
ABSTRACT
Solid tumors are able to establish and sustain an immune suppressive microenvironment, which prevents the infiltration of cytotoxic effector immune cells into the tumor bed. We showed that genetic targeting of the macroautophagy/autophagy gene Becn1/Beclin1 in B16-F10 tumors inhibits their growth by inducing a massive infiltration of functional natural killer (NK) cells into the tumor bed. Such infiltration is primarily due to the ability of BECN1-defective tumor cells to overexpress and release CCL5 cytokine in the tumor microenvironment by a mechanism involving the activation of the MAPK8/JNK-JUN/c-Jun signaling pathway. Clinically, we reported a strong positive correlation between the expression of NK cell marker and CCL5 in human melanoma tumors and more importantly, a significant increased survival is found in melanoma patients expressing a high level of CCL5. Overall, these findings highlight the impact of targeting autophagy in breaking the immunosuppressive tumor microenvironment barrier, thus allowing the trafficking of cytotoxic NK cells into the tumor bed. This study underscore the importance of autophagy inhibition in tumors as a novel therapeutic strategy to fully exploit NK cells antitumor properties in clinical settings.
The infiltration of cytotoxic effector immune cells such as natural killer and T cells into tumors is associated with a good clinical outcome for many solid tumors. Therefore, driving effector immune cells into the tumor bed represents one of the major challenges in the field of immunotherapy. As NK cells are infrequently observed in tumor biopsies, strategies able to increase the infiltration of NK into tumors would be a plausible approach to benefit from the tremendous progress of NK-based antitumor immunotherapies.
Although the role of targeting autophagy in the inhibition of tumor growth in diverse cancer models is well established, the impact of autophagy inhibition on the immune landscape of tumors, and more specifically on the infiltration of cytotoxic effector immune cells, is largely unknown. We therefore evaluated the impact of targeting the autophagy gene Becn1 in melanoma B16-F10 cells on the infiltration of NK cells into B16-F10 tumors after syngeneic transplantation in C57BL/6J mice. We showed a significant inhibition in the tumor growth of BECN1-defective tumor-bearing mice compared with controls. Interestingly, immunohistochemical analysis performed on tumor sections revealed a significantly higher number of functional NK cells in BECN1 tumors ().
Figure 1. Targeting autophagy decreases the tumor volume of syngeneic transplanted B16-F10 melanoma by inducing the infiltration of natural killer (NK) cells into the tumor bed. Up: Targeting BECN1 in tumor cells induces the overexpression and the release of CCL5 involved in the traffic of NK cells to the tumor microenvironment. Down: The molecular mechanism underlying the overexpression of CCL5 in BECN1-defective cancer cells. Targeting BECN1 by a genetic approach (1) leads to a decrease in the phosphatase activity of PPP2/PP2A by a mechanism not yet understood (2). Such a decrease induces the phosphorylation of MAPK8/JNK (p-MAPK8/JNK) on Thr185 and Tyr183 residues (3). Subsequently, p-MAPK8/JNK phosphorylates JUN/c-Jun (p-JUN/c-Jun) on Ser63 and Ser73 residues, (4) which binds to the promoter of Ccl5 and induces its transcription (5). CCL5 released by BECN1-defective tumor cells binds to CCL5 receptors, expressed on the surface of NK cells, and induces their infiltration (6). Functional NK cells recruited to the tumor site kill cancer cells and thereby reduce the tumor volume.
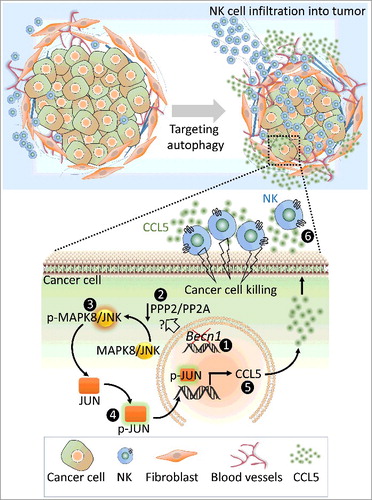
It is now well recognized that the infiltration of immune cells into tumors is orchestrated by an elaborate network of cytokines and chemokines in the tumor microenvironment. Such mediators act in autocrine and/or paracrine manner(s) allowing the communication between several types of cells within the tumor microenvironment in order to control and shape tumor growth. Cytokine profiling revealed significantly higher level of CCL5/RANTES (chemokine [C-C motif] ligand 5) released by BECN1− tumor cells in vitro and in the microenvironment of BECN1− tumors. CCL5 displays chemotaxis activity because it is described to induce the migration of several leucocytes into inflammation sites. CCL5 is released by tumor and immune cells including NK and T cells. We therefore postulated that the high level of CCL5 secreted by BECN1− tumors cells is presumably responsible for the massive infiltration of NK cells into the tumor bed. This assumption was supported by data showing that CCL5 inhibition in BECN1− tumors completely abrogates both the infiltration of NK cells and the inhibition of tumor growth.
In addition to its well-established role in the autophagy process, BECN1 also has a nonautophagic role. Therefore, an immediate question that arises is whether the increased expression of CCL5 in BECN1-defective cells results from its autophagic or nonautophagic role. Consistent with our finding showing that the expression of CCL5 is also increased in cells following ATG5 or SQSTM1/p62 targeting and by chloroquine treatment, our results argue for a role of autophagy in mediating CCL5 overexpression and subsequent enhancement of NK cell infiltration into the tumor.
CCL5 elicits its signaling pathway by binding to 3 different C-C chemokine receptors, CCR1, CCR3 and CCR5. Conflicting reports are published regarding the role of CCL5 in the tumor microenvironment and its signaling pathway in tumor cells. Indeed, some reports highlight the role of CCL5 in promoting breast cancer metastasis, while others point to the importance of CCL5 in T-cell recruitment into melanomas. The consensus appears to be that the pro- or the anti-oncogenic roles of CCL5 depends on the tumor type, the nature of the cells secreting CCL5 and the different interactions within the tumor microenvironment. We further reported that the expression of CCL5 receptors CCR1, CCR3, and CCR5 on the surface of BECN1− tumor cells isolated from their tumor microenvironment is attenuated. This result implies that the autocrine signaling loop of CCL5 on tumor cells, previously described to promote the invasion and migration of tumor cells, is attenuated in BECN1− tumor cells. Such attenuation may provide an important clue suggesting that CCL5 released by tumor cells primarily acts via paracrine signaling to attract NKs to the tumor bed. The molecular mechanism underlying the decreased expression of CCL5 receptors on the surface of BECN1− cells remains an issue of great interest.
Although much remains to be learned about the molecular link between BECN1 targeting and the overexpression of CCL5, the following clues point to the activation of the MAPK8/JNK-JUN/c-jun signaling pathway, which transcriptionally induces the expression of CCL5 in BECN1− cells: i) MAPK8/JNK is highly phosphorylated on Thr185 and Tyr183 residues (p-MAPK8/JNK) due to a decrease in the phosphatase catalytic activity of PPP2/PP2A; ii) activated MAPK8/JNK phosphorylates JUN/c-Jun, a well-known CCL5 transcription factor; iii) phosphorylated JUN/c-Jun binds to the Ccl5 promoter and transcriptionally induces its expression and release of CCL5 in the tumor microenvironment to subsequently attract NK cells to the tumor ().
The clinical significance of this study is underscored by our data showing a strong positive correlation between the expression of NK cell marker and CCL5 mRNA in 471 skin cutaneous melanoma. Such positive correlation was also found by immunohistochemical staining on formalin-fixed paraffin-embedded melanoma biopsy sections.
Whereas NK cells holds great promise for cancer treatment, only modest clinical success has been achieved so far using NK cell-based therapies in cancer patients. Accumulating evidence suggests that successful antitumor strategies based on NK cells rely predominantly on their efficient trafficking to the tumor site. Therefore, a comprehensive knowledge about tumor microenvironment factors that impede NK cell infiltration is needed. This study provides a cutting-edge approach based on targeting autophagy in tumor cells to improve the infiltration of NK cells to the tumor bed and unleash their effector cytotoxic function against cancer cells within the tumor microenvironment. One issue of great interest that remains to be addressed is to investigate the impact of targeting autophagy on the infiltration of other lymphoid and myeloid immune cell populations into the tumor bed and evaluate the net outcome of inhibiting autophagy on the immune landscape of tumors. Several important mediators of immune tolerance such as myeloid-derived suppressor cells and T regulatory cells should also be studied. Because we have successfully driven NK cells into the tumor bed, ongoing studies should be focused on the development of rational synergistic combination strategies. Such strategies may be based on combining autophagy inhibition with approaches aiming at activating NK cells or targeting NK killer inhibitory receptors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from FNRS Televie (7.6503.16; 7.4664.15; 7.4571.15, and 7.4535.16), Luxembourg Institute of Health (LHCE 2013 11 05), “Fondation Cancer”, Luxembourg (F/2016/01) and the Kriibskrank Kanner Foundation (2016 08 15).