
ABSTRACT
Macroautophagy is induced under various stresses to remove cytotoxic materials, including misfolded proteins and their aggregates. These protein cargoes are collected by specific autophagic receptors such as SQSTM1/p62 (sequestosome 1) and delivered to phagophores for lysosomal degradation. To date, little is known about how cells sense and react to diverse stresses by inducing the activity of SQSTM1. Here, we show that the peroxiredoxin-like redox sensor PARK7/DJ-1 modulates the activity of SQSTM1 and the targeting of ubiquitin (Ub)-conjugated proteins to macroautophagy under oxidative stress caused by TNFSF10/TRAIL (tumor necrosis factor [ligand] superfamily, member 10). In this mechanism, TNFSF10 induces the N-terminal arginylation (Nt-arginylation) of the endoplasmic reticulum (ER)-residing molecular chaperone HSPA5/BiP/GRP78, leading to cytosolic accumulation of Nt-arginylated HSPA5 (R-HSPA5). In parallel, TNFSF10 induces the oxidation of PARK7. Oxidized PARK7 acts as a co-chaperone-like protein that binds the ER-derived chaperone R-HSPA5, a member of the HSPA/HSP70 family. By forming a complex with PARK7 (and possibly misfolded protein cargoes), R-HSPA5 binds SQSTM1 through its Nt-Arg, facilitating self-polymerization of SQSTM1 and the targeting of SQSTM1-cargo complexes to phagophores. The 3-way interaction among PARK7, R-HSPA5, and SQSTM1 is stabilized by the Nt-Arg residue of R-HSPA5. PARK7-deficient cells are impaired in the targeting of R-HSPA5 and SQSTM1 to phagophores and the removal of Ub-conjugated cargoes. Our results suggest that PARK7 functions as a co-chaperone for R-HSPA5 to modulate autophagic removal of misfolded protein cargoes generated by oxidative stress.
Introduction
Oxidative stress is caused by the imbalance between the production and scavenging of reactive oxygen species (ROS) such as O2− (superoxide radical), ●OH (hydroxyl radical), and H2O2 (hydrogen peroxide) [Citation1–Citation4]. ROS are mainly generated by the mitochondrion [Citation5]. The cellular concentration of ROS is normally maintained by anti-oxidizing agents such as glutathione and superoxide dismutase [Citation6]. However, various stresses can cause the dysfunction of mitochondrial enzymes associated with the loss of membrane potential and electron leakage, leading to the excessive accumulation of ROS [Citation7–Citation9]. The resulting peroxides and free radicals damage cellular components such as proteins, excessively generating misfolded and damaged proteins and their aggregates [Citation10]. These protein substrates are normally tagged with ubiquitin (Ub) by the Ub ligases and degraded by the proteasome complex [Citation11–Citation13]. However, some of the Ub conjugates are prone to aggregation and, thus, excessively accumulate beyond the capacity of the Ub-proteasome system (UPS). Once UPS-mediated protein quality control enters crisis, macroautophagy is induced as a second defense line to degrade misfolded and aggregated proteins. The cargoes of macroautophagy are selectively recognized and sorted out through a poorly understood mechanism, which is thought to involve molecular chaperones such as the heat shock protein HSPA1A/HSP70 [Citation12]. Protein cargoes associated with chaperones are subsequently collected by autophagic receptors such as SQSTM1 [Citation14]. Upon binding to cargo-chaperone complexes, SQSTM1 undergoes self-oligomerization, forming cargo-chaperone-SQSTM1 complexes, leading to the targeting to phagophores and lysosomal degradation. To date, little is known about the crosstalk mechanism that links oxidative stress to macroautophagy. Specifically, it remains poorly understood how the activity of SQSTM1 is modulated by molecular chaperones under oxidative stress.
The N-end rule pathway is a proteolytic system in which a single N-terminal amino acid acts as an N-degron that determines the half-life of a given protein [Citation15–Citation17]. N-terminal degradation determinants include arginine (Arg), lysine (Lys), histidine (His), leucine (Leu), phenylalanine (Phe), tyrosine (Tyr), tryptophan (Trp), and isoleucine (Ile) [Citation18–Citation20]. These degrons are recognized by N-recognins such as the Ub ligases UBR1 and UBR2, leading to ubiquitination and proteasomal degradation [Citation18,Citation21]. The Nt-Arg is the primary degron which can be generated through the Nt-arginylation of Asp, Glu, or Cys by ATE1 R-transferases [Citation22–Citation26]. Nt-arginylation of cytosolic short-lived proteins has been shown to facilitate selective proteolysis via the UPS [Citation27,Citation28]. Recently, we and others showed that a subpopulation of molecular chaperones, which are known to reside within the ER, are Nt-arginylated by cytosolic ATE1 at the Nt-Asp or Nt-Glu of their mature proteins [Citation29–Citation32]. These substrates include HSPA5, CALR, and P4HB. The Nt-arginylation of these ER-residing chaperones is induced by crisis in the UPS, associated with excessive accumulation of misfolded proteins and their aggregates. Among these arginylation substrates, the Ca2+-binding molecular chaperone HSPA5 is a functional homolog of cytosolic HSPA/HSP70 proteins and normally assists with the folding of incoming clients destined to the Golgi-secretory pathway, which account for approximately one third of the human proteome. As a component of ER protein quality control, HSPA5/BiP/GRP78 also recognizes and delivers terminally misfolded clients to ERAD (ER-associated protein degradation), leading to substrate ubiquitination and proteasomal degradation. Upon Nt-arginylation, HSPA5 accumulates in the cytosol [Citation30,Citation31]. The Nt-Arg of the arginylated form of HSPA5, R-HSPA5, in the cytosol binds the ZZ domain of SQSTM1, leading to self-polymerization of SQSTM1 and the formation of cargo-R-HSPA5-SQSTM1 complexes. The Nt-Arg of R-HSPA5 also enhances the interaction of SQSTM1 with MAP1LC3/LC3 (microtubule-associated protein 1 light chain 3) on autophagic membranes, facilitating the targeting of autophagic cargoes to phagophores for subsequent lysosomal degradation. To date, it remains unknown how oxidative stress is linked to the activation of the R-HSPA5-SQSTM1 circuit in autophagic protein quality control.
The peroxiredoxin-like redox sensor PARK7/DJ-1 is one of the 50 most abundant proteins in humans and is involved in cell proliferation, differentiation, transcriptional regulation, oxidative stress response, and mitochondrial homeostasis [Citation33–Citation35]. The misregulation of PARK7 underlies the pathogenesis of many human diseases such as Parkinson disease, amyotrophic lateral sclerosis, infertility, and cancer [Citation36,Citation37]. Studies have shown that PARK7 plays a cytoprotective role in these processes in part as a scavenger in antioxidant stress and in part as a molecular co-chaperone in protein quality control [Citation38–Citation41]. As an abundant antioxidant scavenger of ROS, PARK7 can be oxidized at Cys46, Cys53, and Cys106 [Citation42–Citation46]. Although PARK7 can undergo an oxidation-reduction cycle, the oxidation of all 3 Cys residues is irreversible [Citation46–Citation50]; the metabolic fate of fully oxidized PARK7 remains unknown. PARK7 insufficiency causes a progressive accumulation of ROS that increases the risk factor for cellular dysfunction including mitochondrial damage [Citation43,Citation45,Citation51–Citation55]. As a co-chaperone, PARK7 binds to molecular chaperones, including HSPA1A/HSP90 and STUB1/CHIP, and facilitates the transcription of antioxidant genes [Citation56,Citation57]. Moreover, recent studies showed that PARK7 loss impairs chaperone-mediated autophagy associated with mitochondrial damage [Citation57] and causes the accumulation of misfolded SNCA/α-synuclein and its aggregates [Citation58].
This study stems from our investigation into the protective role of PARK7 against the oxidative stress caused by TNFSF10/TRAIL (tumor necrosis factor [ligand] superfamily, member 10), an anti-cancer agent that selectively kills cancer cells [Citation59]. During this study, we found that PARK7 acts as a co-chaperone-like protein that binds the Ca2+-binding chaperone R-HSPA5, an ER counterpart of cytosolic HSPA. Upon binding to PARK7, preferentially its oxidized form, R-HSPA5 binds SQSTM1 through its Nt-Arg, enhancing the activity of SQSTM1 as an autophagic receptor for misfolded protein cargoes. Our results suggest that PARK7 mediates the crosstalk between oxidative stress and SQSTM1-dependendent autophagic protein quality control in macroautophagy.
Results
PARK7 protects cells from TNFSF10-induced oxidative stress
Studies have shown that TNFSF10 induces oxidative stress in various cell types [Citation59,Citation60]. To extend these results, we treated HCT116 cells with TNFSF10 and measured the level of mitochondrial ROS using MitoSOX Red [Citation61]. As expected, TNFSF10 markedly induced the level of mitochondrial ROS in a dose-dependent manner as determined by fluorescence activated cell sorting (FACS) (Figure 1(a,b)) as well as confocal fluorescence analyses (). TNFSF10 also excessively generated cytosolic ROS when measured using CM-H2-DCFDA (, columns 3 vs. 1) [Citation62]. These results confirm that TNFSF10 induces the production of excessive ROS in both the mitochondrion and cytosol in HCT116 cells.
Figure 1. Protective role of PARK7 in TNFSF10-induced oxidative stress. (a) Cells were treated with various concentrations (1–10 ng/ml) of TNFSF10 for 4 h and labeled with MitoSOX Red. The total fluorescence intensity of oxidized MitoSOX Red was measured using flow cytometry. (b) Quantification of A. Error bars represent the mean ± SEM from 3 separate experiments (*p < 0.05). (c) Cells were stably transfected with shScramble or shPARK7 and treated with 10 ng/ml TNFSF10 for 4 h. Mitochondria and cells were stained with MitoTracker Green and MitoSOX Red, respectively. Mitochondria (green) and mitochondrial superoxide anion (red) were detected using confocal microscopy. Scale bar: 10 μm. (d) Cells were stably transfected with shScramble or shPARK7, followed by treatment for 4 h with 2.5 mM NAC, 10 ng/ml TNFSF10 or in combination. Cells were subsequently stained with CM-H2-DCFDA for 30 min. Green signals indicate ROS in the cytosol. Scale bar: 200 μm. (e) Cells were stably transfected with control shRNA (shScram.) or PARK7 shRNA (shPARK7), followed by the treatment with 10 ng/ml TNFSF10 for 4 h. Cells were subsequently labeled with MitoSOX Red, and MitoSOX Red fluorescence was analyzed using flow cytometry. a.u., arbitrary units. (f) Quantification of E. Error bars represent the mean ± SEM from 3 separate experiments (**p < 0.01).
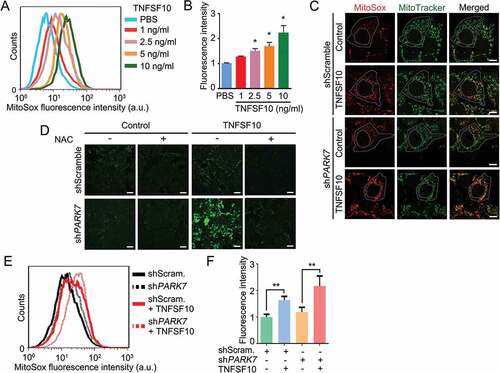
To determine whether PARK7 protects cells from the oxidative stress caused by TNFSF10, we knocked down PARK7 in HCT116 cells using small hairpin RNA (shRNA) and stained the cells with MitoSOX that selectively marks mitochondrial ROS. FACS showed that PARK7-deficient cells spontaneously generated excessive mitochondrial ROS in the absence of TNFSF10 (). ROS was synergistically generated when treated with TNFSF10 (). Confocal microscopy of MitoSOX fluorescence () revealed that PARK7 knockdown cells accumulated excessive mitochondrial O2− (superoxide). Next, we examined the protective role of PARK7 in mitochondrial membrane potential. FACS analysis using the fluorescent cationic dye JC-1 showed that HCT116 cells stably expressing PARK7 shRNA spontaneously lost mitochondrial potential by TNFSF10 treatment (Figure S1(a,b). Thus, PARK7 counteracts TNFSF10-induced excessive formation of ROS as well as mitochondrial dysfunction. Finally, we determined the protective role of PARK7 against excessive cytosolic ROS. An analogous assay using CM-H2-DCFDA also showed that PARK7 knockdown resulted in a striking accumulation of cytosolic ROS (, column 3). Such an induction of ROS was abolished by treatment with the antioxidant N-acetylcysteine (NAC) (, columns 4 vs. 3). These results collectively suggest that PARK7 protects cells from TNFSF10-induced oxidative stress.
Identification of the ER chaperone HSPA5 as an interactor of PARK7
To understand the molecular mechanisms underlying the role of PARK7 in oxidative stress responses, we isolated the proteins that interact with PARK7 by employing tandem affinity purification (TAP) [Citation63]. FLAG-tagged PARK7 (FLAG-PARK7) was overexpressed in HCT116 cells and partially purified using anti-FLAG antibody resin. Co-purified proteins were subjected to liquid chromatography-tandem mass spectrometry (LC-MS/MS). Cells were also treated with TNFSF10 to identify the proteins that selectively interact with PARK7 under oxidative stress. Among the identified proteins, a few were selectively enriched in TNFSF10-treated cells, including the ER-resident chaperone HSPA5 and the cytosolic chaperones HSPA8/HSC70, HSPA1A, and HSP90AA1 as well as the E3 ubiquitin-protein ligase TRIM21 and EIF4B (eukaryotic translation initiation factor 4B) (Figure 2(a), Table S1). In this study, we characterized the interaction of PARK7 with HSPA5, an ER-resident member of the HSPA family, because PARK7 is known as a co-chaperone of HSPA1A [Citation56,Citation64].
To examine the interaction of PARK7 with HSPA5, we performed a co-immunoprecipitation (co-IP) assay using transiently overexpressed FLAG-PARK7 and hemagglutinin (HA)-tagged HSPA5 (HA-HSPA5) in HCT116 cells. The IP of HA-HSPA5 co-precipitated FLAG-PARK7 (). A similar co-IP analysis confirmed that endogenous PARK7 interacted with HSPA5 (). The interaction of endogenous PARK7 with HSPA5 was enhanced when the cells were treated with 5 ng/ml TNFSF10 (). These results suggest that PARK7 interacts with the ER-resident chaperone HSPA5.
Figure 2. PARK7 interacts with R-HSPA5, the Nt-arginylated form of HSPA5. (a) Cells were engineered to stably express either p3x-FLAG or FLAG-tagged PARK7 (FLAG-PARK7) and treated with 5 ng/ml TNFSF10 for 4 h. Cell lysates were immunoprecipitated using anti-FLAG antibody, and the precipitated proteins were visualized using silver staining. (b) Cells were transiently transfected with a plasmid expressing either FLAG-PARK7 or HA-tagged HSPA5. After 48 h, cell lysates were immunoprecipitated with anti-HA antibody, followed by immunoblotting with anti-FLAG or anti-HA antibody (top). The presence of FLAG-PARK7 and HA-HSPA5 in the lysates was verified by immunoblotting (bottom). (c) Cells were treated with 5 ng/ml TNFSF10 for 3 h, and cell lysates were immunoprecipitated with anti-PARK7 antibody or mock antibody (rabbit IgG) followed by immunoblotting with anti-HSPA5 or anti-PARK7 antibody (top). The presence of HSPA5 and PARK7 in the lysates was verified using immunoblotting (bottom). (d) A schematic diagram in which TNFSF10 induces the Nt-arginylation of HSPA5. In this mechanism, newly synthesized HSPA5 translocates into the ER lumen, during which its signal peptide is cleaved off by the signal peptide peptidase, resulting in mature HSPA5. Our results suggest that TNFSF10 induces the cytosolic retrotranslocation and Nt-arginylation of lumenal HSPA5, resulting in cytosolic accumulation of R-HSPA5. (e) HCT116 cells were treated with 10 ng/ml TNFSF10, followed by immunoblotting of R-HSPA5, HSPA5, and ATE1. (f) HCT116 cells were treated with 5 ng/ml TNFSF10 for 4 h. Cell lysates were fractionated to enrich the cytosol, mitochondria, and ER. Fractionated proteins were immunoblotted for R-HSPA5, PARK7, HSPA5, the mitochondrial channel VDAC (voltage dependent anion channel), the ER chaperone CANX (calnexin).
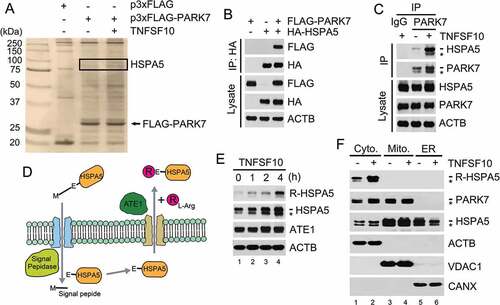
TNFSF10 induces the Nt-arginylation of HSPA5, leading to its cytosolic accumulation and interaction with PARK7
We recently showed that a subpopulation of HSPA5 and other ER-resident molecular chaperones are Nt-arginylated and relocated to the cytosol () [Citation30]. We therefore tested whether PARK7 binds HSPA5 following its Nt-arginylation. Immunoblotting analysis using antibody specific to the Nt-arginylated form of HSPA5, R-HSPA5, showed that HCT116 cells constitutively generated a detectible level of R-HSPA5 (). Moreover, TNFSF10 treatment markedly enhanced the formation of R-HSPA5 (). These results suggest that TNFSF10 induces the Nt-arginylation of HSPA5. Next, to determine whether R-HSPA5 is cytosolic, we fractionated TNFSF10-treated HCT116 cells into the cytosol, mitochondria, and ER using differential centrifugation. R-HSPA5 was mainly retrieved from the cytosol (, lane 1), and its cytosolic level increased by TNFSF10 treatment (, lanes 2 vs. 1). These results suggest that TNFSF10 induces the Nt-arginylation of HSPA5, leading to its cytosolic accumulation.
Given that TNFSF10 induces Nt-arginylation, we assessed the protective role of ATE1 against TNFSF10. Knockdown of ATE1 using siRNA strongly inhibited the formation of R-HSPA5 in TNFSF10-treated cells, which correlated with the increased cleavage of PARP1 (Figure S2(a)). The pro-survival activity of ATE1 was further supported by the MTT (3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide) assay using ATE1 knockdown cells (Figure S2(b)). These results suggest that Nt-arginylation protects the cell from TNFSF10.
To determine whether it is an Nt-arginylated form of HSPA5 that interacts with PARK7, we performed co-IP analyses of HCT116 cells using the R-HSPA5 antibody. The result confirmed that recombinant FLAG-PARK7 interacted with R-HSPA5 (Figure 3(a), lanes 4 vs. 3). The interaction of PARK7 with R-HSPA5 was reproduced in HeLa cells (Figure S3). To further characterize the interaction in vitro, we performed GST affinity-isolation assays using bacterially purified GST-PARK7. The affinity isolation of GST-PARK7 once again co-precipitated R-HSPA5 (, lanes 4 vs 3). These results suggest that PARK7 binds R-HSPA5, possibly as a co-chaperone.
Figure 3. TNFSF10 induces the interaction of oxidized PARK7 with R-HSPA5. (a) HCT116 cells were transfected with a plasmid encoding FLAG or FLAG-PARK7. After 48 h, the cells were treated with 10 ng/ml TNFSF10 for 4 h. Cell lysates were immunoprecipitated with anti-FLAG antibody, followed by immunoblotting with the indicated antibodies. (b) HCT116 cells stably expressing FLAG or FLAG-PARK7 were treated with 5 ng/ml TNFSF10 for 4 h. Cell lysates were incubated with GST-PARK7 proteins for 2 h then immunoprecipitated with glutathione bead, followed by immunoblotting with the indicated antibodies. (c) HCT116 cells were treated with 10 ng/ml TNFSF10 for 4 h. Cell lysates were immunoprecipitated with anti-SQSTM1 antibody, followed by immunoblotting analysis. (d) HCT116 cells were treated with 200 nM bafilomycin A1 (Baf) for 6 h or cultured in the presence of 200 nM bafilomycin A1 for 2 h then additionally treated with 10 ng/ml TNFSF10 for 4 h. Cell lysates were immunoprecipitated with anti-SQSTM1 antibody, followed by immunoblotting analysis. (e) A schematic diagram indicating the interaction between arginylated HSPA5, oxidized PARK7 (oxPARK7), and activated SQSTM1. In this mechanism, HSPA5, PARK7 and SQSTM1 are each modified, and the modification of these 3 components constitutes the complex of 3-way interaction. (f) A schematic diagram in which recombinant Ub-R/V-HSPA5-GFP proteins are processed by a deubiquitination enzyme (DUB). In this mechanism, Ub-R/V-HSPA5-GFP is expressed in HCT116 cells and its ubiquitin is cleaved by DUB and arginine or valine is exposed as a result of cleavage as shown. (g) HCT116 cells were co-transfected with plasmids encoding FLAG-PARK7 and Ub-R-HSPA5-GFP or Ub-V- HSPA5-GFP. After 48 h, the cells were treated with 10 ng/ml TNFSF10 for 4 h. Cell lysates were immunoprecipitated with anti-FLAG antibody followed by immunoblotting with anti-GFP or anti-FLAG antibody.
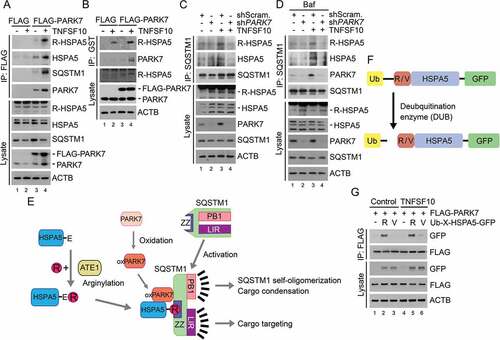
PARK7 enables the chaperone R-HSPA5 to bind SQSTM1
Our earlier work has shown that the Nt-Arg of R-HSPA5 binds the ZZ domain of the autophagic receptor SQSTM1 [Citation30]. We therefore determined whether PARK7 facilitates the binding of R-HSPA5 to SQSTM1. Co-IP analyses showed that the interaction of R-HSPA5 with endogenous SQSTM1 was reinforced by TNFSF10 (, lanes 3 vs. 1). The interaction between endogenous R-HSPA5 and SQSTM1 was reproduced when autophagic flux was blocked using bafilomycin A1 (Baf) (, lanes 3 vs. 1). Given 2 independent interactions between PARK7 and R-HSPA5 () as well as R-HSPA5 and SQSTM1 (), we further asked whether PARK7 also interacts with SQSTM1. Co-IP analyses showed that recombinant FLAG-PARK7 interacted with endogenous SQSTM1 in HCT116 cells (, lanes 4 vs. 3) as well as HeLa cells (Figure S3, lanes 3 vs. 2). A reciprocal co-IP analysis confirmed that the IP of endogenous SQSTM1 pulled down endogenous PARK7 (, lanes 3 vs. 1). These results suggest that TNFSF10 induces a 3-way interaction among PARK7, R-HSPA5, and SQSTM1, possibly forming a stable complex with its protein cargoes ().
The Nt-Arg residue of R-HSPA5 is an anchoring ligand in a 3-way interaction among PARK7, R-HSPA5, and SQSTM1
In the N-end rule pathway, the Nt-Arg of arginylated substrates functions as an N-degron that induces substrate degradation via the UPS or autophagy [Citation16,Citation30,Citation31]. We therefore examined whether the Nt-Arg of R-HSPA5 facilitates the formation of the PARK7-R-HSPA5-SQSTM1 complex. HCT116 cells were engineered to produce HSPA5 proteins bearing Nt-Arg or Nt-Val by expressing Ub-X-HSPA5-GFP (X = Arg or Val). The fusion protein was co-translationally cleaved by deubiquitinating enzymes (DUBs) into the reference Ub and X-HSPA5 () [Citation19]. Co-IP assays showed that PARK7 bound R-HSPA5-GFP (). Importantly, the mutation of Nt-Arg to Nt-Val markedly impaired the interaction between PARK7 and HSPA5 (), suggesting that the Nt-arginylation of HSPA5 facilitates its interaction with PARK7. These results suggest that the Nt-Arg of R-HSPA5 is an anchoring ligand in the formation of the PARK7-R-HSPA5-SQSTM1 complex.
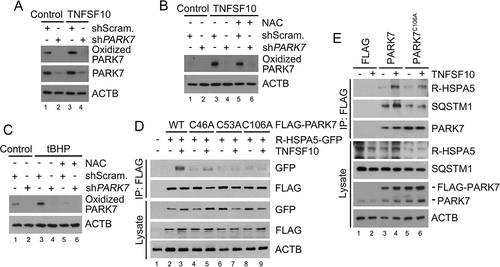
The oxidation of PARK7 facilitates its interaction with R-HSPA5 and SQSTM1
It is known that PARK7 can be oxidized at Cys46, Cys53, and/or Cys106 under oxidative stress, yet its biochemical and physiological importance remain controversial [Citation49]. To determine whether the oxidation of PARK7 is induced by TNFSF10, we used an antibody specific to Cys106-oxidized PARK7. Immunoblotting analyses showed that TNFSF10 induced the oxidation of PARK7 at Cys106 (Figure 4(a)). The oxidation of PARK7 was abolished when ROS was chelated using the anti-oxidant NAC () and facilitated when ROS was excessively generated using tert-butyl hydroperoxide (tBHP) (). These results suggest that PARK7 is oxidized at Cys106 by ROS under oxidative stress. Next, we examined the role of oxidation in the interaction of PARK7 with R-HSPA5. Co-IP analysis showed that the mutation of Cys106 into alanine (Ala) impaired the interaction of FLAG-PARK7 with R-HSPA5-GFP (). A similar impairment in the interaction was observed when Cys46 or Cys53 of PARK7 was mutated into Ala (). Analogous co-IP analyses showed that PARK7C106A failed to properly interact with not only endogenous R-HSPA5 but also endogenous SQSTM1 (). Thus, R-HSPA5 preferentially binds the oxidized form of PARK7. Our results collectively suggest that the 3-way interactions among PARK7, R-HSPA5, and SQSTM1 in a complex are synergistically stabilized by the Nt-arginylation of R-HSPA5 and the oxidation of PARK7.
Figure 4. PARK7 is oxidized by TNFSF10 and the oxidation of PARK7 is required for the interaction of R-HSPA5 and SQSTM1 in HCT116 cells treated with TNFSF10. (a) HCT116 cells were treated with 10 ng/ml TNFSF10 for 4 h, followed by immunoblotting with specific antibodies for oxidized PARK7 and PARK7. (b) HCT116 cells were treated with 10 ng/ml TNFSF10 for 4 h or co-treated with 10 ng/ml TNFSF10 and 2.5 mM NAC, followed by immunoblotting with oxidized PARK7 antibody. (c) HCT116 cells were treated with 250 μM tert-butyl hydroperoxide (tBHP) for 3 h or co-treated with 250 μM tBHP and 2.5 mM NAC, followed by immunoblotting with oxidized PARK7 antibody. (d) HCT116 cells were co-transfected with plasmids encoding Ub-R-HSPA5-GFP and one of the following: FLAG-tagged wild-type PARK7 or its C46A, C53A, and C106A mutants. After 48 h, the cells were treated with 10 ng/ml TNFSF10 for 4 h. Cell lysates were immunoprecipitated with anti-FLAG antibody followed by immunoblotting with anti-GFP or anti-FLAG antibody. (e) HCT116 cells were transfected with a plasmid encoding FLAG, FLAG-PARK7, or FLAG-PARK7C106A. After 48 h, cells were treated with 20 ng/ml TNFSF10 for 2 h. Cell lysates were immunoprecipitated with the anti-FLAG antibody and then immunoblotted with the indicated antibodies.
PARK7 enhances the autophagic activity of SQSTM1
We determined whether PARK7 enhances the interaction of R-HSPA5 with SQSTM1 using co-IP analyses in control and PARK7 knockdown HCT116 cells. The result showed that PARK7 depletion impaired the binding of R-HSPA5 to SQSTM1 (, lanes 4 vs. 3). A similar result was observed under autophagic inhibition (, lanes 4 vs. 3). These results suggest that PARK7 acts as a cofactor (or co-chaperone) that facilitates the interaction of R-HSPA5 with SQSTM1.
Next, we asked whether PARK7 facilitates in vivo the autophagic targeting of R-HSPA5 via SQSTM1. Immunostaining analysis showed that TNFSF10 induced the formation of cytosolic puncta positive for R-HSPA5 (Figure 5(a)). When autophagy flux was blocked, R-HSPA5 formed larger punctate structures with sizes of 0.5–1 μm, most of which colocalized with SQSTM1 puncta. Notably, PARK7 knockdown strongly inhibited the formation of R-HSPA5 puncta but also their colocalization with SQSTM1 puncta (, column 4 vs 3). Whereas approximately 30% of SQSTM1 puncta were positive for R-HSPA5 in control cells, such colocalization was almost non-detectable in PARK7 knockdown cells (). Despite these alternations in SQSTM1 puncta formation, we observed no significant relevance with the mRNA expression of SQSTM1 under TNFSF10 treatment (Figure S4). These results suggest that PARK7 facilitates the autophagic targeting of R-HSPA5 in vivo through its interaction with SQSTM1.
Figure 5. PARK7 is required for the autophagic targeting of R-HSPA5 and SQSTM1 in HCT116 cells treated with TNFSF10. (a) Cells were treated with 200 nM bafilomycin A1 for 6 h or 10 ng/ml TNFSF10 for 4 h. Alternatively, the cells cultured in the presence of 200 nM bafilomycin A1 for 2 h were additionally treated with 10 ng/ml TNFSF10 for 4 h. Immunostaining analysis was performed using antibodies to R-HSPA5 (red) and SQSTM1 (green), followed by confocal microscopy. Scale bar: 10 μm. (b) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with 10 ng/ml TNFSF10 for 4 h or 200 nM bafilomycin A1 (Baf) for 2 h and additionally treated with 10 ng/ml TNFSF10 for 4 h as described in A. (c) Quantification of R-HSPA5 cytosolic puncta in B. Error bars represent the mean ± SEM from each cell (***p < 0.001, n = 50). (d) Quantification of SQSTM1 cytosolic puncta in B. Error bars represent the mean ± SEM from each cell (***p < 0.001, n = 50). (e) Quantification of the colocalization of SQSTM1 cytosolic puncta in B with R-HSPA5 puncta. Error bars represent the mean ± SEM from each cell (***p < 0.001, n = 50). (f) Cells were treated with 10 ng/ml TNFSF10 for 4 h or 200 nM bafilomycin A1 for 6 h. Alternatively, the cells cultured in the presence of 200 nM bafilomycin A1 for 2 h were additionally treated with 10 ng/ml TNFSF10 for 4 h. The SQSTM1 and PARK7 oligomerization assay was followed by nonreducing SDS-PAGE and immunoblotting using antibodies to SQSTM1 and PARK7. (g) Cells were treated with tBHP at the indicated concentration. PARK7 oligomerization assay was followed by nonreducing SDS-PAGE and immunoblotting using an antibody to PARK7.
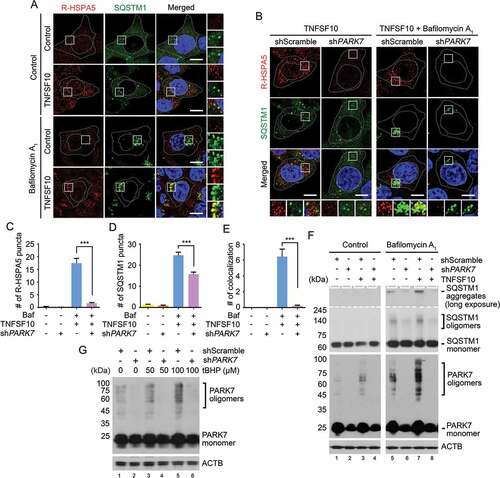
As an autophagic receptor, cargo-loaded SQSTM1 undergoes self-polymerization to facilitate the condensation of cargoes and their delivery to phagophores for lysosomal degradation [Citation65–Citation67]. To determine whether PARK7 induces the self-polymerization of SQSTM1, we performed aggregation analyses. The extracts from TNFSF10-treated HCT116 cells were separated using non-reducing SDS-PAGE, in which disulfide bond-linked oligomers and aggregates migrate as high molecular weight species. The result suggested that a subpopulation of SQSTM1 as well as PARK7 existed as high molecular-weight species (). The formation of PARK7 oligomers was similarly induced by tBHP in a dose-dependent manner (). These oligomeric species accumulated when autophagic flux was blocked, indicating their lysosomal degradation (). Notably, PARK7 knockdown markedly inhibited the formation of not only PARK7 oligomers but also SQSTM1 oligomers (). These results together suggest that PARK7 facilitates the self-polymerization of SQSTM1.
Our results suggest that PARK7 enables R-HSPA5 to activate the autophagic activity of SQSTM1 in macroautophagy. However, as SQSTM1 delivers almost all the cargo types, at least in part independent of R-HSPA5, PARK7 is likely to be responsible for a subset of SQSTM1 functions. We therefore determined to what degree PARK7 loss affects SQSTM1 activities. Although there were variations, there was a clear tendency that SQSTM1 puncta were markedly decreased by PARK7 knockdown in HCT116 cells treated with TNFSF10 (, columns 4 vs. 3), suggesting that the PARK7-R-HSPA5 complex enhances the activity of SQSTM1 in vivo. Consistently, immunostaining analysis with an antibody specific to LC3-II on autophagic membranes revealed that PARK7-deficient cells contained a significantly smaller number of autophagosomes as compared with control cells (Figure 6(a-c)). Moreover, the rarely found LC3-II signals appeared to be abnormal and showed a morphology different from that of typical autophagosomes (). Whereas in control cells, the majority of SQSTM1 puncta colocalized with LC3-II+ autophagosomes, those scarcely found SQSTM1 puncta in PARK7-deficient typically lacked LC3-II signals (). Finally, we also monitored cargo-loaded autophagosomes in the process of fusion with lysosomes by staining PLEKHM1 as a marker of autophagosome-lysosome fusion [Citation68,Citation69]. PARK7-deficient cells contained a significantly reduced number of PLEKHM1 puncta (). These results suggest that PARK7 loss impairs the formation of cargo-loaded autophagosomes.
Figure 6. The autophagic targeting of R-HSPA5 and SQSTM1 is impaired in PARK7-deficient cells. (a) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with 200 nM bafilomycin A1 (Baf) for 6 h and immunostained for LC3-II (red) or PARK7 (green). LC3-II-positive autophagic vacuoles were examined using confocal microscopy. Scale bar: 10 μm. (b) Quantification of the number of LC3-II puncta in A. Error bars represent the mean ± SEM from each cell (***p < 0.001, n = 50). (c) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with 200 nM bafilomycin A1 for 2 h, followed by the treatment with 10 ng/ml TNFSF10 for 4 h. The cells were immunostained for LC3-II (red) or SQSTM1 (green). Puncta formation and colocalization of LC3-II and SQSTM1 were examined using confocal microscopy. Scale bar: 10 μm. (d) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with 200 nM bafilomycin A1 for 2 h, followed by treatment with 10 ng/ml TNFSF10 for 4 h. The cells were immunostained for PLEKHM1 (green). Puncta formation of PLEKHM1 was examined using confocal microscopy. Scale bar: 10 μm. (e) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with 200 nM bafilomycin A1 (Baf) for 6 h and immunoblotted with the indicated antibodies. (f) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with 200 nM bafilomycin A1 for 6 h and immunostained for WIPI2 (red). WIPI2-positive autophagic vacuoles were examined using confocal microscopy. Scale bar: 10 μm.
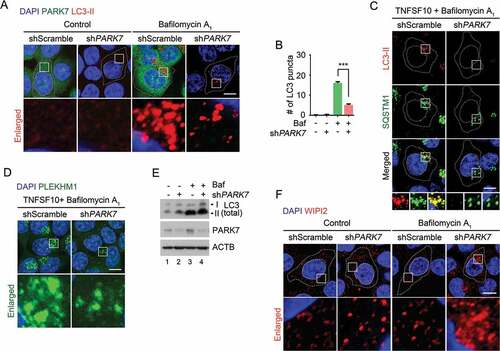
To exclude the possibility that PARK7 loss impairs de novo phagophore biogenesis, we monitored the level of LC3 in control and PARK7-deficient cells. Immunoblotting analysis showed that PARK7 knockdown did not reduce, but even slightly increased, the synthesis of LC3-I and its conversion into LC3-II (). Consistently, immunostaining analysis of WIPI2, a marker of omegasome formation, also showed a comparable, or even enhanced, formation of WIPI2 punctate signals in PARK7-deficient cells (). Our results collectively suggest that PARK7 is not required for phagophore biogenesis but facilitates SQSTM1-dependent delivery of cargoes to phagophores. It is to be determined whether the apparent increases of LC3 synthesis and WIPI2 puncta formation are secondary to oxidative stress or represent the role of PARK7 in the modulation of autophagy.
PARK7 plays a role in autophagic proteolysis
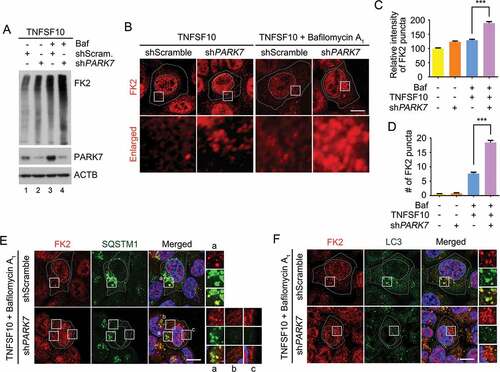
In autophagic protein quality control, SQSTM1 is a general receptor for ubiquitinated protein cargoes destined for lysosomal degradation [Citation30]. We therefore examined the role of PARK7 in autophagic targeting of Ub-conjugated proteins. Immunoblotting analysis of TNFSF10-treated HCT116 cells showed that PARK7 knockdown cells excessively accumulated Ub-conjugated proteins (Figure 7(a), lanes 2 vs. 1). Immunostaining analysis confirmed that unprocessed Ub conjugates accumulated in the cytosol of PARK7 knockdown cells (). (It should be noted that the nuclear signals represent Ub-conjugated histones.) These cytosolic Ub puncta further accumulated upon autophagic inhibition (), indicating their lysosomal degradation. In control cells, the majority (~ 90%) of cytosolic Ub puncta were in the process of autophagic targeting as evidenced by colocalization with SQSTM1 () as well as LC3 (). In sharp contrast, those excessively accumulated Ub puncta were not properly targeted to autophagic vacuoles as judged by poor colocalization with SQSTM1 (, regions i-iii) as well as LC3 (). These results suggest that PARK7 plays a role in the autophagic protein quality control of ubiquitinated proteins destined for lysosomal degradation.
Figure 7. PARK7 is required for the removal of Ub conjugates in HCT116 cells treated with TNFSF10. (a) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with the presence of 200 nM bafilomycin A1 (Baf) for 2 h and were additionally treated with 10 ng/ml TNFSF10 for 4 h. Cell lysates were subjected to immunoblotting analysis using FK2 antibody specific to Ub-conjugated proteins. (b) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with 10 ng/ml TNFSF10 for 4 h alone or were pretreated with bafilomycin A1 (Baf) for 2 h, followed by the treatment with 10 ng/ml TNFSF10 for 4 h, followed by immunostaining analysis using FK2 antibody (red). Scale bar: 10 μm (c) Quantification of the intensity of Ub-positive puncta as visualized using FK2 antibody. Error bars represent the mean ± SEM from each cells (***p < 0.001, n = 50). (d) Quantification of the number of Ub-positive puncta as visualized using FK2 antibody. Error bars represent the mean ± SEM from each cell (***p < 0.001, n = 50). (e) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with 200 nM bafilomycin A1 for 2 h, followed by treatment with 10 ng/ml TNFSF10 for 4 h. The cells were immunostained using FK2 (red) or SQSTM1 (green) antibodies. Puncta formation and colocalization of FK2 and SQSTM1 signals were examined using confocal microscopy. Scale bar: 10 μm. (f) Same as E except that FK2 (red) or LC3 (green) antibodies were used.
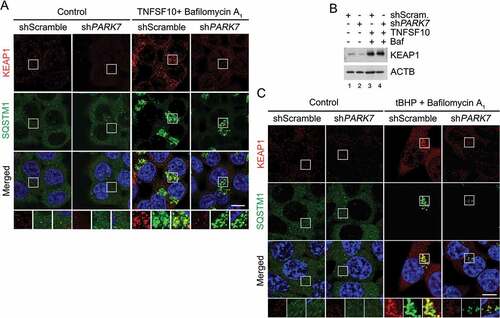
The transcription factor NFE2L2 induces the expression of antioxidant genes in response to oxidative stress [Citation70–Citation72]. In normal conditions, NFE2L2 is physically retained by the E3 ligase KEAP1. Upon oxidative stress, KEAP1 is hijacked by SQSTM1 and degraded by lysosomes, leading to NFE2L2 dissociation and the transcription of its target genes [Citation70–Citation72]. To characterize the role of PARK7 in regulated SQSTM1-dependent proteolysis of normally folded proteins, we monitored the autophagic targeting of KEAP1. Immunostaining colocalization analysis showed that TNFSF10 induced the targeting of KEAP1 to SQSTM1+ autophagic vacuoles (Figure 8(a)). Notably, PARK7 knockdown markedly impaired the formation of KEAP1+ SQSTM1+ puncta () without a marked difference in the steady-state level as determined by immunoblotting analysis (). A similar result, i.e., the failure to form KEAP1+ SQSTM1+ puncta was obtained when PARK7 knockdown cells were treated with tBHP (). These results further support the conclusion that PARK7 plays a role in SQSTM1-dependent autophagic proteolysis.
Figure 8. PARK7 is required for the targeting of KEAP1 to autophagy in HCT116 cells treated with oxidative stressors. (a) HCT116 cells stably expressing scrambled shRNA or shPARK7 were treated with 200 nM bafilomycin A1 for 2 h, followed by treatment with 10 ng/ml TNFSF10 for 4 h. The cells were immunostained using KEAP1 (red) or SQSTM1 (green) antibodies. Puncta formation and colocalization of KEAP1 and SQSTM1 signals were examined using confocal microscopy. Scale bar: 10 μm. (b) HCT116 cells cultured in the presence of 200 nM bafilomycin A1 (Baf) for 2 h were additionally treated with 10 ng/ml TNFSF10 for 4 h. The cells were immunoblotted for anti-KEAP1 antibody. (c) Cells cultured in the presence of 200 nM bafilomycin A1 for 3 h were additionally treated with 250 μM tBHP for 3 h. The cells were immunostained for KEAP1 (red) and SQSTM1 (green), and examined using confocal microscopy. Scale bar: 10 μm.
Discussion
Selective macroautophagy is a cellular defense system that mediates the degradation of cytotoxic materials in response to various stresses. Autophagic cargoes are collected by specific receptors such as SQSTM1 and delivered to the phagophore for lysosomal degradation. An outstanding question in selective macroautophagy is how cells sense different types of stresses and induce autophagy in a timely manner and only when necessary.
In this study, we find that a poorly characterized peroxiredoxin-like protein, PARK7, plays an unexpected role, i.e., the modulation of selective macroautophagy under oxidative stress and other cytotoxicity caused by TNFSF10 (see ). We show that upon TNFSF10 treatment, the ER-resident molecular chaperone HSPA5 was Nt-arginylated, leading to the cytosolic accumulation of its arginylated form, R-HSPA5 (). PARK7 bound cytosolic R-HSPA5 and facilitated the targeting of R-HSPA5 to the phagophore ( and ). As R-HSPA5 is a HSPA family member whose known functions include the recognition and binding of misfolded proteins and their aggregates, we suggest that PARK7 is required for the delivery of R-HSPA5-cargo complexes to phagophores as part of protein quality control (). This possibility agrees with our recent finding that R-HSPA5 plays a role in the autophagic removal of misfolded proteins under proteasomal inhibition [Citation30]. Given that the chaperone functions of HSPA proteins are typically modulated by co-chaperones and other modulators, the modulators of cytosolic R-HSPA5 remain unknown. We propose that PARK7 is one such modulator that regulates autophagic targeting and other functions of R-HSPA5 and facilitates the autophagic targeting of misfolded or damaged proteins excessively generated under TNFSF10-induced stress conditions ().
Figure 9. Hypothetical model for the role of PARK7 in autophagic protein quality control. In this model, TNFSF10 causes mitochondrial misregulation and oxidative stress associated with the excessive generation of ROS. This causes the formation of cytosolic misfolded proteins that are tagged with Ub but cannot be degraded by the UPS (step 1). In response to the proteotoxicity, cells induce autophagic protein quality control, which involves the Nt-arginylation of the ER-resident HSPA5 (step 2). In parallel, PARK7 is oxidized (step 3). The resulting PARK7 binds R-HSPA5 as its cofactor/co-chaperone that facilitates the ability of association with binding Ub-tagged misfolded protein clients (step 4) and enhances the ability of R-HSPA5 to activate SQSTM1 (step 5). Our earlier work [30] has shown that the Nt-Arg of R-HSPA5 binds the ZZ domain of SQSTM1 and allosterically activates the conformation of SQSTM1, exposing PB1 and LIR domains of SQSTM1 (step 6). This enables PB1-mediated self-aggregation of SQSTM1 along with R-HSPA5 and Ub-conjugated misfolded cargoes (step 7) and LIR-mediated interaction with LC3 (step 8), facilitating the autophagic removal of cytotoxic misfolded proteins and their aggregates. In this R-HSPA5-SQSTM1 circuit, PARK7 acts as a cofactor/co-chaperone of R-HSPA5 to modulate SQSTM1-dependent macroautophagy under TNFSF10-induced stresses and possibly other types of stress as well.
![Figure 9. Hypothetical model for the role of PARK7 in autophagic protein quality control. In this model, TNFSF10 causes mitochondrial misregulation and oxidative stress associated with the excessive generation of ROS. This causes the formation of cytosolic misfolded proteins that are tagged with Ub but cannot be degraded by the UPS (step 1). In response to the proteotoxicity, cells induce autophagic protein quality control, which involves the Nt-arginylation of the ER-resident HSPA5 (step 2). In parallel, PARK7 is oxidized (step 3). The resulting PARK7 binds R-HSPA5 as its cofactor/co-chaperone that facilitates the ability of association with binding Ub-tagged misfolded protein clients (step 4) and enhances the ability of R-HSPA5 to activate SQSTM1 (step 5). Our earlier work [30] has shown that the Nt-Arg of R-HSPA5 binds the ZZ domain of SQSTM1 and allosterically activates the conformation of SQSTM1, exposing PB1 and LIR domains of SQSTM1 (step 6). This enables PB1-mediated self-aggregation of SQSTM1 along with R-HSPA5 and Ub-conjugated misfolded cargoes (step 7) and LIR-mediated interaction with LC3 (step 8), facilitating the autophagic removal of cytotoxic misfolded proteins and their aggregates. In this R-HSPA5-SQSTM1 circuit, PARK7 acts as a cofactor/co-chaperone of R-HSPA5 to modulate SQSTM1-dependent macroautophagy under TNFSF10-induced stresses and possibly other types of stress as well.](/cms/asset/685048f2-96c6-4b61-8315-e626f14ea94c/kaup_a_1491212_f0009_c.jpg)
Although the functions of SQSTM1 as an autophagic receptor have been extensively characterized, little is known about how SQSTM1 is activated when autophagic cargoes accumulate in the cell beyond the normal capacity of the UPS [Citation73,Citation74]. We recently showed that the Nt-Arg residue of cytosolic R-HSPA5 binds the ZZ domain of SQSTM1 and allosterically induces a conformational change of SQSTM1, facilitating the self-oligomerization of SQSTM1. In the current study, we showed that PARK7 interacted with SQSTM1, possibly indirectly through R-HSPA5 (). Given the demonstrated function of R-HSPA5 to activate SQSTM1, one would speculate that the PARK7-R-HSPA5 complex functions as an activator of SQSTM1. Consistently, our immunostaining data showed that the formation of SQSTM1 cytosolic puncta, as an indicator of its autophagic targeting, was significantly reduced in the absence of PARK7 (). Not surprisingly, the effect of PARK7 loss in the functions of SQSTM1 is significant but somewhat limited, suggesting that there are other ways to activate SQSTM1. Indeed, recent studies have shown that SQSTM1 can be activated through the phosphorylation of its S349 [Citation75] or S409 residues in response to the accumulation of misfolded proteins and their aggregates [Citation73].
In protein quality control, soluble misfolded proteins are ubiquitinated and degraded by the UPS [Citation14]. However, if ubiquitinated substrates excessively accumulate under stress, autophagy is activated as an alternative proteolytic system. It is therefore reasonable to speculate that PARK7 activates the R-HSPA5-SQSTM1 circuit, contributing to the removal of ubiquitin-conjugated misfolded proteins. Given that the oxidized form of PARK7 preferentially interacts with R-HSPA5 (), the oxidation of PARK7 may be one means to sense oxidative stress in protein quality control. Indeed, we found that PARK7 loss led to the accumulation of ubiquitin-conjugates (). It remains to be determined which step of proteolysis is facilitated by PARK7.
TNFSF10 is an anticancer agent that kills cancer cells through cell surface-associated proapoptotic receptors [Citation76,Citation77] and mitochondrial dysfunction [Citation78,Citation79]. A few isolated studies also reported that mitochondrial dysfunction correlates with oxidative stress as well as the transcriptional induction of ER stress proteins [Citation80]. In this study, we confirmed and extended these results in HCT116 cells by finding that TNFSF10 caused oxidative stress in HCT116 cells, which correlated with its Nt-arginylation and the cytosolic relocalization of the ER lumenal chaperone HSPA5 (). Notably, it is known that PARK7 plays a protective role in all of these stresses caused by TNFSF10, such as apoptosis [Citation81], but a remaining question to be addressed is whether all these protective activities represent the primary functions of PARK7 or secondary effects. It also should be determined to what degree the interaction of PARK7 with R-HSPA5 contributes to the protective activities of PARK7.
PARK7 has been implicated as an atypical peroxiredoxin-like peroxidase whose oxidization contributes to anti-oxidative responses to mitochondrial ROS [Citation45,Citation47,Citation82]. Extensive studies have shown that PARK7 is involved in many key cellular processes, including cell proliferation, differentiation, transcriptional regulation, oxidative stress protection, and mitochondrial function maintenance [Citation33–Citation36,Citation57,Citation83]. Deregulation of PARK7 has been implicated in the pathogenesis of many human diseases such as Parkinson disease, amyotrophic lateral sclerosis, infertility, and cancer [Citation36,Citation37,Citation44]. To date, the molecular mechanisms underlying these functions of PARK7 still remain unclear. In this study, we revealed that the PARK7-R-HSPA5-SQSTM1 circuit plays a role in a seemingly unrelated process, i.e., the modulation of selective autophagy, a major cellular defense system that removes cytotoxic materials such as misfolded proteins and damaged mitochondria. For example, as manifested by its name, the mutation of PARK7 has been linked to Parkinson disease [Citation44]. We suggest that these functions of PARK7 in physiological and pathological processes should be reevaluated with regard to its essential role in the modulation of selective autophagy.
The N-end rule pathway has been characterized as a proteolytic system that mediates selective degradation of short-lived regulatory proteins in normal conditions. In this mechanism, N-recognins recognize and directly bind N-degrons such as Nt-Arg and promote ubiquitination and proteasomal degradation. We recently found that a set of ER-resident proteins are Nt-arginylated under proteasomal inhibition and that the Nt-Arg of R-HSPA5 (and possibly other ER-resident proteins) can induce proteolysis via SQSTM1-dependent proteolysis. This N-end rule pathway now has PARK7 as a cofactor that modulates the activity of R-HSPA5 as well as SQSTM1. Given that SQSTM1 directly binds Nt-Arg, this substrate receptor can be defined as an autophagic N-recognin. How N-degrons modulate biological processes in response to various cellular stresses remains to be further investigated.
Materials and methods
Cell culture
park7−/− and corresponding wild-type mouse embryonic fibroblast (MEF) cell lines were provided by Dr. Mark R. Cookson (NIH, Washington DC, USA). MEFs were maintained in Dulbecco’s modified Eagle’s medium with the addition of 55 µM β-mercaptoethanol (Thermo Fisher Scientific, 21,985,023). Human colon cancer HCT116 cells (Korea Cell Line Bank, 10,247) and human cervical cancer HeLa cells (ATCC®, CCL-2™) were cultured in McCoy’s 5A medium (Thermo Fisher Scientific, 1,600,082) and Dulbecco’s modified Eagle’s minimum essential medium (Thermo Fisher Scientific, 11,965,092), respectively. The media contained 10% fetal bovine serum (Thermo fisher scientific, SH3007003,), 1 mM L-glutamine, and 26 mM sodium bicarbonate. The dishes containing cells were kept in a 37°C humidified incubator with 5% CO2.
Antibodies and reagents
The following primary antibodies were used: anti-PARK7(Abcam, ab76008; Cell Signaling Technology, 5933; Santa Cruz Biotechnology, sc-55,572); anti-MAP1LC3A/B (MBL, M152-3; Sigma, L7543); anti-SQSTM1 (Abcam, ab56416, ab91526; Santa Cruz Biotechnology, sc-25,575); anti-HSPA5 (Abcam, ab21685; Cell Signaling Technology, 3183); anti-WIPI2 (Abcam ab105459); anti-FK2 (Enzo Life Sciences, BML-PW8810); anti-GFP (Cell Signaling Technology, 2555); anti-FLAG (Cell Signaling Technology, 2368; Sigma, F1804); anti-HA (Cell Signaling Technology, 2367); anti-VDAC (Cell Signaling Technology, 4866); anti-CANX (Cell Signaling Technology, 2433); anti-PARP1 (Cell Signaling Technology, 9542); anti-ACTB (MP Biomedicals, 0869100; Sigma, A1978). The following secondary antibodies were used in immunoblotting and immunostaining: anti-mouse-IgG-HRP (Santa Cruz Biotechnology, sc-2005); anti-rabbit-IgG-HRP (Santa Cruz Biotechnology, sc-2004); goat anti-rabbit or mouse IgG with Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific, A-11,034, A-21,429, A-11,029, A-21,424) The autophagy inhibitor bafilomycin A1 was purchased from Sigma (B1793). For production of human TNFSF10, a human TNFSF10 cDNA fragment (amino acids 114–281) obtained by RT-PCR was cloned into a pET-23d (Novagen, 69,748-3CN) plasmid, and His-tagged TNFSF10 protein was purified using the Qiagen express protein purification system (Qiagen, 30,210).
Production of anti-R-HSPA5 antibody
Rabbit polyclonal R-HSPA5 antibody specific for the arginylated form of HSPA5 was raised using the peptide sequence REEEDKKEDVGC corresponding to the N-terminal sequence of the arginylated mature protein through a custom service at AbFrontier Inc. as previously described [Citation30]. R-HSPA5 antibody is now commercially available at AbFrontier Inc. (AR05-PA0001) and EMD Millipore (ABS2103).
Plasmids, cloning, and mutagenesis
HSPA5 tagged with HA, PARK7 tagged with GFP, and PARK7 tagged with GST were cloned. HSPA5 tagged with HA was cloned in pCMV-HA (Sigma, OGS3215), PARK7 tagged with GFP was cloned in pEGFP-N1 (Clontech, 6085–1), and PARK7 tagged with GST was cloned in pGEX (GE Healthcare Life Sciences, 28,954,549). Mutagenesis of PARK7 on the cysteine sites was performed using the QuikChange II XL mutagenesis kit (Agilent Technologies, 200,522). The primers used for mutagenesis on human PARK7 of the 3 cysteine residues in position 46, 53, and 106 were the following: for the mutation of alanine in position 46: 5′- GAAAAGACCCAGTACAGgcTAGCCGTGATGTGGTC-3′ (the mutated nucleotide is lower case); for the mutation of alanine in position 53: 5′- CCGTGATGTGGTCATTgcTCCTGATGCCAGCCTTG-3′ (the mutated nucleotide is in lower case); for the mutation of alanine in position 106: 5′- CCTGATAGCCGCCATCgcTGCAGGTCCTACTGCTC-3′ (the mutated nucleotide is lower case).
Stable transfection and generation of stable cell lines
PARK7 expression plasmids (p3x FLAG-PARK7) were kindly provided by Dr. Guanghui Wang (University of Science and Technology of China). HCT116 cells were transfected with p3x FLAG-PARK7or p3x FLAG-CMV10 mock vector (Sigma, E7658) using Lipofectamine 2000 reagent according to the manufacturer’s protocol (Thermo Fisher Scientific, 11668019). Cells were selected with 1 mg/ml G418 (Merck Millipore, 345810) for 2 weeks, and 5 clones were pooled and then maintained in complete medium containing 500 μg/ml G418. Stable PARK7 knockdown and scramble control cells were produced by transducing HCT116 cells with lentivirus encoding shRNA against PARK7 (Santa Cruz Biotechnology, sc-37080-V) and nonspecific shRNA, respectively (Santa Cruz Biotechnology, sc-108080). Transduced cells were selected with 5 µg/ml puromycin (Merck Millipore, 540411) and maintained in complete media.
PARK7 complex purification and mass spectrometry analysis
A detailed TAP procedure has been described previously [Citation63]. Briefly, FLAG-tagged PARK7 was cloned into p3x FLAG-CMV10 vector. HCT116 cells were used to generate a stable cell line with p3x FLAG-PARK7 expression. A p3x FLAG stable HCT116 cell line was also generated as a control. FLAG-PARK7 and the protein complexes were sequentially immunoprecipitated with anti-FLAG M2 monoclonal antibody (mAb)-conjugated agarose beads (Sigma, A4596). A small portion of FLAG-affinity-purified materials was separated using 12% SDS-PAGE and was silver stained. In-gel digestion using trypsin (Promega, V5280) was performed manually as described by Shevchenko [Citation84]. The digested tryptic peptides were desalted using a Ziptip C18 (Sigma, Z720046). Peptides were eluted from the Ziptip with 0.5 μL of matrix solution (α-cyano-4-hydroxycinnamic acid 5 mg/mL in 50% acetonitrile, 0.1% trifluoroacetic acid, 25 mM ammonium bicarbonate) and spotted on a MALDI plate. MALDI-TOF mass spectra were acquired in reflectron positive ion mode, averaging 4000 laser shots per spectrum. LC-MS/MS analysis was performed on an LTQ mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray source (New Objective,) in the Mass Spectrometry Facility of the University of Pittsburgh. The Scaffold Viewer (Proteome Software) database search engine was used to match tandem mass spectra to the identified peptide sequences.
Subcellular fractionation
A detailed subcellular fractionation procedure is described in a previous study [Citation85]. Cells (109) were harvested, washed by centrifugation at 500 xg for 5 min with phosphate-buffered saline (PBS: 1.55 mM KH2PO4, 155 mM NaCl, 2.7 mM Na2HPO4-7H2O, pH 7.4; supplemented with 2 mM Na3VO4, 2 mM NaF when preservation of protein phosphorylation states was required), resuspended in homogenization buffer (225 mM mannitol [Sigma, M4125], 75 mM sucrose [Sigma, S0389], 30 mM Tris-HCl, pH 7.4, 0.1 mM EGTA, 0.1 mM PMSF), and gently disrupted by Dounce homogenization (Glass/Teflon Potter Elvehjem homogenizers, and 30 strokes). The homogenate was centrifuged twice at 600 xg for 5 min to remove nuclei and unbroken cells, and then the supernatant was centrifuged at 10,300 xg for 10 min to pellet crude mitochondria. The resultant supernatant was centrifuged at 20,000 xg for 30 min at 4°C. The pellet consisted of lysosomal and plasma membrane fractions. Further centrifugation of the obtained supernatant at 100,000 xg for 90 min (70-Ti rotor, Beckman, Milan, Italy) at 4°C resulted in the isolation of ER (pellet) and cytosolic fraction (supernatant). The crude mitochondrial fraction, resuspended in isolation buffer (250 mM mannitol, 5 mM HEPES, pH 7.4, 0.5 mM EGTA), was subjected to Percoll gradient centrifugation (Percoll medium: 225 mM mannitol, 25 mM HEPES, pH 7.4, 1 mM EGTA, 30% vol:vol Percoll [Sigma, P1644]) in a 10-ml polycarbonate ultracentrifuge tube. After centrifugation at 95,000 xg for 30 min (SW40 rotor, Beckman), a dense band containing purified mitochondria was recovered approximately 3/4 down the tube, washed by centrifugation at 6,300 xg for 10 min to remove the Percoll, and finally resuspended in isolation medium. When preservation of protein phosphorylation states was required, immediately after the recovery, 2 mM Na3VO4 and 2 mM NaF were added to each fraction.
Co-immunoprecipitation
Co-immunoprecipitation was carried out using protein G-coated sepharose beads (GE Healthcare, 28,944,008) following the manufacturer’s instructions. For whole-cell extracts, cells were lysed in buffer containing 30 mM Tris-HCl (Duchefa Biochemie, T1501), pH 7.4, 50 mM NaCl (Duchefa Biochemie, S0520), 1% NP-40 (Sigma, 74,385) and cleared by centrifugation. Protein extractions in the ER fraction were carried out by adding 50 mM NaCl, 1% NP-40 to the homogenization buffer. All the buffers were supplemented with protease and phosphatase inhibitors (2 mM Na3VO4 [Sigma, S6508], 2 mM NaF [Sigma, S7920], 1 mM PMSF [Sigma, P7626], protease inhibitor cocktail [Sigma, P8340]). Extracted proteins (1000 μg) were first precleared by incubating lysates with sepharose beads for 1 h at 4°C and the supernatant (referred to as Input) was incubated overnight with antibodies at 4°C. Precipitation of the immune complexes was carried out for 4 h at 4°C. Afterwards, beads were washed with 50 mM Tris-HCl, pH 7.4, 0.1% NP-40 4°C supplemented with phosphatase inhibitors and PMSF. Samples were processed by SDS-PAGE and analyzed by standard western blotting technique.
Immunostaining assay
Cells were washed with PBS twice, fixed in 4% paraformaldehyde at room temperature for 15 min, incubated in blocking solution (0.5% BSA [Sigma, A7906] or 5% goat serum [Abcam, ab7481] in PBS), and followed by incubation with primary antibodies or 200 nM MitoTracker® Green (Thermo Fisher Scientific, M7514). Subsequently, goat anti-rabbit or mouse IgG with Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific) were used. Images were acquired using a 488-nm argon-ion laser and a 555-nm argon-ion laser. For confocal microscopy, a LSM700 laser scanning confocal microscope (Zeiss, Seoul, Korea) was used and processed using ZEN 2012 (Version 1.1.13064.302) under a 63X water immersion objective.
Detection of ROS generation
ROS generation was evaluated using MitoSOX™ Red reagent (Thermo Fisher Scientific, M36008) and CM-H2-DCFDA (Thermo Fisher Scientific, C369), which permeate live cells where they selectively target mitochondrial and cytosolic ROS, respectively, following the manufacturer’s protocol. Cells were placed in Lab-Tek chambered cover glass (Sigma, Z734853) filled with normal medium. For colocalization studies of mitochondria and ROS, cells were loaded with MitoSOX™ Red (5 μM) and MitoTracker® Green (0.2 μM; Thermo Fisher Scientific, M7514). Cells were loaded with MitoSOX™ Red at 5 μM for 30 min at 37°C, and then treated with TNFSF10 for 1 h at 37°C. Using a flow cytometer, MitoSOX™ Red was excited at 488 nm and fluorescence emission at 575 nm was measured. Relative fluorescence intensity was used as a measurement of mitochondrial superoxide production. For cytosolic ROS measurement, 7 μM of CM-H2-DCFDA was used for 30 min at 37°C with pre-treatment of TNFSF10 for 4 h at 37°C.
Western blotting
Western blotting was carried out as previously described [Citation86]. Cells were lysed with Laemmli lysis buffer (Bio-Rad, 1,610,747) and boiled for 7 min. Protein content was measured with BCA Protein Assay Reagent (Thermo Fisher Scientific, 23,225), separated by SDS-PAGE, and electrophoretically transferred to nitrocellulose membrane. Immunoreactive proteins were visualized by using the chemiluminescence protocol (ECL; Amersham, RPN2232).
GST affinity isolation assay
The plasmids pGEX and pGEX-GST-PARK7 were transformed into E. coli DH5α to express GST and GST-PARK7 proteins, respectively. Glutathione agarose resin (Pierce™ GST Protein Interaction Pull-Down Kit; Thermo Fisher Scientific, 21,516) balanced with equilibrium buffer was utilized to capture the proteins into complexes according to the manufacturers’ direction. Then, the complexes were incubated with lysate of HCT116 cells overexpressing FLAG or FLAG-PARK7 treated with 5 ng/ml of TNFSF10 (4 h at 4°C) for 2 h. After that, elution buffer was used to separate the combined proteins and the eluted proteins were analyzed using western blot.
JC-1 mitochondrial membrane potential assay
After human TNFSF10 treatment, cells were stained using a JC-1 Mitochondrial Membrane Potential Detection Kit (Thermo Fisher Scientific, M34152) for 10 min and analyzed using flow cytometry. Fluorescence intensity was measured with the FACScan flow cytometer (Beckman Coulter, Pittsburgh, PA, USA), and the resulting data were analyzed using Flowjo software.
Survival assay
MTT assays were carried out using the Promega CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, G3582). Cells were grown in tissue culture-coated 96-well plates and treated as described in Results. Cells were then treated with MTS/phenazine methosulfate solution for 2 h at 37°C. Absorbance at 490 nm was determined using an enzyme-linked immunosorbent assay plate reader.
RT-PCR analysis
Total RNA was isolated from untreated or TNFSF10-treated cells using the RNAeasy Kit (Qiagen, 74,104) according to the manufacturer’s protocol. Total RNA (2 μg) was used to generate complementary DNA using SuperScript III reverse transcriptase (Thermo Fisher Scientific, 18,090,010). All PCR reactions were performed in triplicate, and PCR products were subjected to a melting curve analysis. The expression of SQSTM1 was detected by TaqMan® Gene Expression Assays 20 (Thermo Fisher Scientific, Hs01061917_g1) according to the manufacturer’s protocol.
Statistical analysis
Statistical analysis was carried out using GraphPad Prism6 software (GraphPad Software, Inc., version 6.07). The results were expressed as the mean of arbitrary values ± SEM. All results were evaluated using an unpaired Student’s t test, where a p-value of less than 0.05 was considered significant.
Abbreviations
| Ala | = | alanine |
| ATE1 | = | arginyltransferase 1 |
| Baf | = | bafilomycin A1 |
| Cys | = | cysteine |
| EGFP | = | enhanced green fluorescent protein |
| EIF4B | = | eukaryotic translation initiation factor 4B, ER, endoplasmic reticulum |
| FACS | = | fluorescence-activated cell sorting |
| FLAG-PARK7 | = | FLAG-tagged PARK7 |
| HA | = | hemagglutinin |
| HA-HSPA5 | = | HA-tagged HSPA5 |
| HSPA1A/HSP70 | = | heat shock protein family A (Hsp70) member 1A |
| HSPA5/BiP/GRP78 | = | heat shock protein family A (Hsp70) member 5 |
| HSPA8/HSC70 | = | heat shock protein family A (Hsp70) member 8 |
| HSP90AA1/HSP90 | = | heat shock protein 90 alpha family class A member 1 |
| LC-MS/MS | = | liquid chromatography-tandem mass spectrometry |
| MAP1LC3B | = | microtubule associated protein 1 light chain 3 beta |
| MEF | = | mouse embryonic fibroblast |
| MTT | = | 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide |
| NAC | = | N-acetylcysteine |
| PARK7 | = | Parkinson associated deglycase |
| PBS | = | phosphate-buffered saline solution |
| PMSF | = | phenylmethane sulfonyl fluoride or phenylmethylsulfonyl fluoride |
| PRDX6 | = | peroxiredoxin 6 |
| RACK1 | = | receptor for activated C kinase 1 |
| R-HSPA5 | = | arginylated HSPA5 |
| ROS | = | reactive oxygen species |
| RPS3 | = | ribosomal protein S3 |
| SDS-PAGE | = | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| ShRNA | = | small hairpin RNA |
| SQSTM1 | = | sequestosome 1 |
| STUB1/CHIP | = | STIP1 homology and U-box containing protein 1 |
| TAP | = | tandem affinity purification; |
| Tbhp | = | tert-butyl hydroperoxide |
| TNFSF10/TRAIL | = | tumor necrosis factor (ligand) superfamily, member 10 |
| TRIM21 | = | tripartite motif containing 21 |
| Ub | = | ubiquitin |
| WIPI2 | = | WD repeat domain, phosphoinositide interacting 2 |
| YWHAZ | = | tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein zeta. |
Supplemental Material
Download Zip (5.5 MB)Acknowledgments
We appreciate Hosun Kim (Seoul National University) for her editorial assistance and Jeong Hun Kim (Sahmyook University) for technical assistance in confocal analyses. This work was supported by National Cancer Institute grants R01CA140554, R03CA205267, and R03CA212125 (to Y.J.L), the Basic Science Research Programs of the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (MSIT) (NRF-2016R1A2B3011389 to Y.T.K., 2015R1D1A1A01058303 to D.H.L., and 2017R1A6A3A11032084 to S.M.S), the Brain Korea 21 PLUS Program (to SNU), SNU Nobel Laureates Invitation Program, and the Bio and Medical Technology Development Program (project no. 2012M3A9B6055305) through the MSIT, the R&D Convergence Program (CAP-16-03-KRIBB) of National Research Council of Science & Technology (NST), and KRIBB Research Initiative Program (to B.Y.K). This project also used the UPCI Core Facility and was supported in part by the award P30CA047904 (to Y.J.L).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Kiffin R, Bandyopadhyay U, Cuervo AM. Oxidative stress and autophagy. Antioxid Redox Signal. 2006 Jan–Feb;8(1–2):152–162. PubMed PMID: 16487049.
- Scherz-Shouval R, Shvets E, Fass E, et al. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007 Apr 04;26(7):1749–1760. PubMed PMID: 17347651; PubMed Central PMCID: PMCPMC1847657.
- Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009 Jul;16(7):1040–1052. PubMed PMID: 19407826.
- Filomeni G, De Zio D, Cecconi F. Oxidative stress and autophagy: the clash between damage and metabolic needs. Cell Death Differ. 2015 Mar;22(3):377–388. PubMed PMID: 25257172; PubMed Central PMCID: PMCPMC4326572.
- Chance B, Boveris A, Oschino N, and Loschen, G. The nature of the catalase intermediate in its biological function. In: King TE, Mason HS, Morrison M, editors. Oxidases and related redox systems. Baltimore: University Park Press; 1973. p. 350–353.
- Dodson M, Darley-Usmar V, Zhang J. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med. 2013 Oct;63:207–221. PubMed PMID: 23702245; PubMed Central PMCID: PMCPMC3729625.
- Altman BJ, Rathmell JC. Metabolic stress in autophagy and cell death pathways. Cold Spring Harb Perspect Biol. 2012 Sep 01;4(9):a008763. PubMed PMID: 22952396; PubMed Central PMCID: PMCPMC3428762.
- Hatsugai N, Perez Koldenkova V, Imamura H, et al. Changes in cytosolic ATP levels and intracellular morphology during bacteria-induced hypersensitive cell death as revealed by real-time fluorescence microscopy imaging. Plant Cell Physiol. 2012 Oct;53(10):1768–1775. PubMed PMID: 22942251.
- Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013 Dec;1833(12):3460–3470. PubMed PMID: 23850759; PubMed Central PMCID: PMCPMC3834229.
- Wang M, Kaufman RJ. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature. 2016 Jan 21;529(7586):326–335. PubMed PMID: 26791723.
- Ciechanover A. The unravelling of the ubiquitin system. Nat Rev Mol Cell Biol. 2015 May;16(5):322–324. PubMed PMID: 25907614.
- Ciechanover A, Kwon YT. Protein Quality Control by Molecular Chaperones in Neurodegeneration. Front Neurosci. 2017;11:185. PubMed PMID: 28428740; PubMed Central PMCID: PMCPMC5382173.
- Kwon YT, Ciechanover A. The Ubiquitin Code in the Ubiquitin-Proteasome System and Autophagy. Trends Biochem Sci. 2017 Nov;42(11):873–886. PubMed PMID: 28947091.
- Ciechanover A. Intracellular protein degradation: from a vague idea through the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Bioorg Med Chem. 2013 Jun 15;21(12):3400–3410. PubMed PMID: 23485445.
- Bachmair A, Finley D, Varshavsky A. In vivo half-life of a protein is a function of its amino-terminal residue. Science. 1986 Oct 10;234(4773):179–186. PubMed PMID: 3018930.
- Sriram SM, Kim BY, Kwon YT. The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat Rev Mol Cell Biol. 2011 Oct 21;12(11):735–747. PubMed PMID: 22016057.
- Tasaki T, Sriram SM, Park KS, et al. The N-end rule pathway. Annu Rev Biochem. 2012;81:261–289. PubMed PMID: 22524314; PubMed Central PMCID: PMCPMC3610525.
- Kwon YT, Reiss Y, Fried VA, et al. The mouse and human genes encoding the recognition component of the N-end rule pathway. Proc Natl Acad Sci U S A. 1998 Jul 7;95(14):7898–7903. PubMed PMID: 9653112; PubMed Central PMCID: PMCPMC20901.
- Kwon YT, Xia Z, Davydov IV, et al. Construction and analysis of mouse strains lacking the ubiquitin ligase UBR1 (E3alpha) of the N-end rule pathway. Mol Cell Biol. 2001 Dec;21(23):8007–8021. PubMed PMID: 11689692; PubMed Central PMCID: PMCPMC99968.
- Tasaki T, Mulder LCF, Iwamatsu A, et al. A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol Cell Biol. 2005 Aug;25(16):7120–7136. PubMed PMID: 16055722; PubMed Central PMCID: PMCPMC1190250.
- Kwon YT, Xia Z, An JY, et al. Female lethality and apoptosis of spermatocytes in mice lacking the UBR2 ubiquitin ligase of the N-end rule pathway. Mol Cell Biol. 2003 Nov;23(22):8255–8271. PubMed PMID: 14585983; PubMed Central PMCID: PMCPMC262401.
- Kwon YT, Kashina AS, Varshavsky A. Alternative splicing results in differential expression, activity, and localization of the two forms of arginyl-tRNA-protein transferase, a component of the N-end rule pathway. Mol Cell Biol. 1999 Jan;19(1):182–193. PubMed PMID: 9858543; PubMed Central PMCID: PMCPMC83877.
- Kwon YT, Kashina AS, Davydov IV, et al. An essential role of N-terminal arginylation in cardiovascular development. Science. 2002 Jul 05;297(5578):96–99. PubMed PMID: 12098698.
- Lee MJ, Tasaki T, Moroi K, et al. RGS4 and RGS5 are in vivo substrates of the N-end rule pathway. Proc Natl Acad Sci U S A. 2005 Oct 18;102(42):15030–15035. PubMed PMID: 16217033; PubMed Central PMCID: PMCPMC1257735.
- Lee MJ, Pal K, Tasaki T, et al. Synthetic heterovalent inhibitors targeting recognition E3 components of the N-end rule pathway. Proc Natl Acad Sci U S A. 2008 Jan 8;105(1):100–105. PubMed PMID: 18162545; PubMed Central PMCID: PMCPMC2224166.
- Lee MJ, Kim DE, Zakrzewska A, et al. Characterization of arginylation branch of N-end rule pathway in G-protein-mediated proliferation and signaling of cardiomyocytes. J Biol Chem. 2012 Jul 6;287(28):24043–24052. PubMed PMID: 22577142; PubMed Central PMCID: PMCPMC3390678.
- Sriram SM, Banerjee R, Kane RS, et al. Multivalency-assisted control of intracellular signaling pathways: application for ubiquitin- dependent N-end rule pathway. Chem Biol. 2009 Feb 27;16(2):121–131. PubMed PMID: 19246002; PubMed Central PMCID: PMCPMC2665046.
- Sriram SM, Kwon YT. The molecular principles of N-end rule recognition. Nat Struct Mol Biol. 2010 Oct;17(10):1164–1165. PubMed PMID: 20924402.
- Lopez Sambrooks C, Carpio MA, Hallak ME. Arginylated calreticulin at plasma membrane increases susceptibility of cells to apoptosis. J Biol Chem. 2012 Jun 22;287(26):22043–22054. PubMed PMID: 22577148; PubMed Central PMCID: PMCPMC3381163.
- Cha-Molstad H, Sung KS, Hwang J, et al. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol. 2015 Jul;17(7):917–929. PubMed PMID: 26075355; PubMed Central PMCID: PMCPMC4490096.
- Cha-Molstad H, Yu JE, Feng Z, et al. p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat Commun. 2017 Jul 24;8(1):102. PubMed PMID: 28740232; PubMed Central PMCID: PMCPMC5524641.
- Shim SM, Choi HR, Sung KW, et al. The endoplasmic reticulum-residing chaperone BiP is short-lived and metabolized through N-terminal arginylation. Sci Signal. 2018 Jan 2;11(511):eaan0630. PubMed PMID: 29295953.
- Hayashi T, Ishimori C, Takahashi-Niki K, et al. DJ-1 binds to mitochondrial complex I and maintains its activity. Biochem Biophys Res Commun. 2009 Dec 18;390(3):667–672. PubMed PMID: 19822128.
- Kim JM, Shin HI, Cha SS, et al. DJ-1 promotes angiogenesis and osteogenesis by activating FGF receptor-1 signaling. Nat Commun. 2012;3:1296. PubMed PMID: 23250426.
- Won KJ, Jung SH, Lee CK, et al. DJ-1/park7 protects against neointimal formation via the inhibition of vascular smooth muscle cell growth. Cardiovasc Res. 2013 Mar 01;97(3):553–561. PubMed PMID: 23230227.
- Lev N, Ickowicz D, Barhum Y, et al. DJ-1 protects against dopamine toxicity. J Neural Transm (Vienna). 2009 Feb;116(2):151–160. PubMed PMID: 18974921.
- Cao J, Lou S, Ying M, et al. DJ-1 as a human oncogene and potential therapeutic target. Biochem Pharmacol. 2015 Feb 01;93(3):241–250. PubMed PMID: 25498803.
- Olzmann JA, Brown K, Wilkinson KD, et al. Familial Parkinson’s disease-associated L166P mutation disrupts DJ-1 protein folding and function. J Biol Chem. 2004 Feb 27;279(9):8506–8515. PubMed PMID: 14665635.
- Shendelman S, Jonason A, Martinat C, et al. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004 Nov;2(11):e362. PubMed PMID: 15502874; PubMed Central PMCID: PMCPMC521177.
- Jeong HJ, Kim DW, Woo SJ, et al. Transduced Tat-DJ-1 protein protects against oxidative stress-induced SH-SY5Y cell death and Parkinson disease in a mouse model. Mol Cells. 2012 May;33(5):471–478. PubMed PMID: 22526393; PubMed Central PMCID: PMCPMC3887734.
- Yamaguchi S, Yamane T, Takahashi-Niki K, et al. Transcriptional activation of low-density lipoprotein receptor gene by DJ-1 and effect of DJ-1 on cholesterol homeostasis. PLoS One. 2012;7(5):e38144. PubMed PMID: 22666465; PubMed Central PMCID: PMCPMC3364227.
- Ariga H, Takahashi-Niki K, Kato I, et al. Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longev. 2013;2013:683920. PubMed PMID: 23766857; PubMed Central PMCID: PMCPMC3671546.
- Mitsumoto A, Nakagawa Y, Takeuchi A, et al. Oxidized forms of peroxiredoxins and DJ-1 on two-dimensional gels increased in response to sublethal levels of paraquat. Free Radic Res. 2001 Sep;35(3):301–310. PubMed PMID: 11697128.
- Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003 Jan 10;299(5604):256–259. PubMed PMID: 12446870.
- Taira T, Saito Y, Niki T, et al. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004 Feb;5(2):213–218. PubMed PMID: 14749723; PubMed Central PMCID: PMCPMC1298985.
- Zhou W, Zhu M, Wilson MA, et al. The oxidation state of DJ-1 regulates its chaperone activity toward alpha-synuclein. J Mol Biol. 2006 Mar 03;356(4):1036–1048. PubMed PMID: 16403519.
- Andres-Mateos E, Perier C, Zhang L, et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007 Sep 11;104(37):14807–14812. PubMed PMID: 17766438; PubMed Central PMCID: PMCPMC1976193.
- Im JY, Lee KW, Junn E, et al. DJ-1 protects against oxidative damage by regulating the thioredoxin/ASK1 complex. Neurosci Res. 2010 Jul;67(3):203–208. PubMed PMID: 20385180; PubMed Central PMCID: PMCPMC2883683.
- Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox Signal. 2011 Jul 01;15(1):111–122. PubMed PMID: 20812780; PubMed Central PMCID: PMCPMC3110098.
- Saito Y. Oxidized DJ-1 as a possible biomarker of Parkinson’s disease. J Clin Biochem Nutr. 2014 May;54(3):138–144. PubMed PMID: 24894116; PubMed Central PMCID: PMCPMC4042152.
- Canet-Aviles RM, Wilson MA, Miller DW, et al. The Parkinson’s disease protein DJ-1 is neuroprotective due to cysteine-sulfinic acid-driven mitochondrial localization. Proc Natl Acad Sci U S A. 2004 Jun 15;101(24):9103–9108. PubMed PMID: 15181200; PubMed Central PMCID: PMCPMC428480.
- Kim RH, Smith PD, Aleyasin H, et al. Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc Natl Acad Sci U S A. 2005 Apr 05;102(14):5215–5220. PubMed PMID: 15784737; PubMed Central PMCID: PMCPMC555037.
- Park J, Kim SY, Cha GH, et al. Drosophila DJ-1 mutants show oxidative stress-sensitive locomotive dysfunction. Gene. 2005 Nov;21(361):133–139. PubMed PMID: 16203113.
- Meulener MC, Xu K, Thomson L, et al. Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc Natl Acad Sci U S A. 2006 Aug 15;103(33):12517–12522. PubMed PMID: 16894167; PubMed Central PMCID: PMCPMC1533799.
- Kahle PJ, Waak J, Gasser T. DJ-1 and prevention of oxidative stress in Parkinson’s disease and other age-related disorders. Free Radic Biol Med. 2009 Nov 15;47(10):1354–1361. PubMed PMID: 19686841.
- Li HM, Niki T, Taira T, et al. Association of DJ-1 with chaperones and enhanced association and colocalization with mitochondrial Hsp70 by oxidative stress. Free Radic Res. 2005 Oct;39(10):1091–1099. PubMed PMID: 16298734.
- Wang B, Cai Z, Tao K, et al. Essential control of mitochondrial morphology and function by chaperone-mediated autophagy through degradation of PARK7. Autophagy. 2016 Aug 2;12(8):1215–1228. PubMed PMID: 27171370; PubMed Central PMCID: PMCPMC4968227.
- Xu CY, Kang WY, Chen YM, et al. DJ-1 inhibits alpha-synuclein aggregation by regulating chaperone-mediated autophagy. Front Aging Neurosci. 2017;9:308. PubMed PMID: 29021755; PubMed Central PMCID: PMCPMC5623690.
- Inoue T, Suzuki-Karasaki Y. Mitochondrial superoxide mediates mitochondrial and endoplasmic reticulum dysfunctions in TRAIL-induced apoptosis in Jurkat cells. Free Radic Biol Med. 2013 Aug;61:273–284. PubMed PMID: 23608466.
- Suzuki-Karasaki M, Ochiai T, Suzuki-Karasaki Y. Crosstalk between mitochondrial ROS and depolarization in the potentiation of TRAIL-induced apoptosis in human tumor cells. Int J Oncol. 2014 Feb;44(2):616–628. PubMed PMID: 24337174.
- Dickinson BC, Srikun D, Chang CJ. Mitochondrial-targeted fluorescent probes for reactive oxygen species. Curr Opin Chem Biol. 2010 Feb;14(1):50–56. PubMed PMID: 19910238; PubMed Central PMCID: PMCPMC2830890.
- Ling LU, Tan KB, Lin H, et al. The role of reactive oxygen species and autophagy in safingol-induced cell death. Cell Death Dis. 2011 Mar;10(2):e129. PubMed PMID: 21390063; PubMed Central PMCID: PMCPMC3101809.
- Nakatani Y, Ogryzko V. Immunoaffinity purification of mammalian protein complexes. Methods Enzymol. 2003;370:430–444. PubMed PMID: 14712665.
- Moore DJ, Zhang L, Troncoso J, et al. Association of DJ-1 and parkin mediated by pathogenic DJ-1 mutations and oxidative stress. Hum Mol Genet. 2005 Jan 1;14(1):71–84. PubMed PMID: 15525661.
- Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009 Jun 12;137(6):1001–1004. PubMed PMID: 19524504; PubMed Central PMCID: PMCPMC3971861.
- Lu K, Psakhye I, Jentsch S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell. 2014 Jul 31;158(3):549–563. PubMed PMID: 25042851.
- Huber LA, Teis D. Lysosomal signaling in control of degradation pathways. Curr Opin Cell Biol. 2016 Apr;39:8–14. PubMed PMID: 26827287.
- Marwaha R, Arya SB, Jagga D, et al. The Rab7 effector PLEKHM1 binds Arl8b to promote cargo traffic to lysosomes. J Cell Biol. 2017 Apr 03;216(4):1051–1070. PubMed PMID: 28325809; PubMed Central PMCID: PMCPMC5379943.
- McEwan DG, Popovic D, Gubas A, et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell. 2015 Jan 08;57(1):39–54. PubMed PMID: 25498145.
- Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010 Mar;12(3):213–223. PubMed PMID: 20173742.
- Ichimura Y, Waguri S, Sou YS, et al. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell. 2013 Sep 12;51(5):618–631. PubMed PMID: 24011591.
- Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015 Dec;282(24):4672–4678. PubMed PMID: 26432171.
- Lim J, Lachenmayer ML, Wu S, et al. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11(2):e1004987. PubMed PMID: 25723488; PubMed Central PMCID: PMCPMC4344198.
- Milan E, Perini T, Resnati M, et al. A plastic SQSTM1/p62-dependent autophagic reserve maintains proteostasis and determines proteasome inhibitor susceptibility in multiple myeloma cells. Autophagy. 2015;11(7):1161–1178. PubMed PMID: 26043024; PubMed Central PMCID: PMCPMC4590585.
- Watanabe Y, Tsujimura A, Taguchi K, et al. HSF1 stress response pathway regulates autophagy receptor SQSTM1/p62-associated proteostasis. Autophagy. 2017 Jan 2;13(1):133–148. PubMed PMID: 27846364; PubMed Central PMCID: PMCPMC5240838.
- Wang S, El-Deiry WS. TRAIL and apoptosis induction by TNF-family death receptors. Oncogene. 2003 Nov 24;22(53):8628–8633. PubMed PMID: 14634624.
- Ziauddin MF, Guo ZS, Me O, et al. TRAIL gene-armed oncolytic poxvirus and oxaliplatin can work synergistically against colorectal cancer. Gene Ther. 2010 Apr;17(4):550–559. PubMed PMID: 20182517; PubMed Central PMCID: PMCPMC3250063.
- Dai Y, Dent P, Grant S. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) promotes mitochondrial dysfunction and apoptosis induced by 7-hydroxystaurosporine and mitogen-activated protein kinase kinase inhibitors in human leukemia cells that ectopically express Bcl-2 and Bcl-xL. Mol Pharmacol. 2003 Dec;64(6):1402–1409. PubMed PMID: 14645670.
- LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003 Jan;10(1):66–75. PubMed PMID: 12655296.
- Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxid Redox Signal. 2014 Jul 20;21(3):396–413. PubMed PMID: 24702237; PubMed Central PMCID: PMCPMC4076992.
- Fu K, Ren H, Wang Y, et al. DJ-1 inhibits TRAIL-induced apoptosis by blocking pro-caspase-8 recruitment to FADD. Oncogene. 2012 Mar 08;31(10):1311–1322. PubMed PMID: 21785459.
- Girotto S, Sturlese M, Bellanda M, et al. Dopamine-derived quinones affect the structure of the redox sensor DJ-1 through modifications at Cys-106 and Cys-53. J Biol Chem. 2012 May 25;287(22):18738–18749. PubMed PMID: 22431735; PubMed Central PMCID: PMCPMC3365698.
- Martinat C, Shendelman S, Jonason A, et al. Sensitivity to oxidative stress in DJ-1-deficient dopamine neurons: an ES- derived cell model of primary Parkinsonism. PLoS Biol. 2004 Nov;2(11):e327. PubMed PMID: 15502868; PubMed Central PMCID: PMCPMC521171.
- Shevchenko A, Tomas H, Havlis J, et al. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1(6):2856–2860. PubMed PMID: 17406544.
- Wieckowski MR, Giorgi C, Lebiedzinska M, et al. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc. 2009;4(11):1582–1590. PubMed PMID: 19816421.
- Lee DH, Sung KS, Bartlett DL, et al. HSP90 inhibitor NVP-AUY922 enhances TRAIL-induced apoptosis by suppressing the JAK2-STAT3-Mcl-1 signal transduction pathway in colorectal cancer cells. Cell Signal. 2015 Feb;27(2):293–305. PubMed PMID: 25446253; PubMed Central PMCID: PMCPMC4276460.