
Macroautophagy, hereafter autophagy, is a catabolic process by which cells remove protein aggregates and damaged organelles for recycling. This process is important for maintaining cellular homeostasis, and its alteration has been linked to various diseases including cancer. Previous studies indicated that autophagy was essential for the progression of benign hepatic tumors into malignant hepatocellular carcinoma (HCC). However, the molecular mechanism by which autophagy promotes hepatocarcinogenesis was not clear. In our recent studies, we found that mitophagy, the selective removal of mitochondria by autophagy, plays a critical role in the maintenance of hepatic cancer stem cells (CSCs). CSCs are a rare type of cancer cells that are capable of self-renewal and extensive proliferation. These properties of CSCs can lead to their accumulation of genetic and epigenetic changes and the production of a heterogeneous population of progeny cells. We found that the induction of mitophagy increases the hepatic CSC population and the inhibition of mitophagy decreases it. This need for mitophagy to maintain hepatic CSCs provides an explanation as to why autophagy is required for the malignant transformation of benign hepatic tumors into HCC.
We had previously studied mice with hepatocyte-specific knockout of Atg5, a gene essential for autophagy, and found that these mice can develop benign hepatic tumors but not HCC, not even after they are treated with the carcinogen diethylnitrosamine (DEN), which induces HCC in control mice. When the tumors isolated from atg5-knockout mice and control mice that had been treated with DEN were analyzed, we found that the population of PROM1/CD133 and ITGA6/CD49f double-positive cells in atg5-knockout mice is significantly reduced. PROM1/CD133 and ITGA6/CD49f are markers of CSCs. This observation indicated that autophagy might be important for maintaining hepatic CSCs. This possibility was confirmed by the analysis of HepG2 cells, a human hepatoblastoma cell line. We found that indeed, the induction of autophagy increases PROM1/CD133+ HepG2 cells, whereas the inhibition of autophagy reduces them. We also confirmed that these PROM1/CD133+ cells posses the properties of CSCs, as, unlike PROM1/CD133− HepG2 cells, they can self renew and proliferate when they are plated on a low-attachment plate and form tumors when they are grafted into nude mice.
Curiously, when the experiments were repeated using Huh7 and Hep3B cells, 2 different human hepatoma cell lines, no such effects of autophagy on PROM1/CD133+ cells are observed. As HepG2 expresses the wild-type tumor suppressor TP53/p53, whereas Huh7 expresses a defective TP53 and Hep3B does not express TP53, it was conceivable that the effect of autophagy on PROM1/CD133+ cells might require a functional TP53. That was indeed the case, as the expression of wild-type TP53 in either Huh7 or Hep3B cells restores the effect of autophagy on PROM1/CD133+ cells of these 2 cell lines. The observation that TP53 is required for autophagy to regulate hepatic CSCs prompted us to further investigate how TP53 mediates the effect of autophagy on CSCs. We found that the inhibition of autophagy can increase the level of TP53 and its phosphorylation at serine 392 (S392), and the induction of autophagy has the opposite effect. The phosphorylation of TP53 at S392 activates TP53 and promotes its nuclear localization. Thus, these findings indicated that autophagy can negatively regulate the TP53 activities. Our further studies indicated that the levels of TP53 and its S392 phosphorylated form negatively correlate with the level of NANOG, a transcription factor critical for the self renewal and for maintaining the stemness of CSCs. For that reason, we further examined the relationship between TP53 and NANOG and found that TP53 phosphorylated at S392 could bind to the promoter of NANOG. This binding prevents the transcription factors POU5F1/OCT4 and SOX2 from binding to and activating the NANOG promoter, resulting in the suppression of NANOG expression. Based on these results, we concluded that autophagy enhances the expression of NANOG and the expansion of hepatic CSCs by restricting the S392 phosphorylation and the activity of TP53.
Interestingly, the induction and the inhibition of mitophagy also reduce and increase the levels of TP53 and its S392 phosphorylated form, respectively. As mitophagy by itself is sufficient to regulate TP53 and hepatic CSCs, the effect of autophagy on TP53 and CSCs is most likely mediated by mitophagy. The examination of the subcellular localization of the S392 phosphorylated form of TP53 revealed that it was localizes to the nucleus when mitophagy is inhibited and to mitochondria when mitophagy is induced. The association of this phosphorylated form of TP53 with mitochondria when mitophagy is induced indicated that it is most likely digested together with its associated mitochondria by mitophagy.
PINK1 is a serine/threonine-protein kinase important for the initiation of mitophagy. To understand the relationship between mitophagy and TP53, we analyzed the effect of PINK1 on TP53 and discovered that the expression level of PINK1 positively correlates with the phosphorylation of TP53 at S392. This observation raised the possibility that PINK1 might be the elusive kinase that phosphorylates TP53 at this serine residue. We subsequently confirmed this possibility by demonstrating that PINK1 immunoprecipitated from hepatoma cells or expressed in E. coli can indeed phosphorylate TP53 at S392 in vitro.
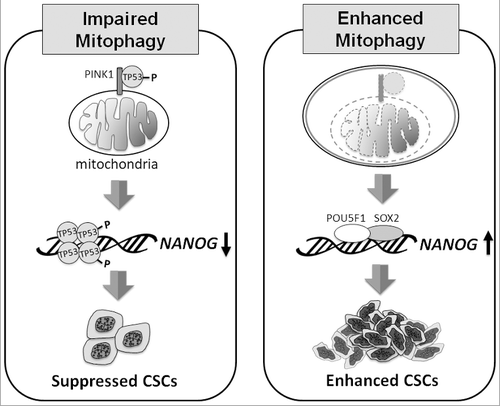
Mitophagy plays an important role in the quality control of mitochondria, but, as discussed above, it can also promote hepatocarcinogenesis by increasing the stemness and the population of CSCs. The model of how mitophagy regulates CSCs is illustrated in . CSCs are resistant to chemotherapy and are often responsible for the recurrence of cancers after chemotherapy. Our discovery that mitophagy is important for maintaining CSCs indicates that it may be possible to target this pathway to deplete CSCs for the treatment of HCC and perhaps also other cancers.
Figure 1. Model of how mitophagy regulates CSCs. When mitophagy is impaired (left panel), PINK1 phosphorylates TP53 at S392, likely on mitochondria, resulting in the activation and nuclear localization of TP53. TP53 then suppresses the expression of NANOG and reduces the self-renewal ability of CSCs. When mitophagy is enhanced (right panel), TP53 associated with PINK1 on mitochondria is degraded with mitochondria in autophagic vacuoles. This leads to the induction of NANOG expression by POU5F1/OCT4 and SOX2, and the enhancement of self-renewal of CSCs.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.