
ABSTRACT
ULK1 (unc51-like autophagy activating kinase 1) is a serine/threonine kinase that plays a key role in regulating macroautophagy/autophagy induction in response to amino acid starvation. Despite the recent progress in understanding ULK1 functions, the molecular mechanism by which ULK1 regulates the induction of autophagy remains elusive. In this study, we determined that ULK1 phosphorylates Ser30 of BECN1 (Beclin 1) in association with ATG14 (autophagy-related 14) but not with UVRAG (UV radiation resistance associated). The Ser30 phosphorylation was induced by deprivation of amino acids or treatments with Torin 1 or rapamycin, the conditions that inhibit MTORC1 (mechanistic target of rapamycin complex 1), and requires ATG13 and RB1CC1 (RB1 inducible coiled-coil 1), proteins that interact with ULK1. Hypoxia or glutamine deprivation, which inhibit MTORC1, was also able to increase the phosphorylation in a manner dependent upon ULK1 and ULK2. Blocking the BECN1 phosphorylation by replacing Ser30 with alanine suppressed the amino acid starvation-induced activation of the ATG14-containing PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3) kinase, and reduced autophagy flux and the formation of phagophores and autophagosomes. The Ser30-to-Ala mutation did not affect the ULK1-mediated phosphorylations of BECN1 Ser15 or ATG14 Ser29, indicating that the BECN1 Ser30 phosphorylation might regulate autophagy independently of those 2 sites. Taken together, these results demonstrate that BECN1 Ser30 is a ULK1 target site whose phosphorylation activates the ATG14-containing PIK3C3 complex and stimulates autophagosome formation in response to amino acid starvation, hypoxia, and MTORC1 inhibition.
Abbreviations
| AMPK | = | 5-AMP-activated protein kinase |
| AKT/protein kinase B | = | AKT serine/threonine kinase |
| ATG5 | = | autophagy-related 5 |
| ATG12 | = | autophagy-related 12 |
| ATG13 | = | autophagy-related 13 |
| ATG14 | = | autophagy-related 14 |
| ATG16L1 | = | autophagy-related 16 like 1 |
| ATG101 | = | autophagy-related 101 |
| BAFA1 | = | bafilomycin A1 |
| BECN1 | = | Beclin 1 |
| CDC37 | = | cell division cycle 37 |
| CRISPR | = | Clustered Regularly Interspaced Short Palindromic Repeats |
| cas9 | = | CRISPR associated protein 9 |
| DMEM | = | Dulbecco's modified Eagle's medium |
| EBSS | = | Earle's balanced salt solution |
| ER | = | endoplasmic reticulum |
| GFP | = | green fluorescence protein |
| GST | = | glutathione S-transferase |
| HSP90 | = | heat shock protein 90 |
| KI | = | kinase-inactive mutant |
| LC3B | = | microtubule associated protein 1 light chain 3 beta |
| LC-MS | = | liquid chromatography-mass spectrometry |
| MAPKAPK | = | mitogen-activated protein kinase-activated protein kinase |
| MEFs | = | mouse embryonic fibroblasts |
| MTOR | = | mechanistic target of rapamycin |
| MTORC1 | = | MTOR complex 1 |
| NAA10 | = | N(alpha)-acetyltransferase 10, NatA catalytic subunit |
| PCR | = | polymerase chain reaction |
| PGK1 | = | phosphoglycerate kinase 1 |
| PIK3C3 | = | phosphoinositide-3-kinase catalytic subunit type 3 |
| PIK3R4 | = | phosphoinositide-3-kinase regulatory subunit 4 |
| PtdIns3P | = | phosphatidylinositol 3-phosphate |
| RB1CC1 | = | RB1 inducible coiled-coil 1 |
| ROCK1 | = | Rho associated coiled-coil containing protein kinase 1 |
| RPS6KB1 | = | ribosomal protein S6 kinase B1 |
| shRNA | = | small hairpin RNA |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| shRNA | = | small hairpin RNA |
| SQSTM1 | = | sequestosome 1 |
| TALEN | = | transcription activator-like effector nuclease |
| TMEM173 | = | transmembrane protein 173 |
| TUBA | = | tubulin alpha |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| ULK2 | = | unc-51 like autophagy activating kinase 2 |
| UVRAG | = | UV radiation resistance associated |
| WIPI2 | = | WD repeat domain, phosphoinositide interacting 2 |
| WT | = | wild type |
Introduction
Autophagy, the evolutionarily conserved process for lysosomal degradation of cellular components, is induced by various stress conditions, such as amino acid starvation [Citation1-6], oxidative stress [Citation7], hypoxia [Citation8] or glucose starvation [Citation9]. Autophagy involves extensive membrane processes of extrusion, invagination, fusion and expansion of lipid bilayer, which result in the formation of phagophores and autophagosomes. Despite the recent progress in understanding the cellular process, the molecular mechanism underlying the initiation of autophagy remains elusive.
ULK1 is a key component upstream of the pathway that regulates autophagy initiation in response to amino acid starvation or MTORC1 inhibition [Citation1-6]. ULK1 triggers the molecular events that lead to the formation of the early autophagic membrane structure called the phagophore through poorly understood mechanisms. ULK1 exists as a large protein complex through association with ATG13, ATG101, and RB1CC1 [Citation2-5,Citation10,Citation11]. The core complex also interacts with HSP90, CDC37, ATG5, Atg8-family proteins, ATG12, and ATG16L1 [Citation12-16]. The current model for ULK1 activation involves phosphorylations of ULK1 and ATG13 by MTORC1 and 5-AMP-activated protein kinase (AMPK) [Citation2-4,Citation11,Citation17-20]. According to the model, MTORC1-mediated phosphorylation of ULK1 suppresses the activation of ULK1 [Citation9], whereas AMPK-mediated phosphorylation of ULK1 releases the suppression [Citation9,Citation21,Citation22], ULK1 is also regulated by acetylation, which positively regulates its activity [Citation23]. Although ULK1-independent autophagy has recently been described [Citation15,Citation24,Citation25], ULK1 is considered mainly to function in the canonical autophagy induction pathway regulated by amino acid starvation and MTORC1.
Once ULK1 is activated by amino acid starvation or MTORC1 inhibition, ULK1 phosphorylates BECN1 at Ser15 and ATG14 at Ser29 to stimulate the lipid kinase activity of PIK3C3 in association with BECN1 [Citation26-28]. ULK1 also phosphorylates PIK3C3 at Ser249 [Citation29]. PIK3C3 is the lipid kinase that interacts with BECN1, ATG14 and PIK3R4 to produce phosphatidylinositol 3-phosphate (PtdIns3P) involved in recruiting autophagic proteins for autophagosome formation [Citation30-34]. By phosphorylating BECN1 and ATG14, ULK1 stimulates the lipid kinase activity of PIK3C3, thus increasing the production of PtdIns3P [Citation35-41]. The PIK3C3 activity is also regulated by AKT, MAPKAPKs, and ROCK1 that phosphorylate BECN1 [Citation42-44]. This indicates that BECN1 might integrate diverse cellular signals to regulate the PIK3C3 activity.
In this study, we determined that BECN1 Ser30 is a target site of ULK1. Lu and colleagues (2017) [Citation45] recently reported that the same serine residue is phosphorylated by PGK1 (phosphoglycerate kinase 1) in response to hypoxia or glutamine deprivation in U87 glioblastoma cells. Although we do not exclude the possibility that PGK1 might phosphorylate BECN1 Ser30 in a certain cellular context, our study revealed that the canonical MTORC1-ULK1 pathway is responsible for BECN1 Ser30 phosphorylation in response to amino acid starvation, MTORC1 inhibition, and hypoxia.
Results
ULK1 phosphorylates BECN1 at Ser30
We have previously reported that the ULK1 complex interacts with the ATG14-containing PIK3C3 complex [Citation27]. When we analyzed the interaction, we observed a mobility shift of BECN1 on SDS-PAGE when BECN1 was coexpressed with ULK1 or ULK2 relative to when coexpressed with RPS6KB1, a cytosolic kinase used as a negative control for immunoprecipitation ((A)). ATG13, a binding partner of ULK1 [Citation27], did not show such a mobility shift, indicating that ULK1 or ULK2 is necessary for the change of the mobility. ULK2 interacted with BECN1 with a lower binding affinity than ULK1, but we were still able to detect the mobility shift of BECN1 in association with ULK2. The mobility change occurred when BECN1 was coexpressed with wild-type (WT) ULK1 relative to kinase-inactive mutant (KI) ULK1, which harbors alanine in replacement of Met92 (M92A) ((B)). We observed a similar result using WT and KI ULK2. The ULK1-dependent mobility change of BECN1 occurred even when BECN1 Ser15, a residue phosphorylated by ULK1 [Citation26], was mutated to alanine ((C)), indicating that ULK1 might phosphorylate BECN1 at sites other than Ser15.
Figure 1. ULK1 phosphorylates BECN1 at Ser30. (A) ULK1 and ULK2 induce a mobility shift of BECN1. The indicated proteins were transiently expressed in HEK293T cells for 2 d. HA-tagged proteins were isolated by immunoprecipitation using anti-HA antibody, and the amounts of MYC-BECN1 in immunoprecipitates and cell lysate were analyzed by western blotting. (B) ULK kinase activity is responsible for the mobility shift of BECN1. MYC-tagged WT or kinase-inactive (KI) mutant ULK1 or ULK2 was transiently expressed with MYC-BECN1 in HEK293T cells. (C) ULK1 kinase induces the mobility shift of BECN1 independently of BECN1 Ser15 phosphorylation. MYC-tagged ULK1 WT or KI was coexpressed with BECN1 WT or S15A mutant in HEK293T cells. (D) Extracted ion chromatograms from capillary LC-MS analyses of tryptic digests of BECN1 show an elution peak at 35.4 min of retention time for the doubly charged, monoisotopic precursor peptide with 644.3193 m/z, which corresponds to LDTS(phos)FKILDR. The chromatographic peak at 35.4 min was not detected for the KI ULK1 sample. (E) Schematic of BECN1 domains and alignment of ULK1 target sequences. BH, BCL2 homology 3 domain; CCD, coiled-coil domain; ECD, evolutionarily conserved domain; CTD, C-terminal domain. (F) Polyclonal antibodies were generated to detect BECN1 Ser30 phosphorylation. HA-tagged WT or KI ULK1 was expressed alone (-) or together with WT or S30A BECN1 in HEK293T cells. BECN1 Ser30 phosphorylation in MYC-BECN1 immunoprecipitates was analyzed by western blotting using the p-Ser30 antibodies we developed. (G) As with ULK1, ULK2 induces the phosphorylation of BECN1 Ser30. The indicated proteins were expressed in HEK293T cells. (H) ULK1 and ULK2 are necessary for the phosphorylation of BECN1 Ser30. Endogenous BECN1 was immunoprecipitated using anti-BECN1 antibody from MEFs and analyzed for the phosphorylation status by western blotting using the BECN1 p-Ser30-specific antibodies. (I) ULK1 phosphorylates BECN1 Ser30 in vitro. ULK1 kinase fragment containing residues 1–649 was incubated with BECN1 purified from E. coli and ATP in vitro. The phosphorylation of BECN1 was analyzed by western blotting. (J) ULK1 kinase activity is responsible for the phosphorylation of BECN1 Ser30 in vitro. The ULK1 kinase activity was assayed in vitro using MYC-ULK1 immunoprecipitates isolated from HEK293T cells as enzyme, and E. coli-purified BECN1 as substrate.
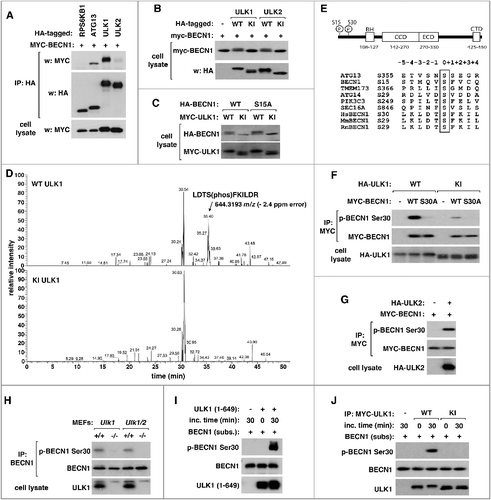
To identify other ULK1 target sites in BECN1, we transiently overexpressed MYC-tagged BECN1 with either WT or KI ULK1 in HEK293T cells. MYC-BECN1 was isolated by immunoprecipitation using anti-MYC antibody, and resolved on SDS-PAGE. The band corresponding to BECN1 was isolated and processed for analysis by mass spectrometry. LC-MS revealed a distinct tryptic peptide peak only in the WT ULK1 sample but not in the KI ULK1 sample ((D)). Through MS/MS spectrum analysis, we identified BECN1 Ser30 as a phosphorylation site (Figure S1). Sequence comparison showed that the surrounding sequence of Ser30 has leucine and an acidic residue at the −3 and −2 positions, respectively, and phenylalanine at the +1 position, which is a pattern seen with a few other ULK1 target sites ((E)). While we were characterizing BECN1 Ser30 phosphorylation, Egan et al. (2015) Citation29] published a paper where they presented evidence for a few serine residues of BECN1, including Ser30, as potential ULK1 target sites based on a mobility shift assay using their alanine mutants. BECN1 Ser30 was also recently reported as a target site of PGK1, a metabolic enzyme involved in glycolysis [Citation45]. For comprehensive analysis of the phosphorylation, we generated polyclonal antibodies specific to the phosphorylation site. The antibodies were able to detect BECN1 at a much higher level when BECN1 was coexpressed with WT ULK1 relative to KI ULK1 ((F)). The antibodies could not detect BECN1 when BECN1 Ser30 was mutated to alanine (S30A), validating their specificity toward the phosphorylated site. Coexpression of BECN1 with ULK2 also induced the phosphorylation of BECN1 Ser30 ((G)), indicating that both ULK1 and ULK2 might be responsible for the phosphorylation.
Using the polyclonal antibodies, we investigated whether ULK1 and ULK2 are responsible for the phosphorylation of endogenous BECN1 Ser30. For this, we used mouse embryonic fibroblasts (MEFs) deficient in ULK1 and/or ULK2, which were developed by the Craig Thompson lab [Citation24,Citation46]. As expected, the BECN1 phosphorylation was not detected in MEFs deficient in ULK1 or both ULK1 and ULK2 ((H)). This result demonstrates that ULK1 and ULK2 are required for the phosphorylation of endogenous BECN1 Ser30. Deficiency of ULK1 alone was sufficient to completely block the phosphorylation in MEFs, indicating that ULK1 may be the major kinase responsible for the phosphorylation at least in MEFs.
Next, we asked whether ULK1 directly phosphorylates BECN1 Ser30. We incubated E. coli-purified BECN1 with an active fragment of ULK1 containing residues 1–649, which was produced and isolated from Sf9 insect cells, in the presence of ATP in vitro. We detected a large induction of the Ser30 phosphorylation at 30 min of the in vitro reaction ((I)). To further clarify if ULK1 directly phosphorylates the residue, we prepared WT or KI ULK1 by immunoprecipitation from HEK293T cells and tested their ability to phosphorylate BECN1 in vitro. We were able to detect the BECN1 phosphorylation only when BECN1 was incubated with WT but not KI ULK1 ((J)). Combined, these results demonstrate that ULK1 is the kinase that phosphorylates BECN1 Ser30.
MTORC1 inhibition and amino acid starvation induce Ser30 phosphorylation of BECN1 in association with ATG14
Knowing that ULK1 phosphorylates BECN1 at Ser30, we investigated whether amino acid starvation or MTORC1 inhibition induces the phosphorylation. The Ser30 phosphorylation gradually increased during the time course of amino acid deprivation in MEFs and HCT116 cells ((A)). This increase was correlated with a reduction in the phosphorylations of RPS6KB1 Thr389 and ULK1 Ser757/758, which reflect the MTORC1 activity, as well as with the changes of SQSTM1 and LC3B-II levels, which reflect autophagy induction. We observed similar increases of the BECN1 phosphorylation when MEFs and HCT116 cells were treated with Torin 1 or rapamycin, which inhibit MTORC1 ((B)). The amino acid starvation-induced phosphorylation of BECN1 Ser30 was almost completely blocked when ULK1 was depleted ((C)). A residual level of the phosphorylation in ulk1 knockout (KO) MEFs was completely eliminated by additional depletion of ULK2. To determine whether the requirement of ULKs for the phosphorylation occurs in different cell types, we generated HCT116 cells deficient in ULK1 and ULK2 by the CRISPR-cas9 technique (Figure S2). Using the cells, we confirmed that ULKs are required for the amino acid starvation-induced phosphorylation of BECN1. Similarly, the BECN1 phosphorylation induced by Torin 1 was completely blocked when ULK1 alone or both ULK1 and ULK2 were depleted in MEFs and HCT116 cells ((D)).
Figure 2. MTORC1 inhibition and amino acid starvation induce the phosphorylation at Ser30 of BECN1 that is associated with ATG14.(A) BECN1 Ser30 phosphorylation is induced by amino acid starvation. MEFs and HCT116 cells were incubated in EBSS, which was supplemented with 10% dialyzed fetal bovine serum and deprived of amino acids (a.a. starvation), for the periods of time as indicated. Western blotting was performed to monitor the total amounts and modifications of the indicated endogenous proteins. To monitor BECN1 p-Ser30, we isolated endogenous BECN1 by immunoprecipitation using anti-BECN1 antibodies. (B) BECN1 Ser30 phosphorylation is induced by MTORC1 inhibition. MEFs and HCT116 cells were treated with Torin 1 (250 nM) or rapamycin (100 nM) for the periods of time as indicated. (C) Starvation-induced phosphorylation of BECN1 Ser30 depends on ULK1 and ULK2. The indicated MEFs and HCT116 cells were incubated in EBSS as described in (A) for 0 or 2 h. (D) ULK1 and ULK2 are necessary for MTORC1-mediated phosphorylation of BECN1 Ser30. MEFs and HCT116 cells were incubated with Torin 1 (250 nM). (E, F) Starvation-induced phosphorylation of BECN1 Ser30 requires ATG13 and RB1CC1. HCT116 cells with ATG13 intact (WT) or deficient (KO) (D) and Rb1cc1+/+ and rb1cc1−/− MEFs (E) were incubated in EBSS as described in (A) for 0 or 2 h. (G) BECN1 Ser30 phosphorylation occurs when BECN1 is associated with ATG14 but not with UVRAG. Endogenous ATG14 and UVRAG were isolated by immunoprecipitation using anti-ATG14 and anti-UVRAG antibodies from MEFs or HEK293T cells incubated in EBSS for the period of time as indicated. (H) Amino acid starvation-induced BECN1 Ser30 phosphorylation requires ATG14. HCT116 cells, where ATG14 is intact (WT) or deficient (KO), were incubated in EBSS for 0 or 2 h. Western blots obtained with short and long exposures are presented for BECN1 and p-BECN1 proteins.
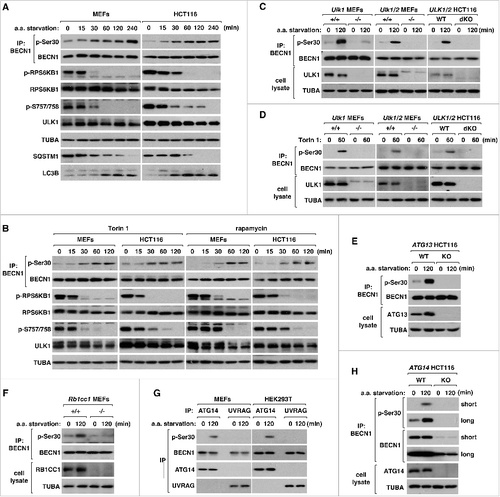
To demonstrate whether the BECN1 phosphorylation depends on the whole ULK complex, we tested the effect of depletion of ATG13 and RB1CC1, which are components of the ULK complex. For this test, we generated ATG13-depleted HCT116 cells by the TALEN technique [Citation25] and obtained RB1CC1-deficient MEFs from the Jun-Lin Guan lab [Citation47]. The ATG13 depletion in HCT116 completely blocked the induction of BECN1 Ser30 phosphorylation in response to amino acid starvation ((E)). Depletion of RB1CC1 in MEFs had a similar effect ((F)). These results demonstrate that the BECN1 phosphorylation requires the integrity of the ULK complex.
Because BECN1 interacts with either ATG14 or UVRAG to form 2 distinct protein complexes [Citation30-34], we wondered whether ULK1 might have any preference for the phosphorylation of BECN1 in either complex. To address this issue, we isolated BECN1 by immunoprecipitation of endogenous ATG14 or UVRAG from MEFs after cells were incubated in the presence or absence of amino acids for 120 min. The phosphorylation of BECN1 was detected only when BECN1 was isolated with ATG14 but not with UVRAG ((G)). We also attempted to confirm the result using HCT116 cells. However, the expression level of UVRAG was very low in HCT116 cells. Thus, we used HEK293T cells to further confirm that the phosphorylation specifically occurs with the ATG14-bound form of BECN1. Consistently, the starvation-induced BECN1 phosphorylation was completely blocked when ATG14 was depleted in HCT116 cells ((H)). This finding is in contrast with what was shown for BECN1 Ser15 phosphorylation that occurs in BECN1 associated with either ATG14 or UVRAG [Citation26].
BECN1 Ser30 phosphorylation is induced by glutamine deprivation and hypoxia in a manner dependent upon ULK
Qian et al. (2017) recently reported that PGK1 phosphorylates BECN1 Ser30 in response to hypoxia or glutamine deprivation [Citation45]. The study exclusively used U87 glioblastoma cells to investigate the PGK1-BECN1 link [Citation45]. They observed that glutamine deprivation does not cause a large induction of the phosphorylation of BECN1 Ser15, a ULK1 target site [Citation26], in U87 cells [Citation45], and assumed that ULK1 might be expressed at a low level in the glioma cells. However, we found that ULK1 is expressed in U87 cells as highly as in HCT116, HEK293T, and HeLa cells ((A)). Furthermore, amino acid deprivation robustly induced the BECN1 Ser30 phosphorylation in U87 cells, and the phosphorylation was completely blocked by SBI-0206965, a ULK1 inhibitor [Citation29] ((B)). Similarly, ATG14 Ser29 phosphorylation was also largely induced by amino acid starvation in U87 cells and inhibited by the ULK1 inhibitor. As with amino acid starvation, Torin 1 also induced the phosphorylations of BECN1 Ser30 and ATG14 Ser29 in U87 cells, which were blocked by the ULK1 inhibitor ((C)). These results suggest that the MTORC1-ULK1 axis is functional in U87 cells to regulate the BECN1 Ser30 phosphorylation.
Figure 3. ULK1 is required for BECN1 Ser30 phosphorylation in response to glutamine deprivation and hypoxia.(A) Comparison of the expression levels of ULK1 in different cell lines. (B-D) BECN1 Ser30 phosphorylation in response to amino acid starvation (B), Torin 1 (C), and glutamine deprivation (D) is completely blocked by SBI-0206965, a ULK1 inhibitor. U87 cells were pre-treated with vehicle (-) or SBI-0206965 at 10 μM (+) for 30 min. Then, the cells were incubated in EBSS supplemented with 10% dialyzed fetal bovine serum (B), treated with Torin 1 (250 nM) (C), or incubated in DMEM deprived of glutamine and supplemented with 10% dialyzed fetal bovine serum (D) for the periods of time as indicated. (E) ULK1 knockdown suppresses the induction of BECN1 Ser30 phosphorylation by glutamine deprivation in U87 cells. Two different lentiviral shRNA constructs were used to knock down ULK1, whereas a GFP shRNA construct was used as control. (F) ULK1 and ULK2 are necessary for glutamine deprivation and hypoxia to induce BECN1 Ser30 phosphorylation. MEFs and HCT116 cells were incubated in DMEM depleted of glutamine or in a hypoxia chamber containing 1% oxygen and 5% CO2 for the periods of time as indicated.
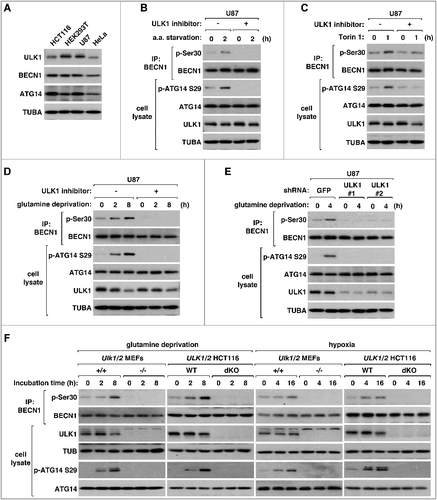
We also tested whether ULK1 is required for the BECN1 Ser30 phosphorylation in response to glutamine deprivation in U87 cells. Because glutamine deprivation inhibits MTORC1 activity [Citation48,Citation49], we predicted that the BECN1 phosphorylation might depend on ULK1, which is downstream of MTORC1. As predicted and also shown by Qian et al. (2017) [Citation45], glutamine deprivation induced the phosphorylation of BECN1 Ser30 in U87 cells. The phosphorylation was completely suppressed by the ULK1 inhibitor ((D)). Similarly, glutamine deprivation induced ATG14 Ser29 phosphorylation, which was completely blocked by the ULK1 inhibitor. To rule out potential off-target effects of the ULK1 inhibitor, we employed an shRNA-mediated knockdown approach to deplete ULK1 in U87 cells. Knockdown of ULK1 in U87 cells using shRNA constructs specific to 2 different shRNA target sequences completely blocked the induction of the phosphorylations of BECN1 Ser30 and ATG14 Ser29 in response to glutamine deprivation ((E)). Furthermore, we were able to detect ULK1 but not PGK1 in BECN1 immunoprecipitates from U87 cells (Figure S3(A)). These results demonstrate that the glutamine deprivation-induced phosphorylation of BECN1 Ser30 requires ULK1 in U87 cells.
Glutamine deprivation also induced the phosphorylations of BECN1 Ser30 and ATG14 Ser29 in MEFs and HCT116 cells. The phosphorylations were completely blocked when both ULK1 and ULK2 were depleted ((F)). In either cell type, knockout of ULK1 and ULK2 did not have effects on the expression level of PGK1 (Figure S3(B and C)). Furthermore, we could not detect PGK1 in BECN1 immunoprecipitates from those cells. As shown by Qian et al. (2017) [Citation45], BECN1 Ser30 phosphorylation was induced by hypoxia (1% oxygen) in MEFs and HCT116 cells ((F)). Hypoxia also induced ATG14 Ser29 phosphorylation, indicating that hypoxia led to ULK activation. The hypoxia-induced phosphorylations of BECN1 and ATG14 were completely blocked when ULK1 and ULK2 were depleted. Because hypoxia is an energy stress condition, we wondered whether BECN1 Ser30 is induced by glucose starvation. Although the increase was much less dramatic than the increase with amino acid starvation, we were able to detect a moderate increase of BECN1 Ser30 phosphorylation during the course of glucose starvation in MEFs and HCT116 cells (Figure S4).
BECN1 Ser30 phosphorylation is required for the activation of the ATG14-containing PIK3C3 complex in response to amino acid starvation
Knowing that ULK1 phosphorylates BECN1 Ser30, we wondered whether the phosphorylation regulates the kinase activity of PIK3C3 that interacts with BECN1 and ATG14. To establish the in vitro kinase assay for PIK3C3, we isolated the ATG14-containing PIK3C3 complex by immunoprecipitation from HCT116 cells using anti-ATG14 antibody. Incubation of the ATG14 immunoprecipitates with phosphatidylinositol (PtdIns) and ATP as substrates in vitro led to a high production of PtdIns3P when the immunoprecipitates were obtained from starved cells. When the ATG14 immunoprecipitates were isolated from BECN1 KO HCT116 cells, we were not able to detect the production of PtdIns3P ((A)). This result demonstrates that the in vitro kinase assay is specific to the ATG14-containing PIK3C3 complex. We noticed that BECN1 KO dramatically reduced the expression level of ATG14. Reciprocally, ATG14 KO dramatically reduced the expression level of BECN1 ((H)). This effect might be because ATG14 and BECN1 depend on each other for their stability.
Figure 4. BECN1 Ser30 phosphorylation stimulates the kinase activity of PIK3C3 that is in association with ATG14. (A) BECN1 Ser30 phosphorylation is important for starvation-induced activation of the ATG14-containing PIK3C3 complex. ATG14 immunoprecipitates were isolated from the indicated cells cultured in either DMEM (full medium) or EBSS (starvation medium [starv.]) for 1 h using anti-ATG14 antibodies. The immunoprecipitates were incubated with PtdIns and ATP for 30 min. The production of PtdIn3P was analyzed by the dot blot assay (see Materials and Methods). (B) Similar results were obtained as in (A) using HCT116 cells modified in the genome to introduce the BECN1 S30A mutation. (C) Quantitative analysis of PtdIn3P production in (A) and (B). The y-axis reflects the amounts of PtdIn3P relative to those from WT cells in full medium, which are marked ‘#’. (*p<0.05; **p<0.01; ****p<0.0001, Student t-test). Mean and SEM are shown as horizontal bars. (D) BECN1 Ser30 phosphorylation regulates the kinase activity of the PIK3C3 complex independently of BECN1 Ser15 and ATG14 Ser29 phosphorylations. HCT116 cells described in (B) were analyzed for the phosphorylation of BECN1 and ATG14.
![Figure 4. BECN1 Ser30 phosphorylation stimulates the kinase activity of PIK3C3 that is in association with ATG14. (A) BECN1 Ser30 phosphorylation is important for starvation-induced activation of the ATG14-containing PIK3C3 complex. ATG14 immunoprecipitates were isolated from the indicated cells cultured in either DMEM (full medium) or EBSS (starvation medium [starv.]) for 1 h using anti-ATG14 antibodies. The immunoprecipitates were incubated with PtdIns and ATP for 30 min. The production of PtdIn3P was analyzed by the dot blot assay (see Materials and Methods). (B) Similar results were obtained as in (A) using HCT116 cells modified in the genome to introduce the BECN1 S30A mutation. (C) Quantitative analysis of PtdIn3P production in (A) and (B). The y-axis reflects the amounts of PtdIn3P relative to those from WT cells in full medium, which are marked ‘#’. (*p<0.05; **p<0.01; ****p<0.0001, Student t-test). Mean and SEM are shown as horizontal bars. (D) BECN1 Ser30 phosphorylation regulates the kinase activity of the PIK3C3 complex independently of BECN1 Ser15 and ATG14 Ser29 phosphorylations. HCT116 cells described in (B) were analyzed for the phosphorylation of BECN1 and ATG14.](/cms/asset/d52050cc-901e-42db-b7cd-3916f46345c3/kaup_a_1422851_f0004_b.gif)
To test the effects of the BECN1 Ser30 phosphorylation on the kinase activity of the ATG14-containing PIK3C3 complex, we reconstituted WT or S30A mutant BECN1 in BECN1 KO HCT116 cells by stable transduction using a lentiviral vector. We isolated the PIK3C3 complex by immunoprecipitation from the reconstituted cells using anti-ATG14 antibody, and analyzed the kinase activity of the complex. The production of PtdIns3P was significantly reduced when ATG14 immunoprecipitates were isolated from BECN1S30A cells compared to the immunoprecipitates from WT cells ((A)). This result suggests that the Ser30 phosphorylation might be important for the activation of the ATG14-containing PIK3C3 complex in response to amino acid starvation. We also tested the kinase activity of ATG14 immunoprecipitates isolated from BECN1 KO HCT116 cells reconstituted with the BECN1S30D mutant, in which BECN1 Ser30 was replaced by aspartate as a potential mimic of the phosphorylation, without seeing any drastic change in the production of PtdIns3P compared to the activity of the WT immunoprecipitates ((A)). This indicates that the Ser30 phosphorylation is required, but not sufficient, for the activation of the PIK3C3 complex in response to amino acid starvation.
To better assess the effects of BECN1 Ser30 phosphorylation in the regulation of the PIK3C3 complex, we used the CRISPR-cas9-assisted genome editing technique [Citation50] to introduce the S30A point mutation into the HCT116 genome (Figure S4). By genotyping and sequencing, we isolated 2 mutant clones that harbor the S30A mutation in the genome. We also isolated 2 WT clones that were prepared at the same time but failed to have the mutation incorporated. Consistent with the result from the reconstitution experiment, ATG14 immunoprecipitates isolated from the genome-edited S30A mutant cells showed a significant reduction in the production of PtdIns3P compared to the immunoprecipitates from WT cells ((B and C)).
Previous studies have shown that ULK1 phosphorylates BECN1 Ser15 and ATG14 Ser29 to stimulate the PIK3C3 activity [Citation26-28]. Thus, we considered a possibility that the S30A mutation might reduce the kinase activity of the PIK3C3 complex by suppressing those other phosphorylations. Excluding the possibility, we could not detect any noticeable difference in the phosphorylation status of BECN1 Ser15 and ATG14 Ser29 in response to amino acid starvation between WT and mutant cells ((D)). This result suggests that the effect of BECN1 Ser30 phosphorylation on the PIK3C3 complex is independent of those other sites. However, we do not exclude a possibility that BECN1 Ser15 or ATG14 Ser29 phosphorylation might affect the phosphorylation status of BECN Ser30 thereby increasing the kinase activity of the PIK3C3 complex. We noticed that BECN1 Ser15 phosphorylation was less sensitive to amino acid starvation compared to BECN1 Ser30 and ATG14 Ser29 phosphorylations ((D)). This finding implies that BECN1 Ser15 phosphorylation might be subject to an unknown mechanism in addition to the regulation by ULK1 [Citation26].
BECN1 Ser30 phosphorylation is important for autophagosome formation and autophagy flux in response to amino acid starvation
Using the genome-edited S30A mutant HCT116 cells, we investigated whether the Ser30 phosphorylation is important for autophagosome formation in response to amino acid starvation. For this, we monitored the formation of autophagosomes by immunostaining endogenous LC3B using anti-LC3B antibodies. Compared to WT cells, the cells harboring the BECN1S30A mutation showed a significant reduction in LC3B puncta formation in response to amino acid starvation ((A and B)). Because the ATG14-containing PIK3C3 complex participates in phagophore formation, we also monitored WIPI2, which is localized to phagophores, by immunostaining endogenous WIPI2. Similar to LC3B, WIPI2 showed puncta formation in response to amino acid starvation in WT cells, which was significantly suppressed in the S30A cells ((A and C)). This result suggests that the BECN1 Ser30 phosphorylation by ULK1 is important for the formation of phagophores and autophagosomes in response to amino acid starvation.
Figure 5. BECN1 Ser30 phosphorylation is important for phagophore and autophagosome formation in response to amino acid starvation.(A) BECN1 Ser30 phosphorylation is important for phagophore and autophagosome formation. HCT116 cells described in (B) were incubated with anti-LC3B or WIPI2 antibodies for immunostaining. Cells were incubated in DMEM (full medium) or EBSS (starvation medium) for 2 h. Nuclei were stained with DAPI (blue). Scale bar: 10 μm. (B, C) Quantitative analysis of (A). Red bars are mean ± SEM (****p< 0.0001 versus the full medium; Student t-test). (D) BECN1 Ser30 phosphorylation is required for optimal autophagy flux induced by amino acid starvation. HCT116 cells reconstituted with BECN1 constructs or edited in the genome to introduce the S30A mutation were incubated in DMEM or EBSS for 2 h in the presence or absence of BAFA1. (E, F) Quantitative analysis of (D). The y-axis reflects values relative to WT cells in the full medium. Error bars are mean ± SEM (*p< 0.05; Student t-test).
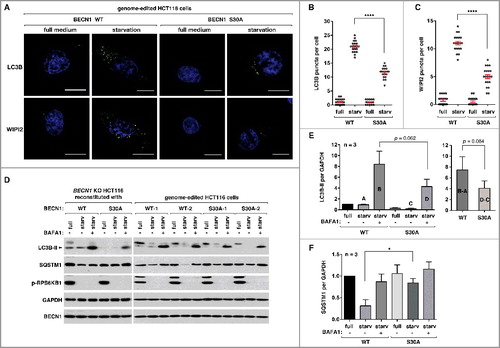
We also analyzed autophagy flux by monitoring the levels of LC3B-II and SQSTM1 in the presence or absence of bafilomycin A1 (BAFA1), a lysosomal inhibitor. Amino acid starvation did not show a drastic increase of LC3B-II level in WT cells ((D)). This might be because LC3B-II formation is balanced by its degradation through autophagy. By adding BAFA1, we were able to detect a large increase in the level of LC3B-II. The increase was much greater in WT cells than in mutant cells ((D and E)). This result indicates that autophagic flux of LC3 might be reduced in mutant cells. SQSTM1, which is a protein degraded in the lysosome, decreased in amount in response to starvation in both WT and mutant cells ((D)). However, the reduction induced by starvation was significantly larger in WT cells than in mutant cells ((F)). This result suggests that the Ser30 phosphorylation might be important for autophagic degradation of SQSTM1. However, because the S30A mutation did not completely block the autophagic activities, it is likely that other mechanisms, perhaps ATG14 Ser29 and BECN1 Ser15 phosphorylations, might play roles to regulate autophagy when BECN1 Ser30 is blocked. This possibility is consistent with the result from the in vitro kinase assay above, which showed that the S30A mutation did not completely block the activation of the ATG14-containing PIK3C3 complex upon amino acid starvation ((A–C)).
Discussion
In this study, we report that ULK1 phosphorylates BECN1 at Ser30 in response to amino acid starvation, MTORC1 inhibition, and hypoxia. We found that the phosphorylation occurs in BECN1 that interacts with ATG14 but not with UVRAG. The phosphorylation stimulates the kinase activity of the ATG14-containing PIK3C3 complex in response to amino acid starvation. The specificity toward the ATG14 complex distinguishes the Ser30 phosphorylation from the Ser15 phosphorylation that occurs in BECN1 that interacts with either ATG14 or UVRAG [Citation26]. Given that the ATG14 complex participates in phagophore formation [Citation30-34], the Ser30 phosphorylation might have a function in the early step of autophagosome formation. Consistent with this speculation, we found that the phosphorylation is important for WIPI2-positive phagophore formation.
BECN1 Ser30 was recently reported as a target site of PGK1 [Citation45], a glycolytic enzyme whose main function is to catalyze the conversion between 1,3-diphosphoglycerate and 3-phosphoglycerate. According to the mechanism proposed by the study, hypoxia or glutamine deprivation suppresses the MTORC1 activity and thereby prevents MTORC1 from phosphorylating and inhibiting NAA10, an enzyme that catalyzes acetylation of PGK1. Once acetylated, PGK1 binds and phosphorylates BECN1 at Ser30. Although this is a compelling model, the study investigated the phosphorylation exclusively using U87 cells. Furthermore, the study assumed that ULK1 expression might be low in the glioma cells, as BECN1 Ser15 phosphorylation was only marginally affected by glutamine deprivation. However, we found that ULK1 was expressed in U87 cells as highly as in other tested cell lines ((A)). Although we do not exclude the possibility that PGK1 might phosphorylate BECN1 Ser30 in a certain condition or specific cell types, the PGK1-BECN1 link must depend on ULK because ULK depletion completely eliminated the BECN1 phosphorylation even in U87 cells where the PGK1-BECN1 link was demonstrated [Citation45]. We also note that hypoxia and glutamine deprivation can enhance the activity of ULK1 via MTORC1 inhibition. Thus, the stimulatory effects of those treatments on BECN1 Ser30 phosphorylation can be mediated through the canonical MTORC1-ULK1 pathway without requiring PGK1.
BECN1 has multiple other phosphorylation sites, such as Ser93, Ser234/295, Ser90, and Thr119, which are phosphorylated by AMPK, AKT, MAPKAPKs, and ROCK1, respectively [Citation42-44,Citation51]. This implies that BECN1 might function as a key node integrating multiple signaling inputs to coordinate autophagy with other cellular processes. While some of those phosphorylations were shown to regulate the activity of the PIK3C3 complex or the interaction with BCL2, it remains unclear how the multiple phosphorylations coordinately regulate BECN1 functions. In the case of BECN1 Ser30 phosphorylation, Qian and colleagues showed that the phosphorylation facilitates binding of PtdIns to PIK3C3 [Citation45]. This implies that the Ser30 phosphorylation might play a role in coupling the ULK1 activation with the production of PtdIns3P by PIK3C3 at the site of autophagosome formation [Citation45].
Recent studies have shown that ULK1 phosphorylates not only autophagy regulators but also non-autophagic proteins, such as TMEM173, a protein involved in immune response [Citation52], and SEC16A that functions in ER-to-Golgi trafficking [Citation53]. These findings indicate that ULK1 has functions beyond the autophagy pathway. Although our knowledge about ULK1 targets has been greatly expanded in recent years, we still have a limited scope of understanding about the functions of ULK1. Identifying more ULK1 targets and their functional characterization may be necessary to better understand the various roles of ULK1 in diverse cellular processes.
Materials and methods
Reagents and antibodies
The sources of antibodies and chemicals used in the experiments are: ULK1 (sc-10900 for immunoprecipitation and sc-33182 for western blotting of the human protein), BECN1 (sc-11427 for western blotting and sc-10086 for immunoprecipitation), ATG14 (sc-164767 for immunoprecipitation), UVRAG (sc-82115), SQSTM1 (sc-28359), and TUBA (sc-12462) antibodies from Santa Cruz Biotechnology; PIK3C3 (3358), phospho (p)-RPS6KB1 Thr389 (9205), RPS6KB1 (9202), p-ATG14 Ser29 (13155), ATG14 (96752 for western blotting), MAP1LC3B/LC3B (2775), and p-BECN1 Ser15 (84966) from Cell Signaling Technology; ATG14 antibody (PD026) for western blotting of mouse protein from MBL International Corporation; MYC (9E10 clone, OP10) and WIPI2 (MABC91) antibodies from EMD Biosciences; HA antibody (clone HA.11, AFC-101P) from Covance; SBI-0206965 and WIPI2 antibody (SAB4200400 for immunostaining), and bafilomycin A1 (B1793-10UG) from Sigma-Aldrich; Torin 1 (4247) from R&D Systems. The polyclonal antibodies for the phosphorylated Ser30 of BECN1 were generated using C-KLDT-(pS)-FKILDKLDT-(pS)-FKILD-amide as an antigenic peptide at AbFrontier Bioscience (Seoul, Korea).
Identification of BECN1 Ser30 phosphorylation
MYC-tagged BECN1 was coexpressed with HA-tagged WT or KI ULK1 in HEK293T cells for 2 days. Cells were lysed in a buffer containing 40 mM HEPES (Sigma-Aldrich, H7006), pH 7.4, 120 mM NaCl, 1 mM EDTA, 50 mM NaF, 1.5 mM Na3VO4, 10 mM β-glycerophosphate, 0.3% CHAPS (Sigma-Aldrich, C3023) supplemented with protease inhibitors (Roche Diagnostics, 11873580001), and MYC-BECN1 was isolated by immunoprecipitation using anti-MYC antibody. The immunoprecipitated BECN1 was resolved by SDS-PAGE, and the BECN1 band was subjected to digestion using trypsin (1:20 of protease to protein ratio, overnight; ThermoFisher Scientific, 90057). Tryptic peptides were purified via MCX columns (Waters Corporation) and phosphopeptides were enriched using immobilized metal ion affinity chromatography as described in Stone et al. (2011) [Citation54]. Enriched peptides were dried by speed vacuum then resuspended in solvent (98:2:0.01, water:acetonitrile:formic acid) and analyzed by an Orbitrap XL mass spectrometer (ThermoFisher Scientific) as described previously [Citation55]. Tandem MS data for the phosphorylated BECN1 peptide was acquired using the ion trap mass analyzer in the ion trap (CID) mode. Peptide spectra were analyzed by MS/MS using Sequest (Thermo Fisher Scientific, Inc., version 27) and the PEAKS software (Bioinformatics Solution Inc.). We validated MS/MS-based peptide and protein identification using Scaffold (version Scaffold 4.2.1, Proteome Software Inc., Portland, OR, USA). Peptide identifications were accepted if the probability was greater than 95% by the Peptide Prophet algorithm [Citation56,Citation57]. Peptides of interest (phosphorylated candidates) were manually verified.
Plasmid construction, mutagenesis, and knockdown
Human full-length cDNA of the BECN1 gene was obtained from Open Biosystems (GE Dharmacon, IMAGE 2901072), amplified by polymerase chain reaction (PCR), and cloned into the pRK5 vector [Citation58] BECN1 S15A, S30A, and S30D mutations were made using a site-directed mutagenesis kit (Agilent Technologies, 200522) using primers 5′-CaccatgcaggtgGCcttcgtgtgccagcgctgc-3′ and 5′-gcagcgctggcacacgaagGCcacctgcatggtG-3′ for S15A, 5′-ctgaaactggacacgGCtttcaagatcctggac-3′ and 5′-gtccaggatcttgaaaGCcgtgtccagtttcag-3′ for S30A, and 5′-CtgaaactggacacgGAtttcaagatcctg-3′ and 5′-caggatcttgaaaTCcgtgtccagtttcaG-3′ for S30D. For knockdown of ULK1, we used the pLKO.1 vector (Addgene, 8453; deposited by Dr. Robert Weinberg). We generated 2 pLKO.1 constructs for 2 different shRNA target sequences: gacttccaggaaatggctaat (ULK1#1) and tacacgccatctcctcaagtt (ULK1#2). GFP-targeting shRNA (tacaacagccacaacgtctat) was cloned into pLKO.1 and used as a control. Lentiviruses harboring the shRNA constructs were prepared as we described previously [Citation59]. U87 cells were incubated with the shRNA lentiviruses, and stably transduced cells were selected under puromycin treatment as we described previously [Citation59]. All other DNA constructs used in this study have been previously described [Citation2,Citation27,Citation60].
Cell culture, transfection, and treatments
MEFs, HEK293T cells, and HCT116 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; ThermoFisher Scientific, 11995) supplemented with 10% fetal bovine serum (ThermoFisher Scientific, 26140079), penicillin and streptomycin at 37°C in 5% CO2. Transient transfection of HEK293T cells with plasmids was conducted using Lipofectamine 3000 (ThermoFisher Scientific, L3000075) following the manufacturer's protocol.Cells were harvested 2 d after the transfection. All the DNAs we used for transfection were prepared in the pRK5 vector to express proteins with a MYC or HA tag at the N terminus. For amino acid starvation, we used Earle's balanced salt solution (EBSS; MilliporeSigma; E2888) supplemented with dialyzed 10% fetal bovine serum (ThermoFisher Scientific, 26400044). For glutamine deprivation, we used DMEM (Lonza 12-707F) supplemented with 10% dialyzed fetal bovine serum. For hypoxia, we used a hypoxia chamber (Biospherix Ltd, Parish, NY, USA) containing 1% oxygen and 5% CO2. For glucose starvation, we used DMEM (11966-025, ThermoFisher Scientific) supplemented with 10% dialyzed fetal bovine serum.
Preparation of BECN1-reconstituted cells
WT and mutant BECN1 cDNAs were cloned into the lentiviral expression vector pCSII-EF-MCS, which was kindly provided by N. Somia (University of Minnesota, MN, USA). The vector was introduced into HEK293FT cells with lentiviral packaging DNAs, ΔNRF-HIV-1-gag-pol (provided by N. Somia) and pCMV-VSV-G (provided by S. Stewart at the Washington University, St Louis, MO), using Lipofectamine 3000. Medium that contains viruses was collected 60 h post-transduction. Cell debris from the viral soup was removed using a 0.45-μm filter. One million HCT116 cells on 6-cm dishes were incubated with the collected viruses twice in the presence of polybrene (8 μg/ml; Sigma-Aldrich, H9268). The first infection was performed using 1.5 ml of viral soup for 4 h. The second infection was performed overnight using 0.5 ml of viral soup combined with 3.5 ml of DMEM culture medium. Two d after the second infection, Zeocin (100 μg/ml; ThermoFisher Scientific, R25005) was added to cells for selection.
Cell lysate preparation, immunoprecipitation, and western blotting
For immunoprecipitation, we used a cell lysis buffer containing 40 mM HEPES, pH 7.4, 120 mM NaCl, 1 mM EDTA, 50 mM NaF, 1.5 mM Na3VO4, 10 mM β-glycerophosphate, 0.3% CHAPS supplemented with protease inhibitors (Roche Diagnostics, 11873580001). For experiments that do not require immunoprecipitation, we used 1% Triton X-100 (Sigma-Aldrich, T8787) instead of CHAPS for a high yield of proteins. Immunoprecipitation was conducted as described in our previous reports using protein G agarose (GenDEPOT, P9202) [Citation2,Citation27]. Proteins were resolved on Novex Tris-Glycine gels (ThermoFisher Scientific). Western blotting was performed as described previously using polyvinylidene difluoride (PVDF) membranes (Bio-Rad, 1620177) and horseradish peroxidase substrate reagents (Advansta Inc., E-119-50).
Immunostaining and fluorescence microscopy
HCT116 cells were fixed with 4% formaldehyde in phosphate-buffered saline (PBS; BioExpress, 0780.50L) for 15 min, and permeabilized with 0.3% Triton X-100 for 30 min at room temperature. Permeabilized cells were incubated in PBS containing 1% BSA for 30 min and subsequently with antibodies overnight at 4°C. Endogenous LC3B and WIPI2 were stained using anti-LC3B antibody (Cell Signaling Technology, 2775) or anti-WIPI2 antibody (EMD-Millipore, MABC91; Sigma-Aldrich, SAB4200400). After the primary antibody binding, cells were incubated with Alexa Flour 647-conjugated anti-rabbit IgG (Invitrogen, A-21443). Nuclei were stained with DAPI (4'-6-diamidino-2-phenylindole; Invitrogen, D-1306). Images from stained cells were obtained using a DeltaVision PersonelDV microscope (Applied Precision). LC3B and WIPI2 puncta were quantified using ImageJ and statistically analyzed by Student ttest using the Prism v6 software (GraphPad Software Inc, La Jolla, CA, USA).
Genome editing to introduce the BECN1 S30A mutation
To introduce the BECN1 S30A mutation in the genome of HCT116 cells, we used the CRISPR-cas9-assisted genome editing technique. We obtained pSpCas9(BB)-2A-GFP (PX458) from Addgene (48138; deposited by Dr. Feng Zhang) and cloned a gRNA sequence fragment (5′-ggacacgagtttcaagatcc-3′) into the vector. The replacement DNA harboring the BECN1 S30A mutation (Figure S4) was synthesized by GenScript USA Inc. (Piscataway, NJ). The replacement DNA and the CRISPR-cas9 construct were introduced into HCT116 cells using the Neon Transfection System (ThermoFisher Scientific, MPK5000). Two d post-transfection, green fluorescence-positive single cells were sorted and plated into 96-well dishes using a FACSAria II SORP (BD Biosciences, San Diego, CA, USA). Single colonies were isolated and expanded. Genomic DNA was isolated from the amplified colonies for genotype screening. In brief, cell pellets were resuspended in 50 mM NaOH and heated at 95°C for 30 min. After being neutralized with 1 M Tris-HCl, the cell extract was centrifuged at 15,200 x g for 10 min at 4°C. Supernatant, which contains genomic DNA, was used for PCR. PCR was performed using a pair of primers to screen WT clones (5′-TGACCCTGGGCTCTAAACTG-3′ and 5′-GGTCCAGGATCTTGAAACT-3′) and a pair of primers to screen mutant clones (5′-TGACCCTGGGCTCTAAACTG-3′ and 5′-GGTCgAGGATCTTGAAAgc-3′). If a clone showed a PCR product only with the mutant primers, the clone was marked as potential homozygotic mutant cells. Selected mutant clones were subject to PCR amplification of the mutated region using a pair of primers (5′-tcgcttctccctaatgttgc-3′ and 5′-gaaaacagggagccagatga-3′). The PCR products were cloned into the pRK5 vector and sequenced to confirm the mutation.
Genome editing to knock out ULK1 and ULK2
To generate HCT116 cells where both ULK1 and ULK2 are depleted, we used the CRISPR-cas9 technique. In brief, we used the website available at the http://crispr.mit/edu to search candidate gRNA sequences for ULK1 and ULK2. Through the search, we chose 5′-cgggctgaccgatcttgacg-3′ and 5′-gacctcgcagattatttgca-3′ as gRNAs for the ULK1 and ULK2 genes, respectively. The gRNA sequences were cloned into the pSpCas9(BB)-2A-GFP vector. The vector DNA was introduced into HCT116 cells using the Neon Transfection System. Two d post-transfection, we used a FACSAria II SORP to sort GFP-positive single cells and plate them into 96-well dishes. Single colonies were amplified for genomic DNA prep and western blotting. ULK1 depletion was confirmed by western blotting, whereas ULK2 depletion was confirmed by sequencing. All other cell lines obtained by genome editing have been described in our previous report [Citation27].
In vitro kinase assay for ULK1 activity
To determine if ULK1 directly phosphorylates BECN1, we used a commercially available ULK1 kinase (SignalChem, U01-11G) or ULK1 immunoprecipitates as enzyme source.We purified recombinant BECN1 from E. coli as described previously [Citation2,Citation27,Citation61]. To prepare ULK1 immunoprecipitates, MYC-tagged WT or M92A mutant ULK1 was expressed in HEK293T cells for 2 d. MYC-ULK1 was isolated by immunoprecipitation using anti-MYC antibody (EMD Biosciences, 9E10).The in vitro reaction was conducted in a buffer containing 25 mM MOPS, pH 7.5, 1 mM EGTA, 0.1 mM Na3VO4, 15 mM MgCl2, 100 μM ATP (Sigma-Aldrich, A7699). We used 400 ng of purified BECN1 for each reaction.
In vitro kinase assay for PIK3C3 activity
The PIK3C3 activity was assayed as described previously [Citation62]. In brief, the PIK3C3 complex was obtained by immunoprecipitation using anti-ATG14 antibody in a lysis buffer containing 50 mM Tris, pH 7.4, 7.5% glycerol, 150 mM NaCl, 1 mM EDTA, and protease inhibitors. The immunoprecipitates were washed 3 times with lysis buffer and further washed once with 2.5× substrate buffer containing 75 mm Tris, pH 7.5, 125 mm NaCl, 12.5 mm MnCl2. The immunoprecipitates were resuspended and incubated in 1x substrate buffer containing 30 mm Tris, pH 7.5, 50 mm NaCl, 5 mm MnCl2, PtdIns (250 μg/ml; Avanti Polar Lipids, 840042P) on ice for 10 min, then followed by 5 additional minutes of incubation at room temperature. The PIK3C3 kinase reaction was initiated by adding ATP (10 µM) and incubated for 30 min at room temperature. All the kinase reaction products were spotted onto nitrocellulose membrane. The membrane was blocked with 1% fat milk in PBS for 1 h, and incubated with 0.5 μg/ml of recombinant glutathione S-transferase (GST)-tagged p40PX domain (Echelon, G0302), which specifically binds to PtdIns3P, in PBS containing 3% BSA (EMD-Millipore, 2960-500G) and 0.1% Tween 20 (Sigma, 274348-4L) for 2 h. After multiple times of extensive washing, the amount of GST-p40PX remaining on the nitrocellulose membrane was analyzed by western blotting using anti-GST antibody.
Statistical analysis
Western blot protein bands and PtdIns3P dots were quantitatively analyzed for their intensities using ImageJ software. Puncta images were quantitatively analyzed by counting the number in each cell, setting the same threshold for control and experimental groups using ImageJ. Outcomes were summarized by using the mean and the standard error of the mean (SEM) as indicated in the figure legends. Statistical significance of difference between groups was analyzed by the Student t test using Prism 6 (Version 6.0d, GraphPad Software).
Acknowledgements
We thank M. Kundu for Ulk1 and Ulk2 MEFs; J.-L. Guan for Rb1cc1 KO MEFs; U87 cells for Z. Dong; L. Anderson in the Center for Mass Spectrometry and Proteomics for sample prep and analysis; R. Foncea at the Minnesota Obesity Center for lentivirus preparation; Kim lab and Neufeld lab members for discussion and helpful comments.
Disclosure of potential conflicts of interest
There is no potential conflict of interest to be disclosed.
supplemental_materials_rev.pdf
Download PDF (1.6 MB)2017AUTO0421R2-s03.xlsx
Download MS Excel (220.3 KB)Additional information
Funding
Related Research Data
References
- Kamada Y, Sekito T, Ohsumi Y. Autophagy in yeast: a TOR-mediated response to nutrient starvation. Curr Top Microbiol Immunol. 2004;279:73–84. PMID:14560952
- Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi:10.1091/mbc.E08-12-1249. PMID:19225151
- Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. doi:10.1091/mbc.E08-12-1248. PMID:19211835
- Ganley IG, Lam du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. doi:10.1074/jbc.M900573200. PMID:19258318
- Chang YY, Neufeld TP. An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol Biol Cell. 2009;20(7):2004–2014. doi:10.1091/mbc.E08-12-1250. PMID:19225150
- Jung CH, Ro SH, Cao J, et al. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295. doi:10.1016/j.febslet.2010.01.017. PMID:20083114
- Lee J, Giordano S, Zhang J. Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling. Biochem J. 2012;441(2):523–540. doi:10.1042/BJ20111451. PMID:22187934
- Bellot G, Garcia-Medina R, Gounon P, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol. 2009;29(10):2570–2581. doi:10.1128/MCB.00166-09. PMID:19273585
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi:10.1038/ncb2152. PMID:21258367
- Hosokawa N, Sasaki T, Iemura S, et al. Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009;5:973–979. doi:10.4161/auto.5.7.9296. PMID:19597335
- Hara T, Takamura A, Kishi C, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181(3):497–510. doi:10.1083/jcb.200712064. PMID:18443221
- Joo JH, Dorsey FC, Joshi A, et al. Hsp90-cdc37 chaperone complex regulates ulk1- and atg13-mediated mitophagy. Mol Cell. 2011;43(4):572–585. doi:10.1016/j.molcel.2011.06.018. PMID:21855797
- Kraft C, Kijanska M, Kalie E, et al. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J. 2012;31(18):3691–3703. doi:10.1038/emboj.2012.225. PMID:22885598
- Nishimura T, Kaizuka T, Cadwell K, et al. FIP200 regulates targeting of Atg16L1 to the isolation membrane. EMBO Rep. 2013;14(3):284–291. doi:10.1038/embor.2013.6. PMID:23392225
- Gammoh N, Florey O, Overholtzer M, et al. Interaction between FIP200 and ATG16L1 distinguishes ULK1 complex-dependent and -independent autophagy. Nat Struct Mol Biol. 2013;20(2):144–149. doi:10.1038/nsmb.2475. PMID:23262492
- Alemu EA, Lamark T, Torgersen KM, et al. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem. 2012;287(47):39275–39290. doi:10.1074/jbc.M112.378109. PMID:23043107
- Ohsumi Y. Molecular mechanism of autophagy in yeast, Saccharomyces cerevisiae. Philos Trans R Soc Lond B Biol Sci. 1999;354(1389):1577–1580. doi:10.1098/rstb.1999.0501. PMID:10582243
- Kamada Y, Funakoshi T, Shintani T, et al. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000;150(6):1507–1513. doi:10.1083/jcb.150.6.1507. PMID:10995454
- Chang YY, Juhasz G, Goraksha-Hicks P, et al. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem Soc Trans. 2009;37(Pt 1):232–236. doi:10.1042/BST0370232. PMID:19143638
- Chan EY, Longatti A, McKnight NC, et al. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol Cell Biol. 2009;29(1):157–171. doi:10.1128/MCB.01082-08. PMID:18936157
- Lee JW, Park S, Takahashi Y, et al. The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010;5(11):e15394. doi:10.1371/journal.pone.0015394. PMID:21072212
- Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461. doi:10.1126/science.1196371. PMID:21205641
- Lin SY, Li TY, Liu Q, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science. 2012;336(6080):477–481. doi:10.1126/science.1217032. PMID:22539723
- Cheong H, Lindsten T, Wu J, et al. Ammonia-induced autophagy is independent of ULK1/ULK2 kinases. Proc Natl Acad Sci USA. 2011;108(27):11121–11126. doi:10.1073/pnas.1107969108. PMID:21690395
- Alers S, Loffler AS, Paasch F, et al. Atg13 and FIP200 act independently of Ulk1 and Ulk2 in autophagy induction. Autophagy. 2011;7(12):1423–1433. doi:10.4161/auto.7.12.18027. PMID:22024743
- Russell RC, Tian Y, Yuan H, et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat Cell Biol. 2013;15(7):741–750. doi:10.1038/ncb2757. PMID:23685627
- Park JM, Jung CH, Seo M, et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy. 2016;12(3):547–564. doi:10.1080/15548627.2016.1140293. PMID:27046250
- Wold MS, Lim J, Lachance V, et al. ULK1-mediated phosphorylation of ATG14 promotes autophagy and is impaired in Huntington's disease models. Mol Neurodegener. 2016;11(1):76. doi:10.1186/s13024-016-0141-0. PMID:27938392
- Egan DF, Chun MG, Vamos M, et al. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015;59(2):285–297. doi:10.1016/j.molcel.2015.05.031. PMID:26118643
- Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 6(6):764–776. doi:10.4161/auto.6.6.12709. PMID:20639694
- Matsunaga K, Saitoh T, Tabata K, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol. 2009;11(4):385–396. doi:10.1038/ncb1846. PMID:19270696
- Zhong Y, Wang QJ, Li X, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–476. doi:10.1038/ncb1854. PMID:19270693
- Itakura E, Kishi C, Inoue K, et al. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19(12):5360–5372. doi:10.1091/mbc.E08-01-0080. PMID:18843052
- Sun Q, Fan W, Chen K, et al. Identification of Barkor as a mammalian autophagy-specific factor for Beclin 1 and class III phosphatidylinositol 3-kinase. Proc Natl Acad Sci USA. 2008;105(49):19211–19216. doi:10.1073/pnas.0810452105. PMID:19050071
- Yamamoto A, Masaki R, Tashiro Y. Characterization of the isolation membranes and the limiting membranes of autophagosomes in rat hepatocytes by lectin cytochemistry. J Histochem Cytochem. 1990;38(4):573–580. doi:10.1177/38.4.2319125. PMID:2319125
- Kovacs AL, Rez G, Palfia Z, et al. Autophagy in the epithelial cells of murine seminal vesicle in vitro. Formation of large sheets of nascent isolation membranes, sequestration of the nucleus and inhibition by wortmannin and 3-ethyladenine. Cell Tissue Res. 2000;302(2):253–261. PMID:11131136
- Mizushima N, Yamamoto A, Hatano M, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152(4):657–668. doi:10.1083/jcb.152.4.657. PMID:11266458
- Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 12(9):831–835. doi:10.1038/ncb0910-831. PMID:20811355
- Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. doi:10.1083/jcb.200803137. PMID:18725538
- Simonsen A, Stenmark H. Self-eating from an ER-associated cup. J Cell Biol. 2008;182(4):621–622. doi:10.1083/jcb.200807061. PMID:18725534
- Hayashi-Nishino M, Fujita N, Noda T, et al. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11(12):1433–1437. doi:10.1038/ncb1991. PMID:19898463
- Wang RC, Wei Y, An Z, et al. Akt-mediated regulation of autophagy and tumorigenesis through Beclin 1 phosphorylation. Science. 2012;338(6109):956–959. doi:10.1126/science.1225967. PMID:23112296
- Wei Y, An Z, Zou Z, et al. The stress-responsive kinases MAPKAPK2/MAPKAPK3 activate starvation-induced autophagy through Beclin 1 phosphorylation. Elife. 2015;4:e05289. doi:10.7554/eLife.05289.
- Gurkar AU, Chu K, Raj L, et al. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nature communications. 2013;4:2189. doi:10.1038/ncomms3189. PMID:23877263
- Qian X, Li X, Cai Q, et al. Phosphoglycerate Kinase 1 Phosphorylates Beclin1 to Induce Autophagy. Mol Cell. 2017;65(5):917–931. doi:10.1016/j.molcel.2017.01.027. PMID:28238651
- Kundu M, Lindsten T, Yang CY, et al. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood. 2008;112(4):1493–1502. doi:10.1182/blood-2008-02-137398. PMID:18539900
- Gan B, Peng X, Nagy T, et al. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol. 2006;175(1):121–133. doi:10.1083/jcb.200604129. PMID:17015619
- Duran RV, Oppliger W, Robitaille AM, et al. Glutaminolysis activates Rag-mTORC1 signaling. Mol Cell. 2012;47(3):349–358. doi:10.1016/j.molcel.2012.05.043. PMID:22749528
- Jewell JL, Kim YC, Russell RC, et al. Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science. 2015;347(6218):194–198. doi:10.1126/science.1259472. PMID:25567907
- Cong L, Ran FA, Cox D, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi:10.1126/science.1231143. PMID:23287718
- Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303. doi:10.1016/j.cell.2012.12.016. PMID:23332761
- Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155(3):688–698. doi:10.1016/j.cell.2013.09.049. PMID:24119841
- Joo JH, Wang B, Frankel E, et al. The noncanonical role of ULK/ATG1 in ER-to-Golgi trafficking is essential for cellular homeostasis. Mol Cell. 2016;62(4):491–506. doi:10.1016/j.molcel.2016.04.020. PMID:27203176
- Stone MD, Chen X, McGowan T, et al. Large-scale phosphoproteomics analysis of whole saliva reveals a distinct phosphorylation pattern. J Proteome Res. 2011;10(4):1728–1736. doi:10.1021/pr1010247. PMID:21299198
- Bandhakavi S, Stone MD, Onsongo G, et al. A dynamic range compression and three-dimensional peptide fractionation analysis platform expands proteome coverage and the diagnostic potential of whole saliva. J Proteome Res. 2009;8(12):5590–5600. doi:10.1021/pr900675w. PMID:19813771
- Keller A, Nesvizhskii AI, Kolker E, et al. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74(20):5383–5392. doi:10.1021/ac025747h. PMID:12403597
- Nesvizhskii AI, Keller A, Kolker E, et al. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75(17):4646–4658. doi:10.1021/ac0341261. PMID:14632076
- Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002; 110:163–175. doi:10.1016/S0092-8674(02)00808-5. PMID:12150925
- Vander Haar E, Lee SI, Bandhakavi S, et al. Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol. 2007;9(3):316–323. doi:10.1038/ncb1547. PMID:17277771
- Kim YM, Jung CH, Seo M, et al. mTORC1 phosphorylates UVRAG to negatively regulate autophagosome and endosome maturation. Mol Cell. 2015;57(2):207–218. doi:10.1016/j.molcel.2014.11.013. PMID:25533187
- Jung CH, Seo M, Otto NM, et al. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7(10):1212–1221. doi:10.4161/auto.7.10.16660. PMID:21795849
- Farkas T, Daugaard M, Jaattela M. Identification of small molecule inhibitors of phosphatidylinositol 3-kinase and autophagy. J Biol Chem. 2011;286(45):38904–38912. doi:10.1074/jbc.M111.269134. PMID:21930714