
ABSTRACT
Phosphorylation is a major type of post-translational modification, which can influence the cellular physiological function. ATG4B, a key macroautophagy/autophagy-related protein, has a potential effect on the survival of tumor cells. However, the role of ATG4B phosphorylation in cancers is still unknown. In this study, we identified a novel phosphorylation site at Ser34 of ATG4B induced by AKT in HCC cells. The phosphorylation of ATG4B at Ser34 had little effect on autophagic flux, but promoted the Warburg effect including the increase of L-lactate production and glucose consumption, and the decrease of oxygen consumption in HCC cells. The Ser34 phosphorylation of ATG4B also contributed to the impairment of mitochondrial activity including the inhibition of F1Fo-ATP synthase activity and the elevation of mitochondrial ROS in HCC cells. Moreover, the phosphorylation of ATG4B at Ser34 enhanced its mitochondrial location and the subsequent colocalization with F1Fo-ATP synthase in HCC cells. Furthermore, recombinant human ATG4B protein suppressed the activity of F1Fo-ATP synthase in MgATP submitochondrial particles from patient-derived HCC tissues in vitro. In brief, our results demonstrate for the first time that the phosphorylation of ATG4B at Ser34 participates in the metabolic reprogramming of HCC cells via repressing mitochondrial function, which possibly results from the Ser34 phosphorylation-induced mitochondrial enrichment of ATG4B and the subsequent inhibition of F1Fo-ATP synthase activity. Our findings reveal a noncanonical working pattern of ATG4B under pathological conditions, which may provide a scientific basis for developing novel strategies for HCC treatment by targeting ATG4B and its Ser34 phosphorylation.
Abbreviations
| AKT1/PKB | = | AKT serine/threonine kinase 1 |
| ATG4A | = | autophagy-related 4A cysteine peptidase |
| ATG4B | = | autophagy-related 4B cysteine peptidase |
| ATG4D | = | autophagy-related 4D cysteine peptidase |
| ATG5 | = | autophagy-related 5 |
| ATG7 | = | autophagy-related 7 |
| ATP5A1 | = | ATP synthase, H+ transporting, mitochondrial F1 complex, alpha subunit 1, cardiac muscle |
| ATP5C1 | = | ATP synthase, H+ transporting, mitochondrial F1 complex, gamma polypeptide 1 |
| Baf A1 | = | bafilomycin A1 |
| CRISPR | = | clustered regularly interspaced short palindromic repeats |
| DGc | = | differential gradient centrifugation |
| GABARAPL1 | = | GABA type A receptor associated protein like 1 |
| GFP | = | green fluorescent protein |
| HCC | = | hepatocellular carcinoma |
| IP | = | immunoprecipitation |
| MAP1LC3/LC3 | = | microtubule associated protein 1 light chain 3 |
| MEF | = | mouse embryonic fibroblast |
| mitoROS | = | mitochondrial reactive oxygen species |
| MTP | = | mitochondrial targeting peptide |
| OMM | = | outer mitochondrial membrane |
| OXPHOS | = | oxidative phosphorylation |
| PK | = | proteinase K |
| PTEN | = | phosphatase and tensin homolog |
| siRNA | = | small interfering RNA |
| SMPs | = | submitochondrial particles |
| WCL | = | whole cell lysate |
Introduction
Hepatocellular carcinoma (HCC), a primary malignancy of liver, has become a main cause of cancer-related death worldwide with an incidence of approximately 850,000 new cases per year [Citation1]. HCC is a highly aggressive neoplasm characterized by frequent metastasis and therapeutic resistance. Although the treatments for HCC patients have improved in the past decades, there are still major needs for effective prevention at an early stage and new therapies at the intermediate and advanced stage. Therefore, it is necessary and urgent to understand the more detailed mechanisms about the occurrence and progression of HCC so as to discover novel targets for its management.
Mitochondria, as the powerhouses of the cells, produce the most energy-rich molecules, adenosine triphosphates (ATPs), via oxidative phosphorylation (OXPHOS) under normal physiological conditions. Previous reports have shown that the dysfunction of mitochondria often results in many disorders [Citation2].– [Citation6] In 1926, Otto Warburg reported that tumor cells could produce ATPs mainly via glycolysis (i.e., not by OXPHOS) even under aerobic conditions, which may be due to the impairment of mitochondrial respiration. Lately, more and more studies have indicated that the impairment of mitochondrial metabolism can enhance glycolysis and play a key role in metabolic remodeling in many kinds of tumors [Citation7–Citation9]. This metabolic transformation of tumors from mitochondrial respiration to aerobic glycolysis is also named the Warburg effect, which can obviously promote occurrence, progression and chemoresistance of malignancies via different mechanisms [Citation10]. It has been reported that the Warburg effect is also a typical characteristic in most HCC cases. PKM (pyruvate kinase M1/2), a key enzyme in regulating the Warburg effect, is upregulated in HCC cells and is associated with tumorigenesis, aggressive progression, and poor prognosis [Citation11–Citation13]. SLC2A1/glucose transporter 1, a rate-limiting transporter for glucose uptake, is also elevated in some HCC patients and affects tumorigenicity of HCC [Citation14]. In addition, the microRNA-mediated inhibition of the Warburg effect can also influence HCC progression [Citation15]., [Citation16] Moreover, Cuezva et al. have demonstrated that carcinogens in liver are related to a decrease of mitochondrial content, a selective downregulation of ATP5B/beta-F1-ATP synthase level and an increase of the glycolytic GAPDH (glyceraldehyde-3-phosphate dehydrogenase), which supports the hypothesis of the Warburg effect [Citation17]. Recently, targeting the Warburg effect has emerged as a promising option for HCC treatment [Citation18]. Therefore, investigating the detailed mechanisms of the Warburg effect in HCC will be beneficial to develop novel related therapeutic strategies.
ATG4B (autophagy-related 4B cysteine peptidase), a member of C54 peptidases, plays a key role in the regulation of autophagy in mammalian cells. Previous studies have demonstrated that ATG4B regulates autophagy through either cleaving pro-MAP1LC3/LC3 for its further lipidation or delipidating LC3-II from the autophagosome, leading to a change of the autophagy level [Citation19–Citation28]. Growing evidence has shown that ATG4B participates in certain physiological and pathological processes including tumors through affecting the autophagic flux [Citation29–Citation39]. For example, ATG4B can promote tumor progression or chemoresistance via its canonical role of autophagy regulation. Meanwhile, a recent study in colon cancer cells has revealed that ATG4B can enhance cell proliferation independent of its role in autophagy [Citation40], which opens new insight for the noncanonical roles of ATG4B. Besides the regulation of protein level, protein modifications such as phosphorylation, oxidative modification and O-GlcNAcylation are also important mechanisms to affect the function of ATG4B [Citation41–Citation44]. Previous research using high-resolution mass spectrometry-based proteomics has indicated that ATG4B could be phosphorylated at Thr7, Tyr8, Ser34, Ser44, Tyr122, Tyr123, Ser316, Ser383 and Ser392 (http://www.phosphosite.org). Moreover, recent studies have shown that the phosphorylation of ATG4B at Ser383 and Ser392 or Ser316 can influence ATG4B protease activity [Citation42,Citation45] suggesting that the phosphorylation of ATG4B would be an important aspect to regulate its function. However, it is still unclear as to the role of ATG4B phosphorylation in HCC cells.
In the present study, we identified a novel phosphorylation site (Ser34) on ATG4B mediated by AKT in HCC cells. The phosphorylation of ATG4B at Ser34 presented little effect on basal autophagic flux of HCC cells, but dramatically contributed to the inhibition of F1Fo-ATP synthase activity, the increase of mitochondrial reactive oxygen species (mitoROS) and the Warburg effect. Moreover, the phosphorylation of ATG4B at Ser34 promoted its mitochondrial enrichment and the subsequent colocalization with the F1Fo-ATP synthase in HCC cells. Furthermore, recombinant human ATG4B protein suppressed the activity of the F1Fo-ATP synthase in MgATP submitochondrial particles (SMPs) from patient-derived HCC tissues in vitro. In summary, our results demonstrate for the first time that the phosphorylation of ATG4B at Ser34 participates in the metabolic reprogramming of HCC cells via inhibiting mitochondrial function, which possibly results from the Ser34 phosphorylation-induced enrichment of mitochondrial ATG4B and the subsequent repression of F1Fo-ATP synthase activity. Our findings reveals a noncanonical working model of ATG4B under pathological conditions and the role of ATG4B in HCC malignance, which may provide a scientific basis for developing novel strategies for HCC treatment by targeting ATG4B and its Ser34 phosphorylation.
Results
Activation of AKT induces the phosphorylation of ATG4B at Ser34 in HCC cells
It has been reported that overactivation of protein kinases including AKT is a typical phenotype of HCC, so we explored whether the activated AKT could influence the phosphorylation of ATG4B in HCC cells. As shown in A, co-immunoprecipitation (Co-IP) assays using the ATG4BCRISPR HepG2 cells revealed that the ectopically expressed AKT1/PKB and ATG4B appeared in 1 complex, suggesting the probability of interaction between the 2 proteins. Here, the ATG4BCRISPR HepG2 cells were ATG4B hemizygous knockout cells generated by a CRISPR/Cas9-mediated genome editing system (Figure S1A and S1B). Then the effect of AKT1 on the phosphorylation of endogenous ATG4B was detected with Phos-tag technology. As shown in B, overexpression of AKT1 in HepG2 cells significantly increased the phosphorylated ATG4B (p-ATG4B), which was reversed by phosphatase, suggesting that AKT1 may induce the phosphorylation of endogenous ATG4B in HCC cells. Meanwhile, we noticed that there were different positions of bands corresponding to ATG4B (i.e., phosphorylated bands of ATG4B) in the gel. As the migration rate of a protein in a Phos-tag gel can be affected by the number of phosphorylated sites, the different sites of gel shift may arise from different phosphorylation types of ATG4B in this condition.
Figure 1. Activation of AKT induces the phosphorylation of ATG4B at Ser34 in HCC cells. (A) ATG4BCRISPR HepG2 cells (ATG4B hemizygous knockout cells) were transfected with the indicated expression plasmids. Then the whole cell lysates (WCL) were separately used for immunoblotting and immunoprecipitation assays with the corresponding antibodies. (B) HepG2 cells were transfected with MYC-AKT1 expression plasmid or control empty vector (EV). Then the cell lysates were obtained and treated with or without lambda phosphatase. Subsequently, the cell lysates were loaded onto SDS-PAGE gels with or without Phos-tag acrylamide and MnCl2 for immunoblotting assays. (C) The potential phosphorylation sites in ATG4B were predicted with motifscan (http://scansite.mit.edu/motifscan_seq.phtml), and the potential AKT1 phosphorylation motif 31RKYS34 in human ATG4B is shown. The red label represents the location of Ser34 in the 3D structure of the ATG4B protein. (D) ATG4BCRISPR HepG2 cells were transfected with Flag-ATG4BWT or Flag-ATG4BS34A expression plasmid in the presence or absence of MYC-AKT1WT expression vector. Then the cell lysates were prepared and loaded onto SDS-PAGE gels with or without Phos-tag acrylamide and MnCl2 for immunoblotting assays. The average ratio of F-p-ATG4B to t-ATG4B from 3 independent experiments is shown on the right. (E) ATG4BCRISPR HepG2 cells were transfected with the Flag-ATG4BWT expression plasmid in the presence or absence of MYC-AKT1WT expression vector. Then the cells were treated with 3 μM MK2206 or vehicle control (DMSO). Subsequently, the cell lysates were obtained for immunoblotting assays. (F) HepG2 cells were transfected with control siRNA or PTEN siRNA, and then the cell lysates were prepared and loaded onto SDS-PAGE gels with or without Phos-tag acrylamide and MnCl2 for immunoblotting assays. Data are mean ± SD from 3 independent experiments. *, P < 0.05; ns, no significance. MYC-AKT1WT, 1 × MYC-tagged wild-type AKT1 expression plasmid; 3 × Flag-ATG4BWT, 3 × Flag-tagged wild-type ATG4B expression plasmid; p-ATG4B, phosphorylated ATG4B; non-p-ATG4B, non phosphorylated ATG4B; t-ATG4B, total ATG4B; phos, SDS-PAGE gel containing Phos-tag acrylamide and MnCl2; Flag-ATG4BWT, 1 × Flag-tagged wild-type ATG4B expression plasmid; Flag-ATG4BS34A, 1 × Flag-tagged mutant ATG4B expression plasmid (in which Ser34 of ATG4B was mutated to Ala); F-p-ATG4B, the first band of phosphorylated ATG4B; S-p-ATG4B, the second band of phosphorylated ATG4B; LE, long exposure; SE, short exposure; p-ATG4B (S34), Ser34-phosphorylated ATG4B; p-AKT (S473), Ser473-phosphorylated AKT.
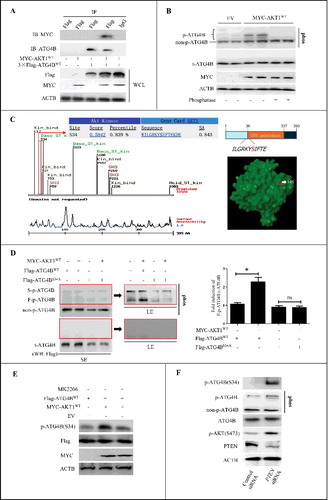
Next, the potential phosphorylation sites in ATG4B were predicted with motifscan (http://scansite.mit.edu/motifscan_seq.phtml). As shown in C, ATG4B (Homo sapiens) contains a potential AKT1 phosphorylation motif 31RKYS34 in its N terminus. Sequence homology alignment showed that several other species including S. cerevisiae, C. elegans, Gallus gallus, Mus musculus, Bos taurus, C. l. familiaris and O. r. rosmarus also contain this “R × × S” motif (Figure S1C). Moreover, the results from NCBI protein blast suggested that the Ser34 site in human ATG4B may be relatively specific compared to human ATG4A, ATG4C and ATG4D (Figure S1D). In addition, previous studies have demonstrated that the Ser34 of endogenous ATG4B can be phosphorylated in mouse liver [Citation46], 3T3-L1 fibroblasts [Citation47] and HEK293T cells [Citation42]. Therefore, we further investigated whether the Ser34 of ATG4B was the phosphorylation target of AKT1 in HCC cells. As shown in D, overexpresion of AKT1 dramatically elevated the level of F-p-ATG4B (the first presumably phosphorylated band of ATG4B from the bottom to the top of the gel) in the HCC cells transfected with a wild-type ATG4B expression plasmid (Flag-ATG4BWT), but not in the cells transfected with the mutant ATG4B expression plasmid (Flag-ATG4BS34A, in which the Ser34 of ATG4B was mutated to Ala). Meanwhile, the ratio of F-p-ATG4B to t-ATG4B (total ATG4B) was markedly increased in the cells co-overexpressing ATG4BWT and AKT1, indicating that the phosphorylation of Ser34 was an important type of ATG4B phosphorylation in AKT1-expressing HCC cells. Co-IP assays also showed that overexpression of AKT1 significantly enhanced the Ser/Thr phosphorylation of ATG4BWT (but not ATG4BS34A)-containing immunoprecipitates using immunoblotting with the antibody against the AKT target motif (“RxRxxS/T” or “RxxS/T”), further suggesting that the Ser34 in the motif 31RKYS34 of ATG4B may be phosphorylated by AKT1 (Figure S2A).
Subsequently, we prepared an antibody against the phosphorylated Ser34 of ATG4B with a titer more than 1:32000 using KLH-C-LGRKY(p-S)IF-NH2 as an immunogen (Figure S2B), and verified the specificity of this antibody with immunoblotting assays (Figure S2C and S2D). Using this antibody, we found that ectopic expression of AKT1 remarkably increased the Ser34 phosphorylation of ATG4B, which was dramatically attenuated by the AKT inhibitor MK2206 (E). Furthermore, we evaluated the phosphorylation state of ATG4B by activation of endogenous AKT in the HCC cells with PTEN knockdown. As shown in Figure S2E, an optimal PTEN siRNA (PTEN siRNA-1) was identified for the subsequent experiments, which resulted in an efficient interference of PTEN and activation of AKT (S473). F revealed that knockdown of PTEN obviously promoted the phosphorylation of ATG4B at Ser34.
Additionally, we also verified the specificity of ATG4B antibody so as to rule out the possibility of a wrong recognition against its homolog ATG4A (Figure S2F), as ATG4A and ATG4B showed an identity of 55% (NP_443168.2 vs NP_037457.3) in NCBI protein blast analysis. Moreover, motifscan showed that human ATG4A also contains a potential AKT1 phosphorylation motif “RKFS” (Figure S3A), so we examined the effect of AKT-mediated phosphorylation on ATG4A. As shown in Figure S3B, activation of AKT had little effect on ATG4A phosphorylation.
Taken together, the above results demonstrate that activation of AKT induces the phosphorylation of ATG4B at Ser34 in HCC cells.
The Ser34 phosphorylation of ATG4B has little effect on autophagic flux in ATG4B-overexpressing HCC cells
To investigate whether the phosphorylation of ATG4B at Ser34 can influence autophagic flux, autophagy-related markers were detected in the HCC cells overexpressing ATG4BWT or ATG4BS34A. As shown in Figure S4A, overexpression of ATG4BWT or ATG4BS34A dramatically decreased LC3-II and increased SQSTM1/p62. Moreover, overexpression of ATG4BWT or ATG4BS34A in GFP-LC3 stably expressing HepG2 cells markedly decreased the amount of GFP-LC3 puncta (Figure S4B). These results suggested that excess exogenous ATG4B may impair the lipidation of LC3 and negatively regulate autophagic flux in HCC cells, which was consistent with the previous studies in HeLa and MCF-7 cells [Citation23,Citation26]. Meanwhile, this effect was not influenced by the mutation of Ser34 under basal conditions (without extra stress, such as AKT overactivation).
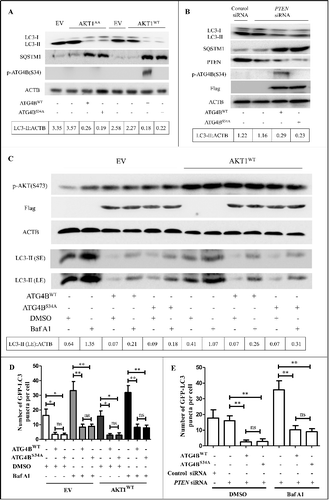
As the phosphorylated degree of ATG4B under the basal condition would be limited, we further examined the effect of ATG4B Ser34 phosphorylation on autophagy in AKT-overactivated HCC cells. As shown in A, co-overexpression of ATG4BWT or ATG4BS34A with AKT1WT or AKT1 kinase-dead mutant (AKT1AA, in which both Thr308 and Ser473 of AKT1 were mutated to Ala) significantly reduced LC3-II and elevated SQSTM1. Similarly, ectopic expression of ATG4BWT or ATG4BS34A combined with PTEN knockdown also dramatically decreased LC3-II and increased SQSTM1 (B). As the decrease of LC3-II level may arise from the impairment of autophagy flux or the enhancement of autophagosome turnover [Citation48], we further used bafilomycin A1 (Baf A1, a compound that blocks the fusion of autophagosomes with lysosomes) in the subsequent experiments to detect the ATG4B-mediated effect on autophagy. As shown in C, Baf A1 treatment significantly upregulated the level of LC3-II, which was largely reversed by overexpression of ATG4BWT or ATG4BS34A, whether or not there was AKT overactivation. In addition, Baf A1 treatment obviously increased the amount of GFP-LC3 puncta in GFP-LC3 stably expressing HepG2 cells, which was reversed by transfection with a plasmid expressing exogenous ATG4BWT or ATG4BS34A, whether or not there was AKT overexpression (D). Similar results were also obtained in the cells with PTEN knockdown (E). The assays with Baf A1 treatment suggested that the decrease of LC3-II level and GFP-LC3 puncta by excess exogenous ATG4BWT or ATG4BS34A may not be attributable to the enhancement of autophagosome turnover, whether or not there was AKT overactivation. In brief, the above data indicate that overexpression of ATG4B impairs autophagic flux in HCC cells, which is independent of ATG4B Ser34 phosphorylation.
Figure 2. The Ser34 phosphorylation of ATG4B has little effect on autophagic flux in ATG4B-overexpressing HCC cells. (A and B) ATG4BCRISPR HepG2 cells were transfected with the indicated expression plasmids in the presence or absence of PTEN siRNA. Then the cell lysates were obtained for immunoblotting assays. The corresponding ratios of LC3-II to ACTB are listed below. (C) ATG4BCRISPR HepG2 cells were transfected with the indicated expression plasmids. Then the cells were treated with 10 nM bafilomycin A1 (Baf A1) or vehicle control for 3 h. Subsequently, the cell lysates were obtained for immunoblotting assays. The corresponding ratios of LC3-II (LE) to ACTB are listed below. (D and E) HepG2 cells stably expressing GFP-LC3 were transfected with the indicated expression plasmids or siRNAs, followed by treatment with 10 nM Baf A1 or vehicle control. Then the GFP-LC3 puncta in the cells were counted under a fluorescence microscope. The statistical diagrams represented the average numbers of GFP-LC3 puncta per cell (50 cells per sample were observed). Data are mean±SD from 3 independent experiments. *, P < 0.05; **, P < 0.01; ns, no significance. AKT1AA, 1 × MYC-tagged mutant AKT1 expression plasmid (in which both Thr308 and Ser473 were mutated to Ala); AKT1WT, 1 × MYC-tagged wild-type AKT1 expression plasmid; ATG4BWT, 1 × Flag-tagged wild-type ATG4B expression plasmid; ATG4BS34A, 1 × Flag-tagged mutant ATG4B expression plasmid (in which Ser34 of ATG4B was mutated to Ala); EV, empty vector; p-ATG4B (S34), Ser34-phosphorylated ATG4B; p-AKT (S473), Ser473-phosphorylated AKT; LC3-II (SE), LC3-II band with short exposure; LC3-II (LE), LC3-II band with long exposure.
The phosphorylation of ATG4B at Ser34 promotes the Warburg effect in HCC cells
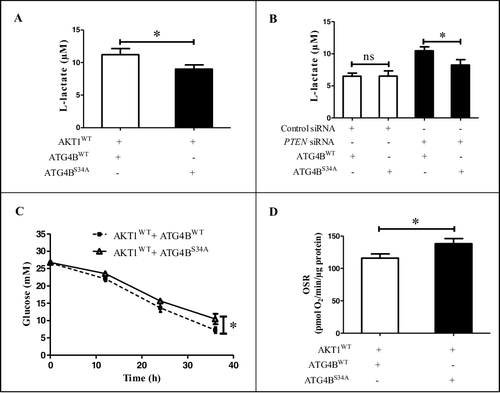
Although the phosphorylation of ATG4B at Ser34 did not influence its role in the regulation of autophagic flux in HCC cells, we interestingly found that the medium color was different between ATG4BWT- and ATG4BS34A-expressing cells under the condition of AKT1 overexpression (Figure S5A). As the color of the medium could reflect the pH, the results indicated that the cells with ATG4BWT and AKT1WT expressing plasmid cotransfection produced more acidic material than that with ATG4BS34A and AKT1WT co-expression. It has been demonstrated that L-lactate is a main metabolic product of HCC cells, especially in the cells with a high Warburg effect [Citation49], so we detected the level of L-lactate in the culture supernatant. As shown in A, the ATG4BWT- and AKT1WT-co-expressing cells produced more L-lactate compared to the ATG4BS34A- and AKT1WT-co-expressing cells. This phenomenon was also found in PTEN-knockdown cells (B). However, the difference of L-lactate levels between ATG4BWT-expressing and ATG4BS34A-expressing cells disappeared when AKT1AA was co-expressed (Figure S5B). These results suggested that the phosphorylation of ATG4B at Ser34 may play an important role in AKT1-induced L-lactate production and glycolytic potential. As the increase of glycolysis will consume more glucose and less oxygen, we further examined the changes of glucose and oxygen consumption. As shown in C and D, under the condition of AKT1WT overexpression, the glucose consumption rate (C) was higher but oxygen consumption rate (D) was lower in ATG4BWT-expressing cells compared to ATG4BS34A-expressing cells, while these differences were abolished under the condition of AKT1AA overexpression (Figure S5C and S5D). These data indicate that the phosphorylation of ATG4B at Ser34 by AKT promotes the Warburg effect in HCC cells.
Figure 3. The phosphorylation of ATG4B at Ser34 promotes the Warburg effect in HCC cells. (A) ATG4BCRISPR HepG2 cells were transfected with the indicated expression plasmids. Then the concentrations of L-lactate in the culture media were measured. (B) ATG4BCRISPR HepG2 cells were transfected with the indicated expression plasmids and siRNAs, and the concentrations of L-lactate in the culture media were determined. (C) ATG4BCRISPR HepG2 cells were transfected as in (A). Next, the concentrations of glucose in the culture media at different time points were detected separately. The time after 6 h of plasmids transfection was considered as the time “0 h” in the diagram. (D) ATG4BCRISPR HepG2 cells were seeded into 24-well microplates and treated as in (A). Then the oligomycin-sensitive respiration (OSR) was assayed. Data are mean ± SD from 3 independent experiments. *, P < 0.05; ns, no significance. AKT1WT, ATG4BWT and ATG4BS34A were the corresponding expression plasmids as described in .
The Ser34 phosphorylation of ATG4B contributes to the inhibition of F1Fo-ATP synthase activity and the elevation of mitoROS in HCC cells
As the oxygen consumption rate was decreased in ATG4BWT- and AKT1WT-co-expressing cells, we deduced that the mitochondrial function may be suppressed or impaired in this condition. Previous reports have shown that the F1Fo-ATP synthase plays a key role in mitochondrial OXPHOS, and downregulating the expression of ATP synthase or inhibition of ATP synthase activity can induce dysfunction of mitochondrial respiration, which finally contributes to the Warburg effect [Citation50,Citation51]. Therefore, we investigated the influence of Ser34 phosphorylation of ATG4B on F1Fo-ATP synthase activity in HCC cells. As shown in A, the activity of the F1Fo-ATP synthase was inhibited in ATG4BWT- and AKT1WT-co-expressing cells, but not in ATG4BS34A- and AKT1WT (or ATG4BWT- and AKT1AA or ATG4BS34A- and AKT1AA)-co-expressing cells. Similarly, the F1Fo-ATP synthase activity was also repressed in ATG4BWT (but not ATG4BS34A)-expressing cells with PTEN knockdown (B). These results indicated that the AKT-mediated phosphorylation of ATG4B at Ser34 contributed to the inhibition of F1Fo-ATP synthase activity in ATG4B-overexpressing HCC cells.
Figure 4. The Ser34 phosphorylation of ATG4B contributes to the inhibition of F1Fo-ATP synthase activity and the elevation of mitoROS in HCC cells. (A-C) ATG4BCRISPR HepG2 cells were transfected with the indicated expression plasmids and siRNAs. Then the activity of the F1Fo-ATP synthase was examined. (D-F) ATG4BCRISPR HepG2 cells were transfected with the indicated expression plasmids and siRNAs. Then the level of mitoROS was detected with a fluorescence microplate reader. Data are mean ± SD from 3 independent experiments. *, P < 0.05; ns, no significance. a.u., arbitrary units; ATG4BC74A, 1 × Flag-tagged mutant ATG4B expression plasmid (in which Cys74 of ATG4B was mutated to Ala); ATG4BS34A,C74A, 1 × Flag-tagged mutant ATG4B expression plasmid (in which both Ser34 and Cys74 were mutated to Ala); AKT1AA, AKT1WT, ATG4BWT and ATG4BS34A were the corresponding expression plasmids as described in .
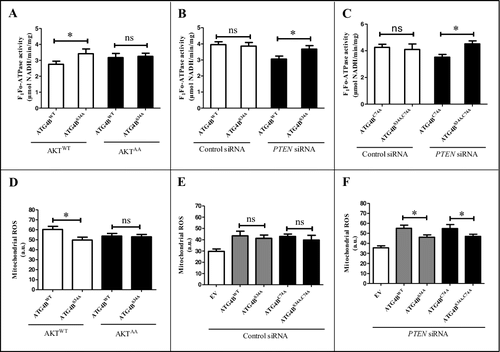
To further determine whether this effect is involved in the proteolytic activity of ATG4B on LC3, we constructed ATG4BC74A (Cys74 was mutated to Ala) and ATG4BS34A,C74A (both Ser34 and Cys74 were mutated to Ala) expression plasmids. As shown in C, the activity of the F1Fo-ATP synthase was suppressed in ATG4BC74A (but not ATG4BS34A,C74A)-expressing HCC cells with PTEN knockdown, indicating that the inhibitory effect of ATG4B on F1Fo-ATP synthase activity was dependent on its Ser34 phosphorylation, but independent of its proteolytic activity on LC3. Additionally, as mitochondria are the major source of cellular ROS, the dysfunction of mitochondria usually results in the increase or decrease of mitoROS. Accordingly, we detected the role of ATG4B Ser34 phosphorylation on mitoROS generation in HCC cells. As shown in D, the level of mitoROS was dramatically upregulated in the cells cotransfected with plasmids expressing ATG4BWT and AKT1WT, but not in the cells co-expressing ATG4BS34A and AKT1WT (or either ATG4BWT and AKT1AA or ATG4BS34A and AKT1AA). Moreover, the mitoROS levels had no significant difference between ATG4BWT- and ATG4BS34A-expressing cells, or between ATG4BC74A- and ATG4BS34A,C74A-transfected cells in the presence of control siRNA (E). However, upon PTEN knockdown, the level of mitoROS in ATG4BWT- or ATG4BC74A-expressing cells was higher compared to ATG4BS34A- or ATG4BS34A,C74A-expressing cells (F), indicating that the Ser34 phosphorylation of ATG4B was important for ATG4B-induced mitoROS generation. Taken together, the above results indicate that the Ser34 phosphorylation of ATG4B contributes to the ATG4B-mediated inhibition of F1Fo-ATP synthase activity and elevation of mitoROS, which may repress or impair mitochondrial function in AKT-activated HCC cells.
The Ser34 phosphorylation of ATG4B enhances its mitochondrial translocation in HCC cells
A previous report has shown that a small amount of ATG4B can be detected in mitochondrial-enriched fractions of RNF5−/− macrophages [Citation52]. Moreover, ATG4D, a homolog of ATG4B, can translocate into the interior of mitochondria after removing 63 amino acid residues in the N terminus [Citation53], and influence mitochondrial structure and function. Additionally, some proteins that do not locate in mitochondria under a normal physiological state can translocate into mitochondria in pathological condition, such as FOXP3 and TERT in HCC cells [Citation54–Citation58]. Therefore, we explored whether the mitochondrial dysfunction mediated by ATG4B Ser34 phosphorylation was involved in the mitochondrial translocation of ATG4B in HCC cells.
First, the mitochondrial fractions from 3 HCC patient-derived tumor samples (#1 to #3, Table S1) were separately isolated using a differential gradient centrifugation (DGc) strategy (#1 and #2) and anti-TOMM22 magnetic beads technique (#3) according to the methods described previously [Citation59,Citation60] As shown in Figures S6A and S6B, ATG4B could be detected in the DGc-enriched and magnetic beads-sorted mitochondrial fractions from HCC samples. Furthermore, ATG4B (but not ATG4A) could be detected in the magnetic beads-sorted mitochondrial fractions from HepG2 cells (A). Meanwhile, we noticed that LC3-II was also contained in the mitochondrial fractions in this condition, which was consistent with the previous finding in RNF5−/− macrophages [Citation52]. These data suggested that ATG4B in mitochondrial fractions of HCC cells may result from (1) ATG4B directly located in mitochondria, or (2) ATG4B copurified via mitochondria that contain LC3-II.
Figure 5. The phosphorylation of ATG4B at Ser34 promotes its mitochondrial translocation in HCC cells. (A) Cytosol and mitochondria fractions were separately isolated from HepG2 cells using a differential centrifugation strategy and an anti-TOMM22 magnetic beads method. Then the proteins in the cytosol and mitochondria were analyzed by immunoblotting assays, taking the total cellular proteins from the whole cell lysate (WCL) as a control. (B) HepG2 cells were transfected with the control siRNA or ATG7 siRNA. Then the WCL and mitochondrial proteins were detected by immunoblotting assays. (C) The mitochondrial fractions obtained from HepG2 cells using the differential gradient centrifugation strategy were treated with or without proteinase K (PK), and then subjected to immunoblotting assays. (D) The mitoplasts separated from HepG2 cells were treated with or without PK, and then subjected to immunoblotting assays. (E) The distributions of ATG4B in ATG4BCRISPR HepG2 cells and wild-type HepG2 cells were examined with immunogold-labeling electron microscopy assays (bar: 200 nm), and the ATG4B-positive dots were counted. Sixty random fields were observed from 3 samples (20 fields per sample), and the statistical diagram represents the average number of ATG4B-positive dots per field. (F and G) HepG2 cells were transfected with the indicated siRNAs or expression plasmids. Then the distributions of ATG4B in the cytosol and mitochondria were determined with immunogold-labeling electron microscopy assays, and the average ATG4B-positive dots in one field were calculated and statistically analyzed. (H) ATG4BCRISPR HepG2 cells were transfected with the indicated expression plasmids in the presence or absence of PTEN siRNA. Then the distributions of ATG4B in mitochondria were detected with immunogold-labeling electron microscopy assays, and the average ATG4B-positive dots in one field were calculated and statistically analyzed. (I) ATG4BCRISPR HepG2 cells were transfected with the indicated expression plasmids. Then the cells were stained with MitoTracker Red (MT-Red) and observed under a fluorescence confocal microscope (bar: 5 μm). The colocalization of GFP-puncta with mitochondria was evaluated. The data in the statistical diagram were from 30 random fields of 3 samples (10 fields per sample). Data are mean±SD from 3 independent experiments. *, P < 0.05; **, P < 0.01; ns, no significance. Mito, mitochondria; OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane; GFP-ATG4BWT, 1 × GFP-tagged wild-type ATG4B expression plasmid; GFP-ATG4BS34A, 1 × GFP-tagged mutant ATG4B expression plasmid (in which Ser34 of ATG4B was mutated to Ala); AKT1AA, AKT1WT, ATG4BWT and ATG4BS34A were the corresponding expression plasmids as described in .
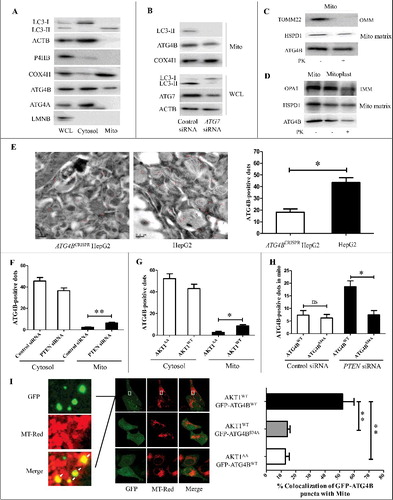
To further investigate the source of ATG4B in the mitochondrial fraction in HCC cells, ATG7 siRNA was used to inhibit the formation of LC3-II, and the level of ATG4B in magnetic beads-sorted mitochondrial fractions was examined. As shown in B, in the presence of ATG7 siRNA, the LC3-II level in the whole cell lysate (WCL) was significantly decreased, and little LC3-II was detected in the mitochondrial fractions, but ATG4B was still present in the mitochondrial fractions with a mild decrease compared to control siRNA treatment, which indicated that most ATG4B could be enriched in mitochondria of HCC cells in a manner that was independent of LC3-II binding.
Second, the mitochondrial fractions and mitoplast fractions (including mitoplast, inner mitochondrial membrane and mitochondrial matrix) were isolated for proteinase K (PK) protection assays. As shown in C, treatment of the mitochondrial fractions with PK effectively degraded the outer mitochondrial membrane (OMM) protein TOMM22, but not ATG4B and matrix-resident HSPD1/HSP60, suggesting that ATG4B may locate within the mitochondria. D showed that ATG4B could still be detected in the PK-treated mitoplast fractions, indicating that ATG4B could (at least partially) be transloacted into the mitochondrial matrix. Moreover, the subcellular localization of ATG4B was observed using immunogold-labeling electron microscopy. As shown in E, the ATG4B-positive dots in wild-type HepG2 cells were much more evident than in ATG4BCRISPR HepG2 cells. Most of the labeled ATG4B distributed in the cytoplasm including cytosol, mitochondria, autophagosomes and endoplasmic reticulum (E). The majority of mitochondrial ATG4B was located inside of mitochondria, while a small part localized at the circumference (E). These results indicated that a small portion of ATG4B could probably be imported into the mitochondrial matrix in HCC cells.
Third, the effect of ATG4B Ser34 phosphorylation on its mitochondrial import was examined. Using immunogold-labeling electron microscopy, we counted the average ATG4B-positive dots per visual field in the cytosol and mitochondria. As shown in F and G, activation of AKT (overexpression of AKT1 or knockdown of PTEN) markedly promoted mitochondrial translocation of ATG4B. Meanwhile, the amount of overexpressed Flag-ATG4BWT in mitochondria was much more than that of overexpressed Flag-ATG4BS34A under the condition of PTEN knockdown (H). In order to observe the cellular localization of ATG4B via fluorescent tracing, the GFP-ATG4BWT and GFP-ATG4BS34A expression plasmids were constructed. As shown in Figure S6C, the green fluorescence signals distributed throughout the cell with small aggregates of GFP-ATG4B (hereafter referred to as ATG4B-puncta) in the cytoplasm, while the percentage of ATG4B-puncta in mitochondria was quite low in GFP-ATG4BWT- or GFP-ATG4BS34A-expressing cells and presented no statistical difference between the 2 groups. However, the amount of ATG4B puncta and the percentage of ATG4B puncta in mitochondria were dramatically increased in AKT1WT- and GFP-ATG4BWT- co-expressing cells, but not in the cells co-expressing AKT1WT and GFP-ATG4BS34A or AKT1AA and GFP-ATG4BWT (I). Additionally, activation of AKT significantly increased the amount of ATG4B in DGc-enriched mitochondrial fractions in ATG4BWT (but not ATG4BS34A)-expressing cells (Figure S6D).
In brief, the above data demonstrate that a small portion of ATG4B can be imported to mitochondria in HCC cells, whereas overactivation of AKT further enhances the enrichment of ATG4B in mitochondria via phosphorylation of Ser34.
ATG4B is present in one complex with the subunits of the mitochondrial F1Fo-ATP synthase in HCC cells
As mitophagy is an important mechanism for maintaining mitochondrial homeostasis, and ATG4B can interact with mitophagy-related proteins such as LC3-II, we investigated whether the increased mitochondrial translocation of ATG4B in AKT-activated cells could influence mitochondrial function via mitophagy. As shown in A and S7A, overexpression of Flag-ATG4BWT or Flag-ATG4BS34A significantly inhibited the formation of GFP-LC3 puncta and its colocalization with mitochondria, whether or not there was AKT activation, suggesting that the effect of ATG4B Ser34 phosphorylation on mitochondrial function may not attribute to mitophagy in the condition of ATG4B overexpression. Moreover, an immunofluorescence assay was used to examine the colocalization of ATG4B-puncta and LC3 or GABARAPL1 in ATG4B-overexpressing HCC cells. As shown in Figure S7B, knockdown of ATG7 remarkably decreased the amount of LC3 or GABARAPL1 puncta, demonstrating the utility of LC3 or GABARAPL1 antibody for immunofluorescence.
Figure 6. ATG4B is present in one complex with the subunits of the mitochondrial F1Fo-ATP synthase in HCC cells. (A) HepG2 cells stably expressing GFP-LC3 were transfected with the indicated siRNAs and expression plasmids. Then the cells were labeled with the indicated antibodies using immunofluorescence assays and observed under a fluorescence confocal microscope. Representative images are shown (bar: 10 μm). (B) ATG4BCRISPR HepG2 cells were transfected with the 3 × Flag-ATG4BWT expression plasmid. Then the whole cell lysate (WCL) was obtained for immunoprecipitation assays. Subsequently, the ATG4B antibody-precipitated complex was further subjected to mass spectrometry, taking the IgG-precipitated complex as a control. Three independently prepared lysates were used for immunoprecipitation assays. Only the proteins pulled down in all 3 independent IP-MS assays were selected as candidates, and the mitochondrial candidates in the ATG4B antibody-precipitated complex are listed. (C) HepG2 cells were transfected with the 3 × Flag-ATG4BWT expression plasmid, and then the cell lysates were subjected to immunoprecipitation assays. (D) ATG4BCRISPR HepG2 cells were transfected with the 3 × Flag-ATG4BWT expression plasmid. Then the cell lysates were obtained for immunoprecipitation and immunoblotting assays. 3 × Flag-ATG4BS34A, 3 × Flag-tagged mutant ATG4B expression plasmid (in which Ser34 of ATG4B was mutated to Ala); Flag-ATG4BWT, Flag-ATG4BS34A and 3 × Flag-ATG4BWT were the corresponding expression plasmids as described in .
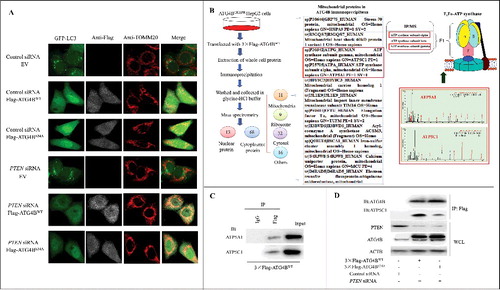
Figure S7C showed that the ATG4B puncta displayed minor colocalization with LC3 or GABARAPL1 puncta in GFP-ATG4B-overexpressing cells, suggesting that most of the GFP puncta may not be autophagy-protein positive dots under this condition. To further explore the function of mitochondrial ATG4B, we screened the potential mitochondrial binding partners of ATG4B in HCC cells using the combined methods of IP and mass spectroscopy (IP-MS). As shown in B, 81 proteins from 3 independent experiments were screened out (only the proteins pulled down in all 3 IP-MS assays were selected), including nuclear proteins and cytoplasmic proteins (mitochondrial proteins, ribosomal proteins, cytosolic proteins, etc.). Some known ATG4B-binding proteins including GABARAPL2, MAP1LC3B and MAP1LC3A were among the candidates, further indicating the efficiency of the affinity isolation in this experiment. Interestingly, we found that ATP5C1 and ATP5A1 (2 components of the mitochondrial F1Fo-ATP synthase) were also candidates with a high score. The Co-IP assay using the lysate of 3 × Flag-ATG4BWT-expressing HCC cells showed that ATP5A1 and ATP5C1 were present in the immunoprecipitates obtained with anti-Flag antibody (C). Furthermore, the Co-IP assay in Figure S7D showed that ATG4B present in the immunoprecipitates resulted from the anti-ATP synthase immunocapture antibody that could recognize multiple subunits of the F1Fo-ATP synthase, indicating that ATG4B could be present in 1 complex with the F1Fo-ATP synthase in HCC cells.
Subsequently, the effect of ATG4B on the activity of the F1Fo-ATP synthase in vitro was evaluated. As shown in Figure S7E, recombinant human ATG4B (but not ATG4A) suppressed the activity of the F1Fo-ATP synthase in MgATP SMPs from patient-derived HCC tissue (#4, Table S1). Additionally, the Co-IP assay in D showed that the level of ATP5C1 was much higher in ATG4BWT-immunoprecipitates than that in ATG4BS34A-immunoprecipitates under the condition of PTEN knockdown, indicating that the enhanced interaction of ATP5C1 and the ATG4B-containing complex was dependent on ATG4B Ser34 phosphorylation. The data above, taken together with the results in A-C and F-I, indicate that the AKT-induced Ser34 phosphorylation of ATG4B can promote its mitochondrial import and the subsequent interaction with ATP5C1-containing complex, leading to the inhibition of F1Fo-ATP synthase activity in HCC cells.
Discussion
To eliminate the influence of endogenous ATG4B, we intended to generate a homozygous ATG4B−/− HCC cell line using the CRISPR/Cas9 system at the beginning of this study. However, ATG4B−/− clones were not obtained in 4 consecutive rounds of CRISPR/Cas9 screening in HepG2 cells. We ultimately got a hemizygous knockout cell line, and its ATG4B level was approximately 50% of that in wild-type HepG2 cells. It has been reported that atg4b−/− mice [Citation61], atg4b−/− mouse embryonic fibroblast (MEF) cells [Citation42] and immortalized atg4b−/− 3T3 cells [Citation40] have been successfully generated. Nevertheless, to our knowledge, no ATG4B−/− cancer cells have been generated until now. Meanwhile, Liu et al. have revealed that only knockdown of ATG4B presents a significant inhibitory effect on colorectal cancer cell growth in vitro and in vivo [Citation40]. Therefore, we deduce that a certain level of ATG4B is required for the survival and/or growth of some cancer cells including HCC cells. In this study, we used the assays of overexpressing ATG4BWT or ATG4BS34A in this hemizygous knockout HCC cell line to investigate the Ser34 phosphorylation of ATG4B in order to reduce the influence of endogenous ATG4B as much as possible.
Previous reports have shown that AKT can either improve or impair OXPHOS through multiple mechanisms, and the final effect may be determined by different situations [Citation62–Citation64] Here, we found that the AKT-mediated phosphorylation of ATG4B at Ser34 dramatically enhanced the mitochondrial location of ATG4B and inhibition of mitochondrial function, which may favor the initiation of the Warburg effect. Therefore, suppressing the AKT-mediated Ser34 phosphorylation of ATG4B may be beneficial to attenuate cellular metabolic remodeling and delay HCC progression. Additionally, Yang et al. have revealed that inhibition of the AKT-downstream target MTOR in MEF or HEK293T cells by rapamycin or incubation in Hank's balanced salt solution induces autophagy via phosphorylating ATG4B at Ser383 and Ser392 [Citation42]. It is interesting that the phosphorylation pattern of ATG4B can be changed according to the different states of the phosphoinositide 3-kinase-AKT-MTOR pathway or diverse cell backgrounds. In future work, we will thoroughly investigate the patterns of ATG4B phosphorylation in HCC cells under different situations.
Generally, the proteins that can locate within the mitochondria often contain a typical mitochondrial targeting peptide (MTP) in the N terminus. The MTP (usually 20∼30 amino acid residues) is cleaved by mitochondrial processing peptidases after the proteins are imported into mitochondria, leading to a decrease of approximately 2∼3 kDa of the protein molecular mass, accordingly. Of course, some proteins such as TST/rhodanese, HADHAP1 and BCS1L can be imported to mitochondria via a noncleavable mechanism [Citation65–Citation67], Moreover, Ruan et al. have reported that the cytosolic proteins prone to aggregation are imported into the mitochondrial intermembrane space and matrix in yeast after heat shock [Citation68]. Meanwhile, Berendzen et al. have revealed that the aggregation-prone proteins expressed in the neurons of C. elegans bind to mitochondria, eliciting a global induction of mitochondrial stress and affecting whole-animal physiology [Citation69]. In this study, we found that ATG4B did not contain a typical MTP in the N terminus, and the molecular weight of mitochondrial ATG4B was almost equivalent to that of cytoplasmic ATG4B, indicating a nonclassical pattern of ATG4B mitochondrial import. In addition, the results from GFP-ATG4B-expressing cells (I) suggested that the Ser34 phosphorylation of ATG4B may enhance the formation of ATG4B aggregates, and the aggregates that presented minor colocalization with LC3- or GABARAPL1-puncta were prone to mitochondrial import in the AKT-activated cells. Here, we guess that the Ser34 phosphorylation of ATG4B may alter its conformation or the binding partners, which facilitates the aggregation of ATG4B and the subsequent mitochondrial import. Of course, more studies are needed to explore the detailed mechanisms.
It has been reported that some autophagy-related proteins (such as BECN1/Beclin 1 [Citation70], ATG5 [Citation71,Citation72] ATG7 [Citation73] and ATG4D [Citation53,Citation74]) can function in an autophagy-independent manner. As a key protease in autophagy process, ATG4B has aroused great interesting in the field of autophagic research. However, a recent study has revealed that ATG4B can promote the proliferation of colorectal cancer cells independent of autophagy [Citation40]. In this study, we demonstrated that ATG4B could influence metabolic actions in HCC cells in a macroautophagy-independent manner, opening a new door for the research on the relationship between autophagy and mitochondrial function under some pathological conditions. Additionally, the ATG4B that modulates autophagy and the ATG4B that targets mitochondria may compete with each other because of the limitation of endogenous ATG4B in the cells. In the present study, maybe a portion of exogenous ATG4B is enough to regulate macroautophagy under basal conditions, or other ATG4s could compensate for the autophagy-regulating role of ATG4B, so the mitochondria translocation of ATG4B presented little effect on the macroautophagic signaling. The balance between the canonical and noncanonical roles of ATG4B in some physiological and pathological contexts is really interesting, and needs to be further studied.
As to the mechanism by which ATG4B regulates mitochondrial activity, we propose the following 3 preferential possibilities based on the previous reports and our data. (1) ATG4B regulates macroautophagy via cleaving LC3/GABARAP proteins and influences the clearance of mitochondria. (2) ATG4B is translocated into mitochondria and functions as a mitophagy receptor, which participates in the mitophagy-mediated mitochondrial degradation. (3) ATG4B is imported to mitochondria and regulates mitochondrial activity via directly affecting the function of mitochondrial proteins. In this study, we found that macroautophagy/mitophagy had no significant difference between ATG4BWT- and ATG4BS34A-overexpressing HCC cells whether or not AKT was activated, whereas the mitochondrial activity was impaired in ATG4BWT-overexpressing cells with AKT activation. Therefore, we favor that the third possibility above by which ATG4B Ser34 phosphorylation regulates mitochondrial activity. Of course, the detailed mechanisms need to be further studied. It has been reported that chemotherapy and irradiation can upregulate ATG4B protein in several tumor cells including HCC cells [Citation29,Citation32,Citation75] In addition, our unpublished data also indicated that the protein level of ATG4B was dramatically increased in HCC tissues (even more than a 5-fold increase in some samples), and the Ser34 phosphorylation of ATG4B could be detected in some clinical HCC samples. Therefore, we used the strategy of ATG4B overexpression so as to relate to the relevant biology in this study. Moreover, compared to the site directed mutation strategy for targeting endogenous ATG4B at Ser34, overexpression of ATG4B (WT and S34A) would induce little off-target effect, making it relatively specific for investigating the role of ATG4B Ser34 phosphorylation. Of course, the mutation of endogenous ATG4B at Ser34 will be more in line with the actual physiological condition compared to the overexpression strategy, and we will perform the corresponding experiments in the future.
Due to the key role of the ATP synthase in mitochondrial OXPHOS, the inhibition of ATP synthase activity or downregulation of ATP synthase subunits can obviously induce dysfunction of mitochondrial respiration, which finally contributes to the Warburg effect [Citation17,Citation50,Citation51,Citation76] Moreover, ATP synthase can also affect the production of mitochondrial superoxide radicals through regulating mitochondrial transmembrane potential hyperpolarization mediated by H+-translocation [Citation77–Citation80]. According to the findings in this study, we deduced that ATG4B may bind to the mitochondrial F1Fo-ATP synthase and suppress its activity, leading to an increase of mitoROS in AKT-activated HCC cells. However, we also noticed that the percentage of mitochondria that colocalized with ATG4B was not high in our model, so we conjectured that the effect of mitochondrial ATG4B on a portion of F1Fo-ATP synthase in HCC cells may be very sensitive, strong and cumulative, leading to a mild increase (even not detectable) of ROS production. This mild alternation of ROS may further influence mitochondrial function persistently and ultimately drives a detectable change in ROS level. Of course, more studies are needed to verify this hypothesis. Meanwhile, we cannot exclude F1Fo-ATP synthase-independent mechanisms by which ATG4B affects mitochondrial activity, so more research is required to clarify the details. In addition, the mitochondria-targeting ATG4D can also lead to disruption of mitochondrial cristae architecture and sensitize cells to ROS-associated damage in differentiating erythroid cells [Citation53]. These results indicate that ATG4s can influence ROS level via mitochondrial import. Furthermore, due to the diverse roles of ROS on cell biology, the mitochondrial ATG4s-mediated increase of ROS may present distinct functions under different situations, which needs to be further investigated.
In summary, we identify a novel phosphorylation site (Ser34) on ATG4B by AKT. Moreover, the ATG4B Ser34 phosphorylation participates in the metabolic reprogramming of HCC cells via suppressing mitochondrial function, which is possibly due to the Ser34 phosphorylation-mediated increase of ATG4B mitochondrial location and the subsequent inhibition of F1Fo-ATP synthase activity. Our findings reveals a noncanonical working model of ATG4B under pathological conditions, which may provide a scientific basis for developing novel strategies for HCC treatment by targeting ATG4B and its Ser34 phosphorylation.
Materials and methods
Antibodies
The information of antibodies is as follows: TOMM20 (Santa Cruz Biotechnology, sc-17764); P4HB (Proteintech, 11245-1-AP); PEX1 (Proteintech, 13669-1-AP); ACTB (Proteintech, 60008-1-Ig); COX4I1 (Cell Signaling Technology, 6014); ATG4B (Abcam, ab70867); ATP synthase (Abcam, ab109867); HSPD1 (Abcam, ab46798); TOMM22 (Abcam, ab57523); ATG4A (Abcam, ab108322); Flag-tag (Proteintech, 66008-2-Ig, 20543-1-AP); ATP5A1 (Proteintech, 14676-1-AP, 66037-1-Ig); ATP5C1 (Proteintech, 10910-1-AP, 60284-1-Ig); Alexa Fluor 647 anti-rabbit IgG (Invitrogen, A21245); Alexa Fluor 594 anti-mouse IgG (Invitrogen, A21203); SQSTM1 (Cell Signaling Technology, 8025); LC3B-I/II (Cell Signaling Technology, 2775); GABARAPL1 (Abcam, ab86497); p-AKT (Cell Signaling Technology, 4060); MYC-tag (Cwbiotech, CW0089); phospho-AKT substrate (RXXS*/T*) (Cell Signaling Technology, 9614).
Plasmid construction
3 × Flag-ATG4BWT, Flag-ATG4BWT, Flag-ATG4BS34A, Flag-ATG4BC74A, Flag-ATG4BS34A,C74A, MYC-AKT1WT, MYC-AKT1AA, GFP-ATG4BWT, GFP-ATG4BS34A and Flag-ATG4AWT expression plasmids were prepared in our lab. To obtain the cDNAs of ATG4B and AKT1, total RNA was extracted from HepG2 cells and reverse-transcribed to cDNA by M-MLV Reverse Transcriptase (Invitrogen, 2641A) with the primer oligo(dT)18 (Takara, 3806). The human ATG4A cDNA was purchased from Guangzhou Genecopoeia Technology Co. Ltd in China. The cDNA fragments were separately cloned into pCMV5 (YouBio,VT2114) or pcDNA3.1 (YouBio,VT1001) vector. The mutant constructs were obtained from site-directed mutagenesis by overlap extension PCR, taking the corresponding wild-type constructs as templates.
siRNA and plasmid transfections
siRNAs were purchased from Genepharma Technology Co. Ltd in China. Transfections were performed using Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer's protocol. Briefly, cells were seeded in culture plates and cultured overnight. Then siRNA or plasmid and Lipofectamine 2000 were separately diluted in Opti-MEM (Gibco, 31985070). After 5-min incubation, the diluted siRNA or plasmid and the diluted Lipofectamine 2000 were mixed gently and the mixtures were incubated for 20 min at room temperature. Subsequently, the cell culture medium was removed and the corresponding mixture was added into culture plates. After incubation for 6 h, the mixture was removed and the cells were cultured in fresh culture medium.
Purification of recombinant human ATG4B and ATG4A
The recombinant His-tagged human ATG4B (rhATG4B) and ATG4A (rhATG4A) proteins were prepared as previously described [Citation81].
Isolation of subcellular fractions
The isolation of cytosol was performed as previously described [Citation82]. The isolation of mitochondria was separately performed using a differential gradient centrifugation strategy [Citation60] and anti-TOMM22 magnetic beads method [Citation59] with minor revision. The corresponding detailed protocols were described in supplementary Materials and methods. The isolation of mitochondrial subfractions was performed according to the methods described previously [Citation83].
Measurement of L-lactate and glucose
Cells were seeded into 6-well plates and transfected with different plasmids. Then the medium was subjected to the detection of L-lactate and glucose separately using the L-lactate assay kit (Eton Bioscience, 1200051002) and glucose colorimetric/fluorometic assay kit (BioVision, K606).
Determination of oligomycin-sensitive respiration
The detection of oligomycin-sensitive respiration (OSR) was performed as previously described [Citation50]. Briefly, cells were seeded into XF24-well microplates and given different treatments. Then the cellular oxygen consumption was detected using an extracellular flux analyzer (Seahorse Bioscience, Seahorse XFe24) with the injected substances including oligomycin (1 μM; Seahorse Bioscience, 101706-100), FCCP (1 μM; Seahorse Bioscience, 101706-100), rotenone (0.5 μM; Seahorse Bioscience, 101706-100) and antimycin (0.5 μM; Seahorse Bioscience, 101706-100).
SMPs isolation
The isolation of MgATP SMPs was performed according to the previous method with minor modification [Citation84]. Briefly, mitochondria separated from human HCC tissues were exposed to ultrasonics for 60 sec in the buffer containing 1 mM ATP, 15 mM MgSO4 and 0.25 M sucrose (pH 7.6). Then the sonicate was centrifuged at 12,000 g for 15 min. Subsequently, the supernatant was centrifuged at 105,000 g for 30 min, and the pellet was washed and stored using 0.25 M sucrose (Santa Cruz Biotechnology, sc-204311). To activate the SMPs, a buffer containing 125 mM sucrose, 25 mM Tris-acetate and 2 mM EDTA (pH 7.8) was added and incubated at room temperature for 30 min. Then the SMPs were used to detect the activity of ATP synthase.
MitoROS detection
MitoROS was measured using the mitoROS-sensitive probe MitoTracker Red (Invitrogen, M7512) as described previously [Citation85]. The fluorescence values of the cells stained with MitoTracker Red were detected at excitation/emission wavelength 575 nm/595 nm using a fluorescence microplate reader.
Co-IP assays
The cell extracts were diluted in lysis buffer (Beyotime, P0013) and incubated with anti-ATP synthase, anti-Flag or the corresponding mouse/rat monoclonal antibodies overnight at 4°C. Then protein-A/G agarose (Santa Cruz Biotechnology, sc-2003) was added to the mixtures and adequately resuspended. After incubation at room temperature for 90 min, the mixtures were centrifuged at 1,100 g for 3 min and washed with lysis buffer 7 times. Next, the immunoprecipitates were dissolved in lysis buffer containing loading buffer and heated at 100°C for 8 min, followed by SDS-PAGE for immunoblotting assays. In addition, for the IP-MS experiment, the immunoprecipitate was washed with 0.2 M glycine-HCl and the supernatant was subjected to MALDI TOF mass spectrometry for protein identification. The mass spectrometry was performed by Shanghai Applied Protein Technology Co. Ltd in China.
Immuno-electron microscopy
The samples of cultured HEK293 cells were prepared using a high-pressure freezing technique. Then the samples were stained with 1% uranyl acetate and dehydrated, infiltrated and embedded with the polymerization of Lowicryl resins (Polyscience, 14330). Subsequently, the ultra-thin sections of embedded cells with 80-nm thickness were prepared and incubated with specific antibody against ATG4B or a negative IgG control, followed by the incubation with gold colloid-conjugated goat anti-rabbit IgG (Sigma-Aldrich, G7402). Next, the immuno-protein complexes were stabilized with gluteraldehyde, and then the sections were imaged using a JEOL 1200 electron microscope.
Detection of ATP synthase activity
ATP synthase activity was detected with an F1Fo-ATPase activity spectrophotometric assay kit (JianCheng, S50083) according to the manufacturer's instructions. The absorbance at 340 nm was measured continuously for 5 min. Additionally, the protein concentration was quantified by Bradford to normalize the reaction system. The unit of ATP synthase activity was defined as that required for oxidation of 1 μmol NADH/min/mg.
Immunoblotting assays
Immunoblotting assays were performed as described previously [Citation81]. Briefly, the protein extracts were subjected to SDS-PAGE at 30∼80 µg/lane and transferred to PVDF membranes. Then the membranes were incubated with 5% milk in PBST (Boster, AR0194) or 3% BSA (Solarbio, A8020) in TBST (Boster, AR0195; for detection of phosphorylated proteins) at 37°C for 60 min, followed by incubation with different primary antibodies at 4°C overnight. After washing with PBST or TBST, the membranes were incubated with the corresponding secondary antibodies at 37°C for 60 min. Finally, the immunoreactive bands on the PVDF membranes were detected using enhanced chemiluminescence reagents (Thermo Scientific, 34076). Additionally, in order to separate the phosphorylated and nonphosphorylated proteins, the SDS polyacrylamide gel was prepared according to the manufacturer's instructions (Wako, 300–93523).
Statistical analysis
All data were displayed as mean ± SD (standard deviation) from at least 3 independent experiments. The difference between 2 groups was analyzed by Student t test, while the analysis of difference among 3 or more groups was performed by one-way ANOVA with the post hoc Tukey test. P < 0.05 was considered statistically significant.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Suup_data1407887.zip
Download Zip (1.1 MB)Acknowledgments
This work was supported by the National Natural Science Foundation of China (31671464, 81572375 and 31501061), National Postdoctoral Program for Innovative Talents (BX201600023), and Natural Science Foundation Project of Chongqing (cstc2017jcyjBX0079 and cstc2014jcyjA10110).
Related Research Data
References
- Llovet JM, Zucman-Rossi J, Pikarsky E, et al. Hepatocellular carcinoma. Nat Rev Dis Primers. 2016;2: 16018. doi:10.1038/nrdp.2016.18. PMID:27158749
- Newmeyer DD, Ferguson-Miller S. Mitochondria: releasing power for life and unleashing the machineries of death. Cell. 2003;112:481–490. doi:10.1016/S0092-8674(03)00116-8. PMID:12600312
- Pessayre D. Role of mitochondria in non-alcoholic fatty liver disease. J Gastroenterol Hepatol. 2007;22(Suppl 1):S20–S27. doi:10.1111/j.1440-1746.2006.04640.x. PMID:17567459
- Sas K, Robotka H, Toldi J, et al. Mitochondria, metabolic disturbances, oxidative stress and the kynurenine system, with focus on neurodegenerative disorders. J Neurol Sci. 2007;257:221–239. doi:10.1016/j.jns.2007.01.033. PMID:17462670
- Mathupala SP, Ko YH, Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta. 2010;1797:1225–1230. doi:10.1016/j.bbabio.2010.03.025. PMID:20381449
- Weinberg SE, Sena LA, Chandel NS. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42:406–417. doi:10.1016/j.immuni.2015.02.002. PMID:25786173
- Vyas S, Zaganjor E, Haigis MC. Mitochondria and cancer. Cell. 2016;166:555–566. doi:10.1016/j.cell.2016.07.002. PMID:27471965
- Senyilmaz D, Teleman AA. Chicken or the egg: Warburg effect and mitochondrial dysfunction. F1000Prime Rep. 2015;7:41. doi:10.12703/P7-41. PMID:26097714
- Chen X, Qian Y, Wu S. The Warburg effect: evolving interpretations of an established concept. Free Radic Biol Med. 2015;79:253–263. doi:10.1016/j.freeradbiomed.2014.08.027. PMID:25277420
- Wallace DC. Mitochondria and cancer: Warburg addressed. Cold Spring Harb Symp Quant Biol. 2005;70:363–374. doi:10.1101/sqb.2005.70.035. PMID:16869773
- Zhu H, Luo H, Zhu X, et al. Pyruvate kinase M2 (PKM2) expression correlates with prognosis in solid cancers: a meta-analysis. Oncotarget. 2017;8:1628–1640. PMID:27911861
- Xu Q, Liu X, Zheng X, et al. PKM2 regulates Gli1 expression in hepatocellular carcinoma. Oncol Lett. 2014;8:1973–1979. PMID:25289083
- Dong T, Yan Y, Chai H, et al. Pyruvate kinase M2 affects liver cancer cell behavior through up-regulation of HIF-1alpha and Bcl-xL in culture. Biomed Pharmacother. 2015;69:277–284. doi:10.1016/j.biopha.2014.12.010. PMID:25661370
- Amann T, Hellerbrand C. GLUT1 as a therapeutic target in hepatocellular carcinoma. Expert Opin Ther Targets. 2009;13:1411–1427. doi:10.1517/14728220903307509. PMID:19874261
- Jia YY, Zhao JY, Li BL, et al. miR-592/WSB1/HIF-1alpha axis inhibits glycolytic metabolism to decrease hepatocellular carcinoma growth. Oncotarget. 2016;7:35257–35269. doi:10.18632/oncotarget.9135. PMID:27153552
- Han H, Li W, Shen H, et al. microRNA-129-5p, a c-Myc negative target, affects hepatocellular carcinoma progression by blocking the Warburg effect. J Mol Cell Biol. 2016;8:400–410. doi:10.1093/jmcb/mjw010.
- Cuezva JM, Krajewska M, de Heredia ML, et al. The bioenergetic signature of cancer: a marker of tumor progression. Cancer Res. 2002;62:6674–6681. PMID:12438266
- Savic LJ, Chapiro J, Duwe G, et al. Targeting glucose metabolism in cancer: new class of agents for loco-regional and systemic therapy of liver cancer and beyond? Hepat Oncol. 2016;3:19–28. doi:10.2217/hep.15.36. PMID:26989470
- Hemelaar J, Lelyveld VS, Kessler BM, et al. A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J Biol Chem. 2003;278:51841–51850. doi:10.1074/jbc.M308762200. PMID:14530254
- Tanida I, Ueno T, Kominami E. Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met121 to expose Gly120 for lipidation and targeting to autophagosomal membranes. J Biol Chem. 2004;279:47704–47710. doi:10.1074/jbc.M407016200. PMID:15355958
- Sugawara K, Suzuki NN, Fujioka Y, et al. Structural basis for the specificity and catalysis of human Atg4B responsible for mammalian autophagy. J Biol Chem. 2005;280:40058–40065. doi:10.1074/jbc.M509158200. PMID:16183633
- Satoo K, Suzuki NN, Fujioka Y, et al. Crystallization and preliminary crystallographic analysis of human Atg4B-LC3 complex. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:99–102. doi:10.1107/S1744309106056429. PMID:17277449
- Fujita N, Hayashi-Nishino M, Fukumoto H, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19:4651–4659. doi:10.1091/mbc.E08-03-0312. PMID:18768752
- Satoo K, Noda NN, Kumeta H, et al. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. EMBO J. 2009;28:1341–1350. doi:10.1038/emboj.2009.80. PMID:19322194
- Kabeya Y, Mizushima N, Yamamoto A, et al. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004;117:2805–2812. doi:10.1242/jcs.01131. PMID:15169837
- Tanida I, Sou YS, Ezaki J, et al. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem. 2004;279:36268–36276. doi:10.1074/jbc.M401461200. PMID:15187094
- Kumanomidou T, Mizushima T, Komatsu M, et al. The crystal structure of human Atg4b, a processing and de-conjugating enzyme for autophagosome-forming modifiers. J Mol Biol. 2006;355:612–618. doi:10.1016/j.jmb.2005.11.018. PMID:16325851
- Li M, Hou Y, Wang J, et al. Kinetics comparisons of mammalian Atg4 homologues indicate selective preferences toward diverse Atg8 substrates. J Biol Chem. 2011;286:7327–7338. doi:10.1074/jbc.M110.199059. PMID:21177865
- Peng WX, Wan YY, Gong AH, et al. Egr-1 regulates irradiation-induced autophagy through Atg4B to promote radioresistance in hepatocellular carcinoma cells. Oncogenesis. 2017;6:e292. doi:10.1038/oncsis.2016.91. PMID:28134935
- Dower CM, Bhat N, Wang EW, et al. Selective reversible inhibition of autophagy in hypoxic breast cancer cells promotes pulmonary metastasis. Cancer Res. 2017;77:646–657. doi:10.1158/0008-5472.CAN-15-3458. PMID:28115361
- Zhang L, Li J, Ouyang L, et al. Unraveling the roles of Atg4 proteases from autophagy modulation to targeted cancer therapy. Cancer Lett. 2016;373:19–26. doi:10.1016/j.canlet.2016.01.022. PMID:26805760
- Wu Y, Ni Z, Yan X, et al. Targeting the MIR34C-5p-ATG4B-autophagy axis enhances the sensitivity of cervical cancer cells to pirarubicin. Autophagy. 2016;12:1105–1117. doi:10.1080/15548627.2016.1173798. PMID:27097054
- Wu S, Su J, Qian H, et al. SLC27A4 regulate ATG4B activity and control reactions to chemotherapeutics-induced autophagy in human lung cancer cells. Tumour Biol. 2016;37:6943–6952. doi:10.1007/s13277-015-4587-4. PMID:26662804
- Liao H, Xiao Y, Hu Y, et al. Methylation-induced silencing of miR-34a enhances chemoresistance by directly upregulating ATG4B-induced autophagy through AMPK/mTOR pathway in prostate cancer. Oncol Rep. 2016;35:64–72. doi:10.3892/or.2015.4331. PMID:26499184
- Bortnik S, Choutka C, Horlings HM, et al. Identification of breast cancer cell subtypes sensitive to ATG4B inhibition. Oncotarget. 2016;7:66970–66988. doi:10.18632/oncotarget.11408. PMID:27556700
- Toshima T, Shirabe K, Matsumoto Y, et al. Autophagy enhances hepatocellular carcinoma progression by activation of mitochondrial beta-oxidation. J Gastroenterol. 2014;49:907–916. doi:10.1007/s00535-013-0835-9. PMID:23702609
- Rothe K, Lin H, Lin KB, et al. The core autophagy protein ATG4B is a potential biomarker and therapeutic target in CML stem/progenitor cells. Blood. 2014;123:3622–3634. doi:10.1182/blood-2013-07-516807. PMID:24755409
- Akin D, Wang SK, Habibzadegah-Tari P, et al. A novel ATG4B antagonist inhibits autophagy and has a negative impact on osteosarcoma tumors. Autophagy. 2014;10:2021–2035. doi:10.4161/auto.32229. PMID:25483883
- Tran E, Chow A, Goda T, et al. Context-dependent role of ATG4B as target for autophagy inhibition in prostate cancer therapy. Biochem Biophys Res Commun. 2013;441:726–731. doi:10.1016/j.bbrc.2013.10.117. PMID:24184480
- Liu PF, Leung CM, Chang YH, et al. ATG4B promotes colorectal cancer growth independent of autophagic flux. Autophagy. 2014;10:1454–1465. doi:10.4161/auto.29556. PMID:24991826
- Jo YK, Park NY, Park SJ, et al. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget. 2016;7:57186–57196. doi:10.18632/oncotarget.11083. PMID:27527864
- Yang Z, Wilkie-Grantham RP, Yanagi T, et al. ATG4B (Autophagin-1) phosphorylation modulates autophagy. J Biol Chem. 2015;290:26549–26561. doi:10.1074/jbc.M115.658088. PMID:26378241
- Qiao S, Dennis M, Song X, et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. 2015;6: 7014. doi:10.1038/ncomms8014. PMID:25916556
- Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi:10.1038/sj.emboj.7601623. PMID:17347651
- Pengo N, Agrotis A, Prak K, et al. A reversible phospho-switch mediated by ULK1 regulates the activity of autophagy protease ATG4B. Nat Commun. 2017;8: 294. doi:10.1038/s41467-017-00303-2. PMID:28821708
- Villen J, Beausoleil SA, Gerber SA, et al. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci U S A. 2007;104:1488–1493. doi:10.1073/pnas.0609836104. PMID:17242355
- Humphrey SJ, Yang G, Yang P, et al. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013;17:1009–1020. doi:10.1016/j.cmet.2013.04.010. PMID:23684622
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. doi:10.1080/15548627.2015.1100356. PMID:26799652
- Faloppi L, Bianconi M, Memeo R, et al. Lactate dehydrogenase in hepatocellular carcinoma: something old, something new. Biomed Res Int. 2016;2016: 7196280. doi:10.1155/2016/7196280. PMID:27314036
- Sanchez-Cenizo L, Formentini L, Aldea M, et al. Up-regulation of the ATPase inhibitory factor 1 (IF1) of the mitochondrial H+-ATP synthase in human tumors mediates the metabolic shift of cancer cells to a Warburg phenotype. J Biol Chem. 2010;285:25308–25313. doi:10.1074/jbc.M110.146480. PMID:20538613
- Willers IM, Cuezva JM. Post-transcriptional regulation of the mitochondrial H(+)-ATP synthase: a key regulator of the metabolic phenotype in cancer. Biochim Biophys Acta. 2011;1807:543–551. doi:10.1016/j.bbabio.2010.10.016. PMID:21035425
- Kuang E, Okumura CY, Sheffy-Levin S, et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 2012;8:e1003007. doi:10.1371/journal.pgen.1003007. PMID:23093945
- Betin VM, MacVicar TD, Parsons SF, et al. A cryptic mitochondrial targeting motif in Atg4D links caspase cleavage with mitochondrial import and oxidative stress. Autophagy. 2012;8:664–676. doi:10.4161/auto.19227. PMID:22441018
- Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi:10.1016/S1097-2765(03)00050-9. PMID:12667443
- Rojas J, Teran-Angel G, Barbosa L, et al. Activation-dependent mitochondrial translocation of Foxp3 in human hepatocytes. Exp Cell Res. 2016;343:159–167. doi:10.1016/j.yexcr.2016.04.008. PMID:27068374
- Yan J, Zhou Y, Chen D, et al. Effects of mitochondrial translocation of telomerase on drug resistance in hepatocellular carcinoma cells. J Cancer. 2015;6:151–159. doi:10.7150/jca.10419. PMID:25561980
- Ling X, Wen L, Zhou Y. Role of mitochondrial translocation of telomerase in hepatocellular carcinoma cells with multidrug resistance. Int J Med Sci. 2012;9:545–554. doi:10.7150/ijms.4648. PMID:22991493
- Chen J, Siddiqui A. Hepatitis B virus X protein stimulates the mitochondrial translocation of Raf-1 via oxidative stress. J Virol. 2007;81:6757–6760. doi:10.1128/JVI.00172-07. PMID:17428866
- Franko A, Baris OR, Bergschneider E, et al. Efficient isolation of pure and functional mitochondria from mouse tissues using automated tissue disruption and enrichment with anti-TOM22 magnetic beads. PLoS One. 2013;8:e82392. doi:10.1371/journal.pone.0082392. PMID:24349272
- Hartwig S, Kotzka J, Lehr S. Isolation and quality control of functional mitochondria. Methods Mol Biol. 2015;1264:9–23. doi:10.1007/978-1-4939-2257-4_2. PMID:25630999
- Marino G, Fernandez AF, Cabrera S, et al. Autophagy is essential for mouse sense of balance. J Clin Invest. 2010;120:2331–2344. doi:10.1172/JCI42601. PMID:20577052
- Shaik ZP, Fifer EK, Nowak G. Akt activation improves oxidative phosphorylation in renal proximal tubular cells following nephrotoxicant injury. Am J Physiol Renal Physiol. 2008;294:F423–F432. doi:10.1152/ajprenal.00463.2007. PMID:18077599
- Li C, Li Y, He L, et al. PI3K/AKT signaling regulates bioenergetics in immortalized hepatocytes. Free Radic Biol Med. 2013;60:29–40. doi:10.1016/j.freeradbiomed.2013.01.013. PMID:23376468
- De Rosa V, Iommelli F, Monti M, et al. Reversal of Warburg effect and reactivation of oxidative phosphorylation by differential inhibition of EGFR signaling pathways in non-small cell lung cancer. Clin Cancer Res. 2015;21:5110–5120. doi:10.1158/1078-0432.CCR-15-0375. PMID:26216352
- Liu J, Fang H, Chi Z, et al. XPD localizes in mitochondria and protects the mitochondrial genome from oxidative DNA damage. Nucleic Acids Res. 2015;43:5476–5488. doi:10.1093/nar/gkv472. PMID:25969448
- Stan T, Brix J, Schneider-Mergener J, et al. Mitochondrial protein import: recognition of internal import signals of BCS1 by the TOM complex. Mol Cell Biol. 2003;23:2239–2250. doi:10.1128/MCB.23.7.2239-2250.2003. PMID:12640110
- Hammen PK, Gorenstein DG, Weiner H. Structure of the signal sequences for two mitochondrial matrix proteins that are not proteolytically processed upon import. Biochemistry. 1994;33:8610–8617. doi:10.1021/bi00194a028. PMID:7913339
- Ruan L, Zhou C, Jin E, et al. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature. 2017;543:443–446. doi:10.1038/nature21695. PMID:28241148
- Berendzen KM, Durieux J, Shao LW, et al. Neuroendocrine coordination of mitochondrial stress signaling and proteostasis. Cell. 2016;166:1553–1563 e10. doi:10.1016/j.cell.2016.08.042. PMID:27610575
- Wirawan E, Lippens S, Vanden Berghe T, et al. Beclin1: a role in membrane dynamics and beyond. Autophagy. 2012;8:6–17. doi:10.4161/auto.8.1.16645. PMID:22170155
- Maskey D, Yousefi S, Schmid I, et al. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat Commun. 2013;4: 2130. doi:10.1038/ncomms3130. PMID:23945651
- Ma T, Li J, Xu Y, et al. Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat Cell Biol. 2015;17:1379–1387. doi:10.1038/ncb3256. PMID:26502054
- Lee IH, Kawai Y, Fergusson MM, et al. Atg7 modulates p53 activity to regulate cell cycle and survival during metabolic stress. Science. 2012;336:225–228. doi:10.1126/science.1218395. PMID:22499945
- Betin VM, Lane JD. Atg4D at the interface between autophagy and apoptosis. Autophagy. 2009;5:1057–1059. doi:10.4161/auto.5.7.9684. PMID:19713737
- Liao H, Xiao Y, Hu Y, et al. Methylation-induced silencing of miR-34a enhances chemoresistance by directly upregulating ATG4B-induced autophagy through AMPK/mTOR pathway in prostate cancer. Oncol Rep. 2016;35:64–72. doi:10.3892/or.2015.4331. PMID:26499184
- Sanchez-Arago M, Formentini L, Cuezva JM. Mitochondria-mediated energy adaption in cancer: the H(+)-ATP synthase-geared switch of metabolism in human tumors. Antioxid Redox Signal. 2013;19:285–298. doi:10.1089/ars.2012.4883. PMID:22901241
- Ni R, Zheng D, Xiong S, et al. Mitochondrial calpain-1 disrupts ATP synthase and induces superoxide generation in type-1 diabetic hearts: a novel mechanism contributing to diabetic cardiomyopathy. Diabetes. 2015;65:255–268. doi:10.2337/db15-0963. PMID:26470784
- Formentini L, Sanchez-Arago M, Sanchez-Cenizo L, et al. The mitochondrial ATPase inhibitory factor 1 triggers a ROS-mediated retrograde prosurvival and proliferative response. Mol Cell. 2012;45:731–742. doi:10.1016/j.molcel.2012.01.008. PMID:22342343
- Bensinger SJ, Christofk HR. New aspects of the Warburg effect in cancer cell biology. Semin Cell Dev Biol. 2012;23:352–361. doi:10.1016/j.semcdb.2012.02.003. PMID:22406683
- Zanotti F, Gnoni A, Mangiullo R, et al. Effect of the ATPase inhibitor protein IF1 on H+ translocation in the mitochondrial ATP synthase complex. Biochem Biophys Res Commun. 2009;384:43–48. doi:10.1016/j.bbrc.2009.04.046. PMID:19379707
- Ni Z, Gong Y, Dai X, et al. AU4S: a novel synthetic peptide to measure the activity of ATG4 in living cells. Autophagy. 2015;11:403–415. doi:10.1080/15548627.2015.1009773. PMID:25831015
- Song Y, Hao Y, Sun A, et al. Sample preparation project for the subcellular proteome of mouse liver. Proteomics. 2006;6:5269–5277. doi:10.1002/pmic.200500893. PMID:16941572
- Perciavalle RM, Stewart DP, Koss B, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nat Cell Biol. 2012;14:575–583. doi:10.1038/ncb2488. PMID:22544066
- Beltran C, de Gomez-Puyou MT, Gomez-Puyou A, et al. Release of the inhibitory action of the natural ATPase inhibitor protein on the mitochondrial ATPase. Eur J Biochem. 1984;144:151–157. doi:10.1111/j.1432-1033.1984.tb08443.x. PMID:6236977
- Ni Z, Wang B, Dai X, et al. HCC cells with high levels of Bcl-2 are resistant to ABT-737 via activation of the ROS-JNK-autophagy pathway. Free Radic Biol Med. 2014;70:194–203. doi:10.1016/j.freeradbiomed.2014.02.012. PMID:24576507