
ABSTRACT
In recent years, the role of autophagy in the pathogenesis of most neurodegenerative diseases has transitioned into a limbo of protective or detrimental effects. Genetic evidence indicates that mutations in autophagy-regulatory genes can result in the occurrence of amyotrophic lateral sclerosis (ALS), suggesting a physiological role of the pathway to motoneuron function. However, experimental manipulation of autophagy in ALS models led to conflicting results depending on the intervention strategy and the disease model used. A recent work by the Maniatis group systematically explored the role of cell-specific autophagy in motoneurons at different disease stages, revealing surprising and unexpected findings. Autophagy activity at early stages may contribute to maintaining the structure and function of neuromuscular junctions, whereas at later steps of the disease it has a pathogenic activity possibly involving cell-nonautonomous mechanisms related to glial activation. This new study adds a new layer of complexity in the field, suggesting an intricate interplay between proteostasis alterations, the time-differential function of autophagy in neurons, and muscle innervation in ALS.
Motoneurons are the most polarized mammalian cells. Due to their complex morphology and post-mitotic status, motoneurons are highly dependent on efficient energy supply, axonal retrograde transport of vesicles through long distances, and the proper clearance of damaged organelles and abnormal protein aggregates. Amyotrophic lateral sclerosis (ALS) is the most common motoneuron disease characterized by the progressive loss of upper and lower motor neurons [Citation1]. Among the almost 40 genetic factors that can cause ALS and its related disease frontotemporal dementia (FTD), several genes affected encode proteins involved in macroautophagy (hereafter, called autophagy), including mutations in TBK1, SQSTM1/p62, OPTN, UBQLN2, VCP, and C9orf72, the most common cause of ALS-FTD [Citation1-3]. Autophagy is a central catabolic and recycling process in the cell, and in general, it is predicted that most of the ALS-linked mutations frequently deregulate the activity of the pathway [Citation3-5]. This observation led to the idea of an overall weakening of clearance processes during the disease course that may contribute to its pathogenesis through the abnormal accumulation of cargo. The presence of dysfunctional organelles and abnormal protein aggregates in affected neurons, in addition to the increased levels of autophagy markers in human postmortem samples [Citation6,Citation7] suggest that autophagy dysfunction represents an important pathogenic mechanism in ALS-FTD. Thus, autophagy-handling mechanisms are emerging as an attractive therapeutic target in the field of ALS and neurodegeneration [Citation8].
However, in the context of experimental ALS, pharmacological and genetic modulation of autophagy levels could result in diverse and even opposite consequences to the survival of mouse models and disease progression depending on the intervention strategy and the animal model employed. For example, treating Tardbp/Tdp-43 mutant mice with rapamycin or drugs that trigger MTOR-independent autophagy (i.e., spermidine and carbamazepine) protects against disease development [Citation9]. In sharp contrast, administration of rapamycin to mutant SOD1 transgenic mice can exacerbate disease progression due to enhanced apoptosis [Citation10] or may have no effects at all [Citation11]. However, activation of autophagy through the administration of trehalose (an MTOR-independent inducer of the pathway) also protects against SOD1 pathogenesis as demonstrated in various studies [Citation12-14]. Unexpectedly, we previously reported that genetic ablation of the central autophagy regulator BECN1/Beclin 1 through the use of haploinsufficient mice results in the increased life span of ALS mice despite increased protein aggregation [Citation15]. However, another study found opposite results, with SOD1G127X and SOD1G93A mice heterozygous for Becn1, developing a more aggressive phenotype compared to control animals [Citation7]. These observations may be related in part to differences in the animal models used, the nonspecific nature of the drugs tested, the stage of the disease in which the treatment begins, and possibly differential roles of the pathway in neurons and glial cells. Overall, most of the available data suggest the idea that (i) autophagy activity may affect ALS progression, and (ii) the specific effects of targeting the pathway should be studied in more detail in a temporal and cell-specific manner and possibly by comparing multiple disease parameters.
These points were addressed in a recent study by Maniatis's group showing the first specific and time-dependent characterization of the autophagy process in spinal cord motoneurons in a mouse model of ALS [Citation16]. The authors solved some of the previous controversies and depicted a complex scenario where the autophagy pathway may influence distinct aspects of ALS pathogenesis, where it has (i) beneficial effects to sustain early muscle innervation, but (ii) it exacerbates disease severity during late disease stages, unraveling the physiological and pathological role of autophagy in a temporal (and cell-specific) way.
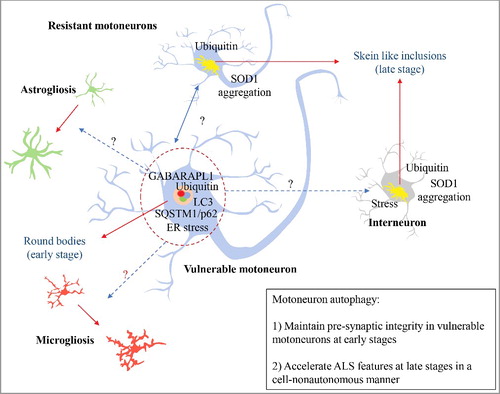
The authors first performed a detailed description of the activation of autophagy in motoneurons during the disease progression of using mutant SOD1 transgenic mice. The course of neurodegeneration in ALS encompasses early denervation of specific muscles that drive the manifestation of the first motor problems, followed by neuronal cell death and muscle paralysis with the advance of the disease. ALS is characterized by a differential neuronal vulnerability, where a subpopulation of large motoneurons that innervate fast-twitch muscles (i.e., tibialis anterior muscle) dies very early in the disease process, whereas other motoneurons that innervate slow-twitched muscles (i.e., soleus muscle) maintain their functionality until late stages (reviewed in detail in ref [Citation17].). For instance, at the pre-symptomatic stage (P30 in a SOD1 mouse model), the neuromuscular junction (NMJ) innervation in fast-twitch muscles is already reduced by 40% [Citation18]. Maniatis and co-authors reported the abundance of 2 types of inclusions in vulnerable and resistant motoneurons, in addition to interneurons [Citation16]. At early stages, vulnerable motoneurons present abundant round body inclusions that are positive for ubiquitin and SQSTM1/p62, in addition to the autophagosome marker GABARAPL1 [Citation16], suggesting that these cells can recruit the autophagic machinery. However, the inclusions of mutant SOD1 are not detected in these structures. The resistant motoneurons and interneurons are almost negative for round body protein inclusions at the same early stages. However, at late stages, when almost all fast vulnerable motoneurons are lost, the slow resistant motoneurons contain skein-like inclusions, that are positive to ubiquitin, SQSTM1/p62, and mutant SOD1, but negative to LC3 or GABARAPL1. The authors propose that these resistant cells are unable to recruit and activate the autophagy process, resulting in the accumulation of these cargos over time [Citation16]. The ability to recognize cargos and activate the autophagy machinery might be correlated to the motoneuron vulnerability to degeneration.
To dissect the cell type-specific role of autophagy, the authors developed and characterized a conditional knockout mouse for Atg7 specifically in motoneurons (Atg7cKO). The loss of Atg7 is not sufficient to cause neurodegeneration in these mice. However, targeting autophagy in motoneurons results in spontaneous phenotypes that affect selective muscle connectivity. Atg7cKO mice develop electrophysiological alterations associated with a disrupted morphology of the NMJ of tibialis anterior muscle, innervated by the motoneurons that degenerate early in ALS. These results uncovered a physiological role of the autophagy and proteostasis network in the maintenance of motoneuron-muscle.
In a parallel work, Wilhelm and colleagues found that silencing genes from the initial steps of the autophagy machinery in neurons, such as bec-1/BECN1, in adult worms promotes an enhancement of health and life span in older animals compared to wild-type worms [Citation19]. Interestingly, they showed that the muscle and pharynx in aged C. elegans lacking bec-1 from adulthood maintain the youthful characteristics [Citation19]. These apparently contradictory effects of autophagy deletion in muscle physiology in both models illustrate, among others possible reasons, the outcome of time-dependent gene inactivation. Autophagy has been demonstrated to be indispensable to the early period of life and the NMJ connection development. However, there is a progressive reduction in the last steps of autophagy during aging, including the optimal lysosomal activity (reviewed by refs [Citation20-23]). In a cell with almost null mitotic activity such as neurons, the input of new autophagosomes to the pathway is continuous to handle the accumulation of dysfunctional organelles and protein aggregates. With the later steps blocked, the permanent autophagy induction becomes an additional stressor for the cell. Thus, the silencing of genes from the beginning of the pathway in neurons from adulthood, as evaluated in worms, relieves the pressure on a highly demanding and, in several cases, dysfunctional late-stage pathway [Citation19].
Using the Atg7cKO model, the authors then moved forward to assess the consequences of disruption of autophagy in experimental ALS. Despite expectations that targeting an essential autophagy gene should exacerbate disease progression, the generation of a double mutant mouse (Atg7cKO SOD1G93A transgenics) increases the life span of SOD1G93A animals, despite NMJ impairment, consistent with our results in mutant SOD1 mice lacking Becn1 [Citation15]. We showed an improvement in life span in SOD1 mutant mice haploinsufficient for Becn1, despite an increase in high molecular weight species of SOD1 [Citation15]. At the molecular level, Atg7cKO SOD1G93A mice showed anticipated electrophysiological and muscle connectivity alterations [Citation16], confirming that basal autophagy is a critical process in the maintenance of NMJ integrity (). The study elegantly demonstrates that autophagy—in early stages—is functioning as a physiological mechanism to sustain the vulnerable motoneuron-muscle connectivity. Conversely, at late stages, motoneuron autophagy operates as a detrimental mechanism that accelerates ALS progression possibly through cell-nonautonomous mechanisms. The role of autophagy at late stages of ALS progression revealed intriguing results. Despite showing accelerated NMJ dysfunction at early stages, canonical features of ALS pathogenesis are delayed in Atg7cKO SOD1G93A mice, including SOD1 aggregation, astrogliosis, and microgliosis. Their results suggest the occurrence of cell-nonautonomous mechanisms when Atg7 is targeted in motoneurons, involving abnormal activation of glial cells.
Figure 1. Autophagy and ALS. In early stages of ALS progression, vulnerable motoneurons accumulate active round bodies and depict the presence of autophagy markers. This phenomenon is proposed to sustain the connectivity of the neuromuscular junction (NMJ) of this neuronal population. However, this pool of motoneurons is the first to degenerate during the disease course. Conversely, resistant motoneurons show skein like inclusions accumulation that may reflect an inability to recruit and activate autophagy. Surprisingly, autophagy inactivation in motoneurons increases survival in mutant SOD1 mice, associated with reduced gliosis, ER stress markers, and SOD1 aggregation. Blue dotted-arrows represent the potential cell-nonautonomous effects due to motoneuron autophagy; red-dotted arrows represent the cellular changes influenced by motoneuron autophagy; black arrows present protein inclusions.
In ALS, the molecular basis of the selective vulnerability of motoneurons involves a combination of morphological and cellular stress response features: they are large, present fast-conducting axons, with more presynaptic NMJ per motorneuron, and enhanced excitability early in life (reviewed in ref [Citation17]). Also, they are more susceptible to endoplasmic reticulum (ER) stress and the unfolded protein response, presenting upregulated markers as the earliest pathological signature identified in ALS mice [Citation24,Citation25]. Moreover, a cell type-specific ribosome profiling analysis also indicated that chronic ER stress is a salient feature of gene expression alterations in motoneurons in ALS [Citation26]. We previously reported that targeting a central component of the ER stress signaling pathway also protects against ALS, associated with altered autophagy levels [Citation6]. Additionally, it is already known that genetic alteration in ER foldases operate as a risk factor to develop ALS [Citation27], affecting the integrity of NMJ by regulating the composition and quality of this synaptic structure [Citation28,Citation29]. Interestingly, the appearance of ER markers in vulnerable motoneurons coincides in time with the local activation of microglia and with the first presence of unfolded protein response markers in resistant motoneurons and interneurons, proposing an intercellular stress activation spread [Citation25]. The results from Rudnick and coworkers show that deletion of Atg7 in motoneurons reverts the occurrence of stress responses in Atg7cKO SOD1G93A mice, highlighting markers of ER stress and inflammation, suggesting a crosstalk between autophagy and other components of the proteostasis network [Citation16]. Taken together, a new concept is emerging where proteostasis balance is essential to maintain the integrity of NMJ, a critical component underlying the manifestation of early ALS disease features. The recent works also improve our understanding of fundamental molecular mechanisms associated with motoneuron vulnerability: large and fast motoneurons are dependent on a high autophagy activity rate to generate enough energy supply to maintain their functions. By increasing the input of new autophagosomes to the pathway, the backward step of the process is saturated earlier, increasing the susceptibility to stress and, over time, acting over other cells by a cell-nonautonomous mechanism. Also, it has been shown that the lysosomal activity in vulnerable motoneurons is progressively decreased in mouse models of ALS [Citation30,Citation31] possibly altering global proteostasis control.
The work from Maniatis and co-authors confirm the idea of a negative role of autophagy in ALS progression at the level of motoneurons. The results found in Atg7cKO SOD1G93A mice regarding anticipated tremor onset and delayed survival illustrate the complex nature of ALS pathogenesis, which may also be a result of the differential vulnerability of motoneurons and a sophisticated interplay among different cell types at distinct disease stages. A more in-depth study of available data, including the gene expression profile analysis of Atg7cKO SOD1G93A mice and the use of cell-type specific studies [Citation16], may provide essential clues about signaling pathways activated differentially in astrocytes, microglia, oligodendrocytes, and specific neuronal populations. This information will help to elucidate some of the pathogenic mechanisms underlying cell-autonomous and nonautonomous effects in ALS [Citation25,Citation26,Citation32].
The development of genetic manipulations in a cell-type and time-specific manner has proved to be fundamental to reveal the complex involvement of autophagy in ALS. The next challenge in the field is the development of cell-type specific strategies to target the autophagy pathway. As a therapeutic approach, gene therapy using adeno-associated viruses (AAV) may overcome the problems of systemic administration of autophagy-targeting drugs, as already demonstrated in various studies [Citation8]. Many serotypes of AAVs are available now that can transduce specific cell types of the brain and other tissues (i.e., muscle) and even subpopulations of neurons [Citation33]. Even strategies to deliver AAVs selectively to motoneurons in adult mice are now available [Citation34]. With the progress in the understanding of the genetic differences between vulnerable and resistant motoneurons—such as the specific expression of MMP9/metalloproteinase-9 in the former [Citation35]—targeting genes specifically in vulnerable motoneurons will be possible. The therapeutic consequences of such interventions need to be experimentally tested in preclinical models of ALS. Identifying cell type-specific and temporal defects in the autophagy pathway in ALS is a current challenge in the field to develop therapeutic strategies because future treatments may aim to restore autophagy impairment and also stimulate the dynamic range that is not pathogenic [Citation36].
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors
Acknowledgments
This work was funded by FONDECYT under grant 11160288 (MN) and FONDECYT under grant 3170622 and ALSA 17-PDF-362 (VV), FONDECYT under grant 1140549, FONDAP program under grant 15150012, Millennium Institute under grant P09-015-F, European Commission R&D MSCA-RISE under grant #734749 (CH). We also thank the support from Michael J Fox Foundation for Parkinson´s Research – Target Validation grant No 9277, FONDEF ID16I10223, FONDEF D11E1007, US Office of Naval Research-Global (ONR-G) N62909-16-1-2003, U.S. Air Force Office of Scientific Research FA9550-16-1-0384, ALSRP Therapeutic Idea Award AL150111, Muscular Dystrophy Association 382453, and CONICYT-Brazil 441921/2016-7 (CH).
Additional information
Funding
Related Research Data
References
- Taylor JP, Brown RH Jr., Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016;539(7628):197–206. doi:10.1038/nature20413. PubMed PMID: 27830784.
- Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2016;13(2):96–104. doi:10.1038/nrneurol.2016.182. PubMed PMID: 27982040.
- Nassif M, Woehlbier U, Manque PA. The enigmatic role of C9ORF72 in Autophagy. Front Neurosci. 2017;11:442. doi:10.3389/fnins.2017.00442. PubMed PMID: 28824365; PubMed Central PMCID: PMC5541066.
- Sellier C, Campanari ML, Julie Corbier C, et al. Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin-2 to induce motor neuron dysfunction and cell death. EMBO J. 2016;35(12):1276–1297. doi:10.15252/embj.201593350. PubMed PMID: 27103069; PubMed Central PMCID: PMC4910533.
- Ugolino J, Ji YJ, Conchina K, et al. Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet. 2016;12(11):e1006443. doi:10.1371/journal.pgen.1006443. PubMed PMID: 27875531.
- Hetz C, Thielen P, Matus S, et al. XBP-1 deficiency in the nervous system protects against amyotrophic lateral sclerosis by increasing autophagy. Genes Dev. 2009;23(19):2294–2306. doi:10.1101/gad.1830709. PubMed PMID: 19762508; PubMed Central PMCID: PMC2758741.
- Tokuda E, Brannstrom T, Andersen PM, et al. Low autophagy capacity implicated in motor system vulnerability to mutant superoxide dismutase. Acta Neuropathol Commun. 2016;4:6. doi:10.1186/s40478-016-0274-y. PubMed PMID: 26810478; PubMed Central PMCID: PMC4727314.
- Vidal RL, Matus S, Bargsted L, et al. Targeting autophagy in neurodegenerative diseases. Trends Pharmacol Sci. 2014;35(11):583–591. doi:10.1016/j.tips.2014.09.002. PubMed PMID: 25270767.
- Wang IF, Guo BS, Liu YC, et al. Autophagy activators rescue and alleviate pathogenesis of a mouse model with proteinopathies of the TAR DNA-binding protein 43. Proc Natl Acad Sci USA. 2012;109(37):15024–15029. doi:10.1073/pnas.1206362109. PubMed PMID: 22932872; PubMed Central PMCID: PMC3443184.
- Zhang X, Li L, Chen S, et al. Rapamycin treatment augments motor neuron degeneration in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Autophagy. 2011;7(4):412–425. PubMed PMID: 21193837. doi:10.4161/auto.7.4.14541.
- Bhattacharya A, Bokov A, Muller FL, et al. Dietary restriction but not rapamycin extends disease onset and survival of the H46R/H48Q mouse model of ALS. Neurobiol Aging. 2012; 33(8):1829–1832. doi:10.1016/j.neurobiolaging.2011.06.002. PubMed PMID: 21763036.
- Castillo K, Nassif M, Valenzuela V, et al. Trehalose delays the progression of amyotrophic lateral sclerosis by enhancing autophagy in motoneurons. Autophagy. 2013;9(9):1308–1320. doi:10.4161/auto.25188. PubMed PMID: 23851366.
- Li Y, Guo Y, Wang X, et al. Trehalose decreases mutant SOD1 expression and alleviates motor deficiency in early but not end-stage amyotrophic lateral sclerosis in a SOD1-G93A mouse model. Neuroscience. 2015;298:12–25. doi10.1016/j.neuroscience.2015.03.061. PubMed PMID: 25841320.
- Zhang X, Chen S, Song L, et al. MTOR-independent, autophagic enhancer trehalose prolongs motor neuron survival and ameliorates the autophagic flux defect in a mouse model of amyotrophic lateral sclerosis. Autophagy. 2014;10(4):588–602. doi:10.4161/auto.27710. PubMed PMID: 24441414; PubMed Central PMCID: PMC4091147.
- Nassif M, Valenzuela V, Rojas-Rivera D, et al. Pathogenic role of BECN1/Beclin 1 in the development of amyotrophic lateral sclerosis. Autophagy. 2014;10(7):1256–1271. doi:10.4161/auto.28784. PubMed PMID: 24905722; PubMed Central PMCID: PMC4203551.
- Rudnick ND, Griffey CJ, Guarnieri P, et al. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc Natl Acad Sci USA. 2017;114(39):E8294–E8303. doi:10.1073/pnas.1704294114. PubMed PMID: 28904095.
- Kanning KC, Kaplan A, Henderson CE. Motor neuron diversity in development and disease. Ann Rev Neurosci. 2010;33:409–440. doi:10.1146/annurev.neuro.051508.135722. PubMed PMID: 20367447.
- Vinsant S, Mansfield C, Jimenez-Moreno R, et al. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain Behav. 2013;3(4):431–457. doi:10.1002/brb3.142. PubMed PMID: 24381813; PubMed Central PMCID: PMC3869683.
- Wilhelm T, Byrne J, Medina R, et al. Neuronal inhibition of the autophagy nucleation complex extends life span in post-reproductive C. elegans. Genes Dev. 2017;31(15):1561–1572. doi:10.1101/gad.301648.117. PubMed PMID: 28882853; PubMed Central PMCID: PMC5630021.
- Carmona-Gutierrez D, Hughes AL, Madeo F, et al. The crucial impact of lysosomes in aging and longevity. Ageing Res Rev. 2016;32:2–12. doi:10.1016/j.arr.2016.04.009. PubMed PMID: 27125853; PubMed Central PMCID: PMC5081277.
- Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24(12):604–612. doi:10.1016/j.tig.2008.10.002. PubMed PMID: 18992957; PubMed Central PMCID: PMC2745226.
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19(8):983–997. doi:10.1038/nm.3232. PubMed PMID: 23921753.
- Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell. 2011;146(5):682–695. doi:10.1016/j.cell.2011.07.030. PubMed PMID: 21884931.
- Hetz C, Saxena S. ER stress and the unfolded protein response in neurodegeneration. Nat Rev Neurol. 2017;13(8):477–491. doi:10.1038/nrneurol.2017.99. PubMed PMID: 28731040.
- Saxena S, Cabuy E, et al. A role for motoneuron subtype-selective ER stress in disease manifestations of FALS mice. Nature Neurosci. 2009;12(5):627–636. doi:10.1038/nn.2297. PubMed PMID: 19330001.
- Sun S, Sun Y, Ling SC, et al. Translational profiling identifies a cascade of damage initiated in motor neurons and spreading to glia in mutant SOD1-mediated ALS. Proc Natl Acad Sci USA. 2015;112(50):E6993–E7002. doi: 10.1073/pnas.1520639112. PubMed PMID: 26621731; PubMed Central PMCID: PMC4687558.
- Gonzalez-Perez P, Woehlbier U, Chian RJ, et al. Identification of rare protein disulfide isomerase gene variants in amyotrophic lateral sclerosis patients. Gene. 2015;566(2):158–165. doi:10.1016/j.gene.2015.04.035. PubMed PMID: 25913742; PubMed Central PMCID: PMC5553116.
- Bernard-Marissal N, Sunyach C, Marissal T, et al. Calreticulin levels determine onset of early muscle denervation by fast motoneurons of ALS model mice. Neurobiol Dis. 2015;73:130–136. doi:10.1016/j.nbd.2014.09.009. PubMed PMID: 25277755.
- Woehlbier U, Colombo A, Saaranen MJ, et al. ALS‐linked protein disulfide isomerase variants cause motor dysfunction. EMBO J. 2016;35(8):845–865. doi:10.15252/embj.201592224. PMID:26869642.
- Xie Y, Zhou B, Lin MY, et al. Progressive endolysosomal deficits impair autophagic clearance beginning at early asymptomatic stages in fALS mice. Autophagy. 2015;11(10):1934–1936. doi:10.1080/15548627.2015.1084460. PubMed PMID: 26290961.
- Xie Y, Zhou B, Lin MY, et al. Endolysosomal deficits augment mitochondria pathology in spinal motor neurons of asymptomatic fALS Mice. Neuron. 2015;87(2):355–370. doi:10.1016/j.neuron.2015.06.026. PubMed PMID: 26182418; PubMed Central PMCID: PMC4511489.
- Brockington A, Ning K, Heath PR, et al. Unravelling the enigma of selective vulnerability in neurodegeneration: motor neurons resistant to degeneration in ALS show distinct gene expression characteristics and decreased susceptibility to excitotoxicity. Acta Neuropathol. 2013;125(1):95–109. doi:10.1007/s00401-012-1058-5. PubMed PMID: 23143228; PubMed Central PMCID: PMC3535376.
- Dirren E, Towne CL, Setola V, et al. Intracerebroventricular injection of adeno-associated virus 6 and 9 vectors for cell type-specific transgene expression in the spinal cord. Hum Gene Ther. 2014;25(2):109–120. doi: 10.1089/hum.2013.021. PubMed PMID: 24191919.
- Deverman BE, Pravdo PL, Simpson BP, et al. Cre-dependent selection yields AAV variants for widespread gene transfer to the adult brain. Nat Biotechnol. 2016;34(2):204–209. doi: 10.1038/nbt.3440. PubMed PMID: 26829320; PubMed Central PMCID: PMC5088052.
- Kaplan A, Spiller KJ, Towne C, et al. Neuronal matrix metalloproteinase-9 is a determinant of selective neurodegeneration. Neuron. 2014;81(2):333–348. doi:10.1016/j.neuron.2013.12.009. PubMed PMID: 24462097.
- Nassif M, Hetz C. Targeting autophagy in ALS: a complex mission. Autophagy. 2011;7(4):450–453. doi:10.4161/auto.7.4.14700. PubMed PMID: 21252621.