
ABSTRACT
Macroautophagy/autophagy is a fundamental cellular degradation mechanism that maintains cell homeostasis, regulates cell signaling, and promotes cell survival. Its role in promoting tumor cell survival in stress conditions is well characterized, and makes autophagy an attractive target for cancer therapy. Emerging research indicates that autophagy also influences cancer metastasis, which is the primary cause of cancer-associated mortality. However, data demonstrate that the regulatory role of autophagy in metastasis is multifaceted, and includes both metastasis-suppressing and -promoting functions. The metastasis-suppressing functions of autophagy, in particular, have important implications for autophagy-based treatments, as inhibition of autophagy may increase the risk of metastasis. In this review, we discuss the mechanisms and context underlying the role of autophagy in metastasis, which include autophagy-mediated regulation of focal adhesion dynamics, integrin signaling and trafficking, Rho GTPase-mediated cytoskeleton remodeling, anoikis resistance, extracellular matrix remodeling, epithelial-to-mesenchymal transition signaling, and tumor-stromal cell interactions. Through this, we aim to clarify the context-dependent nature of autophagy-mediated metastasis and provide direction for further research investigating the role of autophagy in cancer metastasis.
Abbreviations
| ACTA2 | = | actin, alpha 2, smooth muscle, aorta |
| AIM2 | = | absent in melanoma 2 |
| AKT | = | AKT serine/threonine kinase |
| AMPK | = | 5’-adenosine monophosphate-activated protein kinase |
| ARHGEF2 | = | Rho/Rac guanine nucleotide exchange factor 2 |
| ARPC | = | Actin related protein 2/3 complex |
| ATF4 | = | activating transcription factor 4 |
| ATG | = | autophagy-related |
| BCL2 | = | BCL2, apoptosis regulator |
| BCL2L1 | = | BCL2 like 1 |
| BCL2L11 | = | BCL2 like 11 |
| BECN1 | = | Beclin 1 |
| BMF | = | Bcl2 modifying factor |
| BNIP3 | = | BCL2 interacting protein 3 |
| BNIP3L | = | BCL2 interacting protein 3 like |
| CAFs | = | cancer-associated fibroblasts |
| CALCOCO2 | = | calcium binding and coiled-coil domain 2 |
| CAV1 | = | caveolin 1 |
| CBLC | = | Cbl proto-oncogene C |
| CCAR2 | = | cell cycle and apoptosis regulator 2 |
| CD47 | = | CD47 molecule |
| CDC42 | = | cell division cycle 42 |
| CDH1 | = | cadherin 1 |
| CDH2 | = | cadherin 2 |
| CFLAR | = | CASP8 and FADD like apoptosis regulator |
| CLIC3 | = | chloride intracellular channel 3 |
| COL | = | collagens |
| COL6A | = | collagen type VI alpha chain |
| COL18A1 | = | collagen type XVIII alpha 1 chain |
| CTC | = | circulating tumor cell |
| CTGF | = | connective tissue growth factor |
| DCN | = | decorin |
| ECM | = | extracellular matrix |
| EGFR | = | epidermal growth factor receptor |
| EIF2A | = | eukaryotic translation initiation factor 2A |
| EIF2AK3 | = | eukaryotic translation initiation factor 2 alpha kinase 3 |
| EIF4EBP1 | = | eukaryotic translation initiation factor 4E binding protein 1 |
| EMT | = | epithelial-to-mesenchymal transition |
| ER | = | endoplasmic reticulum |
| FERMT | = | fermitin |
| FN1 | = | fibronectin 1 |
| GABARAP | = | GABA type A receptor-associated protein |
| H2O2 | = | hydrogen peroxide |
| HIF1A | = | hypoxia inducible factor 1 alpha subunit |
| HMOX1 | = | heme oxygenase 1 |
| HRE | = | hypoxia response element |
| HSPG2 | = | heparin sulfate proteoglycan 2 |
| IL1A | = | interleukin 1 alpha |
| IL18 | = | interleukin 18 |
| IL1B | = | interleukin 1 beta |
| ILVs | = | intralumenal vesicles |
| IKBKB | = | inhibitor of nuclear factor kappa B kinase subunit beta |
| IKK | = | IkB kinase complex |
| ITGA | = | integrin subunit alpha |
| ITGB | = | integrin subunit beta |
| KDR | = | kinase insert domain receptor |
| LAM | = | laminin subunit |
| LAMA2 | = | laminin subunit alpha 2 |
| LIR | = | LC3 interacting region |
| LOX | = | lysyl oxidase |
| Lyz2/LysM | = | lysozyme 2 |
| MAP1LC3/LC3 | = | microtubule associated protein 1 light chain 3 |
| MAP1S | = | microtubule associated protein 1S |
| MCTs | = | monocarboxylate transporters |
| MEFs | = | mouse embryonic fibroblasts |
| MET | = | MET proto-oncogene, receptor tyrosine kinase |
| MMH | = | MET murine hepatocyte |
| MMTV-PyMT | = | mouse mammary tumor virus-polyomavirus middle T-antigen |
| MMP | = | matrix metallopeptidase |
| MTOR | = | mechanistic target of rapamycin kinase |
| MTORC1 | = | mechanistic target of rapamycin kinase complex 1 |
| mtROS | = | mitochondrial reactive oxygen species |
| MVB | = | multivesicular body |
| NBR1 | = | NBR1, autophagy cargo receptor |
| NFKB | = | nuclear factor kappa B subunit |
| NFE2L2 | = | nuclear factor, erythroid 2 like 2 |
| NLRP3 | = | NLR family pyrin domain containing 3 |
| OCLN | = | occludin |
| OPTN | = | optineurin |
| P4HA1/2 | = | prolyl 4-hydroxlyase subunit alpha 1/2 |
| PLG | = | plasminogen |
| PLOD1 | = | procollagen-lysine,2-oxoglutarate 5-dioxygenase 1 |
| PNRC | = | perinuclear recycling complex |
| PtdIns3K | = | class III phosphatidylinositol 3-kinase |
| PTK2 | = | protein tyrosine kinase 2 |
| PXN | = | paxillin |
| RAB | = | RAB, member RAS oncogene family |
| RAC1 | = | Rac family small GTPase 1 |
| RB1CC1 | = | RB1 inducible coiled-coil 1 |
| RET | = | ret proto-oncogene |
| RHOA | = | ras homolog family member A |
| ROS | = | reactive oxygen species |
| ROCK1 | = | Rho associated coiled-coil containing protein kinase 1 |
| RUBCN | = | RUN and cysteine rich domain containing beclin 1 interacting protein |
| S100A4 | = | S100 calcium binding protein A4 |
| SERPINE1 | = | serpin family E member 1 |
| SH3GLB1 | = | SH3 domain containing GRB2 like, endophilin B1 |
| SIRT1 | = | sirtuin 1 |
| SLC16A1 | = | solute carrier family 16 member 1 |
| SLC16A4 | = | solute carrier family 16 member 4 |
| SMAD | = | SMAD family member |
| SNAI1 | = | snail family transcriptional repressor 1 |
| SNAI2 | = | snail family transcriptional repressor 2 |
| SQSTM1/p62 | = | sequestosome 1 |
| SRC | = | SRC proto-oncogene, non-receptor tyrosine kinase |
| STX17 | = | syntaxin 17 |
| TAM | = | tumor-associated macrophage |
| TBC1D14 | = | TBC1 domain family member 14 |
| TFRC | = | transferrin receptor |
| TGFB1 | = | transforming growth factor beta 1 |
| TIGAR | = | TP53 induced glycolysis regulatory phosphatase |
| TLN | = | talin |
| TLR4 | = | toll like receptor 4 |
| TME | = | tumor microenvironment |
| TWIST | = | twist family bHLH transcription factor |
| UBB | = | ubiquitin B |
| ULK1 | = | unc-51 like autophagy activating kinase 1 |
| UPR | = | unfolded protein response |
| UVRAG | = | UV radiation resistance associated |
| VIM | = | vimentin |
| WASHC1 | = | WASH complex subunit 1 |
| ZEB1 | = | zinc finger E-box binding homeobox 1 |
| ZYX | = | zyxin |
Introduction
Tumor metastasis is the primary cause of cancer-associated mortality, and effective treatments remain limited [Citation1,Citation2]. Understanding the mechanisms that regulate metastasis is critical for the development of effective therapeutic agents. Metastasis is divided into several steps [Citation1,Citation3]. First, tumor cells migrate locally and invade into surrounding tissue to gain vasculature access. Once there, invading tumor cells can intravasate through the basal membrane and detach from the extracellular matrix (ECM) to become circulating tumor cells (CTCs) [Citation4,Citation5]. CTCs must then survive harsh conditions within the bloodstream, including shear stress, ECM detachment-induced apoptosis (i.e., anoikis), and immune attack [Citation6]. Finally, CTCs extravasate from the bloodstream and engraft in secondary tissue sites that are conducive to tumor growth. To successfully metastasize, tumor cells must overcome challenges associated with each of these steps. The biological factors that influence metastasis include both intrinsic properties of the cancer cell, such as the propensity to migrate, invade, and survive using intracellular signaling pathways, as well as extrinsic properties of the microenvironment, such as the composition of the ECM, interactions with other cell types, and access to vasculature, which dictates nutrient and oxygen availability [Citation1]. Thus, a complex network that integrates both intrinsic and extrinsic factors regulates metastasis.
Autophagy is an evolutionarily conserved degradation process whereby double membrane vesicles, called autophagosomes, deliver engulfed cytoplasmic cargo to the lysosome [Citation7]. Unlike the ubiquitin-proteasome system, autophagy can engulf cellular material in bulk, allowing for the degradation of diverse cargo types that can consist of single proteins, protein aggregates, whole organelles, and energy stores, such as lipid droplets and glycogen [Citation7,Citation8]. Basal levels of autophagy maintain cellular homeostasis through the regular turnover of dysfunctional proteins and organelles [Citation8,Citation9]. However, autophagy is strongly upregulated by cellular stress and nutrient deprivation, and provides an important cell survival mechanism by facilitating the recycling of essential nutrients, preventing the accumulation of misfolded proteins and reactive oxygen species (ROS), maintaining organelle function, and regulating intracellular signaling pathways [Citation8–10].
Mechanistically, more than 30 autophagy-related (ATG) genes regulate autophagy, which is separated into 4 discrete steps: (1) the initiation of autophagosome biogenesis, (2) nucleation of the phagophore, (3) the expansion of the phagophore to a mature autophagosome, and (4) the fusion of the autophagosome to the lysosome, followed by breakdown of the cargo and efflux of the resulting macromolecules () [Citation7,Citation10,Citation11]. Inhibition of particular ATGs selectively inhibits autophagy at specific steps during autophagosome biogenesis. Furthermore, autophagy selectively targets specific types of cellular cargo for degradation using autophagy cargo receptors, which contain a binding site for the target cargo as well as a MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) interacting region (LIR) motif that mediates their binding to the phagophore [Citation7,Citation12,Citation13]. For example, autophagy cargo receptors SQSTM1/p62 (sequestosome 1) and NBR1 (NBR1, autophagy cargo receptor) promote autophagy-mediated clearance of poly-ubiquitinated aggregated proteins (i.e., aggrephagy), while BNIP3L (BCL2 interacting protein 3 like) and BNIP3 (BCL2 interacting protein 3) promote selective autophagic degradation of mitochondria (i.e., mitophagy) [Citation7,Citation12]. Thus, modulation of specific autophagy cargo receptors can dictate the type of cargo degraded through autophagy. In the context of cancer, autophagy serves as a mechanism of survival in established tumors, generating metabolic fuel and reducing oxidative stress to promote growth and metastasis [Citation14–16]. However, reports conflict as to whether autophagy is truly a metastasis-suppressing or -promoting pathway (). The role of autophagy in metastasis is multifaceted and dependent on cell type and the tumor microenvironment (TME). Specifically, autophagy regulates several mechanisms that contribute to metastasis, including: (1) focal adhesion dynamics, (2) integrin signaling and trafficking, (3) Rho GTPase-mediated cytoskeleton remodeling, (4) anoikis resistance, (5) composition of the ECM, (6) epithelial-mesenchymal transition (EMT) signaling, and (7) tumor-stromal cell interactions. Here, we discuss how autophagy regulates each of these mechanisms to clarify the context-dependent nature of autophagy in metastasis.
Figure 1. The core autophagy machinery. Autophagy-related genes regulate autophagy [Citation8,Citation10], which is divided into four distinct steps: (1) initiation of autophagy by cellular stress, (2) phagophore nucleation, (3) phagophore elongation and closure, and (4) autophagosome fusion with the lysosome for the degradation and recycling of intravesicular material. Initiation of autophagy is predominantly mediated by the ULK1 complex with multiple regulatory subunits including ATG13 and RB1CC1. This complex is negatively regulated by MTORC1 through hyper-phosphorylation of ULK1 in nutrient-rich environments, whereas AMPK binds and phosphorylates ULK1 to activate autophagy under oxygen- and nutrient-deprivation [Citation200,Citation201]. Hypoxia induces autophagy through upregulation of HIF1A, which activates autophagy through BNIP3, AMPK, and EIF2AK3 signaling. Upon activation, ULK induces phagophore nucleation by phosphorylating the BECN1 complex, which consists of BECN1, ATG14 and PtdIns3K. BECN1 acts as a nexus point between autophagy, hypoxia, endosomal, and cell death pathways [Citation202]. Several effector molecules, including BNIP3, BCL2, BCL2L1/BCL-XL, UVRAG, RUBCN, and SH3GLB1/BIF-1, modulate BECN1 to positively and negatively regulate PtdIns3K activity [Citation202,Citation203]. Activation of PtdIns3K generates phosphatidylinositol-3-phophate (PtdIns3P) on the phagophore, which subsequently recruits other autophagy-related proteins to begin the process of elongation. Phagophore elongation is carried out by 2 ubiquitin-like conjugation processes. The first involves the formation of the ATG12–ATG5-ATG16L1 complex, which is mediated by the E1-like enzyme ATG7 and the E2-like enzyme ATG10 [Citation204]. The second involves the cleavage of LC3 by the protease ATG4B to make LC3-I, which is conjugated to phosphatidylethanolamine (PE) on the growing phagophore membrane by ATG7, the E2-like enzyme ATG3, and the E3-like ATG12–ATG5-ATG16L1 complex, generating LC3-II (LC3–PE). After completion of the autophagosome, the SNARE protein STX17 facilities fusion of the autophagosome with the lysosome, generating a hybrid vesicle called an autolysosome. Hydrolytic enzymes degrade the inner autophagosome membrane and its cargo, and the degradation products are released from lysosomes and recycled into metabolic and biosynthetic pathways.
![Figure 1. The core autophagy machinery. Autophagy-related genes regulate autophagy [Citation8,Citation10], which is divided into four distinct steps: (1) initiation of autophagy by cellular stress, (2) phagophore nucleation, (3) phagophore elongation and closure, and (4) autophagosome fusion with the lysosome for the degradation and recycling of intravesicular material. Initiation of autophagy is predominantly mediated by the ULK1 complex with multiple regulatory subunits including ATG13 and RB1CC1. This complex is negatively regulated by MTORC1 through hyper-phosphorylation of ULK1 in nutrient-rich environments, whereas AMPK binds and phosphorylates ULK1 to activate autophagy under oxygen- and nutrient-deprivation [Citation200,Citation201]. Hypoxia induces autophagy through upregulation of HIF1A, which activates autophagy through BNIP3, AMPK, and EIF2AK3 signaling. Upon activation, ULK induces phagophore nucleation by phosphorylating the BECN1 complex, which consists of BECN1, ATG14 and PtdIns3K. BECN1 acts as a nexus point between autophagy, hypoxia, endosomal, and cell death pathways [Citation202]. Several effector molecules, including BNIP3, BCL2, BCL2L1/BCL-XL, UVRAG, RUBCN, and SH3GLB1/BIF-1, modulate BECN1 to positively and negatively regulate PtdIns3K activity [Citation202,Citation203]. Activation of PtdIns3K generates phosphatidylinositol-3-phophate (PtdIns3P) on the phagophore, which subsequently recruits other autophagy-related proteins to begin the process of elongation. Phagophore elongation is carried out by 2 ubiquitin-like conjugation processes. The first involves the formation of the ATG12–ATG5-ATG16L1 complex, which is mediated by the E1-like enzyme ATG7 and the E2-like enzyme ATG10 [Citation204]. The second involves the cleavage of LC3 by the protease ATG4B to make LC3-I, which is conjugated to phosphatidylethanolamine (PE) on the growing phagophore membrane by ATG7, the E2-like enzyme ATG3, and the E3-like ATG12–ATG5-ATG16L1 complex, generating LC3-II (LC3–PE). After completion of the autophagosome, the SNARE protein STX17 facilities fusion of the autophagosome with the lysosome, generating a hybrid vesicle called an autolysosome. Hydrolytic enzymes degrade the inner autophagosome membrane and its cargo, and the degradation products are released from lysosomes and recycled into metabolic and biosynthetic pathways.](/cms/asset/20bc6371-6952-48a5-af71-e95c9aa66cc1/kaup_a_1450020_f0001_c.jpg)
Figure 2. Comparison of publications reporting autophagy is a metastasis promoting [Citation27,Citation28,Citation32,Citation59,Citation83,Citation92,Citation109,Citation125,Citation166,Citation205–210] or suppressing [Citation35,Citation72,Citation75,Citation76,Citation79,Citation93,Citation167,Citation169, Citation170,Citation211–215] mechanism, or both [Citation199]. The number of papers that include an in vivo mouse model is also included. Only publications explicitly reporting whether autophagy promotes or suppresses migration or metastasis were included.
![Figure 2. Comparison of publications reporting autophagy is a metastasis promoting [Citation27,Citation28,Citation32,Citation59,Citation83,Citation92,Citation109,Citation125,Citation166,Citation205–210] or suppressing [Citation35,Citation72,Citation75,Citation76,Citation79,Citation93,Citation167,Citation169, Citation170,Citation211–215] mechanism, or both [Citation199]. The number of papers that include an in vivo mouse model is also included. Only publications explicitly reporting whether autophagy promotes or suppresses migration or metastasis were included.](/cms/asset/fa891e4b-81a8-4f49-9e2c-1962d4e06e2a/kaup_a_1450020_f0002_c.jpg)
Autophagy regulates focal adhesion dynamics and focal adhesion-associated kinases
Cell migration is critical during the early stages of metastasis, including local invasion and intravasation. Mechanistically, cell migration is characterized by a series of distinct steps [Citation17–19]. A migrating cell first establishes front-rear polarity based on chemotactic and haptotactic factors. An actin-rich leading edge is then generated at the front end of the cell, creating cytoplasmic projections termed lamellipodia and filopodia [Citation20]. Importantly, integrins present in the plasma membrane of the leading edge adhere to the ECM and mature into focal adhesion signaling complexes that stimulate cytoskeleton contractility and activate intracellular signaling cascades [Citation20,Citation21]. Finally, the cell disassembles these integrin adhesion complexes to facilitate forward movement. There are 25 known integrin heterodimers comprised of 18 ITGA/α-subunits and 8 ITGB/β-subunits, which allow particular integrins to bind specifically to different ECM ligands, including FN1 (fibronectin 1), COL/collagen, and LAM/laminin [Citation21–23]. The binding of ECM ligands to integrin heterodimers promotes tension-induced conformational changes in the integrin cytoplasmic tail, leading to the recruitment of adaptor proteins, such as TLN (talin) and PXN (paxillin) [Citation24,Citation25]. As tension increases and focal adhesions mature, PTK2 (protein tyrosine kinase 2) and SRC (SRC proto-oncogene, non-receptor tyrosine kinase) kinase are recruited, which provide the enzymatic kinase activity to promote downstream signal transduction, including Rho GTPase signaling, anoikis signaling, mitogenic signaling, and ECM turnover [Citation21]. Thus, the mechanisms that mediate integrin focal adhesion formation and disassembly are involved in both cell migration and in processes that occur throughout the metastatic cascade. Several publications recently established that autophagy regulates cell migration through selective degradation of focal adhesion proteins (A) [Citation26–28].
Figure 3. Autophagy regulates multiple metastasis-related signaling pathways. (A) Autophagy mediates the degradation of focal adhesion proteins to promote focal adhesion disassembly and migration. The autophagy protein LC3-II mediates the targeted degradation of several focal adhesion proteins, including ubiquitinated (UBB) focal adhesion (FA) proteins through NBR1, phosphorylated SRC (SRC p-Y416) through CBLC, and SRC-mediated phosphorylated PXN. (B) Autophagy negatively regulates Rho GTPases. Autophagy is activated by RHOA-ROCK signaling activity to target ARHGEF2 and RHOA for SQSTM1-dependent degradation through a negative feedback mechanism. Loss of autophagy can promote metastasis through increased RHOA activity. Autophagy and RAC negatively regulate one another, whereas CDC42 promotes autophagy. (C) Autophagy promotes anoikis-resistance. In detached cells and CTCs, autophagy is stimulated to suppress anoikis through several mechanisms, including EIF2AK3-ATF4-mediated increases in ATG gene expression, EIF2AK3-mediated suppression of MTORC1, and ROS-CCAR2-mediated IKK activation. (D) Autophagy suppresses EMT and fibrosis. EMT and fibrosis promote metastasis and exhibit mechanistic overlap. TGFB1 signals through SMAD, which promotes SNAIL- and TWIST-induced EMT and fibrosis. Autophagy negatively regulates EMT through SQSTM1-mediated degradation of SNAIL and by reducing SQSTM1-mediated stabilization of TWIST. Autophagy reduces FN1 and fibrosis by suppressing ROS to inhibit IL1B- and NFKB-induced fibrosis, and through MAP1S-dependent autophagic degradation of FN1.
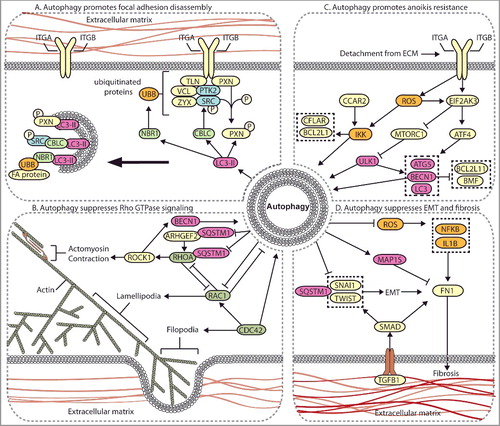
SRC kinase transduces signals from activated integrins to downstream cytoskeletal proteins, is frequently overexpressed in solid tumors, and is associated with increased metastasis [Citation29]. The autophagy-regulating proteins ATG7, ATG12, and LC3 colocalize with total-SRC and SRC p-Y416 at both focal adhesion sites and in cytoplasmic vesicles in squamous cell carcinoma cells (SCCs) [Citation26]. As these ATG proteins are involved in autophagosome formation, their colocalization with SRC suggests that SRC is targeted for autophagic degradation. Furthermore, both SRC p-Y416 and total SRC immunoprecipitate with LC3, confirming a direct connection between autophagy and focal adhesion proteins [Citation26]. Interestingly, disruption of focal adhesion formation through PTK2 knockout increases CBLC-mediated cytoplasmic colocalization of LC3 and ATG7 with SRC. Short interfering RNA (siRNA)-mediated knockdown of ATG5 and ATG12 reverses this phenotype, leading to SRC relocalization in the cell periphery within PTK2-null SCCs. Autophagic targeting of SRC also promotes degradation of RET/receptor tyrosine kinase, a focal adhesion-associated kinase that activates PTK2 [Citation30,Citation31]. Together, these data identify selective targeting of SRC p-Y416 and RET from focal adhesion sites to phagophores and provide an important link between autophagy and focal adhesion signaling. Autophagy-mediated degradation of SRC may facilitate focal adhesion disassembly to promote migration. Conversely, autophagy may reduce pro-migratory signaling by degrading SRC and RET in the absence of integrin-PTK2 signaling. However, evidence demonstrating that autophagic degradation of SRC and RET reduces migration is absent. These data demonstrate that the loss of cell adhesion promotes autophagy, targeting of SRC p-Y416 to LC3 puncta, and cell survival, suggesting that autophagy may function as an anoikis-resistance mechanism in detached cells [Citation26].
Autophagic degradation of focal adhesion proteins promotes focal adhesion disassembly and turnover, and loss of autophagy results in reduced cell migration and metastasis [Citation27,Citation28,Citation32]. Inhibition of ATG7, ATG12, or ATG5 reduces invasion and migration of 4T1, MDA-MB-231, B16-F10, and MCF10A cells [Citation27,Citation28,Citation32], and increases cell spreading [Citation27]. Knockdown of ATG7, ATG12, or ATG5 increases the size and number of focal adhesions, as assessed by PXN and ZYX (zyxin) immunofluorescent staining, while LC3-positive autophagosomes localize to focal adhesions in autophagy-competent cells [Citation27,Citation28]. Assessment of focal adhesion assembly and disassembly rates by time-lapse imaging of fluorescently-labeled PXN indicates that both assembly and disassembly are significantly reduced in autophagy-deficient cells, resulting in fewer cycles of focal adhesion assembly and disassembly [Citation27,Citation28]. However, autophagy is particularly important for focal adhesion disassembly, as disassembly rates are reduced to a greater extent compared to assembly rates in ATG7- or ATG12-deficient MCF10A-HRASG12V transformed cells [Citation27], and assembly rates are unaffected in ATG5-deficient 4T1 cells [Citation28].
The selective autophagy cargo receptor [Citation7,Citation12,Citation13] NBR1 regulates autophagosome recruitment to focal adhesions in HRAS-transformed MCF10A cells [Citation27]. Inhibition of NBR1 reduces focal adhesion turnover and cell migration, whereas other autophagy cargo receptors, including SQSTM1, OPTN (optineurin), and CALCOCO2 (calcium binding and coiled-coil domain 2) are dispensable [Citation27]. However, inhibition of NBR1 in other breast cancer cell lines has no effect on migration, suggesting that NBR1 is not required for focal adhesion disassembly in all cell types [Citation28]. Furthermore, the loss of ATG7 and ATG12 results in PXN accumulation in vitro and in syngeneic 4T1 tumors [Citation28]. This effect is also observed in 4T1, MDA-MB-231, and B16-F10 cell lines following treatment with bafilomycin A1 [Citation28], which inhibits autophagy by preventing lysosome acidification and autophagosome-lysosome fusion [Citation33]. Although increased levels of SRC p-Y416 would be expected in these autophagy-deficient cells, because data indicate that autophagy promotes active SRC degradation [Citation26], no changes in PTK2 or SRC protein levels or phosphorylation are detected in this system [Citation28]. Importantly, PXN is directly targeted to LC3-positive puncta in the cytosol and at focal adhesion sites through a LIR motif [Citation28]. LC3 binding of PXN is stimulated by constitutively active SRCY527F, suggesting that activated SRC stimulates autophagic turnover of focal adhesions. Taken together, these results indicate that autophagy targets focal adhesions and promotes disassembly through degradation of focal adhesion complex proteins, including PXN and SRC p-Y416 (A).
In addition to focal adhesion degradation, autophagy affects focal adhesion dynamics through the ULK1-RB1CC1-PTK2 signaling axis. RB1CC1/FIP200/Atg17 (RB1 inducible coiled-coil 1) directly binds to and inhibits the kinase domain of PTK2, reducing cell spreading, migration, and cell cycle progression [Citation34,Citation35]. RB1CC1 is also an essential component of the ULK1 (unc-51 like autophagy activating kinase 1) complex and is required for autophagosome formation () [Citation36]. Importantly, ULK1 activity negatively regulates PTK2 activity by mediating RB1CC1-PTK2 interactions [Citation35]. Reducing 5’-adenosine monophosphate-activated protein kinase (AMPK) and ULK1 activity dissociates RB1CC1 from PTK2, which increases PTK2 activity and promotes MDA-MB-231 cell motility [Citation35]. Consistently, overexpression of ULK1 or activated AMPK reduces metastasis in MDA-MB-231 and H460 cell xenograft models, and inactive ULK1 correlates with reduced overall survival in human non-small cell lung cancer [Citation35]. These data suggest that RB1CC1 mediates an inverse relationship between autophagy and PTK2-induced migration, where the ULK1-RB1CC1 complex sequesters and inhibits PTK2 in low-energy conditions to promote autophagy and reduce migration. Taken together, this suggests that inhibition of ULK1 or RB1CC1 will increase metastasis. However, complete knockout of RB1CC1 has also been reported to suppress autophagy, tumor initiation, tumor progression, and lung metastasis in mouse mammary tumor virus-polyomavirus middle T-antigen (MMTV-PyMT) mammary tumor mouse models [Citation37]. Although the reduced metastases reported here may be secondary to primary tumor growth defects, these data demonstrate that the role of RB1CC1 in autophagy initiation and autophagy-mediated tumor growth may counterbalance its role in the negative regulation of PTK2-mediated metastasis. Thus, further research is needed to determine the degree to which RB1CC1 mediates PTK2-induced cell migration.
Taken together, these data suggest that starvation-induced autophagy may suppress migration and metastasis through ULK1-RB1CC1-mediated inhibition of PTK2. However, basal autophagy is required for migration in normal nutrient conditions by promoting focal adhesion turnover.
Shared machinery in endosomal integrin trafficking and autophagy
The machinery that controls endosomal trafficking and integrin degradation plays a critical role in regulating focal adhesion turnover to facilitate cell migration [Citation22,Citation38,Citation39]. In addition to mediating focal adhesion assembly and disassembly, integrin trafficking contributes to migration through Rho GTPase signaling [Citation40], EGFR (epidermal growth factor receptor) signaling [Citation38,Citation41], and KDR/VEGFR (kinase insert domain receptor) [Citation42], as well as through ECM modulation via FN1 polymerization and degradation [Citation43]. Integrin trafficking, recycling, and degradation depend on several factors, including the integrin conformational status, the activation of integrins by ECM proteins, and the binding of effector proteins, such as TLNs and FERMTs/kindlins, to the integrin cytoplasmic tail to transduce “inside-out” signals [Citation22,Citation44–47]. The conformational status of an integrin heterodimer dictates the method by which it is trafficked and recycled by endocytic machinery () [Citation48]. Endocytosis of non-activated integrins into RAB5 (RAB5, member RAS oncogene family)-positive early endosomes results in rapid shuttling back to the plasma membrane via RAB4-positive endosomes, which is referred to as ‘short loop’ recycling [Citation48]. This shuttling mechanism results in localization of inactive integrins in the plasma membrane of the leading edge, which is necessary for the formation of new adhesions to drive directional migration and invasion [Citation40,Citation48,Citation49]. Conversely, active, or ligand-bound, integrins exhibit increased internalization via endocytosis compared to inactive complexes, and are recycled to the plasma membrane via a RAB11-mediated ‘long loop’ pathway that involves the perinuclear recycling complex (PNRC) [Citation48]. Active integrins are prone to reside intracellularly within the RAB11-positive PNRC, where they can be sorted for degradation, recycled back to the plasma membrane, or maintained as a reservoir that can mobilize to facilitate cell movement in response to hypoxia or certain growth factors [Citation50,Citation51].
Figure 4. Autophagy contributes to integrin recycling and degradation. Cell migration is dependent on integrin recycling, where integrins are endocytosed, recycled back to the plasma membrane, or degraded in the lysosome. Canonical integrin recycling is depicted, including short-loop integrin recycling (pink arrows), long-loop integrin recycling (blue arrows), and endosome-mediated trafficking of integrins to the lysosome or to the plasma membrane for exosome secretion (black arrows). The autophagy pathway (green arrows) converges with and contributes to endosomal integrin trafficking pathways at several points, including in the formation of amphisomes, vesicle trafficking from RAB11 recycling endosomes to growing phagophores, and autophagy-mediated delivery of integrins to lysosomes.
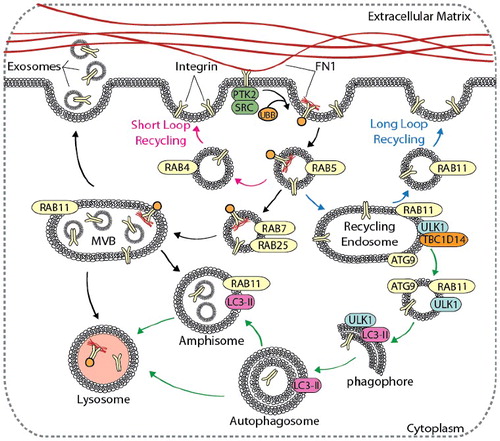
Regulation of the integrin ITGA5-ITGB1/α5β1 heterodimer highlights the complicated mechanism by which integrin trafficking facilitates cell migration. Binding of FN1 to the integrin ITGA5-ITGB1 heterodimer induces cytoplasmic tail ubiquitination, leading to clathrin-mediated endocytosis of FN1-bound integrins [Citation47,Citation52]. Endocytosed integrin-FN1 complexes first traffic to RAB25-positive late endosomes, then to the lysosome [Citation47,Citation53]. Lysosomal degradation of the integrin ITGA5-ITGB1-FN1 complex enhances cell migration, as inhibition of degradation suppresses migration [Citation47]. Highlighting the complex role of integrin recycling in cell movement, transport of integrin ITGA5-ITGB1 heterodimers from CLIC3 (chloride intracellular channel 3)-positive lysosomes back to the plasma membrane increases SRC signaling and migration, whereas the absence of CLIC3 results in integrin ITGA5-ITGB1 degradation and reduced motility [Citation53]. This suggests that integrin-mediated regulation of cell migration and invasion is dependent on endosome-mediated trafficking and integrin degradation, but may be context specific. As a degradative process, autophagy may play an important role in integrin trafficking and integrin-mediated migration. Although crosstalk between the endocytic and autophagy pathways is well-characterized [Citation54,Citation55], the role of autophagy in endosomal trafficking and lysosomal degradation of integrins is largely unknown. However, growing evidence links these pathways.
RAB11-positive recycling endosomes are a key compartment in which the autophagy and integrin recycling pathways intersect. The essential autophagy proteins ULK1 and ATG9 localize to RAB11-positive recycling endosomes and regulate delivery of membrane to expanding phagophores () [Citation56]. Here, TBC1D14 (TBC1 domain family member 14) negatively regulates trafficking of ULK1 from RAB11 recycling endosomes [Citation56]. Starvation promotes TBC1D14 dissociation from ULK1 on recycling endosomes, leading to ULK1 activation and induction of autophagy [Citation56]. Importantly, the TFRC (transferrin receptor) traffics from RAB11 recycling endosomes to phagophores under starvation conditions [Citation56]. This suggests that ULK1 activation may target integrins present within recycling endosomes for autophagic degradation in a similar fashion, and that the loss of ULK1 may enhance cell motility by promoting long-loop integrin recycling. Similarly, components of the BECN1 (beclin 1) autophagosome nucleation complex, including the class III phosphatidylinositol 3-kinase (PtdIns3K) and RUBCN (RUN and cysteine rich domain containing beclin 1 interacting protein), are present on early endosomes and may regulate integrin recycling [Citation57,Citation58]. Furthermore, localization of MET/c-MET and integrin ITGB1 on LC3-positive autophagy-related endosomes (AREs) in suspended NIH3T3 mouse embryonic fibroblasts (MEFs) promotes tumor progression and invasion in a manner dependent on ATG5 and BECN1, but not on ATG13 [Citation59]. This indicates that ATG5 and BECN1 may influence integrin recycling and metastasis through a non-canonical autophagy pathway [Citation59].
During endosomal maturation, inward budding of the limiting membrane of the late endosome leads to the formation of intralumenal vesicles (ILVs) within the endosome, forming the multivesicular body (MVB). MVBs can fuse with autophagosomes to generate hybrid vesicles, called amphisomes, which subsequently fuse with lysosomes for degradation [Citation60–62]. Induction of autophagy promotes fusion of MVBs with autophagosomes [Citation60]. Activated integrins are present within ILVs, suggesting that induction of autophagy may promote integrin degradation through amphisome formation [Citation47,Citation60]. Alternatively, fusion of the MVB with the plasma membrane leads to the release of exosomes, which are 30- to 100-nm bilayer lipid vesicles that contain a variety of protein, DNA, and RNA. Cell-to-cell communication mediated by exosome uptake is implicated in both cancer progression and metastasis [Citation63–66]. Integrins are present on the surface of secreted exosomes in a cell type-specific manner, and are involved in directed organotropic metastasis and priming of the pre-metastatic niche [Citation63–66]. Uptake of exosomes in pre-metastatic tissues increases the expression of pro-inflammatory genes, and inhibition of these integrin heterodimers decreases lung and liver metastases in vivo [Citation65]. Integrin complexes on the surface of secreted exosomes also play a critical role in directional cell motility and adhesion to the extracellular matrix [Citation67]. Secreted exosomes adhere to the ECM and stabilize leading edge cellular protrusions, facilitating directional cell migration that is inhibited in the absence of exosome secretion [Citation67]. Importantly, induction of autophagy decreases MVB fusion with the plasma membrane and reduces exosome secretion [Citation60], suggesting that the inhibition of autophagy may enhance secretion of integrin-laden exosomes to facilitate directional migration and organotropic metastasis. However, data supporting the relationship between autophagy and exosome-mediated metastasis remain limited, warranting further investigation.
Integrin recycling and autophagy in the context of starvation and hypoxia
Induction of autophagy is tightly regulated by nutritional status and cellular metabolism through MTORC1 (mechanistic target of rapamycin kinase complex 1), AMPK, and HIF1A (hypoxia inducible factor 1 alpha subunit) (). Integrin signaling is also connected to the AMPK, MTOR, and HIF1A cell metabolism and growth pathways [Citation68,Citation69]. Signaling pathways activated by integrin-ECM interactions are generally mitogenic and pro-survival [Citation68]. For example, the activation of PTK2 through interactions between integrins and the ECM induces the phosphoinositide 3-kinase-AKT-MTOR signaling pathway and suppresses autophagy induction [Citation70,Citation71]. This suggests that autophagic activity may be reduced during conditions that enhance integrin-mediated migration. In support of this, starvation-induced autophagy promotes transport of ITGB1 from the plasma membrane to autophagosomes and suppresses cell migration in HeLa cells, whereas inhibition of ATG7 promotes migration and invasion, suggesting that autophagy-mediated integrin degradation may reduce cell motility [Citation72]. However, lysosomal degradation of ITGB1 can also support migration, highlighting the complex relationship between autophagy and integrin trafficking [Citation47]. Speculatively, tumor cell heterogeneity may alter the normal coupling of these processes, such that migration is stimulated more strongly by nutrient deprivation, promoting metastasis. Further investigation into the specific mechanisms of autophagy-mediated integrin degradation will help resolve these discrepancies.
Hypoxia, which also induces autophagy, promotes invasion and metastasis of MDA-MB-231 breast cancer cells through integrin recycling [Citation50]. While nutrient starvation increases integrin internalization, hypoxia promotes recycling of specific integrins, such as integrin ITGA6-ITGB4/α6β4, from RAB11-positive endosomes to the plasma membrane [Citation50]. Interestingly, the ITGB4 subunit is associated with increased metastasis through SRC and S100A4 (S100 calcium binding protein A4) signaling, and may be regulated by autophagy [Citation65,Citation73,Citation74]. The effect of hypoxia-induced autophagy on breast cancer growth and metastasis was assessed using the hypoxia response element (HRE) to drive expression of a dominant-negative form of ULK1, allowing for reversible, hypoxia-dependent suppression of autophagy within hypoxic regions of a tumor in vivo [Citation75]. Surprisingly, targeted inhibition of ULK1 within the hypoxic tumor microenvironment enhances the metastatic potential of MDA-MB-231 breast cancer cells both in vivo and in vitro [Citation75]. Functional proteomic and transcriptome analyses reveal that the loss of hypoxia-regulated ULK1 may promote metastasis by increasing SRC activation, enhancing FN1 deposition, and increasing ITGB4 and S100A4 expression [Citation75]. Moreover, low expression of autophagy genes predicts a worse prognosis in human breast cancer [Citation75,Citation76]. Given these data, loss of autophagy in hypoxic conditions may contribute to integrin-mediated metastasis, but the mechanisms by which this occurs are unclear.
Proteins involved in hypoxia-induced autophagy may dictate whether autophagy promotes or suppresses metastasis. The loss of Bnip3, a mediator of hypoxia-induced autophagy [Citation77,Citation78], increases migration and metastasis in a MMTV-PyMT mouse model of breast cancer [Citation79]. This effect is attributed to the loss of mitophagy, a form of autophagy that selectively degrades mitochondria [Citation79]. The absence of mitophagy results in increased mitochondrial mass, ROS production, and HIF1A target gene expression [Citation79]. Furthermore, mitochondrial ROS (mtROS) production and HIF1A activity both stimulate cancer cell proliferation and motility [Citation80,Citation81], and can enhance integrin ITGB5-mediated migration, although ROS also promotes anoikis [Citation82]. Conversely, shRNA-mediated knockdown of Bnip3 in B16-F10 melanoma cells reduces cell migration and increases mitochondrial mass and ROS production in vitro [Citation83]. However, the reduced migration observed here is attributed to Bnip3-mediated alterations in vascular mimicry, the actin cytoskeleton, and focal adhesion signaling [Citation83]. Specifically, the loss of Bnip3 increases ITGA5 and PTK2 p-Y397 protein levels and localization at focal adhesion sites [Citation83]. This coincides with decreases in CD47, RAC1 (Rac family small GTPase 1), and CDC42 (cell division cycle 42) activity, indicating reduced GTPase activity. As ROS production, PTK2 signaling, and integrin expression stimulate migration [Citation80–82,Citation84], it would be interesting to determine how inhibition of Bnip3 affects melanoma metastasis in vivo.
Taken together, these data suggest that autophagy is an important regulator of integrin recycling and degradation, which affects integrin availability at the plasma membrane and focal adhesion dynamics to facilitate cell motility. Moreover, the context of nutrient deprivation and hypoxia, 2 hallmarks of the tumor microenvironment, regulate autophagy-mediated integrin recycling and metastasis in distinct ways. As integrin trafficking influences several metastasis-related mechanisms, further investigation into how autophagy regulates integrin trafficking may provide insight into the overall role of autophagy in cancer metastasis. This issue is key to understanding how microenvironmental stress induces cancer cell exit from the primary tumor.
Reciprocal regulation of autophagy and GTPase signaling
Rho GTPases are members of the Ras superfamily of proteins that regulate cytoskeletal dynamics to facilitate cell migration. Of the 23 known Rho GTPase family members, the roles of RHOA (ras homolog family member A), RAC1, and CDC42 in actin organization are the best characterized [Citation85]. Briefly, activated RAC1 on the leading edge promotes lamellipodia formation, which facilitates cell spreading and focal adhesion formation [Citation17,Citation86]. Similarly, CDC42 activation at the leading edge promotes the formation of actin-rich filopodia, which protrude out from the leading edge and provide an exploratory function [Citation17,Citation86,Citation87]. Conversely, activated RHOA is typically present in the trailing edge of the cell, where it promotes stress-fiber formation and actomyosin contractility through ROCK1/2 (Rho associated coiled-coil containing protein kinase 1/2) signaling [Citation86,Citation88]. Coordination between actin polymerization, actomyosin contraction, and cytoskeletal turnover is necessary for efficient cell motility. An inverse relationship between RAC1 and RHOA activity balances actin polymerization with contraction [Citation89,Citation90].
Autophagy contributes to the coordination between RAC1, RHOA, and CDC42 signaling (B). In renal and lung cancer cells, SQSTM1 promotes autophagic degradation of RHOA [Citation91]. Here, loss of ATG5 increases RHOA levels, resulting in chromosomal instability, aneuploidy, and enhanced motility [Citation91,Citation92]. Consistently, high RHOA expression positively correlates with autophagy defects in lung carcinoma, suggesting that the loss of autophagy may promote lung cancer progression through RHOA dysregulation [Citation91]. The loss of ATG7, ATG5, or ULK1 increases RHOA activity in MEFs, resulting in increased migration [Citation93]. Mechanistically, autophagy reduces RHOA activity via SQSTM1-dependent degradation of an upstream RHO guanine nucleotide exchange factor, ARHGEF2 (Rho/Rac guanine nucleotide exchange factor 2) [Citation93]. Furthermore, knockdown of RHOA by siRNA in HeLa cells decreases starvation-induced autophagosome formation, which is dependent on ROCK1 kinase activity [Citation94]. Under starvation, activated ROCK1 promotes autophagy by binding to and phosphorylating BECN1 to prevent BECN1-BCL2L1 interaction [Citation95]. This demonstrates a negative feedback mechanism where RHOA and ROCK1 regulate BECN1-mediated autophagy. RHOA and ROCK1 expression is also associated with increased migration and metastasis, particularly in hypoxia [Citation96,Citation97]. Therefore, RHOA- and ROCK1-induced autophagy negatively regulates RHOA and ARHGEF2, preventing chromosomal instability, tumor progression, and metastasis. The Rho GTPase CDC42 also positively regulates autophagy induction, as CDC42 inhibition reduces autophagosome formation and autophagy-mediated delivery of cargo to the lysosome in Huh7 hepatocellular carcinoma cells (HCCs) [Citation98]. However, research demonstrating how autophagy regulates metastasis through CDC42 is absent.
Conversely, RAC1 has an inhibitory effect on autophagy, as its depletion activates autophagy [Citation94]. Expression of active RAC1 interferes with basal and starvation-induced autophagic flux by directly binding to LC3 [Citation99], and inhibition of RAC1 through treatment with simvastatin increases autophagy in coronary arterial myocytes [Citation100]. Similarly, knockdown of RAC3, which is involved in the formation of actin networks, induces autophagy in multiple cell lines [Citation101]. Autophagy also negatively regulates RAC1 activity, as starvation-induced autophagy inactivates RAC1 in human keratinocytes [Citation99]. Moreover, autophagy-deficiency increases lamellipodia formation and cell spreading in several breast cancer cell types [Citation27,Citation75], suggesting that autophagy may decrease lamellipodia formation by inactivating RAC1. However, more research is needed to fully elucidate how RAC1 coordinates with the autophagy pathway to influence cell motility.
Taken together, these results indicate that Rho GTPase activity and autophagy are tightly linked and that their reciprocal regulation generates a feedback network (B). Evidence demonstrates that autophagy may suppress metastasis through targeted RHOA and ARHGEF2 degradation. However, the complex spatial and temporal effects of GTPases on migration make it difficult to conclude how autophagy regulates Rho GTPase-mediated migration. Furthermore, tumor cells exhibit different modes of cell movement, including mesenchymal- and amoeboid-type movement, which depend on the interplay between RHOA and RAC activity [Citation90]. Therefore, autophagy-mediated Rho GTPase dysregulation may affect different modes of tumor cell movement. Several other GTPase-related proteins, including ARPC (Actin related protein 2/3 complex) and WASHC1 (WASH complex subunit 1), are also involved in the formation and trafficking of autophagosomes [Citation102]. More research is necessary to fully elucidate the mechanisms by which Rho GTPases and their effector proteins coordinate with the autophagy pathway to regulate cell movement and metastasis.
Autophagy and anoikis resistance
One of the most recognized relationships between autophagy and integrin signaling involves resistance to anoikis, a form of apoptotic cell death that occurs when cells experience prolonged detachment from the ECM [Citation103–105]. Integrin-mediated attachment and autophagy demonstrate an inverse relationship, where the loss of integrin-ECM adhesions induces autophagy in both cancerous and noncancerous epithelial cells to promote anoikis resistance, which enables survival of CTCs and may enhance metastatic potential [Citation59,Citation106–109].
Several mechanisms regulate detachment-induced autophagy, including those involving EIF2AK3/PERK (eukaryotic translation initiation factor 2 alpha kinase 3). EIF2AK3 is involved in the prosurvival ER unfolded protein response (UPR) [Citation110], by reducing protein translation through EIF2A/eIF2α phosphorylation [Citation111] and activating an NFE2L2 (nuclear factor, erythroid 2 like 2)-mediated antioxidant response [Citation112]. EIF2AK3 regulates the transcription factors ATF4 (activating transcription factor 4) and DDIT3 (DNA damage inducible transcript 3), which induce the autophagy genes LC3 and ATG5 in response to hypoxia (C) [Citation113,Citation114]. Mammary cell detachment induces autophagy and BECN1, ATG5, and LC3 expression through the EIF2AK3-ROS-ATF4 signaling axis, which promotes anoikis resistance [Citation107]. EIF2AK3 can also promote autophagy-mediated anoikis resistance in detached cells through activation of AMPK and inhibition of MTORC1-SERPINE1 (serpin family E member 1) signaling [Citation107]. Taken together, EIF2AK3 induces autophagy following ECM detachment through ATF4-mediated upregulation of ATG5, BECN1, and LC3, as well as through MTORC1 and AMPK signaling. This supports a role of autophagy as a prosurvival mechanism in detached cells.
Interestingly, stimulation of MTORC1 during mammary epithelial cell (MEC) detachment fails to suppress autophagy, indicating that detachment-induced autophagy can be induced in an MTORC1-AMPK-independent manner [Citation115]. Alternatively, loss of ECM adhesion promotes autophagy through the IKK complex, as IKK inhibition suppresses autophagy and promotes anoikis in 3D culture models [Citation115]. Similarly, antibody-mediated inhibition of ITGB1 and ITGA3 in MECs induces IKK activation, autophagy, and anoikis resistance [Citation115]. This demonstrates a direct connection between integrin-mediated adhesion and autophagy induction. This effect is not observed upon inhibition of ITGA6 or ITGB4, indicating that the ITGA3-ITGB1/α3β1 complex may be particularly important for regulating ECM detachment-induced autophagy in MECs. A novel cofactor for IKBKB (inhibitor of nuclear factor kappa B kinase subunit beta), CCAR2/DBC1 (cell cycle and apoptosis regulator 2), protects MECs from anoikis [Citation116]. Interestingly, CCAR2 inhibits SIRT1 (sirtuin 1), a major regulator of autophagy, suggesting that CCAR2 may be a novel regulator of autophagy-mediated anoikis resistance [Citation117].
The mechanisms by which autophagy suppresses anoikis remain poorly characterized. As CTCs are not deprived of nutrients or oxygen in circulation, the contribution of autophagy to CTC survival may be independent of autophagy's typical role as a nutrient recycling mechanism. Instead, autophagy-mediated anoikis resistance is primarily attributed to its regulation of apoptosis-inducing BCL2 family proteins. In attached cells, the pro-apoptotic BH3-only protein BMF (Bcl2 modifying factor) localizes to actin filaments via interaction with MYO5/myosin V motors, and stabilizes the interaction between BECN1 and BCL2 to inhibit autophagy [Citation118–120]. Upon cell detachment, BMF detaches from the cytoskeleton, which releases BECN1 and destabilizes the BECN1-BCL2 interaction, promoting autophagy [Citation119,Citation121]. Similarly, under nutrient-rich conditions, the pro-apoptotic protein BCL2L11/BIM (BCL2 like 11) inhibits autophagy by sequestering BECN1 to microtubules, whereas BCL2L11 and BECN1 dissociate and induce autophagy during starvation [Citation118]. BCL2L11 activation and localization to the mitochondria is critical for inducing anoikis [Citation122–124], an effect that is inhibited following binding to BECN1, indicating that the BCL2L11-BECN1 interaction may contribute to both autophagy modulation and anoikis induction. These data suggest that interactions between BCL2 family proteins and BECN1 act as molecular switches between anoikis and autophagy-mediated survival. However, the precise mechanism by which autophagy promotes anoikis resistance remains unclear.
Although evidence demonstrates a pro-survival role of autophagy following ECM detachment in vitro, there are limited data demonstrating that this significantly affects metastasis in vivo. Suppression of autophagy through shRNA-mediated knockdown of BECN1 or ATG5 in HCCLM3 and HCCLM3-R HCC cell lines mildly reduces pulmonary metastasis in an orthotopic mouse model, as quantified by histological scoring of lung tissue [Citation109]. Inhibition of autophagy in these cell lines does not affect cell migration, invasion, or EMT marker expression in vitro. Instead, the observed decrease in metastasis is attributed to an increase in anoikis, as BECN1 and ATG5 deficiency increases apoptosis in HCC cells in suspension [Citation109]. However, a subsequent study obscures these findings. Here, immunohistochemical analysis of human and mouse HCC tissues demonstrates increased LC3 expression in metastatic tumors compared to primary tumors, suggesting higher levels of autophagic activity at the metastatic site [Citation125]. However, autophagy induction is not detected during cell detachment and dissemination in HCCLM3 cells in vitro, as quantified through LC3 lipidation and GFP-LC3 puncta formation [Citation125]. Furthermore, no alterations in autophagy are demonstrated during dynamic monitoring of cell migration and invasion [Citation125]. These data suggest that autophagy may contribute to metastatic colonization in this liver cancer model through mechanisms independent of cell invasion, migration, and anoikis resistance. However, further analysis of autophagic activity during cell detachment using established autophagy flux assays [Citation126] would provide stronger evidence that autophagy does not contribute to anoikis resistance in HCC cell lines. Taken together, strong in vitro data suggest that autophagy likely promotes CTC survival and metastasis through anoikis resistance. However, the contribution of autophagy-mediated anoikis resistance in cancer metastasis warrants further investigation.
Reciprocal regulation between autophagy and the ECM
The ECM provides the structural framework for cancer cell adherence, proliferation, and migration. Although there are many ECM components [Citation127], FN1 is an abundant and important ECM protein that is organized into fibrillar networks and mediates tumor rigidity, vascularity, and metastasis [Citation128–130]. FN1 is secreted from different cell types as a 230- to 270-kDa disulfide-bonded dimer that is assembled into a fibrotic network through interactions with cell surface receptors, particularly the ITGA5-ITGB1 heterodimer [Citation131–133]. To maintain homeostasis, components of the ECM are continuously turned over and remodeled through several mechanisms, including MMP/matrix metalloproteinase-dependent cleavage, integrin-mediated endocytosis, and lysosomal degradation [Citation39,Citation43,Citation47,Citation134]. Interestingly, RAB11B-mediated integrin ITGA5-ITGB1 internalization and recycling not only degrades FN1, but can also promote fibrillogenesis by facilitating FN1 secretion and vascular morphogenesis [Citation135]. Thus, FN1 matrix assembly is a cell-dependent process that is regulated by secretory and integrin recycling pathways, as well as by integrin surface expression. Importantly, dysregulation of FN1 signaling results in fibrosis and increased tumor stiffness, which is associated with increased cell invasion and metastasis [Citation75,Citation136–143]. Thus, understanding how autophagy regulates the ECM and fibrosis will provide important insight into the role of autophagy in metastasis.
Growing evidence indicates that autophagy significantly affects ECM composition and can reduce tumor fibrosis. Autophagy reduces FN1 levels and dampens TGFB1 (transforming growth factor beta 1)-induced fibrosis, whereas siRNA-mediated inhibition of either BECN1 or LC3 promotes TGFB1-induced FN1 expression [Citation144]. TGFB1 treatment activates MTOR signaling to inhibit autophagy in human lung fibroblasts [Citation144]. Similarly, components of the MTOR signaling pathway, including PTK2, AKT, MTOR, and EIF4EBP1 (eukaryotic translation initiation factor 4E binding protein 1), are activated in FN1-treated gallbladder cancer cells, which promotes proliferation and metastasis [Citation142]. Treatment with rapamycin, an MTOR inhibitor, reverses this metastatic phenotype, suggesting that induction of autophagy may reduce fibrosis-associated metastasis. Moreover, nutrient depletion promotes endocytosis and degradation of ligand-engaged ITGA5-ITGB1 integrins from subnuclear regions [Citation69], indicating that starvation induces both autophagy and ECM degradation. Similarly, inhibition of ULK1 in the hypoxic TME promotes tumor fibrosis and metastases in vivo, indicating that the loss of autophagy promotes fibrosis and metastasis under stress conditions [Citation75]. In hepatocarcinogenesis, a mtROS-NFKB-IL1A/B mechanism promotes fibrosis in ATG5-deficient Kupffer cells [Citation145]. A similar effect is observed in ATG5-deficient macrophages, which induce liver fibrosis in a ROS-IL1A/B-dependent manner [Citation146]. Activation of inflammatory pathways resulting from autophagy deficiency may drive fibrosis, and hypoxia may exacerbate this phenotype [Citation75,Citation147].
Autophagy regulates fibrosis through other mechanisms in addition to inflammation. LC3 positively regulates FN1 mRNA translation by facilitating its sorting and trafficking to the rough ER [Citation148]. Overexpression of LC3 in MEFs significantly increases FN1 protein levels without altering the level of FN1 mRNA [Citation149]. LC3 overexpression also promotes FN1 degradation, demonstrating that LC3 fine-tunes total FN1 levels [Citation149]. Inhibition of the autophagy-related protein MAP1S (microtubule associated protein 1S), which interacts with LC3 and positively regulates autophagosome biogenesis, reduces FN1 degradation in HCC cells [Citation150]. Loss of MAP1S decreases FN1 degradation, resulting in increased FN1 protein levels, organ fibrosis, and HCC tumorigenesis in vivo [Citation149,Citation151,Citation152]. Similarly, increased FN1 levels are correlated with decreased MAP1S expression in patient samples of renal fibrosis [Citation152]. Although MAP1S promotes FN1 turnover, the role of autophagy in this process is not defined. Inhibition of canonical autophagy genes, such as ATG5, ATG7, BECN1, or ULK1, within this model would provide stronger evidence that autophagy regulates FN1 turnover.
Finally, the ECM regulates autophagy through outside-in signaling independent of nutrient status [Citation153]. Matrix-derived molecules, including DCN (decorin), COL6A/collagen VI, LAMA2/laminin α2, COL18A1/endostatin, the endorepellin fragment of HSPG2, HSPG2/perlecan, and the kringle V fragment of PLG, can positively and negatively regulate autophagy [Citation153,Citation154]. How these interactions affect metastasis is not well characterized, further demonstrating the complex interplay between autophagy and the ECM. Similarly, the relationship between autophagy and key ECM remodeling proteins, such as LOX (lysyl oxidase), P4HA1 (prolyl 4-hydroxlyase subunit alpha 1), and PLOD1 (procollagen-lysine,2-oxoglutarate 5-dioxygenase 1), is not well understood. Generally, increased expression of these genes results in increased tumor stiffness, metastatic niche formation, and metastasis, particularly in hypoxic conditions [Citation155–158]. How autophagy regulates these ECM remodeling molecules under stress conditions is unknown, and research into this area may provide new insight into how autophagy regulates metastasis.
Autophagy regulates EMT
EMT is a process whereby tumor cells are reprogrammed from an epithelial phenotype to a mobile and invasive mesenchymal-like phenotype through rearrangement of the cytoskeleton, loss of cell junctions, resistance to anoikis, and remodeling of the ECM [Citation159–162]. EMT is a dynamic process subject to transitional states and microenvironmental influence, and no single signaling pathway governs this transition. Mechanisms known to regulate EMT include transcriptional control, epigenetic modification, alternative splicing, miRNAs, protein stability, and subcellular localization, and autophagy modulates several of these factors [Citation160,Citation163].
A recent study investigating the role of autophagy in hepatocyte EMT found that liver-specific knockout of Atg7 (Alb-Cre;Atg7fl/fl) increases mesenchymal markers, particularly VIM (vimentin) and SNAI1 (snail family transcriptional repressor 1) [Citation164]. Furthermore, siRNA-mediated suppression of both BECN1 and ATG7 in MET murine hepatocytes (MMHs) decreases expression of the epithelial markers CDH1/e-cadherin and OCLN (occludin), and increases expression of the mesenchymal markers VIM, FN1, ACTA2 (actin, alpha 2, smooth muscle, aorta), MMP9, and SNAI1 [Citation164]. Treatment of wild-type MMH hepatocytes with TGFB1 suppresses autophagy, whereas starvation-induced autophagy inhibits TGFB1-mediated EMT [Citation164]. Importantly, SNAI1 is degraded through SQSTM1-dependent autophagy, demonstrating a direct mechanism of autophagy-mediated regulation of EMT [Citation164]. In support of this, autophagy-mediated degradation of SNAI1 reduces hypoxia-induced EMT in human cardiac microvascular endothelial cells (HCMECs) [Citation165]. In contrast, starvation-induced autophagy activates EMT and is required for HepG2 and BEL7402 HCC cell invasion in vitro [Citation166]. Knockdown of ATG7 or ATG3 in these cells suppresses EMT and invasion, and decreases expression of FN1, TGFB1, and activated SMAD3 (SMAD family member 3). However, EMT and invasion increase dramatically following treatment of these autophagy-deficient HCC cells with exogenous TGFB1, suggesting that the presence of TGFB1 may regulate whether autophagy promotes or suppresses EMT in liver tissues.
Recent findings establish that SQSTM1 induces EMT by stabilizing the transcription factor TWIST (twist family bHLH transcription factor) [Citation167,Citation168]. Loss of autophagy increases TWIST in a SQSTM1-dependent manner, in which SQSTM1 binds directly to TWIST to prevent its proteasomal degradation and to promote EMT-mediated cell migration and metastasis in vivo [Citation167]. A subsequent study confirms these findings, as accumulated SQSTM1 stabilizes TWIST and activates TGFB1-SMAD signaling, which promotes EMT-associated junction remodeling [Citation168]. These data provide a strong link between autophagy and EMT, and demonstrate a direct mechanism by which autophagy-deficiency induces EMT and metastasis.
In glioblastoma cells, starvation- or rapamycin-induced autophagy reduces cell migration and invasion, and downregulates SNAI2 and SNAI1 [Citation169]. Silencing of BECN1, ATG7, or ATG5 in these cells enhances migration and invasion, suggesting that autophagy may suppress EMT-mediated metastasis in glioblastoma [Citation169]. In papillary thyroid carcinoma cells (PTCs), inhibition of CDH6 (cadherin 6) suppresses autophagy to promote EMT and metastasis through direct interaction with the autophagy proteins BNIP3, BNIP3L, and GABARAP (GABA type A receptor-associated protein) [Citation170]. In Skov-3 ovarian cancer cells, low basal autophagy levels correlate with an increased propensity for migration and invasion compared to cells with high basal autophagy [Citation171]. Furthermore, starvation-induced autophagy reduces migration, invasion, and expression of the mesenchymal markers VIM, CDH2/n-cadherin, and ZEB1 (zinc finger E-box binding homeobox 1), while the opposite effect is observed following siRNA-mediated knockdown of ATG7 [Citation171]. Increased levels of ROS and HMOX1 (heme oxygenase 1) regulate the EMT phenotype in these cells, demonstrating a role for autophagy in the ROS-HMOX1-EMT signaling axis [Citation171]. Taken together, these data indicate that autophagy suppresses EMT-induced metastasis in most contexts through reduced TGFB1-SMAD signaling, SQSTM1-dependent degradation of SNAI1, and SQSTM1-mediated stabilization of TWIST (D).
Autophagy in tumor-associated stromal cells
The tumor microenvironment is composed of a variety of stromal cells, including cancer-associated fibroblasts (CAFs) and tumor-associated macrophages (TAMs). Crosstalk between tumor and stromal cells mediated by secreted ECM proteases, metabolic fuel, and growth factors influences tumor growth and metastatic potential [Citation172,Citation173]. As autophagic activity within stromal cells affects tumor growth, survival, and metastasis, consideration of autophagy-mediated crosstalk between the tumor and stroma is necessary.
Autophagy is involved in metabolic coupling within stromal cells, a process by which catabolic CAFs generate metabolites, such as lactate and ketones, that fuel anabolic tumor cell growth and survival () [Citation172,Citation174,Citation175]. Overexpression of BNIP3 or ATG16L1 in human fibroblasts enhances autophagic flux, increasing L-lactate and ketone body production [Citation176]. Co-injection of ATG16L1- or BNIP3-overexpressing fibroblasts and MDA-MB-231 breast cancer cells into the circulation of nude mice results in increased lung colonization compared to co-injections with empty-vector fibroblasts [Citation176], indicating that autophagic activity in CAFs promotes tumor growth and metastasis. Conversely, overexpression of ATG16L1 in MDA-MB-231 cells inhibits tumor growth in nude mice [Citation176], indicating that the role of autophagy in tumor progression is compartment specific. This metabolic synergy between tumor and stroma is exemplified by the compartment-specific expression of monocarboxylate transporters (MCTs), which are key transporters of monocarboxylates, such as ketones and lactate. Here, stromal cells express higher levels of the lactate exporter, SLC16A4, while carcinoma cells exhibit higher expression of the primary lactate and ketone importer, SLC16A1, suggesting that CAFs export monocarboxylate metabolites for uptake by SLC16A1-expressing cancer cells [Citation172,Citation177]. Oxidative stress within the TME drives autophagy-mediated metabolic coupling, as ROS, cancer cell-derived H2O2, and hypoxia stimulate autophagy, glycolysis, and SLC16A4 expression in CAFs through HIF1A and NFKB signaling, promoting tumor growth and metastasis in MDA-MB-231 xenograft models [Citation177–180]. TGFB1 and its downstream target gene, CTGF (connective tissue growth factor), also promote autophagy and catabolic activity in stromal cells, which drives metabolic coupling and tumor growth of MDA-MB-231 xenografts [Citation181,Citation182]. Interestingly, TGFB1- and HIF1A-induced autophagy in MDA-MB-231 tumor cells suppresses xenograft tumor growth, further demonstrating that autophagy may play different roles within the stromal and tumor compartments [Citation179,Citation182].
Figure 5. Autophagy compartmentalization promotes metabolic coupling between cancer cells and cancer-associated fibroblasts (CAFs). Autophagy-mediated metabolic coupling between cancer cells and stromal cells promotes tumor cell growth and metastasis. Loss of CAV1 promotes autophagy and catabolic metabolism through increased NFKB and HIF1A signaling, generating metabolic fuel, such as ketones and lactate, which are used to drive anabolism. Metabolite transport is mediated by increased expression of SLC16A4 and SLC16A1 on CAFs and cancer cells, respectively. Autophagy-mediated metabolic coupling suppresses apoptosis in cancer cells through increased TIGAR expression. CAV1 also promotes fibrosis by increasing SERPINE1 and FN1 expression.
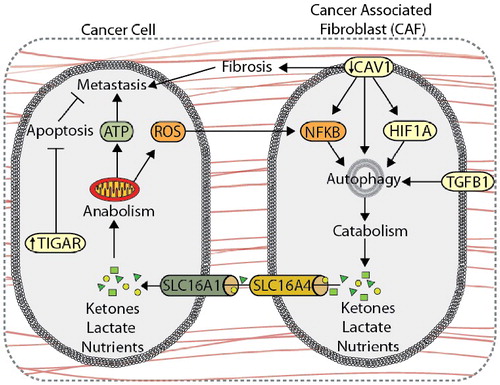
CAV1 (caveolin 1), a plasma membrane lipid raft protein that is degraded through autophagy, is a key marker of tumor-stroma compartmentalization. Reduced CAV1 expression in the stroma promotes autophagy and metabolic compartmentalization, and is associated with poor patient outcome and metastasis in breast cancer [Citation174,Citation183–186]. In addition to providing nutrients, induction of autophagy via CAV1 deletion in CAFs protects against tumor cell apoptosis through upregulation of TIGAR (TP53 induced glycolysis regulatory phosphatase) in adjacent cancer cells [Citation178]. Moreover, the loss of CAV1 in CAFs increases expression of SERPINE1 and FN1, which promotes tumor fibrosis and metastasis [Citation187].
Recruited TAMs contribute to tumor cell invasion and intravasation by promoting extracellular remodeling, angiogenesis, and inflammation [Citation188]. Macrophage-specific autophagy regulates the secretion of inflammatory cytokines, such IL1B and IL18, and enhances the processing and presentation of tumor antigens [Citation189,Citation190]. For example, IL1B is reduced in human monocytes following autophagic degradation of AIM2 (absent in melanoma 2) and NLRP3 (NLR family pyrin domain containing 3) inflammasomes, which are protein oligomers involved in interleukin maturation [Citation189]. Deletion of ATG16L1 in macrophages potentiates TLR4 (toll like receptor 4) induced inflammasome activation and IL1B production [Citation191]. Moreover, mitophagy reduces inflammasome activation by decreasing cellular ROS and free mitochondrial DNA [Citation192–194]. Autophagy in TAMs may suppress tumor-promoting inflammation and enhance immune surveillance to reduce tumor growth. However, evidence demonstrating the role of TAM-specific autophagy in cancer metastasis is limited.
Recently, B16-F10 melanoma tumor metastasis was found to increase, independent of primary tumor growth, in Lyz2/LysM-Cre/Atg5flox/flox mice with a myeloid cell-specific targeted Atg5 deletion [Citation195]. The loss of Atg5 in myeloid cells also suppresses TGFB1 secretion and reduces EMT in adjacent MC38 colon cancer cells [Citation195]. While these data suggest that myeloid-specific autophagy promotes tumor metastasis, the role of TAM-specific autophagy remains limited and warrants further investigation.
The regulatory role of autophagy in cancer metastasis differs across cell types within the TME, and cellular compartmentalization should be considered when developing autophagy-based cancer therapeutics. Autophagic compartmentalization between different cell populations within a tumor may also stimulate tumor cell cooperation to promote metastasis. Future research should incorporate compartment and lineage-specific autophagy knockouts to differentiate the discrete roles of autophagy across cell types in the TME. Immunocompetent models of in vivo metastasis should also be used to investigate interactions between tumor, stromal, and immune cells.
Concluding remarks and future directions
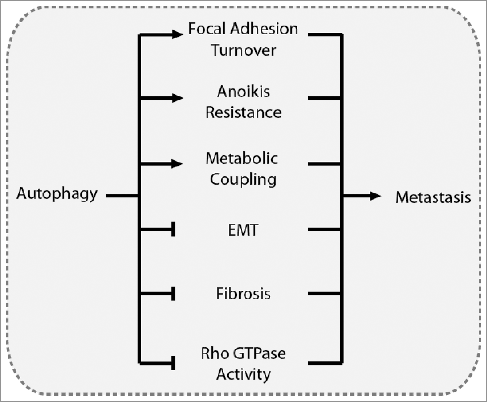
Although the mechanism by which autophagy promotes or suppresses metastasis is context-specific, several themes emerged in this review that provide insight into how autophagy regulates metastasis. Prominently, coordination between autophagy and integrin signaling influences several metastasis-related mechanisms, including focal adhesion dynamics and signaling, anoikis resistance, and ECM remodeling, among others. Due to the involvement of integrins throughout the metastatic cascade, further elucidation of how autophagy regulates integrin activity may clarify the role of autophagy in metastasis. Autophagy contributes to focal adhesion formation and disassembly through targeted degradation of focal adhesion proteins, as well as through ULK1-RB1CC1-PTK2 interactions. Under normal nutrient conditions, basal autophagy supports focal adhesion turnover, which is necessary for cell spreading and migration. However, starvation-induced autophagy may suppress migration and metastasis through ULK1-RB1CC1-mediated inhibition of PTK2. Autophagy promotes anoikis resistance, although further research is needed to better define the underlying mechanisms. Autophagy also suppresses EMT-induced metastasis through SQSTM1-mediated degradation of SNAI1, reduced SQSTM1-mediated stabilization of TWIST, and reduced TGFB1-SMAD signaling. Autophagy suppresses metastasis by reducing FN1 levels and tumor fibrosis in many tissue types, and by negatively regulating Rho GTPase activity. Despite conflicting conclusions, several publications report increased cell spreading and lamellipodia formation in autophagy-deficient cells, suggesting that autophagy may also be an important regulator of RAC activity. Finally, autophagic activity in stromal cells often supports tumor cell growth and metastasis, indicating that autophagy can have a metastasis-promoting role in heterogeneous cell populations. Collectively, the role of autophagy in metastasis is multifaceted: autophagy suppresses metastasis by preventing EMT, fibrosis, and Rho GTPase activity, and promotes metastasis by contributing to focal adhesion turnover, anoikis resistance, and tumor-stromal metabolic coupling ().
Figure 6. Overview of the driving mechanisms underlying the role of autophagy in metastasis. Autophagy can promote metastasis by contributing to focal adhesion turnover, anoikis resistance, and metabolic coupling with the tumor stroma. Conversely, autophagy can suppress metastasis by inhibiting EMT, tumor fibrosis, and Rho GTPase activity.
The question remains as to which of these mechanisms most critically dictates the role of autophagy in metastasis. Unfortunately, the weight of each autophagy-regulated mechanism in cancer metastasis remains unclear. Autophagy-mediated focal adhesion turnover enhances migration, particularly in 2D culture models. Together with the established role of autophagy as a nutrient recycling pathway, this indicates that autophagy can promote metastasis by increasing cell migration and promoting tumor cell survival. However, the intricacy of the physiological tumor microenvironment obscures this inference, as autophagy suppresses metastasis under conditions that include hypoxia, nutrient deprivation, altered ECM dynamics, and enhanced EMT signaling. Furthermore, specific genetic alterations may influence the role of autophagy in cancer metastasis. For instance, cancer cells with overactive RAS are heavily dependent on autophagy for growth [Citation196,Citation197], and autophagy facilities oncogenic RAS-driven invasion [Citation32]. Although speculative, this suggests that autophagy promotes metastasis in cells with activating RAS mutations. Further investigation is warranted to determine how specific genetic mutations influence the role of autophagy in metastasis
In future research, several experimental factors should be considered to further clarify the role of autophagy in metastasis. First, attention should be given to which autophagy genes are inhibited when generating autophagy-deficient cell lines. Inhibition of ATG genes involved in LC3 processing and lipidation, such as ATG4, ATG5, and ATG7, may result in different phenotypes compared to inhibition of upstream or downstream autophagy regulators, such as ULK1, ATG13, RB1CC1, BNIP3, BECN1, or STX17 (syntaxin 17). Similarly, inhibition of genes that regulate mitophagy, such as BNIP3, may produce different results than inhibition of ATG genes in similar cancer models [Citation28,Citation79]. In addition, noncanonical and pleiotropic functions of ATG proteins [Citation198] may influence migration independent of autophagic degradation (e.g., ULK1-RB1CC1-mediated regulation of PTK2, or the regulation of endosomal trafficking by ULK1, ATG9, and BECN1 complex components). Thus, targeting multiple genes throughout the autophagy pathway within a model system will provide more conclusive data and distinguish between phenotypes resulting from autophagic degradation verses noncanonical functions of autophagy genes. For example, it would be interesting to determine whether inhibition of ULK results in PXN and SRC accumulation and reduced focal adhesion turnover, as is observed in ATG7-, ATG5-, and ATG12-deficient cells [Citation26,Citation28]. As ULK1 activity suppresses migration [Citation35,Citation75] and regulates migration through RB1CC1-PTK2 interactions [Citation35], this would determine whether non-canonical roles of ULK1 outweigh its role in promoting the degradation of focal adhesion proteins. Partial suppression versus complete inhibition of autophagy may also result in different phenotypes. For example, partial loss of allelic ATG5 promotes melanoma metastasis, whereas complete knockout reduces metastasis [Citation199]. In addition, implementing strategies that utilize temporal and spatial regulation of autophagy will further clarify the role of autophagy in specific steps of the metastatic cascade, and will help avoid complications involved in altered signaling pathways when using knockout cell lines. To account for the complexity of the TME, in vivo immunocompetent models should be utilized with rigorous quantitative measurements to provide more physiologically relevant conclusions. To supplement this, the use of in vitro models that incorporate hypoxia, starvation, and co-culture will provide more accurate representations of the TME. Moreover, future research should incorporate tissue-specific and lineage-specific autophagy knockouts to differentiate between the discrete roles of autophagy in specific microenvironments and cell types. As a fundamental cellular degradation and survival pathway, autophagy provides an attractive target to modulate metastasis-related signaling for therapeutic benefit. However, it is clear that autophagy also plays metastasis-suppressing roles, which should be taken into consideration to prevent undesired pro-metastatic outcomes.
Disclosure of potential conflicts of interest
The authors report no conflict of interest.
Additional information
Funding
References
- Gupta GP, Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi:10.1016/j.cell.2006.11.001. PMID:17110329
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904. doi:10.1038/nm1469.PMID:16892035
- Valastyan S, Weinberg RA. Tumor metastasis: Molecular insights and evolving paradigms. Cell. 2011;147:275–292. doi:10.1016/j.cell.2011.09.024. PMID:22000009
- Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi:10.1038/nature17038. PMID:26791720
- Plaks V, Koopman CD, Werb Z. Circulating tumor cells. Science. 2013;341:1186–1188. doi:10.1126/science.1235226. PMID:24031008
- Strilic B, Offermanns S. Intravascular survival and extravasation of tumor cells. Cancer Cell. 2017;32:282–293. doi:10.1016/j.ccell.2017.07.001.PMID:28898694
- Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472. doi:10.1038/nrm4024. PMID:26177004
- Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–1836. doi:10.15252/embj.201796697. PMID:28596378
- Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr. 2007;27:19–40. doi:10.1146/annurev.nutr.27.061406.093749. PMID:17311494
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi:10.1101/gad.1599207. PMID:18006683
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102. doi:10.1038/ncb1007-1102. PMID:17909521
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16:495. doi:10.1038/ncb2979. PMID:24875736
- Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7:279–296. doi:10.4161/auto.7.3.14487. PMID:21189453
- White E. Deconvoluting the context-dependent role for autophagy in cancer. Nat Rev Cancer. 2012;12:401–410. doi:10.1038/nrc3262. PMID:22534666
- Zhi X, Zhong Q. Autophagy in cancer. F1000prime Rep. 2015;7:18. doi:10.12703/P7-18. PMID:25750736
- Kimmelman AC, White E. Autophagy and tumor metabolism. Cell Metab. 2017;25:1037–1043. doi:10.1016/j.cmet.2017.04.004. PMID:28467923
- Vicente-Manzanares M, Horwitz AR. Cell migration: an overview. Methods Mol Biol Clifton NJ. 2011;769:1–24. doi:10.1007/978-1-61779-207-6_1.
- Ridley AJ, Schwartz MA, Burridge K, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–1709. doi:10.1126/science.1092053. PMID:14657486
- Case LB, Waterman CM. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat Cell Biol. 2015;17:955–963. doi:10.1038/ncb3191. PMID:26121555
- Caswell PT, Norman JC. Integrin trafficking and the control of cell migration. Traffic Cph Den. 2006;7:14–21. doi:10.1111/j.1600-0854.2005.00362.x.
- Caswell PT, Vadrevu S, Norman JC. Integrins: masters and slaves of endocytic transport. Nat Rev Mol Cell Biol. 2009;10:843–853. doi:10.1038/nrm2799. PMID:19904298
- Paul NR, Jacquemet G, Caswell PT. Endocytic Trafficking of Integrins in Cell Migration. Curr Biol CB. 2015;25:R1092–R1105. doi:10.1016/j.cub.2015.09.049. PMID:26583903
- Bridgewater RE, Norman JC, Caswell PT. Integrin trafficking at a glance. J Cell Sci. 2012;125:3695–3701. doi:10.1242/jcs.095810. PMID:23027580
- Luo B-H, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. doi:10.1146/annurev.immunol.25.022106.141618. PMID:17201681
- Kim M, Carman CV, Springer TA. Bidirectional transmembrane signaling by cytoplasmic domain separation in integrins. Science. 2003;301:1720–1725. doi:10.1126/science.1084174. PMID:14500982
- Sandilands E, Serrels B, McEwan DG, et al. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol. 2011;14:51–60. doi:10.1038/ncb2386. PMID:22138575
- Kenific CM, Stehbens SJ, Goldsmith J, et al. NBR1 enables autophagy-dependent focal adhesion turnover. J Cell Biol. 2016;212:577–590. doi:10.1083/jcb.201503075. PMID:26903539
- Sharifi MN, Mowers EE, Drake LE, et al. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of Paxillin with LC3. Cell Rep. 2016;15:1660–1672. doi:10.1016/j.celrep.2016.04.065. PMID:27184837
- Morton JP, Karim SA, Graham K, et al. Dasatinib inhibits the development of metastases in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology. 2010;139:292–303. doi:10.1053/j.gastro.2010.03.034. PMID:20303350
- Plaza-Menacho I, Morandi A, Mologni L, et al. Focal adhesion kinase (FAK) binds RET kinase via its FERM domain, priming a direct and reciprocal RET-FAK transactivation mechanism. J Biol Chem. 2011;286:17292–17302. doi:10.1074/jbc.M110.168500. PMID:21454698
- Sandilands E, Serrels B, Wilkinson S, et al. Src-dependent autophagic degradation of Ret in FAK-signalling-defective cancer cells. EMBO Rep. 2012;13:733–740. doi:10.1038/embor.2012.92. PMID:22732841
- Lock R, Kenific CM, Leidal AM, et al. Autophagy-dependent production of secreted factors facilitates oncogenic RAS-driven invasion. Cancer Discov. 2014;4:466–479. doi:10.1158/2159-8290.CD-13-0841. PMID:24513958
- Mauvezin C, Neufeld TP. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy. 2015;11:1437–1438. doi:10.1080/15548627.2015.1066957. PMID:26156798
- Abbi S, Ueda H, Zheng C, et al. Regulation of focal adhesion kinase by a novel protein inhibitor FIP200. Mol Biol Cell. 2002;13:3178–3191. doi:10.1091/mbc.E02-05-0295. PMID:12221124
- Caino MC, Chae YC, Vaira V, et al. Metabolic stress regulates cytoskeletal dynamics and metastasis of cancer cells. J Clin Invest. 2013;123:2907–2920. doi:10.1172/JCI67841. PMID:23921130
- Hara T, Takamura A, Kishi C, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008;181:497–510. doi:10.1083/jcb.200712064. PMID:18443221
- Wei H, Wei S, Gan B, et al. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev. 2011;25:1510–1527. doi:10.1101/gad.2051011. PMID:21764854
- Caswell P, Norman J. Endocytic transport of integrins during cell migration and invasion. Trends Cell Biol. 2008;18:257–263. doi:10.1016/j.tcb.2008.03.004. PMID:18456497
- Valdembri D, Serini G. Regulation of adhesion site dynamics by integrin traffic. Curr Opin Cell Biol. 2012;24:582–591. doi:10.1016/j.ceb.2012.08.004. PMID:22981739
- White DP, Caswell PT, Norman JC. Alpha v beta3 and alpha5beta1 integrin recycling pathways dictate downstream Rho kinase signaling to regulate persistent cell migration. J Cell Biol. 2007;177:515–525. doi:10.1083/jcb.200609004. PMID:17485491
- Caswell PT, Chan M, Lindsay AJ, et al. Rab-coupling protein coordinates recycling of alpha5beta1 integrin and EGFR1 to promote cell migration in 3D microenvironments. J Cell Biol. 2008;183:143–155. doi:10.1083/jcb.200804140. PMID:18838556
- Reynolds AR, Hart IR, Watson AR, et al. Stimulation of tumor growth and angiogenesis by low concentrations of RGD-mimetic integrin inhibitors. Nat Med. 2009;15:392–400. doi:10.1038/nm.1941. PMID:19305413
- Shi F, Sottile J. Caveolin-1-dependent beta1 integrin endocytosis is a critical regulator of fibronectin turnover. J Cell Sci. 2008;121:2360–2371. doi:10.1242/jcs.014977. PMID:18577581
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi:10.1016/S0092-8674(02)00971-6. PMID:12297042
- Moser M, Legate KR, Zent R, et al. The tail of integrins, talin, and kindlins. Science. 2009;324:895–899. doi:10.1126/science.1163865. PMID:19443776
- Legate KR, Wickström SA, Fässler R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009;23:397–418. doi:10.1101/gad.1758709. PMID:19240129
- Lobert VH, Brech A, Pedersen NM, et al. Ubiquitination of alpha 5 beta 1 integrin controls fibroblast migration through lysosomal degradation of fibronectin-integrin complexes. Dev Cell. 2010;19:148–159. doi:10.1016/j.devcel.2010.06.010. PMID:20643357
- Arjonen A, Alanko J, Veltel S, et al. Distinct recycling of active and inactive β1 integrins. Traffic Cph Den. 2012;13:610–625. doi:10.1111/j.1600-0854.2012.01327.x.
- Woods AJ, White DP, Caswell PT, et al. PKD1/PKCmu promotes alphavbeta3 integrin recycling and delivery to nascent focal adhesions. EMBO J. 2004;23:2531–2543. doi:10.1038/sj.emboj.7600267. PMID:15192707
- Yoon S-O, Shin S, Mercurio AM. Hypoxia stimulates carcinoma invasion by stabilizing microtubules and promoting the Rab11 trafficking of the alpha6beta4 integrin. Cancer Res. 2005;65:2761–2769. doi:10.1158/0008-5472.CAN-04-4122. PMID:15805276
- Onodera Y, Nam J-M, Hashimoto A, et al. Rab5c promotes AMAP1-PRKD2 complex formation to enhance β1 integrin recycling in EGF-induced cancer invasion. J Cell Biol. 2012;197:983–996. doi:10.1083/jcb.201201065. PMID:22734003
- Ezratty EJ, Bertaux C, Marcantonio EE, et al. Clathrin mediates integrin endocytosis for focal adhesion disassembly in migrating cells. J Cell Biol. 2009;187:733–747. doi:10.1083/jcb.200904054. PMID:19951918
- Dozynkiewicz MA, Jamieson NB, Macpherson I, et al. Rab25 and CLIC3 collaborate to promote integrin recycling from late endosomes/lysosomes and drive cancer progression. Dev Cell. 2012;22:131–145. doi:10.1016/j.devcel.2011.11.008. PMID:22197222
- Lamb CA, Dooley HC, Tooze SA. Endocytosis and autophagy: Shared machinery for degradation. BioEssays News Rev Mol Cell Dev Biol. 2013;35:34–45. doi:10.1002/bies.201200130.
- Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death Differ. 2014;21:348–358. doi:10.1038/cdd.2013.187. PMID:24440914
- Longatti A, Lamb CA, Razi M, et al. TBC1D14 regulates autophagosome formation via Rab11- and ULK1-positive recycling endosomes. J Cell Biol. 2012;197:659–675. doi:10.1083/jcb.201111079. PMID:22613832
- Gaullier JM, Simonsen A, D'Arrigo A, et al. FYVE fingers bind PtdIns(3)P. Nature. 1998;394:432–433. doi:10.1038/28767. PMID:9697764
- Sun Q, Westphal W, Wong KN, et al. Rubicon controls endosome maturation as a Rab7 effector. Proc Natl Acad Sci U S A. 2010;107:19338–19343. doi:10.1073/pnas.1010554107. PMID:20974968
- Barrow-McGee R, Kishi N, Joffre C, et al. Beta 1-integrin-c-Met cooperation reveals an inside-in survival signalling on autophagy-related endomembranes. Nat Commun. 2016;7:11942. doi:10.1038/ncomms11942. PMID:27336951
- Fader CM, Sánchez D, Furlán M, et al. Induction of autophagy promotes fusion of Multivesicular bodies with Autophagic vacuoles in K562 Cells. Traffic. 2008;9:230–250. doi:10.1111/j.1600-0854.2007.00677.x. PMID:17999726
- Morvan J, Köchl R, Watson R, et al. In vitro reconstitution of fusion between immature autophagosomes and endosomes. Autophagy. 2009;5:676–689. doi:10.4161/auto.5.5.8378. PMID:19337031
- Fader CM, Colombo MI. Multivesicular bodies and autophagy in erythrocyte maturation. Autophagy. 2006;2:122–125. doi:10.4161/auto.2.2.2350. PMID:16874060
- Peinado H, Alečković M, Lavotshkin S, et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat Med. 2012;18:883–891. doi:10.1038/nm.2753. PMID:22635005
- Costa-Silva B, Aiello NM, Ocean AJ, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–826. doi:10.1038/ncb3169. PMID:25985394
- Hoshino A, Costa-Silva B, Shen T-L, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–335. doi:10.1038/nature15756. PMID:26524530
- Grange C, Tapparo M, Collino F, et al. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011;71:5346–5356. doi:10.1158/0008-5472.CAN-11-0241. PMID:21670082
- Sung BH, Ketova T, Hoshino D, et al. Directional cell movement through tissues is controlled by exosome secretion. Nat Commun. 2015;6:7164. doi:10.1038/ncomms8164. PMID:25968605
- Ata R, Antonescu CN. Integrins and Cell Metabolism: An intimate relationship impacting Cancer. Int J Mol Sci. 2017;18:pii: E189. doi:10.3390/ijms18010189. PMID:28106780
- Rainero E, Howe JD, Caswell PT, et al. Ligand-Occupied integrin internalization links nutrient signaling to invasive migration. Cell Rep. 2015; doi:10.1016/j.celrep.2014.12.037. PMID:25600874
- Xia H, Nho RS, Kahm J, et al. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem. 2004;279:33024–33034. doi:10.1074/jbc.M313265200. PMID:15166238
- Yang L, Hou Y, Yuan J, et al. Twist promotes reprogramming of glucose metabolism in breast cancer cells through PI3K/AKT and p53 signaling pathways. Oncotarget. 2015;6:25755–25769. doi:10.18632/oncotarget.4697. PMID:26342198
- Tuloup-Minguez V, Hamaï A, Greffard A, et al. Autophagy modulates cell migration and β1 integrin membrane recycling. Cell Cycle Georget Tex. 2013;12:3317–3328. doi:10.4161/cc.26298.
- Kim TH, Kim HI, Soung YH, et al. Integrin (alpha6beta4) signals through Src to increase expression of S100A4, a metastasis-promoting factor: implications for cancer cell invasion. Mol Cancer Res MCR. 2009;7:1605–1612. doi:10.1158/1541-7786.MCR-09-0102. PMID:19808905
- Chen M, Sinha M, Luxon BA, et al. Integrin alpha6beta4 controls the expression of genes associated with cell motility, invasion, and metastasis, including S100A4/metastasin. J Biol Chem. 2009;284:1484–1494. doi:10.1074/jbc.M803997200. PMID:19011242
- Dower CM, Bhat N, Wang EW, et al. Selective reversible inhibition of autophagy in hypoxic breast cancer cells promotes pulmonary metastasis. Cancer Res. 2017;77:646–657. doi:10.1158/0008-5472.CAN-15-3458. PMID:28115361
- Tang J, Deng R, Luo R-Z, et al. Low expression of ULK1 is associated with operable breast cancer progression and is an adverse prognostic marker of survival for patients. Breast Cancer Res Treat. 2012;134:549–560. doi:10.1007/s10549-012-2080-y. PMID:22585231
- Tracy K, Dibling BC, Spike BT, et al. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol. 2007;27:6229–6242. doi:10.1128/MCB.02246-06. PMID:17576813
- Hanna RA, Quinsay MN, Orogo AM, et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem. 2012;287:19094–19104. doi:10.1074/jbc.M111.322933. PMID:22505714
- Chourasia AH, Tracy K, Frankenberger C, et al. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep. 2015;16:1145–1163. doi:10.15252/embr.201540759. PMID:26232272
- Ma J, Zhang Q, Chen S, et al. Mitochondrial dysfunction promotes breast cancer cell migration and invasion through HIF1α accumulation via increased production of reactive oxygen species. PloS One. 2013;8:e69485. doi:10.1371/journal.pone.0069485. PMID:23922721
- Pelicano H, Lu W, Zhou Y, et al. Mitochondrial dysfunction and reactive oxygen species imbalance promote breast cancer cell motility through a CXCL14-mediated mechanism. Cancer Res. 2009;69:2375–2383. doi:10.1158/0008-5472.CAN-08-3359. PMID:19276362
- Hung W-Y, Huang K-H, Wu C-W, et al. Mitochondrial dysfunction promotes cell migration via reactive oxygen species-enhanced β5-integrin expression in human gastric cancer SC-M1 cells. Biochim Biophys Acta. 2012;1820:1102–1110. doi:10.1016/j.bbagen.2012.04.016. PMID:22561002
- Maes H, Van Eygen S, et al. BNIP3 supports melanoma cell migration and vasculogenic mimicry by orchestrating the actin cytoskeleton. Cell Death Dis. 2014;5:e1127. doi:10.1038/cddis.2014.94. PMID:24625986
- Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014;14:598–610. doi:10.1038/nrc3792. PMID:25098269
- Steffen A, Stradal TEB, Rottner K. Signalling pathways controlling cellular actin organization. Handb Exp Pharmacol. 2017;235:153–178. doi:10.1007/164_2016_35. PMID:27757765
- Hanna S, El-Sibai M. Signaling networks of Rho GTPases in cell motility. Cell Signal. 2013;25:1955–1961. doi:10.1016/j.cellsig.2013.04.009. PMID:23669310
- Campellone KG, Welch MD. A nucleator arms race: cellular control of actin assembly. Nat Rev Mol Cell Biol. 2010;11:237–251. doi:10.1038/nrm2867. PMID:20237478
- Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010;11:633–643. doi:10.1038/nrm2957. PMID:20729930
- Olson MF, Sahai E. The actin cytoskeleton in cancer cell motility. Clin Exp Metastasis. 2009;26:273–287. doi:10.1007/s10585-008-9174-2. PMID:18498004
- Sanz-Moreno V, Gadea G, Ahn J, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–523. doi:10.1016/j.cell.2008.09.043. PMID:18984162
- Belaid A, Cerezo M, Chargui A, et al. Autophagy plays a critical role in the degradation of active RHOA, the control of cell cytokinesis, and genomic stability. Cancer Res. 2013;73:4311–4322. doi:10.1158/0008-5472.CAN-12-4142. PMID:23704209
- Belaid A, Ndiaye PD, Cerezo M, et al. Autophagy and SQSTM1 on the RHOA(d) again. Autophagy. 2014;10:201–208. doi:10.4161/auto.27198. PMID:24300375
- Yoshida T, Tsujioka M, Honda S, et al. Autophagy suppresses cell migration by degrading GEF-H1, a RhoA GEF. Oncotarget. 2016;7:34420–34429. doi:10.18632/oncotarget.8883.
- Aguilera MO, Berón W, Colombo MI. The actin cytoskeleton participates in the early events of autophagosome formation upon starvation induced autophagy. Autophagy. 2012;8:1590–1603. doi:10.4161/auto.21459. PMID:22863730
- Gurkar AU, Chu K, Raj L, et al. Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat Commun. 2013;4:2189. doi:10.1038/ncomms3189. PMID:23877263
- Wang W, Goswami S, Lapidus K, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–8594. doi:10.1158/0008-5472.CAN-04-1136. PMID:15574765
- Gilkes DM, Xiang L, Lee SJ, et al. Hypoxia-inducible factors mediate coordinated RhoA-ROCK1 expression and signaling in breast cancer cells. Proc Natl Acad Sci U S A. 2014;111:E384–E393. doi:10.1073/pnas.1321510111. PMID:24324133
- Till A, Saito R, Merkurjev D, et al. Evolutionary trends and functional anatomy of the human expanded autophagy network. Autophagy. 2015;11:1652–1667. doi:10.1080/15548627.2015.1059558. PMID:26103419
- Carroll B, Mohd-Naim N, Maximiano F, et al. The TBC/RabGAP Armus coordinates Rac1 and Rab7 functions during autophagy. Dev Cell. 2013;25:15–28. doi:10.1016/j.devcel.2013.03.005. PMID:23562278
- Wei Y-M, Li X, Xu M, et al. Enhancement of autophagy by simvastatin through inhibition of Rac1-mTOR signaling pathway in coronary arterial myocytes. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2013;31:925–937. doi:10.1159/000350111.
- Zhu WL, Hossain MS, Guo DY, et al. A role for Rac3 GTPase in the regulation of autophagy. J Biol Chem. 2011;286:35291–35298. doi:10.1074/jbc.M111.280990. PMID:21852230
- Kast DJ, Dominguez R. The Cytoskeleton-autophagy connection. Curr Biol CB. 2017;27:R318–R326. doi:10.1016/j.cub.2017.02.061. PMID:28441569
- Frisch SM, Francis H. Disruption of epithelial cell-matrix interactions induces apoptosis. J Cell Biol. 1994;124:619–626. doi:10.1083/jcb.124.4.619. PMID:8106557
- Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–706. doi:10.1016/S0955-0674(97)80124-X. PMID:9330874
- Paoli P, Giannoni E, Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochim Biophys Acta. 2013;1833:3481–3498. doi:10.1016/j.bbamcr.2013.06.026. PMID:23830918
- Fung C, Lock R, Gao S, et al. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol Biol Cell. 2008;19:797–806. doi:10.1091/mbc.E07-10-1092. PMID:18094039
- Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, et al. PERK Integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol Cell Biol. 2011;31:3616–3629. doi:10.1128/MCB.05164-11. PMID:21709020
- Debnath J. Detachment-induced autophagy during anoikis and lumen formation in epithelial acini. Autophagy. 2008;4:351–353. doi:10.4161/auto.5523. PMID:18196957
- Peng Y-F, Shi Y-H, Ding Z-B, et al. Autophagy inhibition suppresses pulmonary metastasis of HCC in mice via impairing anoikis resistance and colonization of HCC cells. Autophagy. 2013;9:2056–2068. doi:10.4161/auto.26398. PMID:24157892
- Harding HP, Zhang Y, Bertolotti A, et al. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi:10.1016/S1097-2765(00)80330-5. PMID:10882126
- Rutkowski DT, Kaufman RJ. A trip to the ER: coping with stress. Trends Cell Biol. 2004;14:20–28. doi:10.1016/j.tcb.2003.11.001. PMID:14729177
- Cullinan SB, Zhang D, Hannink M, et al. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol. 2003;23:7198–7209. doi:10.1128/MCB.23.20.7198-7209.2003. PMID:14517290
- Rouschop KMA, van den Beucken T, Dubois L, et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J Clin Invest. 2010;120:127–141. doi:10.1172/JCI40027. PMID:20038797
- Ogata M, Hino S, Saito A, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26:9220–9231. doi:10.1128/MCB.01453-06. PMID:17030611
- Chen N, Debnath J. IκB kinase complex (IKK) triggers detachment-induced autophagy in mammary epithelial cells independently of the PI3K-AKT-MTORC1 pathway. Autophagy. 2013;9:1214–1227. doi:10.4161/auto.24870. PMID:23778976
- Park SH, Riley P, Frisch SM. Regulation of anoikis by deleted in breast cancer-1 (DBC1) through NF-κB. Apoptosis Int J Program Cell Death. 2013;18:949–962. doi:10.1007/s10495-013-0847-1.
- Chang C, Su H, Zhang D, et al. AMPK-Dependent Phosphorylation of GAPDH Triggers Sirt1 Activation and Is Necessary for Autophagy upon Glucose Starvation. Mol Cell. 2015;60:930–940. doi:10.1016/j.molcel.2015.10.037. PMID:26626483
- Luo S, Garcia-Arencibia M, Zhao R, et al. Bim inhibits autophagy by recruiting Beclin 1 to microtubules. Mol Cell. 2012;47:359–370. doi:10.1016/j.molcel.2012.05.040. PMID:22742832
- Contreras AU, Mebratu Y, Delgado M, et al. Deacetylation of p53 induces autophagy by suppressing Bmf expression. J Cell Biol. 2013;201:427–437. doi:10.1083/jcb.201205064. PMID:23629966
- Delgado M, Tesfaigzi Y. Is BMF central for anoikis and autophagy? Autophagy. 2014;10:168–169. doi:10.4161/auto.26759. PMID:24225781
- Puthalakath H, Villunger A, O'Reilly LA, et al. Bmf: a proapoptotic BH3-only protein regulated by interaction with the myosin V actin motor complex, activated by anoikis. Science. 2001;293:1829–1832. doi:10.1126/science.1062257. PMID:11546872
- Reginato MJ, Mills KR, Paulus JK, et al. Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat Cell Biol. 2003;5:733–740. doi:10.1038/ncb1026. PMID:12844146
- Buchheit CL, Schafer ZT. BIM-EL localization: The key to understanding anoikis resistance in inflammatory breast cancer cells. Mol Cell Oncol. 2016;3:e1011474. doi:10.1080/23723556.2015.1011474. PMID:27308529
- Buchheit CL, Angarola BL, Steiner A, et al. Anoikis evasion in inflammatory breast cancer cells is mediated by Bim-EL sequestration. Cell Death Differ. 2015;22:1275–1286. doi:10.1038/cdd.2014.209. PMID:25526094
- Peng Y-F, Shi Y-H, Shen Y-H, et al. Promoting colonization in metastatic HCC cells by modulation of autophagy. PloS One. 2013;8:e74407. doi:10.1371/journal.pone.0074407. PMID:24058558
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. doi:10.1080/15548627.2015.1100356. PMID:26799652
- Insua-Rodríguez J, Oskarsson T. The extracellular matrix in breast cancer. Adv Drug Deliv Rev. 2016;97:41–55. doi:10.1016/j.addr.2015.12.017. PMID:26743193
- Cox TR, Erler JT. Molecular pathways: connecting fibrosis and solid tumor metastasis. Clin Cancer Res Off J Am Assoc Cancer Res. 2014;20:3637–3643. doi:10.1158/1078-0432.CCR-13-1059.
- Wang K, Seo BR, Fischbach C, et al. Fibronectin mechanobiology regulates tumorigenesis. Cell Mol Bioeng. 2016;9:1–11. doi:10.1007/s12195-015-0417-4. PMID:26900407
- Singh P, Carraher C, Schwarzbauer JE. Assembly of fibronectin extracellular matrix. Annu Rev Cell Dev Biol. 2010;26:397–419. doi:10.1146/annurev-cellbio-100109-104020. PMID:20690820
- Fogerty FJ, Mosher DF. Mechanisms for organization of fibronectin matrix. Cell Differ Dev Off J Int Soc Dev Biol. 1990;32:439–450. doi:10.1016/0922-3371(90)90061-Z.
- Fogerty FJ, Akiyama SK, Yamada KM, et al. Inhibition of binding of fibronectin to matrix assembly sites by anti-integrin (alpha 5 beta 1) antibodies. J Cell Biol. 1990;111:699–708. doi:10.1083/jcb.111.2.699. PMID:2380248
- Mao Y, Schwarzbauer JE. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol J Int Soc Matrix Biol. 2005;24:389–399. doi:10.1016/j.matbio.2005.06.008.
- Shi F, Sottile J. MT1-MMP regulates the turnover and endocytosis of extracellular matrix fibronectin. J Cell Sci. 2011;124:4039–4050. doi:10.1242/jcs.087858. PMID:22159414
- Mana G, Clapero F, Panieri E, et al. PPFIA1 drives active α5β1 integrin recycling and controls fibronectin fibrillogenesis and vascular morphogenesis. Nat Commun. 2016;7:13546. doi:10.1038/ncomms13546. PMID:27876801
- Kenny HA, Chiang C-Y, White EA, et al. Mesothelial cells promote early ovarian cancer metastasis through fibronectin secretion. J Clin Invest. 2014;124:4614–4628. doi:10.1172/JCI74778. PMID:25202979
- Hsu J-Y, Chang K-Y, Chen S-H, et al. Epidermal growth factor-induced cyclooxygenase-2 enhances head and neck squamous cell carcinoma metastasis through fibronectin up-regulation. Oncotarget. 2015;6:1723–1739. doi:10.18632/oncotarget.2783. PMID:25595899
- Knowles LM, Gurski LA, Engel C, et al. Integrin αvβ3 and fibronectin upregulate Slug in cancer cells to promote clot invasion and metastasis. Cancer Res. 2013;73:6175–6184. doi:10.1158/0008-5472.CAN-13-0602. PMID:23966293
- Huang C-R, Lee C-T, Chang K-Y, et al. Down-regulation of ARNT promotes cancer metastasis by activating the fibronectin/integrin β1/FAK axis. Oncotarget. 2015;6:11530–11546. doi:10.18632/oncotarget.3448. PMID:25839165
- Bae YK, Kim A, Kim MK, et al. Fibronectin expression in carcinoma cells correlates with tumor aggressiveness and poor clinical outcome in patients with invasive breast cancer. Hum Pathol. 2013;44:2028–2037. doi:10.1016/j.humpath.2013.03.006. PMID:23684510
- Malik G, Knowles LM, Dhir R, et al. Plasma fibronectin promotes lung metastasis by contributions to fibrin clots and tumor cell invasion. Cancer Res. 2010;70:4327–4334. doi:10.1158/0008-5472.CAN-09-3312. PMID:20501851
- Cao Y, Liu X, Lu W, et al. Fibronectin promotes cell proliferation and invasion through mTOR signaling pathway activation in gallbladder cancer. Cancer Lett. 2015;360:141–150. doi:10.1016/j.canlet.2015.01.041. PMID:25657110
- Wei SC, Fattet L, Tsai JH, et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat Cell Biol. 2015;17:678–688. doi:10.1038/ncb3157. PMID:25893917
- Patel AS, Lin L, Geyer A, et al. Autophagy in idiopathic pulmonary fibrosis. PloS One. 2012;7:e41394. doi:10.1371/journal.pone.0041394. PMID:22815997
- Sun K, Xu L, Jing Y, et al. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-κB-IL1α/β-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017;388:198–207. doi:10.1016/j.canlet.2016.12.004. PMID:28011320
- Lodder J, Denaës T, Chobert M-N, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11:1280–1292. doi:10.1080/15548627.2015.1058473. PMID:26061908
- Manresa MC, Godson C, Taylor CT. Hypoxia-sensitive pathways in inflammation-driven fibrosis. Am J Physiol Regul Integr Comp Physiol. 2014;307:R1369–R1380. doi:10.1152/ajpregu.00349.2014. PMID:25298511
- Zhou B, Rabinovitch M. Microtubule involvement in translational regulation of fibronectin expression by light chain 3 of microtubule-associated protein 1 in vascular smooth muscle cells. Circ Res. 1998;83:481–489. doi:10.1161/01.RES.83.5.481. PMID:9734470
- Li W, Zou J, Yue F, et al. Defects in MAP1S-mediated autophagy cause reduction in mouse lifespans especially when fibronectin is overexpressed. Aging Cell. 2016. doi:10.1111/acel.12441.
- Xie R, Nguyen S, McKeehan K, et al. Microtubule-associated protein 1S (MAP1S) bridges autophagic components with microtubules and mitochondria to affect autophagosomal biogenesis and degradation. J Biol Chem. 2011;286:10367–10377. doi:10.1074/jbc.M110.206532. PMID:21262964
- Yue F, Li W, Zou J, et al. Spermidine prolongs lifespan and prevents liver fibrosis and hepatocellular carcinoma by activating MAP1S-mediated autophagy. Cancer Res. 2017;77:2938–2951. doi:10.1158/0008-5472.CAN-16-3462. PMID:28386016
- Xu G, Yue F, Huang H, et al. Defects in MAP1S-mediated autophagy turnover of fibronectin cause renal fibrosis. Aging. 2016;8:977–985. doi:10.18632/aging.100957. PMID:27236336
- Neill T, Schaefer L, Iozzo RV. Instructive roles of extracellular matrix on autophagy. Am J Pathol. 2014;184:2146–2153. doi:10.1016/j.ajpath.2014.05.010. PMID:24976620
- Gubbiotti MA, Iozzo RV. Proteoglycans regulate autophagy via outside-in signaling: an emerging new concept. Matrix Biol J Int Soc Matrix Biol. 2015;48:6–13. doi:10.1016/j.matbio.2015.10.002.
- Gilkes DM, Bajpai S, Wong CC, et al. Procollagen lysyl hydroxylase 2 is essential for hypoxia-induced breast cancer metastasis. Mol Cancer Res MCR. 2013;11:456–466. doi:10.1158/1541-7786.MCR-12-0629. PMID:23378577
- Gilkes DM, Chaturvedi P, Bajpai S, et al. Collagen prolyl hydroxylases are essential for breast cancer metastasis. Cancer Res. 2013;73:3285–3296. doi:10.1158/0008-5472.CAN-12-3963. PMID:23539444
- Baker A-M, Bird D, Lang G, et al. Lysyl oxidase enzymatic function increases stiffness to drive colorectal cancer progression through FAK. Oncogene. 2013;32:1863–1868. doi:10.1038/onc.2012.202. PMID:22641216
- Cox TR, Rumney RMH, Schoof EM, et al. The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature. 2015;522:106–110. doi:10.1038/nature14492. PMID:26017313
- Klymkowsky MW, Savagner P. Epithelial-mesenchymal transition: a cancer researcher's conceptual friend and foe. Am J Pathol. 2009;174:1588–1593. doi:10.2353/ajpath.2009.080545. PMID:19342369
- Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–273. doi:10.1038/nrc2620. PMID:19262571
- Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi:10.1007/s10555-008-9169-0. PMID:19169796
- Frisch SM, Schaller M, Cieply B. Mechanisms that link the oncogenic epithelial-mesenchymal transition to suppression of anoikis. J Cell Sci. 2013;126:21–29. doi:10.1242/jcs.120907. PMID:23516327
- Nieto MA, Huang RY-J, Jackson RA, et al. EMT: 2016. Cell. 2016;166:21–45. doi:10.1016/j.cell.2016.06.028. PMID:27368099
- Grassi G, Di Caprio G, Santangelo L, et al. Autophagy regulates hepatocyte identity and epithelial-to-mesenchymal and mesenchymal-to-epithelial transitions promoting Snail degradation. Cell Death Dis. 2015;6:e1880. doi:10.1038/cddis.2015.249. PMID:26355343
- Zou J, Liu Y, Li B, et al. Autophagy attenuates endothelial-to-mesenchymal-transition by promoting Snail degradation in human cardiac microvascular endothelial cells. Biosci Rep. 2017;37:5. doi:10.1042/BSR20171049.
- Li J, Yang B, Zhou Q, et al. Autophagy promotes hepatocellular carcinoma cell invasion through activation of epithelial-mesenchymal transition. Carcinogenesis. 2013;34:1343–1351. doi:10.1093/carcin/bgt063. PMID:23430956
- Qiang L, Zhao B, Ming M, et al. Regulation of cell proliferation and migration by p62 through stabilization of Twist1. Proc Natl Acad Sci U S A. 2014;111:9241–9246. doi:10.1073/pnas.1322913111. PMID:24927592
- Bertrand M, Petit V, Jain A, et al. SQSTM1/p62 regulates the expression of junctional proteins through epithelial-mesenchymal transition factors. Cell Cycle Georget Tex. 2015;14:364–374. doi:10.4161/15384101.2014.987619.
- Catalano M, D'Alessandro G, Lepore F, et al. Autophagy induction impairs migration and invasion by reversing EMT in glioblastoma cells. Mol Oncol. 2015;9:1612–1625. doi:10.1016/j.molonc.2015.04.016. PMID:26022108
- Gugnoni M, Sancisi V, Gandolfi G, et al. Cadherin-6 promotes EMT and cancer metastasis by restraining autophagy. Oncogene. 2017;36:667–677. doi:10.1038/onc.2016.237. PMID:27375021
- Zhao Z, Zhao J, Xue J, et al. Autophagy inhibition promotes epithelial-mesenchymal transition through ROS/HO-1 pathway in ovarian cancer cells. Am J Cancer Res. 2016;6:2162–2177. PMID:27822409
- Martinez-Outschoorn U, Sotgia F, Lisanti MP. Tumor microenvironment and metabolic synergy in breast cancers: critical importance of mitochondrial fuels and function. Semin Oncol. 2014;41:195–216. doi:10.1053/j.seminoncol.2014.03.002. PMID:24787293
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009;9:239–252. doi:10.1038/nrc2618. PMID:19279573
- Martinez-Outschoorn UE, Pavlides S, Howell A, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–1051. doi:10.1016/j.biocel.2011.01.023. PMID:21300172
- Martinez-Outschoorn UE, Lin Z, Whitaker-Menezes D, et al. Ketone body utilization drives tumor growth and metastasis. Cell Cycle. 2012;11:3964–3971. doi:10.4161/cc.22137. PMID:23082722
- Capparelli C, Guido C, Whitaker-Menezes D, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle Georget Tex. 2012;11:2285–2302. doi:10.4161/cc.20718.
- Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle Georget Tex. 2011;10:1772–1783. doi:10.4161/cc.10.11.15659.
- Martinez-Outschoorn UE, Trimmer C, Lin Z, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle Georget Tex. 2010;9:3515–3533. doi:10.4161/cc.9.17.12928.
- Chiavarina B, Whitaker-Menezes D, Migneco G, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle Georget Tex. 2010;9:3534–3551. doi:10.4161/cc.9.17.12908.
- Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle Georget Tex. 2012;11:1108–1117. doi:10.4161/cc.11.6.19530.
- Capparelli C, Whitaker-Menezes D, Guido C, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11:2272–2284. doi:10.4161/cc.20717. PMID:22684333
- Guido C, Whitaker-Menezes D, Capparelli C, et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: connecting TGF-β signaling with “Warburg-like” cancer metabolism and L-lactate production. Cell Cycle Georget Tex. 2012;11:3019–3035. doi:10.4161/cc.21384.
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle Georget Tex. 2010;9:3256–3276. doi:10.4161/cc.9.16.12553.
- Witkiewicz AK, Dasgupta A, Sotgia F, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol. 2009;174:2023–2034. doi:10.2353/ajpath.2009.080873. PMID:19411448
- Sloan EK, Ciocca DR, Pouliot N, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol. 2009;174:2035–2043. doi:10.2353/ajpath.2009.080924. PMID:19411449
- Qian N, Ueno T, Kawaguchi-Sakita N, et al. Prognostic significance of tumor/stromal caveolin-1 expression in breast cancer patients. Cancer Sci. 2011;102:1590–1596. doi:10.1111/j.1349-7006.2011.01985.x. PMID:21585620
- Castello-Cros R, Bonuccelli G, Molchansky A, et al. Matrix remodeling stimulates stromal autophagy, “fueling” cancer cell mitochondrial metabolism and metastasis. Cell Cycle Georget Tex. 2011;10:2021–2034. doi:10.4161/cc.10.12.16002.
- Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–266. doi:10.1016/j.cell.2006.01.007. PMID:16439202
- Shi C-S, Shenderov K, Huang N-N, et al. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. doi:10.1038/ni.2215. PMID:22286270
- Zhong Z, Sanchez-Lopez E, Karin M. Autophagy, inflammation, and immunity: A troika governing cancer and its treatment. Cell. 2016;166:288–298. doi:10.1016/j.cell.2016.05.051. PMID:27419869
- Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–268. doi:10.1038/nature07383. PMID:18849965
- Nakahira K, Haspel JA, Rathinam VAK, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol. 2011;12:222–230. doi:10.1038/ni.1980. PMID:21151103
- Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi:10.1038/nature09663. PMID:21124315
- Zhong Z, Umemura A, Sanchez-Lopez E, et al. NF-κB Restricts inflammasome activation via elimination of damaged mitochondria. Cell. 2016;164:896–910. doi:10.1016/j.cell.2015.12.057. PMID:26919428
- Jinushi M, Morita T, Xu Z, et al. Autophagy-dependent regulation of tumor metastasis by myeloid cells. PLOS ONE. 2017;12:e0179357. doi:10.1371/journal.pone.0179357. PMID:28686632
- Yang S, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. 2011;25:717–729. doi:10.1101/gad.2016111. PMID:21406549
- Kim M-J, Woo S-J, Yoon C-H, et al. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. 2011;286:12924–12932. doi:10.1074/jbc.M110.138958. PMID:21300795
- Lindqvist LM, Simon AK, Baehrecke EH. Current questions and possible controversies in autophagy. Cell Death Discov. 2015;1:15036. doi:10.1038/cddiscovery.2015.36. PMID:26682061
- García-Fernández M, Karras P, Checinska A, et al. Metastatic risk and resistance to BRAF inhibitors in melanoma defined by selective allelic loss of ATG5. Autophagy. 2016;12:1776–1790. doi:10.1080/15548627.2016.1199301. PMID:27464255
- Egan DF, Kim J, Shaw RJ, et al. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy. 2011;7:645–646. doi:10.4161/auto.7.6.15123. PMID:21460623
- Lee JW, Park S, Takahashi Y, et al. The association of AMPK with ULK1 regulates autophagy. PloS One. 2010;5:e15394. doi:10.1371/journal.pone.0015394. PMID:21072212
- Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex–at the crossroads of autophagy and beyond. Trends Cell Biol. 2010;20:355–362. doi:10.1016/j.tcb.2010.03.002. PMID:20356743
- Abrahamsen H, Stenmark H, Platta HW. Ubiquitination and phosphorylation of Beclin 1 and its binding partners: Tuning class III phosphatidylinositol 3-kinase activity and tumor suppression. FEBS Lett. 2012;586:1584–1591. doi:10.1016/j.febslet.2012.04.046. PMID:22673570
- Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. 2013;55:39–50. doi:10.1042/bse0550039. PMID:24070470
- Nitta T, Sato Y, Ren XS, et al. Autophagy may promote carcinoma cell invasion and correlate with poor prognosis in cholangiocarcinoma. Int J Clin Exp Pathol. 2014;7:4913–4921. PMID:25197362
- Macintosh RL, Timpson P, Thorburn J, et al. Inhibition of autophagy impairs tumor cell invasion in an organotypic model. Cell Cycle Georget Tex. 2012;11:2022–2029. doi:10.4161/cc.20424. doi:10.4161/cc.20424.
- Barnard RA, Regan DP, Hansen RJ, et al. Autophagy inhibition delays early but not late-stage metastatic disease. J Pharmacol Exp Ther. 2016;358:282–293. doi:10.1124/jpet.116.233908. PMID:27231155
- Zhan Z, Xie X, Cao H, et al. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy. 2014;10:257–268. doi:10.4161/auto.27162. PMID:24321786
- Kim YH, Baek SH, Kim EK, et al. Uncoordinated 51-like kinase 2 signaling pathway regulates epithelial-mesenchymal transition in A549 lung cancer cells. FEBS Lett. 2016;590:1365–1374. doi:10.1002/1873-3468.12172. PMID:27062295
- Yang S-W, Ping Y-F, Jiang Y-X, et al. ATG4A promotes tumor metastasis by inducing the epithelial-mesenchymal transition and stem-like properties in gastric cells. Oncotarget. 2016;7:39279–39292. doi:10.18632/oncotarget.9827. PMID:27276686
- Zhang H, Guo M, Chen J-H, et al. Osteopontin knockdown inhibits αv,β3 integrin-induced cell migration and invasion and promotes apoptosis of breast cancer cells by inducing autophagy and inactivating the PI3K/Akt/mTOR pathway. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2014;33:991–1002. doi:10.1159/000358670.
- Singh KK, Lovren F, Pan Y, et al. The essential autophagy gene ATG7 modulates organ fibrosis via regulation of endothelial-to-mesenchymal transition. J Biol Chem. 2015;290:2547–2559. doi:10.1074/jbc.M114.604603. PMID:25527499
- Qin W, Li C, Zheng W, et al. Inhibition of autophagy promotes metastasis and glycolysis by inducing ROS in gastric cancer cells. Oncotarget. 2015;6:39839–39854. doi:10.18632/oncotarget.5674. PMID:26497999
- Han Q, Deng Y, Chen S, et al. Downregulation of ATG5-dependent macroautophagy by chaperone-mediated autophagy promotes breast cancer cell metastasis. Sci Rep. 2017;7:4759. doi:10.1038/s41598-017-04994-x. PMID:28684853
- Wang C, Jiang J, Ji J, et al. PKM2 promotes cell migration and inhibits autophagy by mediating PI3K/AKT activation and contributes to the malignant development of gastric cancer. Sci Rep. 2017;7:2886. doi:10.1038/s41598-017-03031-1. PMID:28588255