FITC (LC3, green), CY5 (SQSTM1, red), and DAPI (nucleus, blue) filter sets. Image analysis was conducted using the INCell Analyzer software as described above.
Western blotting and immunoprecipitation
After the various treatments, cells were lysed in RIPA buffer (Enzo Life Sciences, ADI-80-1284) supplemented with protease inhibitors and phosphatase inhibitor cocktail (Roche, 5892791001 and 4906837001). The cell lysates were clarified by centrifugation at 20,000 x g for 15 min, and followed by protein quantification with the BCA assay kit (Thermo Fisher Scientific, 23225). The cell lysates with similar protein concentrations were subsequently applied to Bis-Tris or Tris-Acetate gels for protein separation, and the proteins were transferred from gels to polyvinylidene difluoride (PVDF) membrane by dry transfer (Thermo Fisher Scientific, iBlot 2 Gel Transfer Device) or tank wet transfer. Immunoblot analysis was performed with the indicated antibodies and the chemiluminescence signal was visualized with Luminata Forte Western HRP substrate (EMD Millipore, WBLUF0100) in the BioSpectrum system (UVP, LLC). The chemiluminescence intensities of the bands were quantified in the VisionWorks LS software (UVP, LLC).
For the immunoprecipitation, the Pierce direct magnetic IP/Co-IP kit was used (Thermo Fisher Scientific, 88828). Briefly, the antibodies were purified to remove BSA and gelatin, as well as preservative using the Pierce Antibody Clean-up Kit (Thermo Fisher Scientific, 44600), followed by coupling the purified antibodies to the activated N-hydroxysuccinimide magnetic beads. The antibody-coupled beads were washed to remove any noncovalently bound antibodies and quenched. Cell lysates were subjected to immunoprecipitation by incubation with the antibody-coupled beads, followed by a wash to remove non-bound proteins. The samples were then eluted in a low-pH elution buffer to dissociate the coprecipitated proteins that were analyzed by western blotting.
Cellular thermal shift assay (CETSA)
CETSA was conducted as previously described.Citation38 Cells were harvested and rinsed with PBS, then re-suspended in detergent-free buffer (25 mM HEPES, pH 7.0, 20 mM MgCl2, 2 mM DTT) supplemented with protease inhibitors and phosphatase inhibitor cocktail. The cell suspensions were lysed via 3 freeze-thaw cycles with liquid nitrogen, followed by centrifugation at 20,000 g for 20 min at 4°C to separate the soluble fraction from the cell debris. For the CETSA melting curve experiments, the cell lysates were diluted in detergent-free buffer and divided into 2 aliquots, followed by treatment with or without 300 µM MβCD. After 30-min treatment at room temperature, each sample was divided into 14 small aliquots in 100 μl/tube and heated individually at different temperatures (37-63°C with 2°C interval) for 3 min in a thermal cycler (Eppendorf), followed by immediate cooling for 3 min on ice. The heated cell extracts were centrifuged at 20,000 × g for 20 min at 4°C to separate the soluble fractions from precipitates. After centrifugation, the supernatant was analyzed by western blotting with anti-PRKAB1/2 and PRKAA antibody. The relative chemiluminescence intensity of each sample at different temperatures was used to generate the temperature-dependent melting curve. The apparent aggregation temperature (Tagg) for each AMPK subunit in each cell line was calculated by nonlinear regression. For isothermal dose-response fingerprint (ITDR) experiments, the cell lysates were treated with MβCD at 14-point serial dilutions ranging 300 to 1000 µM and were then heated at 53°C following the procedure described above. DMSO, a solvent, was used as a vehicle control. Western blotting analysis of the supernatant with anti-PRKAB1/2 antibody and anti-ACTB/β-actin was conducted. The relative chemiluminescence intensity of PRKAB1 or PRKAB2 subunit was normalized with ACTB and the binding affinity of a compound with PRKAB1 or PRKAB2 subunit was calculated by nonlinear regression. All data represent mean ± SEM of at least 3 replicates.
TR-FRET assay
A cell-based homogeneous TR-FRET assay kit (Cisbio Bioassays, 64MPKPEG) was used to quantify the levels of phosphorylated threonine 172 (phospho-Thr172) on PRKAA, indicating the activation of AMPK. The phospho-AMPK assay involves 2 specific monoclonal anti-AMPK antibodies, one labeled with Eu3+-cryptate (donor) and the other with d2 (acceptor). While one antibody specifically binds to with the phosphorylated threonine 172 residue, the other antibody binds to a nonphosphorylated portion of PRKAA, bringing the 2 dyes in close proximity for FRET. The FRET signal is directly proportional to levels of phospho-Thr172 on PRKAA. The experiment was conducted following the manufacturer's protocol. Briefly, cells were harvested and plated into white bottom solid 384-well plates (Greiner Bio-One, 781074) with 10 µg/ml rhVTN-N supplemented StemPro NSC SFM (Life Technologies, A1050901) at a density of 20,000 cells/well. Cells were incubated overnight, followed by compound treatment for 90 min. The cells were lysed in assay plates and 2 reagent mixtures from the assay kit were added into the wells sequentially. The assay plates were incubated overnight at RT, followed by TR-FRET detection in an EnVision plate reader (Perkin Elmer) with an excitation of 320 nm and emission-1 of 620 nm and emission-2 of 665 nm. The fluorescence signal ratio of 665 nm to 620 nm was calculated and used for quantification. All data represent mean ± SEM of at least 3 replicates.
Data analysis
Half maximal inhibitory (IC50) or activating (EC50) concentration value was calculated using Prism software (GraphPad Software). Unless otherwise noted, all values are expressed as the mean ± SEM. For the statistical analysis, results were analyzed using one-way or 2-way ANOVA and differences were considered significant if p < 0.05.
Abbreviations
| AMPK | = | AMP-activated protein kinase |
| ATG | = | autophagy related |
| CETSA | = | cellular thermal shift assay |
| hESC | = | human embryonic stem cells |
| HPβCD | = | 2-hydroxypropyl-β-cyclodextrin |
| iPSCs | = | induced pluripotent stem cells |
| ITDRF | = | isothermal dose-response fingerprints |
| MAP1LC3B/LC3B | = | microtubule associated protein 1 light chain 3B |
| MβCD | = | methyl-β-cyclodextrin |
| MEF | = | mouse embryonic fibroblast |
| MTORC1 | = | mechanistic target of rapamycin complex 1 |
| NPC | = | Niemann-Pick disease, type C |
| NSCs | = | neural stem cells |
| PRKAA | = | protein kinase AMP-activated catalytic subunit alpha |
| PRKAB | = | protein kinase AMP-activated non-catalytic subunit beta |
| SNARE | = | soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| SQSTM1 | = | sequestosome 1 |
| Tagg, | = | apparent aggregation temperature |
| TFEB | = | transcription factor EB |
| TR-FRET | = | time-resolved fluorescence energy transfer |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Supplemental Material
Download MS Word (39 MB)Acknowledgments
The authors also thank Rong Li for neuron differentiation, Steve Titus for InCell image data analysis, and DeeAnn Visk for editing the manuscript.
Funding
This work is funded by the Intramural Research Program of NCATS, NIH (W.Z. and J.M.) and NICHD, NIH (F.D.P. and C.W.) and by NIH grant R01 NS081985 (D.S.O.).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Related Research Data
References
- Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis 2010; 5:16; PMID:20525256; https://doi.org/10.1186/1750-1172-5-16
- Carstea ED, Morris JA, Coleman KG, Loftus SK, Zhang D, Cummings C, Gu J, Rosenfeld MA, Pavan WJ, Krizman DB, et al. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science 1997; 277:228-31; PMID:9211849; https://doi.org/10.1126/science.277.5323.228
- Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab 2012; 106:330-44; PMID:22572546; https://doi.org/10.1016/j.ymgme.2012.03.012
- Aqul A, Liu B, Ramirez CM, Pieper AA, Estill SJ, Burns DK, Liu B, Repa JJ, Turley SD, Dietschy JM. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J Neurosci 2011; 31:9404-13; PMID:21697390; https://doi.org/10.1523/JNEUROSCI.1317-11.2011
- Liu B, Li H, Repa JJ, Turley SD, Dietschy JM. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J Lipid Res 2008; 49:663-9; PMID:18077828; https://doi.org/10.1194/jlr.M700525-JLR200
- Maarup TJ, Chen AH, Porter FD, Farhat NY, Ory DS, Sidhu R, Jiang X, Dickson PI. Intrathecal 2-hydroxypropyl-beta-cyclodextrin in a single patient with Niemann-Pick C1. Mol Genet Metab 2015; 116:75-9; PMID:26189084; https://doi.org/10.1016/j.ymgme.2015.07.001
- Chen FW, Li C, Ioannou YA. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS One 2010; 5:e15054; PMID:21124786; https://doi.org/10.1371/journal.pone.0015054
- Rosenbaum AI, Zhang G, Warren JD, Maxfield FR. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc Natl Acad Sci U S A 2010; 107:5477-82; PMID:20212119; https://doi.org/10.1073/pnas.0914309107
- Tortelli B, Fujiwara H, Bagel JH, Zhang J, Sidhu R, Jiang X, Yanjanin NM, Shankar RK, Carillo-Carasco N, Heiss J, et al. Cholesterol homeostatic responses provide biomarkers for monitoring treatment for the neurodegenerative disease Niemann-Pick C1 (NPC1). Hum Mol Genet 2014; 23:6022-33; PMID:24964810; https://doi.org/10.1093/hmg/ddu331
- Soga M, Ishitsuka Y, Hamasaki M, Yoneda K, Furuya H, Matsuo M, Ihn H, Fusaki N, Nakamura K, Nakagata N, et al. HPGCD outperforms HPBCD as a potential treatment for Niemann-Pick disease type C during disease modeling with iPS cells. Stem Cells 2015; 33:1075-88; PMID:25522247; https://doi.org/10.1002/stem.1917
- Maetzel D, Sarkar S, Wang H, Abi-Mosleh L, Xu P, Cheng AW, Gao Q, Mitalipova M, Jaenisch R. Genetic and chemical correction of cholesterol accumulation and impaired autophagy in hepatic and neural cells derived from Niemann-Pick Type C patient-specific iPS cells. Stem Cell Reports 2014; 2:866-80; PMID:24936472; https://doi.org/10.1016/j.stemcr.2014.03.014
- Sarkar S, Carroll B, Buganim Y, Maetzel D, Ng AH, Cassady JP, Cohen MA, Chakraborty S, Wang H, Spooner E, et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep 2013; 5:1302-15; PMID:24290752; https://doi.org/10.1016/j.celrep.2013.10.042
- Yu D, Swaroop M, Wang M, Baxa U, Yang R, Yan Y, Coksaygan T, DeTolla L, Marugan JJ, Austin CP, et al. Niemann-Pick Disease Type C: Induced Pluripotent Stem Cell-Derived Neuronal Cells for Modeling Neural Disease and Evaluating Drug Efficacy. J Biomol Screen 2014; 19:1164-73; PMID:24907126; https://doi.org/10.1177/1087057114537378
- Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy 2012; 8:719-30; PMID:22647656; https://doi.org/10.4161/auto.19469
- Elrick MJ, Lieberman AP. Autophagic dysfunction in a lysosomal storage disorder due to impaired proteolysis. Autophagy 2013; 9:234-5; PMID:23086309; https://doi.org/10.4161/auto.22501
- Fullgrabe J, Klionsky DJ, Joseph B. Histone post-translational modifications regulate autophagy flux and outcome. Autophagy 2013; 9:1621-3; PMID:23934085; https://doi.org/10.4161/auto.25803
- Loos B, du Toit A, Hofmeyr JH. Defining and measuring autophagosome flux-concept and reality. Autophagy 2014; 10:2087-96; PMID:25484088; https://doi.org/10.4161/15548627.2014.973338
- Ko DC, Milenkovic L, Beier SM, Manuel H, Buchanan J, Scott MP. Cell-autonomous death of cerebellar purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet 2005; 1:81-95; PMID:16103921
- Pacheco CD, Kunkel R, Lieberman AP. Autophagy in Niemann-Pick C disease is dependent upon Beclin-1 and responsive to lipid trafficking defects. Hum Mol Genet 2007; 16:1495-503; PMID:17468177; https://doi.org/10.1093/hmg/ddm100
- Meske V, Priesnitz T, Albert F, Ohm TG. How to reduce the accumulation of autophagic vacuoles in NPC1-deficient neurons: a comparison of two pharmacological strategies. Neuropharmacology 2015; 89:282-9; PMID:25446672; https://doi.org/10.1016/j.neuropharm.2014.10.006
- Liao G, Cheung S, Galeano J, Ji AX, Qin Q, Bi X. Allopregnanolone treatment delays cholesterol accumulation and reduces autophagic/lysosomal dysfunction and inflammation in Npc1-/- mouse brain. Brain Res 2009; 1270:140-51; PMID:19328188; https://doi.org/10.1016/j.brainres.2009.03.027
- Ordonez MP, Roberts EA, Kidwell CU, Yuan SH, Plaisted WC, Goldstein LS. Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum Mol Genet 2012; 21:2651-62; PMID:22437840; https://doi.org/10.1093/hmg/dds090
- Tamura A, Yui N. beta-Cyclodextrin-threaded biocleavable polyrotaxanes ameliorate impaired autophagic flux in Niemann-Pick type C disease. J Biol Chem 2015; 290:9442-54; PMID:25713067; https://doi.org/10.1074/jbc.M115.636803
- Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 2005; 64:113-22; PMID:15751225; https://doi.org/10.1093/jnen/64.2.113
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, Agid Y. Apoptosis and autophagy in nigral neurons of patients with Parkinson's disease. Histol Histopathol 1997; 12:25-31; PMID:9046040
- Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, et al. Cargo recognition failure is responsible for inefficient autophagy in Huntington's disease. Nat Neurosci 2010; 13:567-76; PMID:20383138; https://doi.org/10.1038/nn.2528
- Song CY, Guo JF, Liu Y, Tang BS. Autophagy and Its Comprehensive Impact on ALS. Int J Neurosci 2012; 122:695-703; PMID:22827270; https://doi.org/10.3109/00207454.2012.714430
- Nah J, Yuan J, Jung YK. Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol Cells 2015; 38:381-9; PMID:25896254; https://doi.org/10.14348/molcells.2015.0034
- Fenyvesi F, Reti-Nagy K, Bacso Z, Gutay-Toth Z, Malanga M, Fenyvesi E, Szente L, Váradi J, Ujhelyi Z, Fehér P, et al. Fluorescently labeled methyl-beta-cyclodextrin enters intestinal epithelial Caco-2 cells by fluid-phase endocytosis. PLoS One 2014; 9:e84856; PMID:24416301; https://doi.org/10.1371/journal.pone.0084856
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 2004; 117:2805-12; PMID:15169837; https://doi.org/10.1242/jcs.01131
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016; 12:1-222; PMID:26799652; https://doi.org/10.1080/15548627.2015.1100356
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007; 282:24131-45; PMID:17580304; https://doi.org/10.1074/jbc.M702824200
- He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 2009; 43:67-93; PMID:19653858; https://doi.org/10.1146/annurev-genet-102808-114910
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008; 30:214-26; PMID:18439900; https://doi.org/10.1016/j.molcel.2008.03.003
- Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011; 13:132-41; PMID:21258367; https://doi.org/10.1038/ncb2152
- Li J, Wang Y, Wen X, Ma XN, Chen W, Huang F, et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J Mol Cell Cardiol 2015; 86:62-74; PMID:26196303; https://doi.org/10.1016/j.yjmcc.2015.07.010
- Li X, Wang L, Zhou XE, Ke J, de Waal PW, Gu X, Tan MH, Wang D, Wu D, Xu HE, et al. Structural basis of AMPK regulation by adenine nucleotides and glycogen. Cell Res 2015; 25:50-66; PMID:25412657; https://doi.org/10.1038/cr.2014.150
- Jafari R, Almqvist H, Axelsson H, Ignatushchenko M, Lundback T, Nordlund P, Martinez Molina D. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc 2014; 9:2100-22; PMID:25101824; https://doi.org/10.1038/nprot.2014.138
- Polekhina G, Gupta A, van Denderen BJ, Feil SC, Kemp BE, Stapleton D, Parker MW. Structural basis for glycogen recognition by AMP-activated protein kinase. Structure 2005; 13:1453-62; PMID:16216577; https://doi.org/10.1016/j.str.2005.07.008
- Xiao B, Sanders MJ, Carmena D, Bright NJ, Haire LF, Underwood E, Patel BR, Heath RB, Walker PA, Hallen S, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun 2013; 4:3017; PMID:24352254; https://doi.org/10.1038/ncomms4017
- Meske V, Erz J, Priesnitz T, Ohm TG. The autophagic defect in Niemann-Pick disease type C neurons differs from somatic cells and reduces neuronal viability. Neurobiol Dis 2014; 64:88-97; PMID:24412309; https://doi.org/10.1016/j.nbd.2013.12.018
- Geng J, Klionsky DJ. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep 2008; 9:859-64.
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell 2012; 151:1256-69; PMID:23217709; https://doi.org/10.1016/j.cell.2012.11.001
- Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, Fedele AO, Polishchuk R, Sorrentino NC, Simons K, et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. Embo J 2010; 29:3607-20; PMID:20871593; https://doi.org/10.1038/emboj.2010.237
- Sarkar S, Maetzel D, Korolchuk VI, Jaenisch R. Restarting stalled autophagy a potential therapeutic approach for the lipid storage disorder, Niemann-Pick type C1 disease. Autophagy 2014; 10:1137-40; PMID:24879158; https://doi.org/10.4161/auto.28623
- Song W, Wang F, Lotfi P, Sardiello M, Segatori L. 2-Hydroxypropyl-beta-cyclodextrin promotes transcription factor EB-mediated activation of autophagy: implications for therapy. J Biol Chem 2014; 289:10211-22; PMID:24558044; https://doi.org/10.1074/jbc.M113.506246
- Wehrmann ZT, Hulett TW, Huegel KL, Vaughan KT, Wiest O, Helquist P, Goodson H. Quantitative comparison of the efficacy of various compounds in lowering intracellular cholesterol levels in Niemann-Pick type C fibroblasts. PLoS One 2012; 7:e48561; PMID:23144769; https://doi.org/10.1371/journal.pone.0048561
- Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 2011; 13:655-67; PMID:21641547; https://doi.org/10.1016/j.cmet.2011.03.023
- Peake KB, Vance JE. Normalization of cholesterol homeostasis by 2-hydroxypropyl-beta-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J Biol Chem 2012; 287:9290-8; PMID:22277650; https://doi.org/10.1074/jbc.M111.326405
- Peake KB, Vance JE. Defective cholesterol trafficking in Niemann-Pick C-deficient cells. FEBS Lett 2010; 584:2731-9; PMID:20416299; https://doi.org/10.1016/j.febslet.2010.04.047
- Dong F, Mo Z, Eid W, Courtney KC, Zha X. Akt inhibition promotes ABCA1-mediated cholesterol efflux to ApoA-I through suppressing mTORC1. PLoS One 2014; 9:e113789; PMID:25415591; https://doi.org/10.1371/journal.pone.0113789
- Yang J, Maika S, Craddock L, King JA, Liu ZM. Chronic activation of AMP-activated protein kinase-alpha1 in liver leads to decreased adiposity in mice. Biochem Biophys Res Commun 2008; 370:248-53; PMID:18381066; https://doi.org/10.1016/j.bbrc.2008.03.094
- Habegger KM, Hoffman NJ, Ridenour CM, Brozinick JT, Elmendorf JS. AMPK enhances insulin-stimulated GLUT4 regulation via lowering membrane cholesterol. Endocrinology 2012; 153:2130-41; PMID:22434076; https://doi.org/10.1210/en.2011-2099
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441:880-4; PMID:16625205; https://doi.org/10.1038/nature04723
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem 2010; 285:13107-20; PMID:20178983; https://doi.org/10.1074/jbc.M110.100420
- Decressac M, Mattsson B, Weikop P, Lundblad M, Jakobsson J, Bjorklund A. TFEB-mediated autophagy rescues midbrain dopamine neurons from alpha-synuclein toxicity. Proc Natl Acad Sci U S A 2013; 110:E1817-26; PMID:23610405; https://doi.org/10.1073/pnas.1305623110
- Cortes CJ, La Spada AR. The many faces of autophagy dysfunction in Huntington's disease: from mechanism to therapy. Drug Discov Today 2014; 19:963-71; PMID:24632005; https://doi.org/10.1016/j.drudis.2014.02.014
- Li L, Zhang X, Le W. Altered macroautophagy in the spinal cord of SOD1 mutant mice. Autophagy 2008; 4:290-3; PMID:18196963; https://doi.org/10.4161/auto.5524
- Zhang X, Chen S, Song L, Tang Y, Shen Y, Jia L, Le W. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy 2014; 10:588-602; PMID:24441414; https://doi.org/10.4161/auto.27710
- Vingtdeux V, Giliberto L, Zhao H, Chandakkar P, Wu Q, Simon JE, Janle EM, Lobo J, Ferruzzi MG, Davies P, et al. AMP-activated protein kinase signaling activation by resveratrol modulates amyloid-beta peptide metabolism. J Biol Chem 2010; 285:9100-13; PMID:20080969; https://doi.org/10.1074/jbc.M109.060061
- Fornai F, Longone P, Cafaro L, Kastsiuchenka O, Ferrucci M, Manca ML, Lazzeri G, Spalloni A, Bellio N, Lenzi P, et al. Lithium delays progression of amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2008; 105:2052-7; PMID:18250315; https://doi.org/10.1073/pnas.0708022105
- Ward S, O'Donnell P, Fernandez S, Vite CH. 2-hydroxypropyl-beta-cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr Res 2010; 68:52-6; PMID:20357695; https://doi.org/10.1203/PDR.0b013e3181df4623
- Pontikis CC, Davidson CD, Walkley SU, Platt FM, Begley DJ. Cyclodextrin alleviates neuronal storage of cholesterol in Niemann-Pick C disease without evidence of detectable blood-brain barrier permeability. J Inherit Metab Dis 2013; 36:491-8; PMID:23412751; https://doi.org/10.1007/s10545-012-9583-x