
ABSTRACT
Macroautophagy/autophagy failure with the accumulation of autophagosomes is an early neuropathological feature of Alzheimer disease (AD) that directly affects amyloid beta (Aβ) metabolism. Although loss of presenilin 1 function has been reported to impair lysosomal function and prevent autophagy flux, the detailed mechanism leading to autophagy dysfunction in AD remains to be elucidated. The resemblance between pathological hallmarks of AD and Niemann-Pick Type C disease, including endosome-lysosome abnormalities and impaired autophagy, suggests cholesterol accumulation as a common link. Using a mouse model of AD (APP-PSEN1-SREBF2 mice), expressing chimeric mouse-human amyloid precursor protein with the familial Alzheimer Swedish mutation (APP695swe) and mutant presenilin 1 (PSEN1-dE9), together with a dominant-positive, truncated and active form of SREBF2/SREBP2 (sterol regulatory element binding factor 2), we demonstrated that high brain cholesterol enhanced autophagosome formation, but disrupted its fusion with endosomal-lysosomal vesicles. The combination of these alterations resulted in impaired degradation of Aβ and endogenous MAPT (microtubule associated protein tau), and stimulated autophagy-dependent Aβ secretion. Exacerbated Aβ-induced oxidative stress in APP-PSEN1-SREBF2 mice, due to cholesterol-mediated depletion of mitochondrial glutathione/mGSH, is critical for autophagy induction. In agreement, in vivo mitochondrial GSH recovery with GSH ethyl ester, inhibited autophagosome synthesis by preventing the oxidative inhibition of ATG4B deconjugation activity exerted by Aβ. Moreover, cholesterol-enrichment within the endosomes-lysosomes modified the levels and membrane distribution of RAB7A and SNAP receptors (SNAREs), which affected its fusogenic ability. Accordingly, in vivo treatment with 2-hydroxypropyl-β-cyclodextrin completely rescued these alterations, making it a potential therapeutic tool for AD.
Abbreviations
| AD | = | Alzheimer disease |
| Aβ | = | amyloid beta |
| AMPK | = | AMP-activated protein kinase |
| APP | = | amyloid beta (A4) precursor protein |
| ATG | = | autophagy related |
| ATP6V0A1 | = | ATPase, H+ transporting, lysosomal V0 subunit A1 |
| BECN1 | = | Beclin 1, autophagy related |
| CTSD | = | cathepsin D |
| DCF | = | 2’,7’-dichlorofluorescin diacetate |
| DRM | = | detergent-resistant membrane |
| EEA1 | = | early endosome antigen 1 |
| FLOT1 | = | flotillin 1 |
| GABARAPL2 | = | gamma-aminobutyric acid (GABA) A receptor-associated protein-like 2 |
| GSH | = | glutathione |
| GSHee | = | glutathione ethyl ester |
| HFIP | = | hexafluoroisopropanol |
| H-β-CD | = | 2-hydroxypropyl-β-cyclodextrin |
| IGF2R/CI-MPR | = | insulin-like growth factor 2 receptor |
| LAMP | = | lysosomal-associated membrane protein |
| LSD | = | lysosomal storage disorders |
| MAP1LC3B/LC3B | = | microtubule-associated protein 1 light chain 3 beta |
| MAPT/tau | = | microtubule-associated protein tau |
| MFN2 | = | mitofusin 2 |
| ML | = | membrane light |
| MTOR | = | mechanistic target of rapamycin (serine/threonine kinase) |
| MDV | = | mitochondrial-derived vesicles |
| NPC | = | Niemann-Pick type C disease |
| PARK2 | = | Parkinson disease (autosomal recessive, juvenile) 2, parkin |
| PINK1 | = | PTEN induced putative kinase 1 |
| PRKN | = | parkin RBR E3 ubiquitin protein ligase |
| PSEN1 | = | presenilin 1 |
| PtdIns3K | = | phosphatidylinositol 3-kinase |
| qRT-PCR | = | quantitative reverse transcriptase-polymerase chain reaction |
| RAB | = | member RAS oncogene family |
| ROS | = | reactive oxygen species |
| RPS6KB1 | = | ribosomal protein S6 kinase polypeptide 1 |
| SNAP | = | soluble NSF attachment protein |
| SNARE | = | SNAP receptor |
| SQSTM1/p62 | = | sequestosome 1 |
| SREBF2/SREBP2 | = | sterol regulatory element binding factor 2 |
| STX | = | syntaxin |
| ULK | = | unc-51 like kinase |
| VAMP | = | vesicle-associated membrane protein |
| VTI1B | = | vesicle transport through interaction with t-SNAREs 1B |
Introduction
The toxic accumulation of amyloid beta (Aβ) peptide and neurofibrillary tangles of hyperphosphorylated MAPT (microtubule associated protein tau) are the defining pathological hallmarks of Alzheimer disease (AD). The majority of mutations that cause early-onset familial AD increase the ratio of fibrillogenic Aβ, which indicates that the peptide is a primary neuropathological insult in AD progression, preceding MAPT pathology [Citation1].
Aβ is derived from the sequential processing of APP (amyloid beta precursor protein) by BACE1/β-secretase 1, followed by the γ-secretase complex with PSEN1 (presenilin 1) and PSEN2 in the active site. At homeostasis, the level of Aβ is tightly regulated through its elimination, via several processes that operate in parallel [Citation2]. However, when Aβ production exceeds clearance, it accumulates to above optimal concentrations, which subsequently leads to its self-association into neurotoxic aggregations. Remarkably, by quantifying the rates of Aβ degradation vs. synthesis, within the cerebrospinal fluid, it has been shown that late-onset sporadic AD, which accounts for more than 98% of AD cases, exhibited significant defects in the clearance of Aβ peptides [Citation3].
Basal macroautophagy, the major intracellular degradation and recycling system, which will hereafter be referred to as autophagy, is responsible for the clearance of misfolded and aggregated proteins in the nervous system. Deficiencies in this system can lead to neurodegeneration [Citation4]. In particular, the heterozygous deletion of Becn1/Beclin 1, whose expression is decreased during the early stages of AD, increases intracellular and extracellular Aβ deposition [Citation5]. In contrast, the reduction of the Aβ load is associated with improved cognitive function in AD mouse models, following the induction of MTOR (mechanistic target of rapamycin [serine/threonine kinase])-dependent autophagy [Citation6,Citation7]. Remarkably, the beneficial effects of rapamycin are observed exclusively in young animals. In the AD brain, abnormalities of the endosomal-lysosomal system are early pathological features, along with the progressive buildup of autophagic intermediates, which indicate impaired autophagic flux [Citation8,Citation9]. Several studies using PSEN1 knockout cells or cells expressing PSEN1 with familial AD-linked point mutations, have described that PSEN deficiency impairs lysosomal function, either by affecting its proteolytic efficiency or fusion ability [Citation10–12]. However, defective PSEN1 would only account for disruption of autophagy flux in cases of early-onset familial AD that carry these specific genetic mutations; hence, it is likely that other factors contribute to autophagy impairment in AD.
A close connection between impaired lysosomal clearance and neurodegeneration has also been illustrated by lysosomal storage disorders (LSD) [Citation13]. Results from Niemann-Pick type C disease (NPC) research, in which intracellular Aβ and neurofibrillary tangles are present despite absence of mutations in AD-related genes, are particularly interesting [Citation14]. These studies show that defective trafficking of cholesterol due to mutations in Npc1 and Npc2 genes leads to altered endocytic trafficking and accumulation of autophagic vesicles, that like in AD precede neuronal loss [Citation15,Citation16]. Cholesterol load inhibits endosome-lysosome trafficking [Citation17] and reduces the ability of lysosomes to fuse with endocytic and autophagic vesicles [Citation18,Citation19]. Moreover, the compound 2-hydroxypropyl-β-cyclodextrin (HP-β-CD), which stimulates cholesterol egress from endosome-lysosome compartments [Citation20] and whose therapeutic effect has been probed in animal models of NPC [Citation21,Citation22], also exerts a protective effect in AD mouse models [Citation23]. Taken together, and notwithstanding the differences, the striking parallels that exist between both these neurodegenerative disorders strongly suggest that a pathological mechanism involving abnormal cholesterol may explain autophagy impairment in AD.
Cholesterol has been consistently linked to AD. Despite decrease in cholesterol levels in healthy aging brains [Citation24], lipid analysis of the brains of patients with AD reveals increased levels of the sterol in vulnerable regions, including the cortex [Citation25,Citation26], basal ganglia [Citation27], and middle frontal gyrus [Citation28]. Cholesterol accumulation has been described in tangle-bearing and Aβ-immunopositive neurons [Citation25,Citation29,Citation30]. In addition, retrospective epidemiological studies show that hypercholesterolemia in middle age is a risk factor for AD [Citation31]. Further, despite conflicting observations from reports examining statin usage [Citation32,Citation33], a growing number of studies demonstrates that changes in cholesterol homeostasis regulate amyloidogenic processing of APP and Aβ aggregation [Citation34]. In addition, using genetic mouse models of cholesterol loading, we previously show that excess cholesterol in brain mitochondria depletes glutathione/GSH, rendering cells more vulnerable to Aβ cytotoxicity [Citation35,Citation36]. Later, we demonstrate that overexpression of the dominant-positive truncated form of SREBF2 (sterol regulatory element binding factor 2) in APP-PSEN1 mice accelerates and worsens diverse pathological AD hallmarks, including Aβ depositions, oxidative damage, neuroinflammation and the presence of neurofibrillary tangles of hyperphosphorylated MAPT, which are associated with cognitive deficits [Citation37]. Importantly, treatment with GSH ethyl ester (GSHee), a cell-permeable form of GSH, recovers the pool of mitochondrial GSH/mGSH and prevents Aβ-induced oxidative damage, which significantly reduces MAPT aggregates and amyloid plaques without affecting APP processing [Citation37]. This suggests that cholesterol may also regulate Aβ disposal. Faster delivery of Aβ to lysosomes and enhanced degradation has been described in microglia after reducing cellular cholesterol levels [Citation38]; however, the molecular mechanisms by which cholesterol may contribute to the endosomal-lysosomal disturbances described in AD are still to be fully elucidated. Using APP-PSEN1-SREBF2 mice we examined the regulatory role of cholesterol on Aβ-induced autophagy and further analyzed the impact of these alterations on Aβ and MAPT metabolism. Our data demonstrate that cholesterol exerts a dual effect on autophagy. Specifically, high intracellular cholesterol enhances Aβ-induced autophagosome formation, but impairs lysosomal fusion ability by altering RAB7A and SNAP receptors (SNAREs) content and distribution, which results in decreased Aβ and MAPT lysosomal clearance. Moreover, induction of autophagy in conjunction with inhibition of degradative autophagic flux stimulates secretory autophagy of Aβ, which may eventually contribute to extracellular Aβ deposition.
Results
Overexpression of SREBF2 in APP-PSEN1 mice enhances autophagosome formation that is associated with reduced levels of BECN1 and impaired SQSTM1 clearance
To explore whether cholesterol can regulate autophagy in AD we first monitored the presence of autophagy markers in the brains of wild-type (WT) and APP-PSEN1 mice, with and without SREBF2 overexpression, at different ages. The longitudinal analysis of APP-PSEN1 mice showed an increase in the expression levels of lipidated MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta), hereafter referred to as LC3B-II, and BECN1 at 10 mo of age (A and D). It is noteworthy that LC3B was analyzed using an antibody with stronger reactivity for the type II (lipidated) form of the protein in the membrane light (ML) fraction, which displays higher LC3B-II enrichment when autophagy is engaged (Figure S1). We assessed the levels of SQSTM1/p62 (sequestosome 1) protein, a ubiquitin-binding protein that acts as a scaffold for protein aggregates and triggers their autophagic degradation, as an indicator of autophagic resolution. The increase in LC3B-II levels in 10-mo-old APP-PSEN1 mice was associated with a significant reduction in SQSTM1 levels, consistent with proper autophagic induction and subsequent cargo degradation (A and D). In APP-PSEN1-SREBF2 mice, the accumulation of LC3B-positive structures was already evident in hippocampal slices at the age of 7 mo (B). Further, the rise in lipidated LC3B was accompanied by high levels of SQSTM1 protein (C and D). Given that the SQSTM1 protein abundance did not correlate with the corresponding mRNA expression levels (Figure S2), the observed increase most likely reflected defective autophagic degradation.
Figure 1. APP-PSEN1 mice overexpressing SREBF2 show an early accumulation of autophagosomes associated with decreased levels of BECN1 and impaired SQSTM1 clearance. Equal amounts of lysates from ML fractions (for LC3 analysis) or brain homogenates were subjected to SDS-PAGE and western blot analysis with the corresponding antibodies. (A) Representative immunoblots showing levels of lipidated LC3B (LC3B-II), BECN1 and SQSTM1 in brains from 10-mo-old WT mice and APP-PSEN1 mice at the indicated ages (mth, month). (B) Representative photomicrographs of hippocampus from 7-mo-old mice labeled with anti-LC3B and counterstained with DRAQ5 (blue) showing increased presence of LC3B puncta in APP-PSEN1-SREBF2 mice. Scale bar: 50 µm. Graph depicts quantification of the average number of LC3B puncta per cell (40 to 50 cells analyzed per genotype from a pool of at least 4 images). (C and D) Immunoblots of lipidated LC3B (LC3B-II), BECN1, SQSTM1, and PARK2 in soluble (sol., detergent-treated) and insoluble (insol., urea-treated) protein fractions of brains from 7-mo-old mice (C), and from mice at the indicated ages (mth, month) (D). (E) Western blot analysis of lipidated LC3B (LC3B-II), BECN1, and SQSTM1 in brains from 15-mo-old WT and APP-PSEN1 mice. In western blot analyses, densitometric values of the bands representing the specific protein immunoreactivity were normalized with the values of the corresponding ACTB/actin β bands and expressed as relative intensity values. *P< 0.05 and **P< 0.01; n≥3. See Figure S15 for uncropped blots.
The protein level of BECN1 was significantly reduced in the brains of APP-PSEN1-SREBF2 mice at 7 and 10 mo, but not in APP-PSEN1 mice (C and D). Quantitative reverse transcriptase-polymerase chain reaction (qRT-PCR) analysis showed unchanged mRNA levels of Becn1 in all genotypes (Figure S2), indicating again that changes at the protein level were not elicited by transcriptional regulation. Interestingly, 15-mo-old APP-PSEN1 mice, in which we have previously described an age-dependent induction of brain cholesterol synthesis [Citation39], reproduced the pattern of altered expression of BECN1 and the accumulation of SQSTM1 that was displayed by young APP-PSEN1-SREBF2 mice (E). Thus, these observations provide evidence for the link between disturbances in autophagy and changes in cholesterol homeostasis.
Recent studies suggest that PRKN (parkin RBR E3 ubiquitin protein ligase), a key player in mitophagy, can also trigger autophagic removal of Aβ by interacting with BECN1 [Citation40]. A decrease in PRKN solubility impairs this interaction and compromises Aβ clearance [Citation40]. We analyzed PARK2 expression levels in soluble and insoluble protein fractions from brain of WT and mutant mice. The immunoblots revealed similar detergent-soluble PARK2 levels with almost negligible presence of the protein in the insoluble fraction regardless of mouse genotype (C), suggesting that the autophagic defects in APP-PSEN1-SREBF2 mice were not related to PARK2 solubility. However, further studies would be needed to evaluate whether changes in brain cholesterol can regulate mitophagy mediated by the PARK2-PINK1 (PTEN induced putative kinase 1) axis.
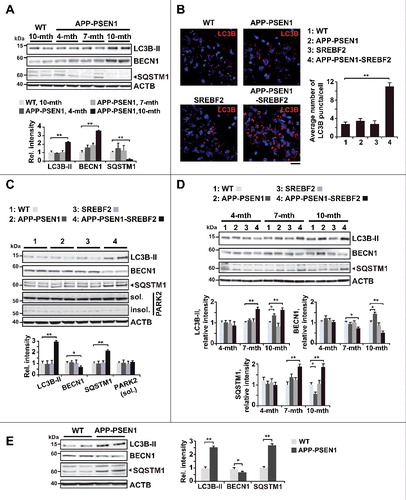
Expression levels of core autophagy proteins and MTOR signaling pathway remain unchanged in APP-PSEN1-SREBF2 mice
Autophagosome biogenesis requires the cooperation between the autophagy-related ATG12–ATG5-ATG16L1 conjugation complex and the processed form of mammalian orthologs of yeast Atg8, which includes the LC3 and the GABARAP (gamma-aminobutyric acid receptor associated protein) subfamilies [Citation41]. We analyzed the expression levels of ATG12, ATG5, and 2 enzymes, ATG7 and ATG10, involved in the conjugation process, by qRT-PCR (A) and western blot (B). There were no changes in any of the components in the brains of 7-mo-old APP-PSEN1 mice, with or without SREBF2 overexpression. In addition, we assessed the expression levels and activity of ULK1 (unc-51 like kinase 1) and ULK2, which are serine-threonine kinases that integrate upstream nutrient and energy signals to coordinate the induction of autophagy. Activity of ULK1 (and ULK2) is oppositely regulated by MTORC1 (MTOR complex 1) and AMPK (AMP-activated protein kinase) [Citation42]. Interestingly, the activity of both enzymatic complexes, as well as the expression levels of ULK1, are altered in response to oxidative stress [Citation43,Citation44]. Nonetheless, although brains from APP-PSEN1-SREBF2 mice displayed exacerbated oxidative damage compared to APP-PSEN1 mice [Citation37], qRT-PCR analysis did not show significant changes in Ulk1 and Ulk2 mRNA levels between both genotypes in the current study (A). Further, ULK1 activation, which was assessed by the level of AMPK-mediated phosphorylation at Ser317, was undetectable in brain extracts from 7-mo-old mice, irrespective of genotype (C). The unchanged levels of phosphorylated RPS6KB1 (ribosomal protein S6 kinase, polypeptide 1) (D), further proved that autophagy in APP-PSEN1-SREBF2 mice was independent of the MTOR signaling pathway. Altogether, these results were consistent with the accumulation of autophagosomes in the triple transgenic mice due to a blockage that occurs late in the autophagic flux pathway.
Figure 2. Expression levels of the ATG12–ATG5 ubiquitin-like conjugation system and activity of the MTOR-dependent pathway remained unchanged in APP-PSEN1-SREBF2 mice. (A) mRNA levels of the indicated autophagy-related proteins in brains from 7-mo-old WT and mutant mice analyzed by qRT-PCR. Absolute mRNA values were determined, normalized to Rn18s ribosomal RNA, and reported as relative levels referred to the expression in WT mice. (B) Protein expression levels were assessed in brain homogenates from 7-mo-old WT and mutant mice. Shown are representative immunoblots for ATG12–ATG5 conjugate, ATG7, and ATG5. (C) Western blot analysis of AMPK-dependent phosphorylation of ULK1 at Ser317. Cellular lysates from SH-SY5Y cells starved in FBS-depleted medium for 24 h were used as positive control (SC). (D) Representative immunoblots showing the MTOR-dependent phosphorylation status of RPS6KB1. In western blot analyses, densitometric values of the bands representing the specific protein immunoreactivity were normalized to the values of the corresponding ACTB bands and expressed as relative intensity values. n=3.
Cholesterol impairs autophagic flux induced by rapamycin treatment and Aβ exposure
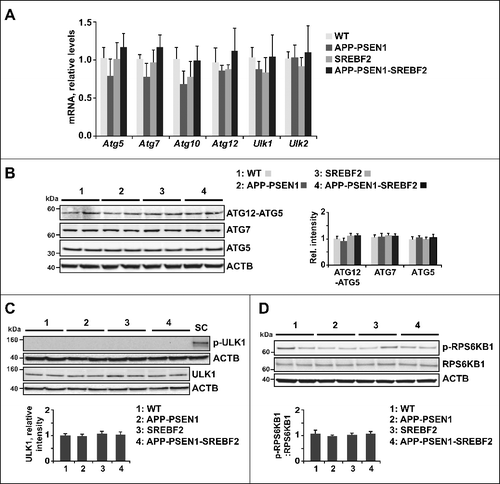
To further explore the role of cholesterol homeostasis on autophagic flux we used both primary neuronal cultures and mice overexpressing SREBF2. First, WT and SREBF2 mice were treated with the MTOR inhibitor rapamycin (5 mg/kg), and the expression of autophagy markers in the brain was evaluated. Inhibition of MTOR activity was confirmed by western blot analysis of phosphorylated RPS6KB1 levels (Figure S3). After 24 h of treatment, only the brain extracts from SREBF2 mice showed significant inhibition of MTOR (Figure S3), which was associated with increased levels of lipidated LC3B (A). The buildup of autophagosomes after treatment in the hippocampus of SREBF2 mice was also assessed by immunofluorescence (B). This accumulation was accompanied with an elevation of BECN1 levels (C). Moreover, consistent with the inhibition of autophagic flux, both western blot (D) and immunofluorescence (E) analyses demonstrated the marked accumulation of SQSTM1 in SREBF2 mice treated with the MTOR inhibitor. Autophagy induction was also observed in WT mice by increasing the dose and time of rapamycin treatment. Rapamycin injection at 10 mg/kg for 2 d significantly reduced the levels of phosphorylated RPS6KB1 in brain homogenates from WT and SREBF2 mice (Figure S4A). Inhibition of MTOR raised the levels of LC3B-II in WT brain extracts to the same extent than in extracts from SREBF2 mice (Figure S4B). However, in contrast to SREBF2 samples, levels of SQSTM1 in brain extracts of WT mice significantly decreased after rapamycin treatment (Figure S4B). As expected, after cotreatment with rapamycin and the lysosomotropic agent chloroquine (50 mg/kg), brains from WT mice displayed a stepwise increase in the LC3B-II:LC3B-I ratio (F). In contrast, the brains of the rapamycin-treated SREBF2 mice showed high LC3B-II:LC3B-I ratio at all time-points that were analyzed, regardless of chloroquine cotreatment (F), which strongly reinforced the evidence for cholesterol-induced impairment of autophagy resolution.
Figure 3. Impaired autophagic flux in SREBF2 mice after in vivo treatment with rapamycin. 7-mo-old WT and SREBF2 mice were treated with the MTOR inhibitor rapamycin (RM) at 5 mg/kg for 24 h. Equal amounts of lysates from ML fractions (for LC3 analysis) or brain homogenates were subjected to SDS-PAGE and western blot analysis with the corresponding antibodies. (A) Representative immunoblots showing levels of lipidated LC3B (LC3B-II). (B) Representative photomicrographs of hippocampus from WT and SREBF2 mice labeled with anti-LC3B and counterstained with DRAQ5 (blue). Scale bar: 50 μm. Graph depicts quantification of the average number of LC3B puncta per cell (80 to 90 cells analyzed per genotype and experimental condition from a pool of at least 4 images). (C and D) Western blot analysis of BECN1 (C), and SQSTM1 (D). (E) Representative photomicrographs of hippocampus from WT and SREBF2 mice labeled with anti-SQSTM1 and counterstained with DRAQ5 (blue). Scale bar: 50 μm. Graph depicts quantification of the average number of SQSTM1 puncta per cell (80 to 90 cells analyzed per genotype and experimental condition from a pool of at least 4 images). (F) To assess changes in autophagic flux mice were cotreated with rapamycin (RM, 5 mg/kg) and chloroquine (CQ, 50 mg/kg) for the indicated times. A representative western blot for LC3B and the corresponding quantification of the LC3B-II:LC3B-I ratio in total brain homogenates are shown. In all western blot analyses, densitometric values of the bands representing the specific protein immunoreactivity were normalized with the values of the corresponding ACTB bands and expressed as relative intensity values. **P< 0.01; n≥3. See Figure S16 for uncropped blots.
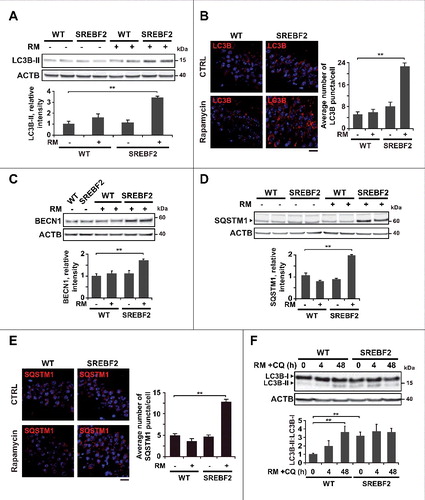
These in vivo studies were complemented with data from primary neuronal cultures. Embryonic cortical and hippocampal neurons from WT and SREBF2 mice were incubated with Aβ at 5 μM for 24 h and double-immunostained with anti-LC3B or anti-SQSTM1 and anti-LAMP2 (lysosomal associated membrane protein 2) antibodies. Confocal images showed a higher accumulation of LC3B-positive structures in the soma of SREBF2 neurons after Aβ treatment, which, unlike WT cells, showed almost no colocalization between LC3B and LAMP2 (A). Similarly, a low degree of colocalization was observed in cells that were incubated with rapamycin at 10 nM (A). SQSTM1 was mainly localized inside lysosomes in WT cells after Aβ incubation (see insets in B). In contrast, the exposure of SREBF2 neurons to Aβ resulted in the accumulation of SQSTM1 in the soma of the cell, which, like LC3B, did not show overt signs of colocalization with the lysosomal marker LAMP2 (B). These results, therefore, recapitulated the autophagic flux defect observed in SREBF2 mice after in vivo treatment with rapamycin. Autophagosome formation in neurons from WT and SREBF2 mice induced by Aβ, was completely abolished by preincubation with the phosphatidylinositol 3-kinase (PtdIns3K) inhibitor wortmannin (C), which meant that class III PtdIns3K complex was involved in Aβ-induced autophagy. Similar results were obtained when cells were preincubated with GSHee. The recovery of the depleted pool of mGSH by GSHee treatment in SREBF2 cells (Figure S5A) paralleled a significantly reduction of reactive oxygen species (ROS) generation after Aβ exposure (Figure S5B) and totally prevented the formation of LC3B-positive vesicles in Aβ-incubated neurons from WT and SREBF2 mice (C), which suggested that mitochondrial oxidative stress is a key player in Aβ-induced autophagy.
Figure 4. SREBF2 overexpression enhances autophagosome accumulation but inhibits autophagy clearance. (A and B) Embryonic cortical and hippocampal neurons isolated from WT and SREBF2 mice were treated with 10 nM rapamycin (RM) or 5 μM Aβ for 24 h. Shown are representative confocal images of neuronal-enriched cultures double immunostained for LC3B (red) and LAMP2 (green) (A) and for SQSTM1 (red) and LAMP2 (green) (B). Insets show a 3-fold magnification of the indicated region. (C) Neurons were pretreated with 5 μM wortmannin or 0.5 mM GSHee for 30 min prior autophagy induction with 5 μM Aβ for 24 h. Shown are representative confocal images of double immunolabeling with antibodies against to LC3B (red) and LAMP2 (green). Nuclei were stained by DRAQ5 (blue fluorescence). Scale bars: 50 μm. Quantification of the average number of LC3B or SQSTM1 puncta per cell was assessed using ImageJ software and depicted in the corresponding graphs (15 to 20 cells analyzed per genotype and experimental condition from a pool of at least 4 images). The Pearson correlation coefficient was used as a measure of colocalization of Alexa fluor 488 signals (LC3B) with Alexa fluor 555 signals (LAMP2). **P< 0.01.
Cholesterol-induced mGSH depletion in APP-PSEN1-SREBF2 mice controls autophagosome formation through regulating the Aβ inhibitory effect on ATG4B
Accumulating evidence supports the idea that a redox imbalance can regulate autophagy, especially during phagophore elongation [Citation45]. Intriguingly, the chemically-induced oxidation of GSH by itself, is able to trigger autophagy, even in the absence of other autophagic stimuli [Citation46]. These data prompted us to analyze whether the depleted mGSH levels can regulate Aβ-induced autophagy in APP-PSEN1-SREBF2 mice. Mice were administered GSHee (1.25 mmol/kg/day) for 2 wk, which recovers the mGSH content [Citation37]. After treatment, APP-PSEN1-SREBF2 mice showed significantly lower levels of lipidated LC3B (A). The decreased autophagosome formation was further confirmed by immunofluorescence and confocal microscopy, which demonstrated reduced LC3B puncta in the hippocampus of APP-PSEN1-SREBF2 mice after GSHee administration (B). In contrast, impaired autophagic degradation of SQSTM1 in the triple transgenic mice did not improve following mGSH recovery (A), which suggested that the regulatory role of mGSH in autophagy is confined to the first steps of the process.
Figure 5. Mitochondrial GSH depletion in APP-PSEN1-SREBF2 mice stimulates autophagosome formation via enhancing the inhibitory effect of Aβ on ATG4B. WT and APP-PSEN1-SREBF2 mice (7-mo-old) were treated with GSHee at 1.25 mmol/kg/d every 12 h for 2 wk. Lysates from ML fractions (for LC3 analysis) or brain homogenates were subjected to western blot analysis. (A) Representative immunoblots showing levels of lipidated LC3B (LC3B-II) and SQSTM1. (B) Representative photomicrographs of hippocampal sections labeled with anti-LC3B and counterstained with DRAQ5 (blue). Scale bar: 50 μm. Graph depicts quantification of the average number of LC3B puncta per cell measured using ImageJ software (80 to 90 cells analyzed per genotype and experimental condition from a pool of at least 4 images). (C) ATG4B activity of brain homogenates from 7-mo-old WT and mutant mice. Lysates were incubated with recombinant HA-GABARAPL2 AMC at 37ºC for 45 min and ATG4B activity was assessed by means of AMC fluorescence. (D) Western blot analysis of ATG4B expression in brain homogenates from 7-mo-old WT and mutant mice. (E) ATG4B activity of neuronal-enriched cultures incubated with 5 μM Aβ for 24 h with or without 30 min preincubation with 2 mM GSHee. Cell lysates were incubated with HA-GABARAPL2 AMC as in C and AMC fluorescence was analyzed. DTT (1 mM) was added to the reaction buffer to assess ATG4B maximum activity. In western blot analyses densitometric values of the bands representing the specific protein immunoreactivity were normalized with the values of the corresponding ACTB bands and expressed as relative intensity values. *P< 0.05 and **P< 0.01; n=3. See Figure S17 for uncropped blots.
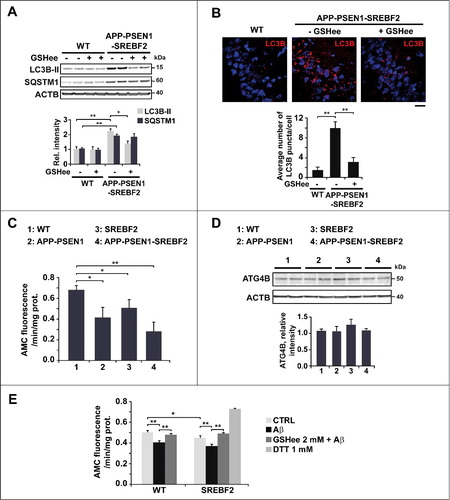
The cysteine protease ATG4B activates Atg8-family proteins by proteolytic clipping, which is a requirement for its subsequent conjugation with phospholipids, during the formation of nascent autophagosomes [Citation41]. ATG4B also exerts an esterase activity that hydrolyzes the link between Atg8-family proteins and phospholipids. Scherz-Shouval et al. describe a local rise in H2O2, upon starvation-induced autophagy, which transiently oxidizes and inactivates ATG4B allowing the conjugation of lipidated Atg8-family proteins to the phagophore membranes [Citation47]. At later stages, ATG4B is reactivated to delipidate and release Atg8-family proteins from the outer membrane of autophagosomes, which facilitates its fusogenic capability [Citation48]. Given the brain oxidative damage displayed by APP-PSEN1-SREBF2 mice [Citation37] together with increased ROS levels in primary neuronal cultures exacerbated by the mitochondrial respiration inhibitor antimycin A (Figure S6), we hypothesized an enhanced inhibitory effect on the ATG4B activity. Using a fluorescence-based assay and the Atg8-family member GABARAPL2/GATE-16 (gamma-aminobutyric acid (GABA) A receptor-associated protein-like 2) as a substrate, we measured the ATG4B activity in brain extracts from 7-mo-old WT and transgenic mice. Enzymatic activity was significantly reduced in the APP-PSEN1 mice compared to WT mice, and overexpression of SREBF2 in the APP-PSEN1 mice further enhanced this inhibition (C). Western blot analysis revealed identical levels of ATG4B protein, which meant that changes in activity were not related to protein expression levels (D). We also assessed the activity of the enzyme in primary neurons from WT and SREBF2 embryos that were exposed to 5 μM Aβ for 24 h (E). In WT neurons, Aβ incubation significantly reduced the cleavage activity of ATG4B. Compared to WT cells, untreated SREBF2 neurons exhibited lower ATG4B activity and exposure to Aβ increased this inhibitory effect (E). Conversely, pretreatment with 2 mM GSHee prevented the Aβ-induced inhibition of ATG4B in both WT and SREBF2 cells (E). Thus, our findings suggest that Aβ promotes autophagosome formation by inhibiting the cleavage activity of ATG4B. The cholesterol-induced depletion of mGSH levels, through stimulating mitochondrial ROS generation triggered by Aβ, increases ATG4B inhibition and subsequent autophagosome formation.
Mitochondrial ROS has also been implicated as a signal for PINK1-PRKN-mediated mitophagy, an event regulated by mitochondrial dynamics [Citation49]. Dysfunctional and depolarized mitochondria lose their fusion capacity, which prevents damaged organelles from incorporating back to the mitochondrial network, targeting them for autophagy clearance. Impaired balance of mitochondrial dynamics has been described in AD, associated with increased mitochondrial fragmentation and reduced mitochondrial density in neuronal processes [Citation50]. In line with these data, confocal analysis of the mitochondrial network in neurons from APP-PSEN1-SREBF2 mice revealed an abnormal mitochondrial distribution pattern and increased mitochondrial fragmentation in neurites, compared to the elongated mitochondria observed in neurons from WT and APP-PSEN1 mice (Figure S7A). Moreover, previous studies in neuronal cells treated with the mitochondrial respiration inhibitor rotenone describe the translocation of cardiolipin to the mitochondrial outer membrane as a key elimination signal for mitophagy [Citation51]. To evaluate cardiolipin externalization, isolated mitochondria from WT and mutant mice were incubated with fluorescently labeled ANXA5/annexin V, which at μM calcium concentrations specifically binds cardiolipin among other anionic phospholipids [Citation51]. The analysis by flow cytometry showed an increased percentage of ANXA5+ events in samples from APP-PSEN1 mice compared to untreated samples from WT mice, which further increased in mitochondria from APP-PSEN1-SREBF2 mice (Figure S7B). Thus, cholesterol likely through regulating Aβ-induced mitochondrial ROS affects mitochondrial dynamics and stimulates cardiolipin externalization, which may prime mitochondria for autophagy.
Loss of autophagic degradation capacity due to high cholesterol levels alters intracellular and extracellular Aβ levels
Once the impact of cholesterol on Aβ-induced autophagy was established, we analyzed whether these alterations in turn regulated Aβ metabolism. Autophagy has been described to influence intracellular Aβ accumulation and plaque formation by regulating the secretion of the peptide to the extracellular space [Citation52]. Using cortical and hippocampal neurons, we determined the levels of Aβ in medium conditioned for 48 h (A). Neurons from APP-PSEN1 mice that overexpressed SREBF2 showed an approximate 2.5-fold increase of extracellular Aβ concentration in comparison with cells from APP-PSEN1 mice (A). Preincubation with wortmannin significantly reduced the extracellular Aβ levels in both APP-PSEN1 and APP-PSEN1-SREBF2 cells, compared to untreated cells (B). However, since the effect was most robust (i.e., close to 60% reduction) in cells that overexpressed SREBF2 (B), the higher Aβ secretion in these cells could be attributed to enhanced autophagy. Intriguingly, an even greater reduction in Aβ extracellular levels was observed on pretreatment with GSHee (B). This decrease in extracellular Aβ levels, which paralleled the reduction of autophagosomes observed after GSHee treatment, underscores a cholesterol-regulated connection between ROS, autophagy and Aβ secretion.
Figure 6. SREBF2 overexpression in APP-PSEN1 mice results in intracellular Aβ accumulation associated with stimulated Aβ secretion. (A and B) Quantitative assessment of extracellular Aβ secretion analyzed in medium conditioned for 48 h of neuronal-enriched cultures isolated from APP-PSEN1 and APP-PSEN1-SREBF2 mice, untreated (A) and treated with 0.2 μM wortmannin (WM) or 4 mM GSHee and expressed as percentage of untreated controls (B). *P< 0.05; n=3. (C and D) Confocal colocalization analysis of Aβ and LC3B (C) or Aβ and LAMP2 (D) in hippocampal slices from 7-mo-old APP-PSEN1 and APP-PSEN1-SREBF2 mice with or without in vivo GSHee treatment. Insets show a 3-fold magnification of the indicated region. Graphs represent fluorescence intensity profiles of Aβ (green) and LC3B or LAMP2 (red) in the regions delineated by a white line. Nuclei were stained with DRAQ5 (blue). Scale bar: 50 μm.
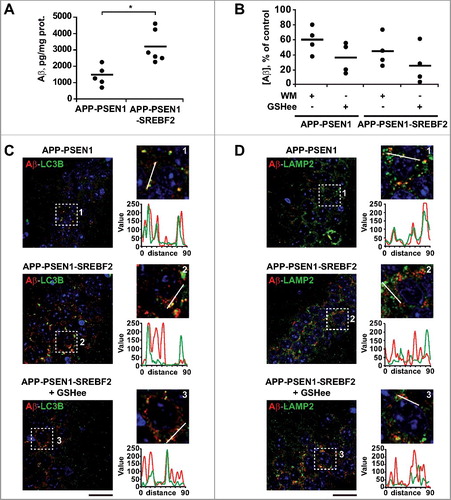
To assess the intracellular localization of Aβ and its relationship with the autophagic system we next performed double immunostaining with antibodies against Aβ and LC3B or LAMP2 (C and D). As expected, confocal microscopy revealed higher presence of intracellular Aβ and autophagosomes (LC3B puncta) in the hippocampus from 7-mo-old APP-PSEN1-SREBF2 mice, in comparison to APP-PSEN1 mice (C). However, while almost all Aβ colocalized with LC3B+ vesicles in hippocampus from APP-PSEN1 mice (C), only a partial colocalization was observed in samples from mice overexpressing SREBF2 (C), which meant that Aβ was located in different intracellular compartments apart from autophagosomes. In vivo GSHee treatment of APP-PSEN1-SREBF2 mice markedly reduced the levels of LC3B puncta; however, it did not ameliorate the degree of colocalization with Aβ that was observed in samples from untreated animals (C). Histological slices from APP-PSEN1 mice showed a high degree of colocalization between Aβ and LAMP2 in lysosomes (D). Conversely, however, almost none of the Aβ+ granules in samples from triple transgenic mice colocalized with the lysosomal marker (D). Further, GSHee treatment did not improve the low levels of colocalization (D). Altogether these findings suggest that the effect of cholesterol on autophagy modifies both intracellular and extracellular Aβ levels. High cholesterol levels impair Aβ lysosomal clearance, which results in intracellular accumulation of the toxic peptide, and via regulation of the mGSH and Aβ-induced ROS axis, cholesterol enhances autophagosome formation that ultimately favors Aβ secretion.
Autophagosomes can be formed de novo or arise from pre-existing membranes derived from different organelles [Citation53]. Autophagosome biogenesis has been described in ZFYVE1 (zinc finger FYVE-type containing 1) positive subdomains of the endoplasmic reticulum (ER). In recent reports, mitochondria and the ER-mitochondrial contact sites have also been proposed as membrane suppliers, showing that disruption of these contact sites by silencing of PACS2 (phosphofurin acidic cluster sorting protein 2) or MFN2 (mitofusin 2), prevents autophagosome synthesis induced by starvation [Citation54]. To analyze the contribution of these ER-mitochondrial contact sites to cholesterol-regulated autophagosome formation in APP-PSEN1-SREBF2 mice we knocked down the expression of MFN2, a protein that tethers the ER to mitochondria (Figure S8). Mouse embryonic fibroblasts were transfected with Mfn2 siRNA or control siRNA for 48 h. Western blot analysis confirmed a marked downregulation of MFN2 in cells derived from APP-PSEN1 and APP-PSEN1-SREBF2 mice after Mfn2 siRNA incubation (Figure S8A). Knockdown of MFN2 expression resulted in a significant reduction of LC3B puncta in cells from APP-PSEN1-SREBF2 mice (Figure S8B). Similarly, the increased presence of LC3B-positive vesicles in cells from SREBF2 mice induced by Aβ was prevented by pretreatment with Mfn2 siRNA (Figure S8C). Furthermore, the quantitative analysis of Aβ levels in conditioned media showed that Mfn2 silencing affected Aβ secretion in both APP-PSEN1 and APP-PSEN1-SREBF2 cells (Figure S8D). These results demonstrate that a correct interplay between ER and mitochondria is required for cholesterol-enhanced autophagosome biogenesis and may play a critical role in regulating Aβ metabolism and secretion.
APP-PSEN1-SREBF2 mice show an enlarged endosomal-lysosomal system with high cholesterol levels but preserved functionality
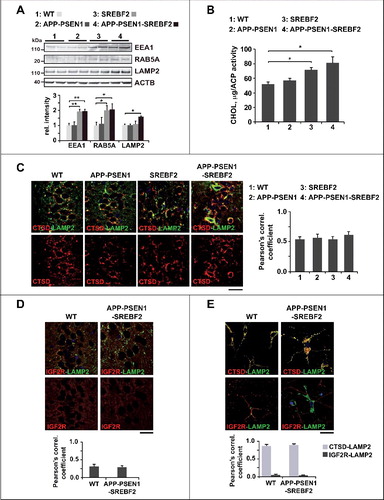
To gain insight into the mechanistic link between high cholesterol levels and the autophagy deficiency observed in the APP-PSEN1-SREBF2 mice we analyzed the endosomal-lysosomal system. Enlarged endosomes in the brain have been reported in patients with sporadic AD at preclinical stages of the disease [Citation9]. This phenotype, which is particularly exacerbated in patients carrying the ϵ4 allele of the APOE gene, has been also observed in neurons and AD animal models after cholesterol loading [Citation17,Citation55–57]. In line with these data, brain homogenates from mice overexpressing SREBF2 showed increased levels of EEA1 (early endosome antigen 1) and the GTPase RAB5A, which has been proposed to be a master regulator of endosome biogenesis [Citation58] (A). In APP-PSEN1-SREBF2 mice, the increased levels of endosomal markers were associated with the high expression of LAMP2, suggesting the synergistic effect between cholesterol and Aβ on the regulation of the endosomal-lysosomal system.
Figure 7. APP-PSEN1-SREBF2 mice show an enlarged endosomal-lysosomal system with high cholesterol levels but preserved functionality. (A) Expression levels of the indicated proteins in brain homogenates from 7-mo-old WT and mutant mice. Densitometric values of the bands representing specific protein immunoreactivity were normalized with the values of the corresponding ACTB bands and expressed as relative intensity values. *P < 0.05 and **P < 0.01; n = 3. (B) Cholesterol levels of endosomes-lysosomes isolated from brain homogenates of the indicated mice at 7 mo of age and expressed as μg of product per ACP (acid phosphatase) activity. *P< 0.05; n=3. (C and D) Hippocampal slices from 7-mo-old WT and transgenic mice. Shown are confocal photomicrographs of CTSD and LAMP2 immunoreactivity (C) or IGF2R and LAMP2 immunoreactivity (D). Scale bar: 50 μm. (G) Embryonic cortical and hyppocampal neurons isolated from WT and APP-PSEN1-SREBF2 mice. Shown are representative confocal images of double immunolabeling with antibodies against LAMP2 (green) and CTSD (red) or IGF2R (red), as indicated. Nuclei were stained with DRAQ5 (blue). Scale bar: 50 μm. The Pearson correlation coefficient was calculated in all the confocal microscopy analysis for quantifying colocalization (15 to 20 cells were analyzed per genotype and experimental condition from a pool of at least 3 images).
Next, we confirmed that the increased cholesterol levels shown in total brain homogenates of mice overexpressing SREBF2 [Citation35,Citation37], resulted in cholesterol-enrichment of the endosomal-lysosomal compartments (B). A similar increase of cholesterol content was observed in endosomes-lysosomes isolated from brains of APP-PSEN1 mice from the age of 10 mo onward (Figure S9A), which also paralleled the rise in cholesterol previously reported in total brain extracts [Citation35]. The lack of cross-contamination in the endosomal-lysosomal fraction by other cellular membranes was estimated from western blot analysis of CALR (calreticulin; marker of endoplasmic reticulum), ATP1A1 (ATPase Na+/K+ transporting, alpha 1 polypeptide; marker of plasma membrane), MAP1LC3B/LC3B (marker of autophagosomes in its LC3B-II lipidated form) and COX4 (cytochrome c oxidase subunit 4; marker of mitochondria) (Figure S9B).
PSEN1 knockdown or defective PSEN1 with AD-linked point mutations such as A246E, M233T, H163Y, M146L, and L392V, have been suggested to inhibit autophagosome clearance by affecting the proteolytic activity of lysosomes [Citation10,Citation11]. However, APP-PSEN1-SREBF2 mice, despite harboring the most severe mutation in PSEN1 that causes 2 amino-acid substitutions and an in-frame deletion of exon 9, did not show signs of impaired lysosomal function. We previously described the unaltered activity of the lysosomal hydrolase CTSD (cathepsin D) in brain homogenates of APP-PSEN1-SREBF2 mice compared with WT counterparts [Citation37]. In addition, and in line with studies showing higher lysosomal membrane stability after cholesterol enrichment [Citation59], confocal analysis of hippocampal slices from transgenic mice revealed a lack of lysosomal leakage (C), with an equal degree of colocalization between CTSD and LAMP2 and total absence of diffuse cytosolic CTSD, in comparison with histological samples from WT mice. In parallel, we evaluated changes in lysosomal acidification. Luminal acidification was assessed by analyzing the degree of colocalization between LAMP2 and the IGF2R/CI-MPR (insulin-like growth factor 2 receptor), taking advantage of the fact that dissociation of IGF2R from cathepsins after their delivery to late endosomes required proper lysosomal acidification. Most LAMP2-positive vesicles were negative for IGF2R in histological samples from both WT and APP-PSEN1-SREBF2 mice (D), which indicated that the dissociation of IGF2R from cathepsins was unaffected. Confocal microscopy analysis of primary neuronal cultures also showed that almost all LAMP2+ vesicles, regardless of cell genotype, were positive for CTSD and negative for IGF2R (E), confirming the lack of lysosomal dysfunction.
Impaired fusion between autophagosomes and lysosomes by high cholesterol levels disrupts Aβ and MAPT autophagic clearance
Fraldi et al [Citation18]. demonstrate that cholesterol accumulation reduces the ability of lysosomes to fuse with endocytic and autophagic vesicles in mouse embryonic fibroblasts derived from 2 different LSD mouse models. In line with this study, impaired autophagy in NPC has been related to defective autophagosome-late endosome fusion, whereas lysosomal proteolytic function remains unaffected [Citation15]. We assessed the fusogenic capability between autophagosomes and lysosomes isolated from brains of 10-mo-old APP-PSEN1 mice, with or without SREBF2 overexpression, and from the brains of rapamycin-primed SREBF2 and WT mice. To characterize whether high cholesterol levels affected fusion dynamics independently of vesicular trafficking, we used an in vitro image-based assay, which has been previously described in detail [Citation19]. Briefly, 3 different fractions corresponding to autophagosomes (F1), autolysosomes (F2), and lysosomes (F3) were isolated from brain homogenates by a discontinuous density Nycodenz gradient. Western blot analysis confirmed marked enrichment of the autophagosome marker LC3B in fraction F1, while CTSD, which was detected in low levels in F1, was mostly enriched in fraction F3 (Figure S10). Autophagosomes (F1) immunolabeled with anti-LC3B antibody, and lysosomes (F3) stained with the fluorescent acidotropic dye LysoTracker Red were incubated together and fusion events were monitored using immunofluorescence microscopy (A). Double labeling of F1 and F3 and the subsequent analysis of individual fluorescence images showed <6% of cross-contamination between fractions (Figure S11). Fusion was calculated as the percentage of total vesicles positive for both fluorophores. Overexpression of SREBF2 significantly impaired the fusogenic capability of vesicles isolated from APP-PSEN1-SREBF2 mice, compared to vesicles from APP-PSEN1 mice (36.0% vs. 64.7%, respectively) (). Fluorescence images from homotypic fusion between autophagosomes and lysosomes from APP-PSEN1-SREBF2 mice displayed differentially labeled vesicles in close proximity but without fusion, unlike samples from APP-PSEN1 mice (A). A similar reduction in the percentage of fusion was observed when the assays were performed using autophagosomes and lysosomes from SREBF2 mice treated with rapamycin, compared with fusion assays of vesicles from WT mice (26.4% vs. 63%, respectively) (). Interestingly, heterotypic fusion assays revealed a comparable decrease in colocalization frequencies regardless of whether the cholesterol-enriched particles were autophagosomes or lysosomes ().
Figure 8. APP-PSEN1 mice overexpressing SREBF2 show impaired autophagosome and endosome-lysosome fusion associated with an accumulation of Aβ and endogenous MAPT in autophagosomes. (A) Autophagosomes (F1) immunolabeled with anti-LC3B antibody and lysosomes (F3) stained with LysoTracker Red were incubated together and fusion events were monitored by immunofluorescence microscopy. Insets show a 3-fold magnification. In inset 2, differentially labeled vesicles in close proximity are indicated by white arrows. Scale bar: 50 μm. (B) Expression levels of Aβ and MAPT in autophagosomes (AP) and endosomes-lysosomes (EL). LC3B and CTSD (intermediate, 45 kDa; and mature form, 34 kDa) were used as markers of AP and lysosomes, respectively. All densitometric values were first normalized to Ponceau S staining to adjust for protein loading. MAPT values were normalized to the corresponding LC3B (I and II) and CTSD (mature form) bands. *P< 0.05 and **P< 0.01; n=3. (C) Proteinase K protection assay. Autophagosomes from APP-PSEN1-SREBF2 mice were exposed to proteinase K (Prot K) with or without Triton X-100 for 30 min. After inhibition of protease activity, levels of Aβ were quantified. To evaluate the presence of Aβ aggregates the same autophagosomal fraction was incubated with HFIP. Disruption of the aggregated forms significantly increased Aβ values, presumably by favoring antibody recognition. *P< 0.05 and **P< 0.01; n=3.
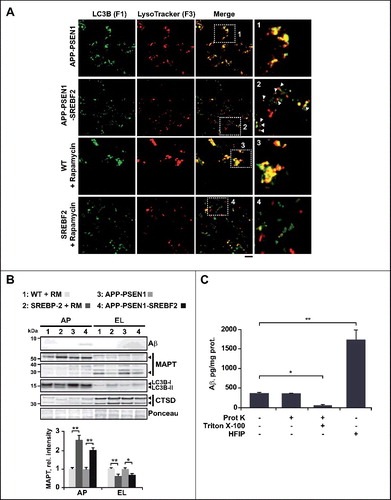
Table 1. Decreased fusion of autophagosomes with endosomes-lysosomes isolated from APP-PSEN1-SREBF2 mice compared to APP-PSEN1 mice.
Table 2. Decreased fusion of autophagosomes with endosomes-lysosomes isolated from SREBF2 mice compared to WT mice.
Next, we analyzed whether cholesterol-impaired fusogenic ability affected the presence of Aβ and MAPT in autophagic vesicles. There was no overt presence of Aβ in lysosomes, regardless of genotype, whereas the toxic peptide accumulated in autophagosomes in 10-mo-old APP-PSEN1-SREBF2 mice (B). Aβ in the autophagosomal fraction was resistant to proteinase K degradation, but it became sensitive when autophagosome membranes were permeabilized by Triton X-100, which indicated that Aβ was completely enclosed by the autophagosome membranes (C). Interestingly, treatment of autophagosomes with HFIP (hexafluoroisopropanol), a compound widely used to break down β-sheet structure, resulted in 3-fold increased Aβ quantification compared with untreated samples (C). This outcome would be consistent with a prominent presence of Aβ aggregates in autophagosomes, whose epitopes became unmasked after HFIP treatment.
Previous studies indicated that autophagy can also enhance the clearance of pathological or total endogenous MAPT [Citation60–63]. Furthermore, it has been shown that coincubation of CTSD and MAPT results in the loss of intact MAPT proteins and production of a 28-kDa fragment [Citation64]. Consistent with an impaired autophagy-mediated MAPT clearance, immunoblotting using an antibody against pan-MAPT (anti-TAU-5) revealed substantially reduced levels of the 28 kDa fragment in lysosomal compartments, accompanied with an accumulation of intact MAPT (55 to 65 kDa) in autophagosomes from mice overexpressing SREBF2 (B).
Mice overexpressing SREBF2 show changes in the expression levels and membrane distribution of key proteins involved in the fusion of autophagosomes and endosomes-lysosomes
The membrane fusion process requires conserved machinery that consists of RAB GTPases, membrane-tethering effectors that mediate the first contact, and SNAREs. SNAREs are membrane-anchored proteins that interact with each other to form a trans-SNARE complex that consists of R-SNAREs (with an arginine [R] residue in the SNARE motif) on the donor membrane and Q-SNAREs (with a glutamine [Q] residue in the SNARE motif) on the acceptor membrane [Citation65]. From the 38 SNAREs identified in humans [Citation65], the Q-SNAREs including STX7 (syntaxin 7), SXT8 (syntaxin 8), STX17(syntaxin 17) and VTI1B (vesicle transport through interaction with t-SNAREs 1B), along with the R-SNAREs VAMP7 (vesicle-associated membrane protein 7) and VAMP8, have been directly or indirectly implicated in the autophagosome and endosome-lysosome fusion process [Citation66]. Remarkably, cholesterol-binding motifs have been identified in some of them [Citation67]. In addition, changes in cellular cholesterol levels have been described to affect the dynamics of SNAREs assembly and disassembly in intracellular membranes, by regulating their location and fusogenic functions [Citation15,Citation18,Citation68]. Based on these data we investigated whether accumulation of cholesterol can modulate the content and distribution of VAMP7, VTI1B and STX7 in endosomes-lysosomes isolated from the brain of mice that overexpress SREBF2. Compared to WT mice, the level of VAMP7, VTI1B, and STX7 were markedly increased in the endosome-lysosome membranes of APP-PSEN1-SREBF2 mice (A). VTI1B was also significantly high in samples from SREBF2 mice. Additionally, as previously reported [Citation69], cholesterol enrichment increased the content of small GTPase RAB7A, while the levels of both the ATP6V0A1 (ATPase, H+ transporting, lysosomal V0 subunit A1), responsible for lysosome acidification, and the lipid raft marker FLOT1 (flotillin 1), remain unaltered (A).
Figure 9. Changes in levels and membrane distribution of key autophagosome and endosome-lysosome fusion proteins in mice overexpressing SREBF2 are prevented by in vivo treatment with 2-hydroxypropyl-β-cyclodextrin resulting in improved MAPT and Aβ clearance. (A) Western blot analysis of the indicated proteins in endosomes-lysosomes isolated from 7-mo-old WT and transgenic mice. (B) Immunoblot analysis of VAMP7 and VTI1B distribution into soluble fractions and DRMs isolated from endosomes-lysosomes of 7-mo-old WT and the indicated transgenic mice. Immunoblots with FLOT1 identified DRMs mainly in fractions 1 and 2. Int-I and Int-II: intermediate fraction I and II, Sol.: soluble fraction. (C) Expression levels of ATG14 and STX17 in autophagosomes isolated from 7-mo-old WT and the indicated mutant mice. To induce autophagy WT and SREBF2 mice were treated with rapamycin (RM, 5 mg/kg) for 24 h. LC3B protein levels were analyzed as a marker of autophagosomes. ATG14 and STX17 were normalized to the corresponding LC3B (I and II) bands. (D to H) Mice were treatment with HP-β-CD (4 g/kg) for 10 wk. To induce autophagy WT and SREBF2 mice were treated with rapamycin (RM, 5 mg/kg) 24 h prior sacrifice. (D) Expression levels of RAB7A and the indicated SNARE proteins in endosomes-lysosomes. LAMP1 was used as a marker of lysosomes. Densitometric values of the bands representing the specific protein immunoreactivity were normalized with the values of the corresponding LAMP1 bands. (E) Immunoblot analysis of VAMP7 and VTI1B distribution into soluble fractions and DRMs isolated from endosomes-lysosomes (EL). Immunoblots with FLOT1 identified DRMs mainly in fractions 1 and 2. Int-I and Int-II: intermediate fractions I and II, Sol.: soluble fraction. (F) Expression levels of ATG14 and STX17 in autophagosomes. LC3B was used as a marker of autophagosomes. ATG14 and STX17 were normalized to the corresponding LC3B (I and II) bands. (G) Expression levels of endogenous MAPT in autophagosomes (AP) and endosomes-lysosomes (EL). LC3B and CTSD (intermediate, 45 kDa; and mature form, 34 kDa) levels were used as autophagosome and lysosome markers, respectively. MAPT values were normalized to the corresponding LC3B (I and II) and CTSD (mature form) bands. (H) Representative immunoblot showing that HP-β-CD treatment prevents the accumulation of Aβ in autophagosomes isolated from APP-PSEN1-SREBF2 mice. In all western blots, densitometric values of the bands representing specific protein immunoreactivity were first normalized to Ponceau S staining to adjust for protein loading. *P< 0.05 and **P< 0.01; n=3. See Figure S18 for uncropped blots.
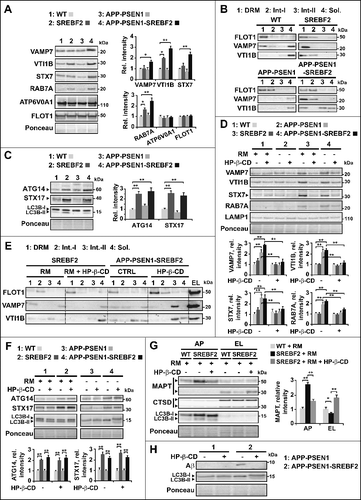
We next analyzed the compartmentalization of SNAREs in endosome-lysosome detergent-resistant membranes (DRMs), cholesterol-enriched domains, which can be isolated through a sucrose-gradient centrifugation and identified by FLOT1 immunostaining [Citation70]. In agreement with previous data from LSD cell models [Citation18], high cholesterol levels in endosome-lysosome membranes from mice overexpressing SREBF2 correlated with an increased presence of VAMP7 and VTI1B in DRMs at the expense of lower amounts of both in the soluble region of the gradient (B). The aberrant sequestration of SNAREs in cholesterol-enriched regions was not observed in samples from WT and APP-PSEN1 mice (B).
ATG14 has been identified as a key player in autophagosome and lysosome fusion, through its binding and subsequent stabilization of the STX17-SNAP28 complex on the autophagosome membranes [Citation71]. Interestingly, our western blot analysis showed increased levels of ATG14 and STX17 in autophagosomes from rapamycin-treated SREBF2 mice and APP-PSEN1-SREBF2 mice (C). The increase in protein expression in both cases was not due to upregulated transcription as qRT-PCR analysis displayed similar Atg14 and Stx17 mRNA levels regardless of mouse genotype (Figure S12). Moreover, the accumulation of ATG14 and STX17 in autophagosomes in SREBF2 mice was not observed unless autophagy was induced (Figure S13). Further, the inhibition of the lysosomal function by chloroquine was not sufficient to promote the accumulation of both proteins in rapamycin-primed WT cells (Figure S13), meaning that ATG14 and STX17 enrichment in samples from SREBF2 mice was not due to the impaired autophagy clearance.
In vivo treatment with 2-hydroxypropyl-β-cyclodextrin in mice overexpressing SREBF2 reduces the endosome-lysosome cholesterol levels, recovering impaired autophagosome-lysosome fusion and restoring Aβ and MAPT autophagy degradation
In npc1 knockout mice, a single brain injection of the hydroxypropyl form of β-cyclodextrin (HP-β-CD), a cyclic polysaccharide compound that modulates the cellular cholesterol content [Citation20], is sufficient to promote cholesterol export from lysosomes [Citation21]. In these mice chronic administration of the drug for 28 d (either by continuous brain infusion or systemically administrated) prevents most of the pathological hallmarks of NPC. Later studies from mice overexpressing mutant App transgenes indicate that HP-β-CD can also be beneficial for AD [Citation23,Citation72]. In these studies, cognitive improvement after chronic HP-β-CD administration is associated with lower phosphorylated MAPT and reduced Aβ burden, suggesting that the therapeutic effect of the drug may in part be mediated by enhanced clearance of Aβ. [Citation23] To analyze the impact of a chronic HP-β-CD treatment on autophagy, mice were administered with the compound (4 g/kg/day) for 10 wk. Compared to WT levels, the treatment significantly reduced the cholesterol content of endosomes-lysosomes of SREBF2 and APP-PSEN1-SREBF2 mice (Figure S14A). The reversal of cholesterol accretion by HP-β-CD prevented the accumulation of RAB7A and SNAREs in endosomes-lysosomes of APP-PSEN1-SREBF2 and rapamycin-primed SREBF2 mice (D). Further, the wrong compartmentalization of SNAREs, which was displayed in the endosomes-lysosomes of mice overexpressing SREBF2, was corrected after HP-β-CD treatment, showing the translocation of VAMP7 and VTI1B from DRMs to more soluble areas of the endosome-lysosome membranes (E). In contrast, the high content of ATG14 and STX17 in autophagosomes from APP-PSEN1-SREBF2 and rapamycin-primed SREBF2 mice remained unchanged, regardless of whether or not the mice were administered the cholesterol-lowering agent (F), indicating that the abnormalities in autophagosome membranes are not recovered by the treatment.
Next, we investigated whether HP-β-CD-induced changes in SNAREs levels and distribution in endosome-lysosome membranes were sufficient to correct the impaired autophagosome-lysosome fusion. Fluorescence images of homotypic incubation of autophagosomes (LC3B+ vesicles) and lysosomes (LysoTracker Red+ vesicles), which were isolated from rapamycin-primed SREBF2 mice, revealed a dramatic increase in the number of colocalization events when mice were previously treated with HP-β-CD (70.3% vs. 31.9%, respectively) ( and Figure S14B). It is noteworthy that the recovery of the fusogenic properties was associated with improved Aβ and MAPT metabolism. SREBF2 mice treated with HP-β-CD showed reduced levels of full-length MAPT in autophagosomes associated with the increased presence of 28-kDa fragments in lysosomes (G), which was indicative of autophagic flux recovery. Similarly, the Aβ accumulation in the autophagosomes of 10-mo-old APP-PSEN1-SREBF2 mice was totally prevented by HP-β-CD treatment (H). Overall, our data demonstrate a dual role of cholesterol in Aβ-induced autophagy, stimulating autophagosomes formation but impairing autophagosome-lysosome fusion, which ultimately reverberates on Aβ and MAPT metabolism ().
Table 3. In vivo treatment with 2-hydroxypropyl-β-cyclodextrin (HP-β-CD) recovers the fusogenic properties of autophagic vesicles isolated from SREBF2 mice.
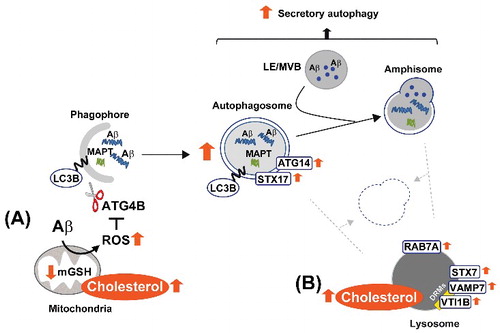
Figure 10. Schema illustrating our proposed model by which high cholesterol levels regulate Aβ-induced autophagy. In mitochondria (A), cholesterol-enrichment enhances ROS generation triggered by Aβ. The increased oxidative stress inhibits the delipidation activity of ATG4B on LC3B, and therefore, favors autophagosome synthesis. ER-mitochondria contact sites contribute to the biogenesis of these vesicles, which mainly enclose Aβ aggregates together with other more soluble forms of the peptide. In contrast, the accumulation of cholesterol in lysosomes (B) affects the levels and distribution of RAB7A and SNARE proteins, which ultimately impairs the ability of lysosomes to fuse with autophagosomes and/or amphisomes. Inhibition of the autophagy flux by high cholesterol levels reduces MAPT and Aβ clearance and stimulates Aβ secretion. DRMs, detergent-resistant membranes; mGSH, mitochondrial glutathione; LE, late endosome; MVB, multivesicular body; ROS, reactive oxygen species.
Discussion
In the present study, we advance the knowledge of the role of cholesterol in AD. We demonstrate that brain cholesterol homeostasis can affect MAPT and Aβ clearance by impairing autophagy. Importantly, according to our findings, the critical factor by which cholesterol influences the metabolism of these 2 key players in AD is most related to increases of the sterol levels in specific compartments within the cell, rather than changes in the total levels (). In mitochondria, cholesterol-mediated depletion of GSH stimulates Aβ-induced oxidative inhibition of ATG4B resulting in increased autophagosome formation. The rise of cholesterol in endosome-lysosome membranes, however, impairs autophagosome and lysosome fusion ability. It is interesting to note that impaired endocytic trafficking of cholesterol and accumulation of the lipid in endosomes-lysosomes have been linked to the apolipoprotein E4 variant, the most prevalent genetic risk factor of sporadic AD [Citation73]. Similarly, in hippocampal neurons of normal mice the knockdown of CYP46A1/cholesterol 24-hydroxylase, i.e., the enzyme that controls cholesterol efflux from the brain, resulted in cognitive deficits and hippocampal atrophy associated with the accumulation of cholesterol and modifications of the endosomal compartment [Citation57]. The affected neurons showed high cholesterol in DRMs, increased levels of RAB5A indicative of early endosomal enlargement, and upregulation of Abca1 and Npc2 mRNA, which could be explained as a compensatory response to the lipid jam in the endosomal-lysosomal system.
In contrast to what was expected, the accumulation of lipidated LC3 in APP-PSEN1-SREBF2 mice was accompanied by a reduction in the levels of BECN1. A similar outcome has been previously reported in the brains of patients with AD and mouse models of the disease [Citation5]. The exact mechanism leading to this alteration, however, still remains unknown. In line with our observations, previous studies demonstrate that APP overexpression alone does not lead to BECN1 decrease unless autophagosomal-lysosomal fusion is inhibited by bafilomycin A1 [Citation74]. In addition, we have found that the cholesterol-enhanced reduction of BECN1 was not observed in SREBF2 mice when autophagy was induced by rapamycin, which indicated that the impairment of autophagy resolution was necessary, but not sufficient to regulate the levels of BECN1. It is known that the translocation of the PtdIns3K complex to autophagic or endocytic membranes is dependent on the interaction between the coiled-coil domain of BECN1 with either ATG14 (complex I) or UVRAG (UV radiation resistance associated) (complex II), thereby specifying its participation in autophagosome formation or endocytic trafficking, as well as autophagosome and endosome-lysosome fusion [Citation75]. It would be worth exploring whether the changes of BECN1 levels may affect these interactions. Furthermore, given the high levels of ATG14 observed in the autophagosomes of APP-PSEN1-SREBF2 mice, it would be also interesting to know whether this enrichment can influence PtdIns3K complex recruitment, favoring the balance towards autophagosome formation.
Alterations in the autophagic flux were also observed in 15-mo-old APP-PSEN1 mice, although this occurred with a temporary lag regarding endosomal-lysosomal cholesterol rise. Differences in the onset of these alterations can be explained by the existence of compensatory systems, which may eventually be affected. Using the same transgenic AD mouse model, a longitudinal analysis of autophagy shows that accumulation of autophagosomes and decreased BECN1 levels correlate with the rise of neuroinflammatory cytokines at 12 mo of age [Citation76], suggesting a link between autophagy impairment and neuroinflammation.
We have found that treatment with GSHee completely prevented the accumulation of LC3-labeled autophagosomes that was induced by Aβ, in both SREBF2 and WT cells, indicating that a certain degree of oxidative stress is required for Aβ-induced autophagy. A similar inhibitory effect has been shown with antioxidants like N-acetyl-L-cysteine and CAT (catalase) under starving conditions [Citation47]. These studies identified the transient oxidative inactivation of ATG4B as the molecular mechanism for redox regulation of the starvation-induced autophagy [Citation47]. More recently, a redox control of ATG4B activity has been reported in response to hypoxia and energy stress via the induction of the pro-oxidant complex REDD1-TXNIP, in which redd1 knockout mice show deregulated ATG4B, impaired autophagic flux, and accumulation of defective mitochondria [Citation77]. Our observations demonstrate the inhibitory effect of Aβ on ATG4B activity for the first time. Furthermore, the fact that the loss of activity was amplified in mice and cells overexpressing SREBF2 (with depleted mGSH), and was prevented by GSHee treatment, which reduces Aβ-induced mitochondrial ROS, strongly suggest the mitochondrial redox control of ATG4B activity. Further, since ATG4B acts as both a conjugating and deconjugating enzyme, although evidence is currently lacking, it would be reasonable to envision a subcellular spatial regulation of ATG4B activity, where a mitochondrial ROS burst would inactivate ATG4B at the site of autophagosome formation. Further work, however, is required to investigate this possibility.
In neurons from APP-PSEN1-SREBF2 mice enhanced ROS generation correlated with increased mitochondrial fragmentation and cardiolipin externalization, both events involved in mitophagy induction. Mitochondrial ROS has been reported as a trigger for PINK1-PRKN-dependent mitophagy [Citation49]. A tight control of ROS homeostasis, by oxidative sensors such as PARK7 or the antioxidant enzyme PRDX6 (peroxiredoxin 6) [Citation78,Citation79], has been shown necessary to promote autophagic mitochondrial clearance and prevent ROS-induced cell death. Oxidative stress has also been linked to cardiolipin function [Citation80]. Whereas externalization of nonoxidized cardiolipin acts as a mitophagy receptor, the presence of peroxidized cardiolipin in the outer mitochondrial membrane potentiates BAX (BCL2 associated X, apoptosis regulator)-mediated mitochondrial apoptosis [Citation80,Citation81]. Previously, we have shown that mGSH depletion in hepatocytes stimulates cardiolipin oxidation, sensitizing cells to tumor necrosis factor-induced apoptosis [Citation82]. Therefore, one would expect that externalized cardiolipin in mitochondria of APP-PSEN1-SREBF2 mice encloses a predominant presence of the oxidized forms, which initially may be more prone to induce apoptosis. Further research would be needed to decipher whether cholesterol by regulating Aβ-induced mitochondrial ROS can modulate the balance between mitophagy (PINK1-PRKN-dependent or -independent) and the mitochondrial apoptotic pathway. Recently, a novel mechanistic pathway for mitochondrial quality control under conditions of oxidative stress has been described, involving the formation of mitochondrial-derived vesicles (MDVs) [Citation83]. PINK1-PRKN-dependent vesicles bud off of damaged mitochondria and are delivered to the endosomal-lysosomal system by a STX17-driven mechanism [Citation84]. Whether MDVs are generated by Aβ-induced mitochondrial ROS is unknown. Studies in that direction might uncover MDVs as a new pathway for targeting mitochondrial Aβ to lysosomal degradation.
A key feature of our findings is that cholesterol-induced changes in autophagy regulate both intracellular and extracellular Aβ levels. Our results suggest that intracellular accumulation of the toxic peptide in brain from APP-PSEN1-SREBF2 mice can be attributed to the enhanced formation of autophagosomes, which have been identified as amyloidogenic sites [Citation85], combined with the disruption of autophagosome-lysosome fusion that is elicited by high endosomal-lysosomal cholesterol levels. Remarkably, treatment with GSHee that failed to restore autophagy clearance (as seen by high SQSTM1 levels) did not normalize the intracellular accumulation of Aβ. Furthermore, confocal analysis only showed a partial colocalization of Aβ with autophagosomes, in line with recent studies that link the sequestration of cholesterol in endosomes-lysosomes with Aβ accumulation mostly in early and late endosomes [Citation86,Citation87]. Moreover, consistent with evidence linking autophagy with Aβ secretion [Citation52], conditioned media from APP-PSEN1-SREBF2 cells displayed increased Aβ levels. The marked reduction of extracellular Aβ levels following wortmannin and GSHee treatment is further evidence for the role that autophagosomes play on Aβ secretion. The induction of autophagy in conjunction with the inhibition of degradative autophagic flux has been also described to induce secretory autophagy of α-synuclein [Citation88]. The mechanisms that drive this unconventional secretory pathway are largely unknown, although the exocytosis of the amphisome and late endosome has been proposed to play a role [Citation89].
The metabolism of MAPT can also be influenced by cholesterol. MAPT is processed by both autophagy and the proteasome; however, unlike proteasomes, autophagy has been described to clear both soluble and insoluble MAPT, regardless of its phosphorylation state [Citation61,Citation62]. Consistent with these reports, activation of autophagy reduces MAPT levels in cultured cells and ameliorates MAPT pathology in MAPT-overexpressing mice [Citation60,Citation63], while the perturbation of lysosomal function induced aggregation of MAPT into tangle-like structures [Citation62]. Once autophagy was engaged in the brains of mice overexpressing SREBF2, full-length MAPT accumulated in autophagosomes, with low amount of cleaved fragments in lysosomal compartments. The reversal effect observed after HP-β-CD treatment highlighted that cholesterol enrichment of endosomes-lysosomes was responsible for the impaired autophagic degradation of endogenous MAPT. Similarly, a buildup of MAPT in autophagic vesicles has been described in the muscles from rats following long-term administration of chloroquine [Citation90]. Therefore, based on these findings it is reasonable to assume that the impaired autophagic clearance may contribute to the full tauopathy observed in APP-PSEN1-SREBF2 mice [Citation37]. Moreover, the induction of autophagy by nutrient deprivation, combined with lysosomal dysfunction has been described to increase MAPT secretion, which has been linked to the propagation of MAPT pathology in the brain [Citation91]. The effect of cholesterol on MAPT secretion has not yet been analyzed; however, given the increase of Aβ secretion in SREBF2 cells, it is likely that a parallel release of MAPT is occurring.
In nerve cells, most autophagosomes are formed distally in the neuritic network. Following their fusion with late endosomes the resulting amphisomes are transported retrogradely along microtubuli toward the centrosome, where most lysosomes reside. Mobility on microtubule tracks is regulated by the late endosomal GTPase RAB7A, which in combination with SNAREs, also plays a central role in the fusion of autophagosomes with endosomes-lysosomes. The cholesterol enrichment of endosome-lysosome membranes has been shown to affect proper sorting and recycling of RAB7A and SNAREs, leading to the inhibition of plus-end motility (anterograde traffic), which impairs the fusogenic properties of endosomes-lysosomes [Citation18,Citation69]. Accordingly, once autophagy is engaged, APP-PSEN1-SREBF2 mice and SREBF2 mice showed perinuclear accumulation of autophagosomes and impaired fusion, which was associated with high levels of RAB7A and aberrant buildup of different SNAREs in DRMs of endosomes-lysosomes. The recovery of the fusion events paralleled the restoration of RAB7A and SNAREs levels in endosome-lysosome membranes after HP-β-CD treatment, which further reinforces the mechanistic link between both alterations. It is noteworthy that VAMP7, VTI1B, and STX7 have been proposed to mediate autophagosome biogenesis by promoting the homotypic fusion of autophagic precursor vesicles [Citation66]. However, whether changes in cholesterol can regulate these events is still unknown. Recently, the RAB7A effector, known as ORP1L, the N-terminally (950 amino acids) longer form of OSBPL1A (oxysterol binding protein-like 1A), has been identified as a cholesterol sensor on mature autophagosomes, which regulates their position and fusion capability [Citation92]. Future studies will be required to evaluate its contribution to cholesterol-mediated autophagy impairment.
The current in vivo studies demonstrate that HP-β-CD treatment significantly reduces Aβ and MAPT accumulation in autophagosomes by recovering the fusion abilities of autophagic vesicles, which indicates the proper clearance of both peptides. HP-β-CD does not cross the blood-brain barrier efficiently; however, given its efficacy in animal models of NPC, the molecule is currently being assayed in different clinical trials for NPC, via intrathecal and i.v. administration. Its exact mechanism of action is unknown. However, in neurons, in contrast to the neurotoxic effect displayed by high HP-β-CD concentrations (> 5 mM), small amounts of the drug are endocytosed and unclog the endosomal-lysosomal system by clearing lipids without significantly affecting total cholesterol levels [Citation21,Citation93]. This has established an important difference regarding other cholesterol-lowering agent such as statins, whose therapeutic use in AD up to date has provided mixed outcomes [Citation32,Citation33]. Overall, our results show that cholesterol exacerbates the pathological potential of Aβ by stimulating autophagy, paradoxically this results in reduced Aβ degradation and increased Aβ secretion due to impaired autolysosome formation. The results further highlight the relevance of specific brain-cholesterol-lowering strategies. Together with former studies showing neuroprotective effects of HP-β-CD in AD mouse model [Citation23,Citation72], and the current use in clinical trials for NPC, HP-β-CD exemplifies the potential for therapeutic approaches in AD, especially in conjunction with autophagy inducers.
Materials and methods
Mice
Breeding pairs of B6C3-Tg(APPswe,PSEN1dE9)85Dbo/J (APP-PSEN1; MMRRC stock 34829) [Citation94] and B6;SJL-Tg(rPEPCKSREBF2)788Reh/J (SREBF2; JAX stock 003311) [Citation95] mice were purchased from The Jackson Laboratory. APP-PSEN1 mice express a chimeric mouse/human amyloid precursor protein (isoform 695) with the Swedish mutation (Mo/HuAPP695swe) and mutant human presenilin 1 (PSEN1dE9), both directed to neurons by the mouse PRNP (prion protein) promoter. Both mutations are associated with early-onset Alzheimer disease. The Swedish mutation is actually a double mutation (K595N/M596L) located immediately before the N terminus of the Aβ domain, which results in 3 to 6 times more Aβ production. Affected individuals meet diagnostic criteria for Alzheimer disease [Citation96]. The PSEN1 sequence encodes human presenilin-1, which models AD-associated mutations in PSEN1 resulting in exclusion of exon 9 (dE9). This domain contains the cleavage site for proteolytic processing, and therefore, PSEN1dE9 accumulates as an uncleaved protein with a specific gain of toxic function for APP processing [Citation94]. In turn, SREBF2 mice express a transgenic construct containing a human SREBF2 cDNA fragment (amino acids 1-468) under the control of the rat PEPCK (phosphoenolpyruvate carboxykinase) promoter. Former studies describe high circulating cholesterol levels and increased cholesterol synthetic rates in liver and adipose tissue of SREBF2 mice [Citation95]. APP-PSEN1-SREBF2 mice were generated from crossbreeding APP-PSEN1 and SREBF2 mice, which were first back-crossed more than 5 generations into the B6SJL background and characterized as previously described [Citation37]. At the time of weaning (21 d), mice were genetically identified by PCR using DNA from ear-tips and following the genotyping protocols provided by the supplier. All procedures involving animals and their care were approved by the animal care committee of the Universitat de Barcelona and were conducted in accordance with institutional guidelines in compliance with national and international laws and policies. Because APP-PSEN1-SREBF2 mice show sex-related differences in brain cholesterol levels, we only used male mice. Autophagy was induced in vivo by intraperitoneal (i.p.) injection of rapamycin (5 mg/kg; Santa Cruz Biotechnology, sc-3504). Rapamycin was reconstituted in DMSO at 25 mg/ml and diluted in phosphate-buffered saline (PBS; Sigma-Aldrich, P3813) containing 5% Tween-80 (Sigma-Aldrich, P4780) and 5% PEG-400 (polyethylene glycol 400; Sigma-Aldrich, 202398). In some cases, autophagic flux was blocked by i.p. administration of chloroquine (50 mg/kg; Sigma-Aldrich, C6628). In mice that overexpress SREBF2, the recovery of mGSH content was accomplished by treatment with GSH ethyl ester (1.25 mmol/kg/d; Sigma-Aldrich, G1404), i.p. injected every 12 h for 2 wk as described before [Citation35]. To reduce brain cholesterol levels, mice were treated with 2-hydroxypropyl-β-cyclodextrin (4 g/kg) (Santa Cruz Biotechnology, sc-203461), subcutaneously injected at the scruff of the neck, twice weekly for 10 wk [Citation23].
Cell culture and treatments
Neuron-rich primary cultures were obtained from cerebral cortex and hippocampus of embryos (d 16) using standard techniques. Dissociated cells were suspended in Neurobasal™ medium (Thermo Fisher Sci., 21103-049) with 2.5% (v:v) B27 supplement (Thermo Fisher Sci., 17504-001), 0.5 mM L-glutamine (Thermo Fisher Sci., 25030-081) and 5 μg/ml plasmocin™ (InvivoGen, ant-mpt), and plated onto poly-D-lysine (Sigma-Aldrich, P6407) and laminin (Sigma-Aldrich, L2020)-coated plates at a density of 2 x 105 cells/cm2. Half of the culture medium was changed every 3 or 4 d. Experiments were performed at 10 to 12 d in vitro (DIV). The cellular composition of the cultures was evaluated at that time by immunocytochemistry using antibodies to neuron- and glial-specific markers and was found to consist of >95% neurons.
Cells were treated with 10 nM rapamycin (Santa Cruz Biotechnology, sc-3504) and 5 μM oligomeric human Aβ (Bachem, H-6466) for 24 h. In some cases, 30 min prior to the treatment, autophagic flux was inhibited by adding 5 μM wortmannin (Sigma-Aldrich, W1628). The recovery of the mGSH content was assessed by preincubation with GSH ethyl ester (from 0.5 mM to 4 mM). To determine the mGSH levels, cells were fractionated into cytosol and mitochondria by digitonin (Santa Cruz Biotechnology, sc-280675) permeabilization and GSH levels were analyzed in the mitochondrial fraction as described previously [Citation35]. ROS generation was monitored fluorometrically using 2′,7′-dichlorofluorescin diacetate (DCF; Sigma-Aldrich, D6883). Cells were incubated with 10 μM DCF for 30 min at 37ºC prior to Aβ or antimycin A exposure. Fluorescence was measured in a SPECTRAmax Gemini XS microplate reader (Molecular Devices LLC., Sunnyvale, CA, USA) at Ex 495 nm and Em 525 nm. In some cases, DCF values were normalized to the fluorescence intensity of the mitochondrial probe MitoTracker® Deep Red FM (5 μM; Invitrogen, M22426), incubated for 30 min at 37ºC (Ex 644 nm and Em 665 nm).
Aβ secretion was analyzed in 48 h media from cultured APP-PSEN1 and APP-PSEN1-SREBF2 neurons using the Aβ42 human ELISA kit (Thermo Fisher Sci., KHB3544). Cells were centrifuged at 600 g for 1 min and supernatants, after adding the protease inhibitor AEBSF (1 mM; Sigma-Aldrich, A8456) were stored at -80ºC until use.
Preparation of Aβ peptides
Human Aβ42 hydrochloride salt (Bachem, H-6466) was dissolved to 1 mM in hexafluoroisopropanol (HFIP; Sigma-Aldrich, 10,522-8) and aliquoted into microcentrifuge tubes, then the HFIP was evaporated and the peptides were stored at -20ºC until use. For oligomeric assembly, concentrated peptides were resuspended by sonication in DMSO at 5 mM concentration and then diluted to 100 μM in phenol red free medium and incubated at 4ºC for 24 h. Oligomeric forms of Aβ were confirmed by western blot as previously described [Citation35,Citation37].
Western blot analysis
Cell or tissue lysates were prepared in RIPA buffer (Sigma-Aldrich, R0278). Samples (30 to 80 μg of protein/lane) were resolved by SDS-PAGE and transferred to nitrocellulose membranes. Blots were probed with the antibodies listed in . After overnight incubation at 4ºC, bound antibodies were visualized using anti-rabbit, mouse or rat IgG horseradish peroxidase-coupled secondary antibodies (Sigma-Aldrich, GENA934, GENA931, GENA935, respectively) and the Pierce ECL western blotting substrate (Thermo Fisher Sci., 32106). Uncropped scans of representative western blots from , , and are shown in Figure S15 to S18. PARK2 levels were analyzed in both soluble and insoluble protein fractions. To extract the soluble fraction, brain tissues were homogenized in 50 mM Tris pH 7.6, 150 mM NaCl, 2 mM EDTA, 0.2% NP-40 (Sigma-Aldrich, NP40), 0.2% BSA (Sigma-Aldrich, A4503), 20 mM PMSF (Sigma-Aldrich, P7626), and complete EDTA-free protease inhibitor cocktail tablets (Sigma-Aldrich, 11873580001), centrifuged at 10,000 g for 20 min at 4ºC, and the supernatants containing the soluble fraction of proteins were collected. To extract the insoluble fraction the pellet was resuspended in 4 M urea and adjusted to pH 7.0 with 1 N NaOH and centrifuged at 10,000 g for 20 min at 4ºC, and the supernatant containing the insoluble fraction was collected.
Table 4. Details of the primary antibodies used in the current study.
Immunofluorescence and laser confocal imaging
Paraffin-embedded blocks were prepared by sequential dehydration in graded ethanol and infiltration in paraffin before embedding. Blocks were serially sectioned between –1.2 mm and –2.4 mm from Bregma at a thickness of 5 μm. Dewaxed and rehydrated hippocampal sections were first boiled in Tris-EDTA buffer, pH 9.0, and incubated with 0.1 M glycine in PBS for 20 min to reduce autofluorescence. Tissues processed for Aβ staining were pretreated with 99% formic acid for 7 min. Sections were then incubated overnight at 4°C with the antibodies listed in . After washing in PBS, the immunoreaction was visualized using Alexa Fluor 488, 555, and 594 conjugated secondary antibodies (1:400; Thermo Fisher Sci., A27012, A27017, A11005, respectively). Nuclei were stained with DRAQ5 fluorescent probe solution (Thermo Fisher Sci., 62251). Neurons were fixed for 15 min with 4% paraformaldehyde (Thermo Fisher Sci., 28908) and permeabilized with 0.1% saponin (Sigma-Aldrich, 47036) in blocking buffer (1% BSA + 20 mM glycine in PBS) for 15 min before proceeding with the immunostaining. Confocal images were collected with a Leica TCS SPE confocal laser scanning microscope (Leica Microsystems, Barcelona, Spain) using a 63x/1.32-0.60 oil PH3 CS objective and a confocal pinhole set at 1 Airy unit. All confocal images shown are single optical sections.
Quantitative RT-PCR
qRT-PCR amplification was carried out using the MyiQ Single-Color RT-PCR detection system (Bio-Rad, 170-9770, Barcelona, Spain) and the iScript one-step RT-PCR kit with SYBR Green (Bio-Rad, 170-8893). The primer sequences used are listed in . PCRs were run in duplicates for each sample. Relative gene expression was quantified by the delta-delta Ct method.
Table 5. Primer sequences used for qRT-PCR.
Cholesterol measurements
Cholesterol levels in lysosomes were determined fluorimetrically using the Amplex Red Cholesterol Assay kit (Thermo Fisher Sci., A12216). Samples (0.02 mg/ml) were extracted with chloroform:isopropanol:IGEPAL CA-630 (Sigma-Aldrich, I8896) (7:11:0.1) and centrifuged at 13,000 g for 10 min to remove insoluble material. Then, the organic phase was transferred to a new tube and dried under vacuum for 30 min. The lipids were dissolved with 120 μl of 1x cholesterol reaction buffer, and vortex until mixture was homogenous. Amount of cholesterol in lysosomes was calculated from the standard curve and expressed as μg of product per acid phosphatase activity.
Acid phosphatase assay
Acid phosphatase activity was determined by using p-nitrophenyl phosphate as a substrate. Briefly, 50 μl of the lysosomal fraction (20 μg protein) were incubated in 200 μl of assay solution (300 mg of 4-nitrophenyl phosphate disodium salt hexahydrate [Sigma-Aldrich, N2765] in 90 mM NaOAc) for 30 min at 37ºC. Then, 0.6 ml of 0.25 M NaOH was added to stop the reaction and samples were microcentrifuged 2 min at 21,000 g. Absorbances were measured at 410 nm against the appropriate blank.
ATG4B activity
Brain homogenates and cells were lysed in 0.05% NP-40, 10 mM HEPES, 10 mM KCl, 1.5 mM MgCl2, pH 8.0. Lysates (10 μg) were incubated with 210 nM of recombinant human HA-GABARAPL2/GATE16 AMC (Boston Biochem, UL-445) in reaction buffer (50 mM Tris-HCl, pH 8.1, 2 M sodium citrate) at 37ºC for 45 min. Release of AMC fluorescence was monitored at Ex 380 nm and Em 460 nm.
Isolation of detergent-resistant membrane (DRM) microdomains
Lysosomal membranes (100 mg of protein) were incubated with 1% of Triton X-114 (Sigma-Aldrich, X114) in 150 mM NaCl, 50 mM Tris-HCl, 5 mM EDTA, pH 7.4, on ice for 30 min [Citation70]. At the end of the incubation, samples were adjusted to 40% sucrose (Sigma-Aldrich, S0389), and loaded at the bottom of a 9 ml step-wise discontinuous sucrose gradient (40, 35, 30, 25, 20, 15, 10 and 5%) and centrifuged at 200,000 g for 19 h. Then, 6 aliquots of 1.5 ml were collected from the top of the gradient corresponding to DRMs (aliquot 1 and 2), intermediate-I (aliquot 3), intermediate-II (aliquot 4) and soluble (aliquot 5 and 6) fractions, and were subjected to acid precipitation with 10% trichloroacetic acid. Precipitates were washed with acetone, resuspended in 2x electrophoresis sample buffer, and subjected to SDS-PAGE and immunoblot.
Endosome-lysosome and autophagosome fractionation
Endosomes-lysosomes were prepared by a discontinuous OptiPrep gradient [Citation97]. Brains were removed of olfactory bulbs, midbrain and cerebellum, and were homogenized in 5 mM Tris-HCl, pH 7.5, 320 mM sucrose, 1 mM MgCl2, 1 mM CaCl2, 1 mM NaHCO3 plus complete EDTA-free protease inhibitor cocktail tablets (Sigma-Aldrich, 11873580001). Homogenates were centrifuged at 1,000 g for 10 min. The supernatant was kept and the pellet was homogenized and centrifuged again. The 2 supernatants were centrifuged at 20,000 g for 20 min. The pellet was designated ML fraction and resuspended in dilution buffer (5 mM MOPS [Sigma-Aldrich, M3183], pH 8.0, 1 mM EDTA and 0.1% ethanol). The ML suspension was further diluted (10 mg of protein/ml) to a solution containing 19% OptiPrep density gradient medium (Sigma-Aldrich, D1556) and osmotically balanced with 2.3 M sucrose (290 mOsm). Different OptiPrep density gradient medium solutions were prepared with dilution buffer, balanced with sucrose, and loaded from bottom to top as follows: 27%, 22.5%, 19% or ML suspension, 16%, 12% and 8%. After ultracentrifugation at 155,500 g for 4 h in an SW40Ti rotor (Beckman Coulter, Barcelona, Spain), 0.5 ml fractions were collected and analyzed for lysosomal markers and total protein. The fraction at the interphase between 8% and 12% of the OptiPrep gradient was taken as endosomes-lysosomes.
Autophagosomes were isolated by centrifugation in a discontinuous Nycodenz gradient, as described previously [Citation98]. The corresponding ML fraction resulting from 5 brains was resuspended in 1ml of 50% Nycodenz and loaded onto 3.5 ml of 26% Nycodenz placed at the bottom of the tubes. Density gradient separation of autophagic vacuoles was achieved after layering above the sample with 1.5 ml of each 24%, 20% and 10% Nycondenz, and ultracentrifugation at 120,000 g for 4 h in a SW40Ti rotor (Beckman Coulter, Barcelona, Spain). The bands were collected from the gradient, diluted 5 times with 0.3 M sucrose and sedimented at 37,000 g for 10 min. Autophagosomes were recovered in the 20% to 10% interface and lysosomes in the 26 to 24% interface.
In vitro vesicular fusion assay
Isolated autophagosomes and lysosomes were carefully resuspended in fusion buffer (10 mM HEPES pH 7.0, 10 mM KCl, 1.5 mM MgCl2, 1 mM DTT, 0.25 M sucrose, and complete EDTA-free protease inhibitor cocktail tablets [Sigma-Aldrich, 11873580001]), and protein content of samples was balanced to 15 mg/ml [Citation98]. Prior to fusion reaction, lysosomes were stained with LysoTracker Red (1 μM; Thermo Fisher Sci., L7528) for 10 min at 37ºC. Labeled lysosomes were then recovered by centrifuging at 21,000 g for 15 min and incubated with autophagosomes at 37ºC for 30 min in the reaction buffer containing 3 mM ATP (Sigma-Aldrich, A2383), 2 mM GTP (Sigma-Aldrich, G8877), 2 mM CaCl2, 8 mM phosphocreatine (Sigma-Aldrich, P6502), 0.16 mg/ml of creatine phosphokinase (Sigma-Aldrich, C7886), and protease inhibitors. To stop the reaction 8% of formaldehyde was added to the suspension. After 10 min fixation at room temperature, samples were rinsed with PBS and immunostained for autophagosomes overnight at 4°C using the anti-LC3B antibody listed in . Finally, the mixture was spotted on a glass slide and visualized under the fluorescence microscope. Images were acquired with a Zeiss Axioplan microscope (Carl Zeiss, Oberkochen, Germany) equipped with a Nikon digital camera DXM1200F (Nikon, Amsterdam, The Netherlands) using a 100x/1.3 N.A. objective. Quantification was performed using ImageJ software [Citation99].
Proteinase K protection assay
Isolated autophagosomal fraction was suspended in 10 mM HEPES pH 7.0, 10 mM KCl, 1.5 mM MgCl2, 0.25 M sucrose and incubated for 30 min on ice with or without 10 μg/ml proteinase K (Sigma-Aldrich, P2308) and 0.5% Triton X-100 (Sigma-Aldrich, X100). Protease activity was stopped by adding 1 mM AEBSF (Sigma-Aldrich, 76307). Samples were stored at −80°C until Aβ quantification. In some cases, samples were evaporated and redissolved in 50 μl of HFIP and incubated for 30 min at room temperature. After treatment, HFIP was removed by vacuum evaporation.
Statistical analysis
Results are expressed as means ± SD. Statistical significance was performed using an analysis of variance (ANOVA) followed by the unpaired 2-tailed Student t test with Tukey post hoc multiple comparison test when required. The Chi-square test of independence was used to test for equality of proportions between populations. All analyses were performed using GraphPad Prism. A P< 0.05 value was considered statistically significant.
Disclosure of potential conflicts of interest
The authors declare that they have no competing interests.
Supplemental Material
Download MS Word (2 MB)Supplemental Material
Download MS Word (96.7 KB)Acknowledgements
The authors are grateful to Dr. R. Trullàs and Dr. G. Mengod, and the respective group members, for their helpful insights and technical support. The technical advices provided by Jordi Creus were greatly appreciated. We also thank the assistance provided by Dr. Teresa Rodrigo and the UB animal facility.
Additional information
Funding
Related Research Data
References
- Rosenberg RN, Lambracht-Washington D, Yu G, et al. Genomics of Alzheimer Disease: A Review. JAMA Neurol. 2016;73:867–874. doi:10.1001/jamaneurol.2016.0301. PMID:27135718
- Tanzi RE, Moir RD, Wagner SL. Clearance of Alzheimer's Abeta peptide: the many roads to perdition. Neuron. 2004;43:605–608. PMID:15339642
- Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science. 2010;330:1774. doi:10.1126/science.1197623. PMID:21148344
- Komatsu M, Waguri S, Chiba T, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi:10.1038/nature04723. PMID:16625205
- Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. PMID:18497889
- Majumder S, Richardson A, Strong R, et al. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One. 2011;6:e25416. doi:10.1371/journal.pone.0025416. PMID:21980451
- Spilman P, Podlutskaya N, Hart MJ, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi:10.1371/journal.pone.0009979. PMID:20376313
- Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64:113–122. doi:10.1093/jnen/64.2.113. PMID:15751225
- Cataldo AM, Peterhoff CM, Troncoso JC, et al. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol. 2000;157:277–286. doi:10.1016/S0002-9440(10)64538-5. PMID:10880397
- Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. doi:10.1016/j.cell.2010.05.008. PMID:20541250
- Neely KM, Green KN, LaFerla FM. Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a gamma-secretase-independent manner. J Neurosci. 2011;31:2781–2791. doi:10.1523/JNEUROSCI.5156-10.2010. PMID:21414900
- Coen K, Flannagan RS, Baron S, et al. Lysosomal calcium homeostasis defects, not proton pump defects, cause endo-lysosomal dysfunction in PSEN-deficient cells. J Cell Biol. 2012;198:23–35. doi:10.1083/jcb.201201076. PMID:22753898
- Lieberman AP, Puertollano R, Raben N, et al. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–730. doi:10.4161/auto.19469. PMID:22647656
- Malnar M, Hecimovic S, Mattsson N, et al. Bidirectional links between Alzheimer's disease and Niemann-Pick type C disease. Neurobiol Dis. 2014;72(Pt A):37–47. doi:10.1016/j.nbd.2014.05.033. PMID:24907492
- Sarkar S, Carroll B, Buganim Y, et al. Impaired autophagy in the lipid-storage disorder Niemann-Pick type C1 disease. Cell Rep. 2013;5:1302–1315. doi:10.1016/j.celrep.2013.10.042. PMID:24290752
- Liao G, Yao Y, Liu J, et al. Cholesterol accumulation is associated with lysosomal dysfunction and autophagic stress in Npc1 -/- mouse brain. Am J Pathol. 2007;171:962–975. doi:10.2353/ajpath.2007.070052. PMID:17631520
- Sobo K, Le Blanc I. Luyet PP, et al. Late endosomal cholesterol accumulation leads to impaired intra-endosomal trafficking. PLoS One. 2007;2:e851. doi:10.1371/journal.pone.0000851. PMID:17786222
- Fraldi A, Annunziata F, Lombardi A, et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010;29:3607–3620. doi:10.1038/emboj.2010.237. PMID:20871593
- Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. FASEB J. 2010;24:3052–3065. doi:10.1096/fj.09-144519. PMID:20375270
- Zidovetzki R, Levitan I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta. 2007;1768:1311–1324. doi:10.1016/j.bbamem.2007.03.026. PMID:17493580
- Aqul A, Liu B, Ramirez CM, et al. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J Neurosci. 2011;31:9404–9413. doi:10.1523/JNEUROSCI.1317-11.2011. PMID:21697390
- Liu B, Turley SD, Burns DK, et al. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1-/- mouse. Proc Natl Acad Sci U S A. 2009;106:2377–2382. doi:10.1073/pnas.0810895106. PMID:19171898
- Yao J, Ho D, Calingasan NY, et al. Neuroprotection by cyclodextrin in cell and mouse models of Alzheimer disease. J Exp Med. 2012;209:2501–2513. doi:10.1084/jem.20121239. PMID:23209315
- Thelen KM, Falkai P, Bayer TA, et al. Cholesterol synthesis rate in human hippocampus declines with aging. Neurosci Lett. 2006;403:15–19. doi:10.1016/j.neulet.2006.04.034. PMID:16701946
- Xiong H, Callaghan D, Jones A, et al. Cholesterol retention in Alzheimer's brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis. 2008;29:422–437. doi:10.1016/j.nbd.2007.10.005. PMID:18086530
- Lazar AN, Bich C, Panchal M, et al. Time-of-flight secondary ion mass spectrometry (TOF-SIMS) imaging reveals cholesterol overload in the cerebral cortex of Alzheimer disease patients. Acta Neuropathol. 2013;125:133–144. doi:10.1007/s00401-012-1041-1. PMID:22956244
- Heverin M, Bogdanovic N, Lutjohann D, et al. Changes in the levels of cerebral and extracerebral sterols in the brain of patients with Alzheimer's disease. J Lipid Res. 2004;45:186–193. doi:10.1194/jlr.M300320-JLR200. PMID:14523054
- Cutler RG, Kelly J, Storie K, et al.. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer's disease. Proc Natl Acad Sci U S A. 2004;101:2070–2075. doi:10.1073/pnas.0305799101. PMID:14970312
- Distl R, Meske V, Ohm TG. Tangle-bearing neurons contain more free cholesterol than adjacent tangle-free neurons. Acta Neuropathol. 2001;101:547–554. PMID:11515782
- Gomez-Ramos P, Asuncion Moran M. Ultrastructural localization of intraneuronal Abeta-peptide in Alzheimer disease brains. J Alzheimers Dis. 2007;11:53–59. doi:10.3233/JAD-2007-11109. PMID:17361035
- Solomon A, Kivipelto M, Wolozin B, et al. Midlife serum cholesterol and increased risk of Alzheimer's and vascular dementia three decades later. Dement Geriatr Cogn Disord. 2009;28:75–80. doi:10.1159/000231980. PMID:19648749
- McGuinness B, Craig D, Bullock R, et al. Statins for the treatment of dementia. Cochrane Database Syst Rev. 2014;8:CD007514. PMID:25004278
- Geifman N, Brinton RD, Kennedy RE, et al. Evidence for benefit of statins to modify cognitive decline and risk in Alzheimer's disease. Alzheimers Res Ther. 2017;9:10. doi:10.1186/s13195-017-0237-y. PMID:28212683
- Maulik M, Westaway D, Jhamandas JH, et al. Role of cholesterol in APP metabolism and its significance in Alzheimer's disease pathogenesis. Mol Neurobiol. 2013;47:37–63. doi:10.1007/s12035-012-8337-y. PMID:22983915
- Fernandez A, Llacuna L, Fernandez-Checa JC, et al. Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J Neurosci. 2009;29:6394–6405. doi:10.1523/JNEUROSCI.4909-08.2009. PMID:19458211
- Fernandez-Checa JC, Fernandez A, Morales A, et al. Oxidative stress and altered mitochondrial function in neurodegenerative diseases: lessons from mouse models. CNS Neurol Disord Drug Targets. 2010;9:439–454. doi:10.2174/187152710791556113. PMID:20522012
- Barbero-Camps E, Fernandez A, Martinez L, et al. APP/PS1 mice overexpressing SREBP-2 exhibit combined Abeta accumulation and tau pathology underlying Alzheimer's disease. Hum Mol Genet. 2013;22:3460–3476. doi:10.1093/hmg/ddt201. PMID:23648430
- Lee CY, Tse W, Smith JD, et al. Apolipoprotein E promotes beta-amyloid trafficking and degradation by modulating microglial cholesterol levels. J Biol Chem. 2012;287:2032–2044. doi:10.1074/jbc.M111.295451. PMID:22130662
- Barbero-Camps E, Fernandez A, Baulies A, et al. Endoplasmic reticulum stress mediates amyloid beta neurotoxicity via mitochondrial cholesterol trafficking. Am J Pathol. 2014;184:2066–2081. doi:10.1016/j.ajpath.2014.03.014. PMID:24815354
- Lonskaya I, Hebron ML, Desforges NM, et al. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol Med. 2013;5:1247–1262. doi:10.1002/emmm.201302771. PMID:23737459
- Lamb CA, Yoshimori T, Tooze SA. The autophagosome: origins unknown, biogenesis complex. Nat Rev Mol Cell Biol. 2013;14:759–774. doi:10.1038/nrm3696. PMID:24201109
- Alers S, Loffler AS, Wesselborg S, et al. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2–11. doi:10.1128/MCB.06159-11. PMID:22025673
- Navarro-Yepes J, Burns M, Anandhan A, et al. Oxidative stress, redox signaling, and autophagy: cell death versus survival. Antioxid Redox Signal. 2014;21:66–85. doi:10.1089/ars.2014.5837. PMID:24483238
- Ci Y, Shi K, An J, et al. ROS inhibit autophagy by downregulating ULK1 mediated by the phosphorylation of p53 in selenite-treated NB4 cells. Cell Death Dis. 2014;5:e1542. doi:10.1038/cddis.2014.506. PMID:25429619
- Filomeni G, Desideri E, Cardaci S, et al. Under the ROS...thiol network is the principal suspect for autophagy commitment. Autophagy. 2010;6:999–1005. doi:10.4161/auto.6.7.12754. PMID:20639698
- Desideri E, Filomeni G, Ciriolo MR. Glutathione participates in the modulation of starvation-induced autophagy in carcinoma cells. Autophagy. 2012;8:1769–1781. doi:10.4161/auto.22037. PMID:22964495
- Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749–1760. doi:10.1038/sj.emboj.7601623. PMID:17347651
- Yu ZQ, Ni T, Hong B, et al. Dual roles of Atg8-PE deconjugation by Atg4 in autophagy. Autophagy. 2012;8:883–892. doi:10.4161/auto.19652. PMID:22652539
- Wang Y, Nartiss Y, Steipe B, et al. ROS-induced mitochondrial depolarization initiates PARK2/PARKIN-dependent mitochondrial degradation by autophagy. Autophagy. 2012;8:1462–1476. doi:10.4161/auto.21211. PMID:22889933
- Wang X, Su B, Lee HG, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer's disease. J Neurosci. 2009;29:9090–9103. doi:10.1523/JNEUROSCI.1357-09.2009. PMID:19605646
- Chu CT, Ji J, Dagda RK, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013;15:1197–1205. doi:10.1038/ncb2837. PMID:24036476
- Nilsson P, Loganathan K, Sekiguchi M, et al. Aβ secretion and plaque formation depend on autophagy. Cell Rep. 2013;5:61–69. doi:10.1016/j.celrep.2013.08.042. PMID:24095740
- Shibutani ST, Yoshimori T. A current perspective of autophagosome biogenesis. Cell Res. 2014;24:58–68. doi:10.1038/cr.2013.159. PMID:24296784
- Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495:389–393. doi:10.1038/nature11910. PMID:23455425
- Marquer C, Laine J, Dauphinot L, et al. Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer's disease early phenotypes. Mol Neurodegener. 2014;9:60. doi:10.1186/1750-1326-9-60. PMID:25524049
- Hui L, Chen X, Geiger JD. Endolysosome involvement in LDL cholesterol-induced Alzheimer's disease-like pathology in primary cultured neurons. Life Sci. 2012;91:1159–1168. doi:10.1016/j.lfs.2012.04.039. PMID:22580286
- Djelti F, Braudeau J, Hudry E, et al. CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer's disease. Brain. 2015;138:2383–2398. doi:10.1093/brain/awv166. PMID:26141492
- Zeigerer A, Gilleron J, Bogorad RL, et al. Rab5 is necessary for the biogenesis of the endolysosomal system in vivo. Nature. 2012;485:465–470. doi:10.1038/nature11133. PMID:22622570
- Appelqvist H, Nilsson C, Garner B, et al. Attenuation of the lysosomal death pathway by lysosomal cholesterol accumulation. Am J Pathol. 2011;178:629–639. doi:10.1016/j.ajpath.2010.10.030. PMID:21281795
- Caccamo A, Magri A, Medina DX, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer's disease and other tauopathies. Aging Cell. 2013;12:370–380. doi:10.1111/acel.12057. PMID:23425014
- Kruger U, Wang Y, Kumar S, et al. Autophagic degradation of tau in primary neurons and its enhancement by trehalose. Neurobiol Aging. 2012;33:2291–2305. doi:10.1016/j.neurobiolaging.2011.11.009. PMID:22169203
- Wang Y, Martinez-Vicente M, Kruger U, et al. Tau fragmentation, aggregation and clearance: the dual role of lysosomal processing. Hum Mol Genet. 2009;18:4153–4170. doi:10.1093/hmg/ddp367. PMID:19654187
- Ozcelik S, Fraser G, Castets P, et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One. 2013;8:e62459. doi:10.1371/journal.pone.0062459. PMID:23667480
- Bednarski E, Lynch G. Cytosolic proteolysis of tau by cathepsin D in hippocampus following suppression of cathepsins B and L. J Neurochem. 1996;67:1846–1855. doi:10.1046/j.1471-4159.1996.67051846.x. PMID:8863489
- Hong W. SNAREs and traffic. Biochim Biophys Acta. 2005;1744:120–144. doi:10.1016/j.bbamcr.2005.03.014. PMID:15893389
- Wang Y, Li L, Hou C, et al. SNARE-mediated membrane fusion in autophagy. Semin Cell Dev Biol. 2016;60:97–104. doi:10.1016/j.semcdb.2016.07.009. PMID:27422330
- Enrich C, Rentero C, Hierro A, et al. Role of cholesterol in SNARE-mediated trafficking on intracellular membranes. J Cell Sci. 2015;128:1071–1081. doi:10.1242/jcs.164459. PMID:25653390
- Reverter M, Rentero C, de Muga SV, et al. Cholesterol transport from late endosomes to the Golgi regulates t-SNARE trafficking, assembly, and function. Mol Biol Cell. 2011;22:4108–4123. doi:10.1091/mbc.E11-04-0332R. PMID:22039070
- Lebrand C, Corti M, Goodson H, et al. Late endosome motility depends on lipids via the small GTPase Rab7. EMBO J. 2002;21:1289–1300. doi:10.1093/emboj/21.6.1289. PMID:11889035
- Kaushik S, Massey AC, Cuervo AM. Lysosome membrane lipid microdomains: novel regulators of chaperone-mediated autophagy. EMBO J. 2006;25:3921–3933. doi:10.1038/sj.emboj.7601283. PMID:16917501
- Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;520:563–566. doi:10.1038/nature14147. PMID:25686604
- Maulik M, Ghoshal B, Kim J, et al. Mutant human APP exacerbates pathology in a mouse model of NPC and its reversal by a beta-cyclodextrin. Hum Mol Genet. 2012;21:4857–4875. doi:10.1093/hmg/dds322. PMID:22869680
- Heeren J, Grewal T, Laatsch A, et al. Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. J Biol Chem. 2004;279:55483–55492. doi:10.1074/jbc.M409324200. PMID:15485881
- Jaeger PA, Pickford F, Sun CH, et al. Regulation of amyloid precursor protein processing by the Beclin 1 complex. PLoS One. 2010;5:e11102. doi:10.1371/journal.pone.0011102. PMID:20559548
- Levine B, Liu R, Dong X, et al. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol. 2015;25:533–544. doi:10.1016/j.tcb.2015.05.004. PMID:26071895
- Francois A, Rioux Bilan A, Quellard N, et al. Longitudinal follow-up of autophagy and inflammation in brain of APPswePS1dE9 transgenic mice. J Neuroinflammation. 2014;11:139. doi:10.1186/s12974-014-0139-x. PMID:25158693
- Qiao S, Dennis M, Song X, et al. A REDD1/TXNIP pro-oxidant complex regulates ATG4B activity to control stress-induced autophagy and sustain exercise capacity. Nat Commun. 2015;6:7014. doi:10.1038/ncomms8014. PMID:25916556
- Gao H, Yang W, Qi Z, et al. DJ-1 protects dopaminergic neurons against rotenone-induced apoptosis by enhancing ERK-dependent mitophagy. J Mol Biol. 2012;423:232–248. doi:10.1016/j.jmb.2012.06.034. PMID:22898350
- Ma S, Zhang X, Zheng L, et al. Peroxiredoxin 6 Is a Crucial Factor in the Initial Step of Mitochondrial Clearance and Is Upstream of the PINK1-Parkin Pathway. Antioxid Redox Signal. 2016;24:486–501. doi:10.1089/ars.2015.6336. PMID:26560306
- Kagan VE, Tyurina YY, Tyurin VA, et al. Cardiolipin signaling mechanisms: collapse of asymmetry and oxidation. Antioxid Redox Signal. 2015;22:1667–1680. doi:10.1089/ars.2014.6219. PMID:25566681
- Korytowski W, Basova LV, Pilat A, et al. Permeabilization of the mitochondrial outer membrane by Bax/truncated Bid (tBid) proteins as sensitized by cardiolipin hydroperoxide translocation: mechanistic implications for the intrinsic pathway of oxidative apoptosis. J Biol Chem. 2011;286:26334–26343. doi:10.1074/jbc.M110.188516. PMID:21642428
- Mari M, Colell A, Morales A, et al. Mechanism of mitochondrial glutathione-dependent hepatocellular susceptibility to TNF despite NF-kappaB activation. Gastroenterology. 2008;134:1507–1520. doi:10.1053/j.gastro.2008.01.073. PMID:18343380
- Sugiura A, McLelland GL, Fon EA, et al. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J. 2014;33:2142–2156. doi:10.15252/embj.201488104. PMID:25107473
- McLelland GL, Lee SA, McBride HM, et al. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016;214:275–291. doi:10.1083/jcb.201603105. PMID:27458136
- Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer's disease. J Cell Biol. 2005;171:87–98. doi:10.1083/jcb.200505082. PMID:16203860
- Maulik M, Peake K, Chung J, et al. APP overexpression in the absence of NPC1 exacerbates metabolism of amyloidogenic proteins of Alzheimer's disease. Hum Mol Genet. 2015;24:7132–7150. PMID:26433932
- Jin LW, Shie FS, Maezawa I, et al. Intracellular accumulation of amyloidogenic fragments of amyloid-beta precursor protein in neurons with Niemann-Pick type C defects is associated with endosomal abnormalities. Am J Pathol. 2004;164:975–985. doi:10.1016/S0002-9440(10)63185-9. PMID:14982851
- Ejlerskov P, Rasmussen I, Nielsen TT, et al. Tubulin polymerization-promoting protein (TPPP/p25alpha) promotes unconventional secretion of alpha-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem. 2013;288:17313–17335. doi:10.1074/jbc.M112.401174. PMID:23629650
- Ponpuak M, Mandell MA, Kimura T, et al. Secretory autophagy. Curr Opin Cell Biol. 2015;35:106–116. doi:10.1016/j.ceb.2015.04.016. PMID:25988755
- Murakami N, Oyama F, Gu Y, et al. Accumulation of tau in autophagic vacuoles in chloroquine myopathy. J Neuropathol Exp Neurol. 1998;57:664–673. doi:10.1097/00005072-199807000-00003. PMID:9690670
- Mohamed NV, Plouffe V, Remillard-Labrosse G, et al. Starvation and inhibition of lysosomal function increased tau secretion by primary cortical neurons. Sci Rep. 2014;4:5715. doi:10.1038/srep05715. PMID:25030297
- Wijdeven RH, Janssen H, Nahidiazar L, et al. Cholesterol and ORP1L-mediated ER contact sites control autophagosome transport and fusion with the endocytic pathway. Nat Commun. 2016;7:11808. doi:10.1038/ncomms11808.
- Rosenbaum AI, Zhang G, Warren JD, et al. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc Natl Acad Sci U S A. 2010;107:5477–5482. doi:10.1073/pnas.0914309107. PMID:20212119
- Jankowsky JL, Fadale DJ, Anderson J, et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi:10.1093/hmg/ddh019. PMID:14645205
- Horton JD, Shimomura I, Brown MS, et al. Activation of cholesterol synthesis in preference to fatty acid synthesis in liver and adipose tissue of transgenic mice overproducing sterol regulatory element-binding protein-2. J Clin Invest. 1998;101:2331–2339. doi:10.1172/JCI2961. PMID:9616204
- Mullan M, Crawford F, Axelman K, et al. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1:345–347. doi:10.1038/ng0892-345. PMID:1302033
- Xiao MF, Xu JC, Tereshchenko Y, et al. Neural cell adhesion molecule modulates dopaminergic signaling and behavior by regulating dopamine D2 receptor internalization. J Neurosci. 2009;29:14752–14763. doi:10.1523/JNEUROSCI.4860-09.2009. PMID:19940170
- Marzella L, Ahlberg J, Glaumann H. Isolation of autophagic vacuoles from rat liver: morphological and biochemical characterization. J Cell Biol. 1982;93:144–154. doi:10.1083/jcb.93.1.144. PMID:7068752
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi:10.1038/nmeth.2089. PMID:22930834