
ABSTRACT
Cystinosis is a lysosomal storage disease due to inactivating mutations in CTNS, the cystinosin transporter that exports cystine out of lysosomes. The lysosomal accumulation of cystine leads to severe dysfunction of the epithelial cells lining the proximal tubule of the kidney, causing defective endocytosis and massive losses of solutes in the urine. The mechanisms linking lysosomal defect and epithelial dysfunction were unknown, preventing the development of disease-modifying therapies. We recently reported that lysosomal alterations in cystinosis lead to defective autophagic clearance of damaged mitochondria, generating oxidative stress. The latter destabilizes tight junctions and activates an abnormal YBX3 (Y box binding protein 3) transcriptional program driving a loss of differentiation and defective apical endocytosis in cystinosis cells. Correction of the primary lysosomal defect, neutralization of mitochondrial oxidative stress, or blockage of tight junction-associated YBX3 signaling rescue epithelial function and endocytic uptake. Our findings suggest a cascade that links lysosomal disease, defective autophagy and epithelial dysfunction, providing new perspectives for cystinosis and lysosomal storage disorders.
The endolysosomal system sustains the function of specialized epithelial cells, including apical receptor-mediated endocytosis, hence their role in homeostasis. Congenital endolysosomal disorders such as cystinosis lead to generalized dysfunction of epithelial cells lining the proximal tubule (PT) of the kidney, causing the loss of essential nutrients in the urine and severe metabolic complications (renal Fanconi syndrome [RFS]).
Cystinosis is a lysosomal storage disease caused by recessive, inactivating mutations in the CTNS gene encoding the proton-driven transporter CTNS (cystinosin, lysosomal cystine transporter) that exports cystine out of lysosomes. The storage of cystine leads to multisystemic defects including kidney disease, diabetes, hypothyroidism, myopathy, and central nervous system deterioration. Cystinosis is the most frequent congenital form of RFS, often causing chronic kidney disease. The only treatment is the oral administration of cysteamine, which allows cystine to exit lysosomes; however, cysteamine has side effects and it does not prevent PT dysfunction. Thus, there is a need to identify novel therapeutic targets for this life-threatening disorder.
Studies using a ctns knockout (KO) mouse model have demonstrated that the loss of CTNS is associated with aberrations of the endolysosomal compartment, with abnormal proliferation and dysfunction of PT cells. Also, the accumulation of distorted mitochondria and of autophagy cargoes/substrates in kidney biopsies and urinary cells from cystinotic patients suggests a possible involvement of autophagy. However, a unifying mechanism linking primary lysosome disease to epithelial transport defects had not been deciphered.
As the lysosomes degrade the cellular material engulfed by phagophores (the precursors to autophagosomes), we hypothesized that cystinosis might skew epithelial function by impairing the autophagy-mediated removal of damaged cellular components. Using primary cell cultures (mPTCs) derived from PT segments of ctns KO mice, we monitored autophagy by quantifying the abundance of the autophagy substrate SQSTM1, the conversion of the non-lipidated form of LC3-I to the lipidated LC3-II and the numbers of punctate LC3 vesicles. Compared with wild-type cells, we noticed in ctns KO cells elevated numbers of punctate LC3 structures and steady-state levels of both LC3-II and SQSTM1, which did not further increase in starved conditions, and more ultrastructural structures compatible with autophagic vacuoles. Moreover, treatment with bafilomycin A1 (BfnA1), which prevents lysosome-based cellular degradation, does not change the already elevated levels of these autophagy proteins in nutrient-deprived ctns KO cells, indicating an abnormal autophagy flux in cystinosis cells. Likewise, accumulation of autophagy cargoes and/or substrates is consistently detected in the PTs of ctns KO kidneys and in cystine-accumulating pronephric tubules of a ctns KO zebrafish model. These studies suggest that cystinosin deficiency perturbs autophagy-mediated clearance both in vitro and in vivo.
One cause of impaired autophagic cargo clearance is defective lysosomal degradation capacity. To substantiate the delayed autophagosome degradation in ctns KO cells, we analyzed trafficking, processing and maturation of lysosomal cathepsins. We observed in ctns KO cells a substantial decrease in proteolytic generation of the mature CTSD (cathepsin D) and in the number of the structures labeled by BODIPY-FL-Pepstatin (PepA), a fluorescent-tagged PepA that binds to the active site of CTSD within acidic lysosomes. These cellular changes, which are also encountered in ctns KO zebrafish, are reverted by exogenously expressing functional CTNS in ctns KO cells. These results demonstrate that CTNS regulates the lysosome response to the arrival of autophagy cargoes and/or substrates in mouse and fish kidney.
The question remained how the primary lysosomal defect and the resulting impairment of autophagy might affect epithelial function in cystinosis. As defective autophagy affects mitochondrial quality control, we investigated the mitochondrial network in cystinosis cells. Correlating with the stalled autophagy, some damaged and/or dysfunctional mitochondria accumulate within enlarged, non-degradative lysosomes in ctns KO cells, overproducing mitochondrial-derived reactive oxygen species (ROS). Similarly, pharmacological or genetic inactivation of autophagy in wild-type PT cells increases damaged/dysfunctional mitochondria, promoting mitochondrial oxidative stress and epithelial dysfunction as shown by defective endocytosis. These results demonstrate the importance of the autophagy-mediated mitochondrial quality control in sustaining the reabsorptive capacity of PT cells.
The final step was to identify the signaling cascade bridging excessive mitochondrial ROS and epithelial dysfunction. Recent advances have shown the role of tight junctions in safeguarding the epithelial cell phenotype. In particular, tight junctions repress the nuclear translocation of YBX3, a transcriptional factor that promotes cell proliferation and represses PT differentiation during kidney development. Because oxidative stress may damage tight junctions, we postulated that excessive mitochondrial ROS might trigger an abnormal tight-junction associated YBX3 signaling which would, in turn, cause epithelial dysfunction in cystinosis cells. In line with this model, we found that the increased levels of mitochondrial ROS enhances GNA12/Gα12-SRC-mediated phosphorylation of TJP1/ZO–1 and its subsequent misrouting to enlarged, non-degradative endolysosomes. The disruption of tight junction integrity promotes YBX3 signaling, with increased proliferation (e.g., ccnd1, pcna) and decreased differentiation (e.g., lrp2) targets, resulting in defective endocytosis in ctns KO cells. Gain- and loss-of-function approaches targeting GNA12 or TJP1-YBX3, or pharmacological interventions impeding activation of the GNA12-SRC signaling axis (e.g., with the mitochondrial-targeted antioxidant MitoTempo or with the SRC inhibitor SU6656) restore epithelial function in ctns KO cells, supporting the biological relevance of the YBX3 signaling. Furthermore, treatment of ctns KO mice and their derived mPTCs with mitochondrial-targeted antioxidants, which are clinically tested in various mitochondrial diseases, not only repairs dysfunctional mitochondria and averts mitochondrial oxidative stress, but also rescues the integrity of tight junctions as well as cell differentiation and endocytic uptake.
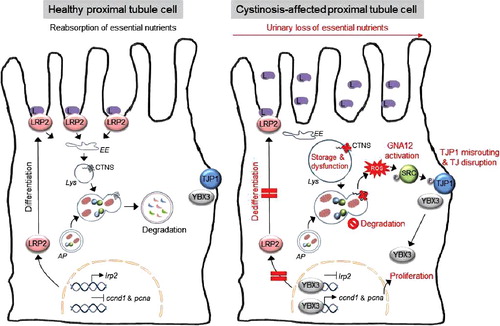
In conclusion, we identified a heretofore unknown signaling cascade linking lysosomal dysfunction, impaired clearance of mitochondria-producing ROS, abnormal tight-junction signaling and loss of transport capacity in epithelial cells (). These findings substantiate the role of lysosomes in preserving the autophagy-mediated quality control of mitochondria that are crucial for the high transport activities performed by specialized epithelial cells. The use of antioxidant compounds targeting mitochondria provides a promising therapeutic approach for treating cystinosis and endolysosome disorders.
Figure 1. A working model that depicts the pathogenic cascade linking the primary lysosomal defect to epithelial dysfunction in cystinosis. Inactivating mutations in the gene coding for the lysosomal transporter CTNS cause a lysosomal storage disease within epithelial cells lining the proximal tubule of the kidney. The ensuing lysosomal dysfunction impairs the cellular degradation of autophagosomes containing SQSTM1+ aggregates and/or damaged mitochondria, promoting the generation of reactive oxygen species (ROS). In turn, the ROS stimulate GNA12-SRC-mediated phosphorylation of the TJP1 adapter protein, resulting in its misrouting to an endolysosomal compartment. The disruption of tight junction integrity releases YBX3, which induces abnormal cell proliferation and represses apical endocytic receptors such as LRP2, causing epithelial dysfunction in cystinosis cells. L, low-molecular weight ligands; EE, early endosome; AP, autophagosome; Lys, lysosome; TJ, tight junction.
Disclosure of potential conflict of interest
The authors declare no competing financial interests.