
ABSTRACT
VCP/p97 (valosin containing protein) is a key regulator of cellular proteostasis. It orchestrates protein turnover and quality control in vivo, processes fundamental for proper cell function. In humans, mutations in VCP lead to severe myo- and neuro-degenerative disorders such as inclusion body myopathy with Paget disease of the bone and frontotemporal dementia (IBMPFD), amyotrophic lateral sclerosis (ALS) or and hereditary spastic paraplegia (HSP). We analyzed here the in vivo role of Vcp and its novel interactor Washc4/Swip (WASH complex subunit 4) in the vertebrate model zebrafish (Danio rerio). We found that targeted inactivation of either Vcp or Washc4, led to progressive impairment of cardiac and skeletal muscle function, structure and cytoarchitecture without interfering with the differentiation of both organ systems. Notably, loss of Vcp resulted in compromised protein degradation via the proteasome and the macroautophagy/autophagy machinery, whereas Washc4 deficiency did not affect the function of the ubiquitin-proteasome system (UPS) but caused ER stress and interfered with autophagy function in vivo. In summary, our findings provide novel insights into the in vivo functions of Vcp and its novel interactor Washc4 and their particular and distinct roles during proteostasis in striated muscle cells.
Introduction
Degradation of proteins as a fundamental module of proteostasis (protein homeostasis) is indispensable for the control of protein quality and quantity in the cell. Protein turnover and removal of misfolded and poly-ubiquitinated proteins are predominately accomplished by 2 degradation machineries, the ubiquitin-proteasome system (UPS) as well as the autophagy and lysosome system [Citation1–Citation3]. Dysfunction of either of these proteolytic machineries has been implicated in the onset and progression of striated muscle as well as neurodegenerative diseases [Citation4,Citation5,Citation6]. In this context, point mutations in VCP/p97 (valosin containing protein) were shown to cause inclusion body myopathy associated with Paget disease of the bone and frontotemporal dementia (IBMPFD) and amyotrophic lateral sclerosis (ALS). Moreover, mutant VCP has been implicated in the pathogenesis of Charcot-Marie-Tooth disease type 2, hereditary spastic paraplegia (HSP) and Parkinson disease (PD) [Citation7–Citation12]. VCP is an evolutionarily highly conserved type II AAA+-ATPase involved in numerous important cellular processes such as membrane dynamics, cell cycle, DNA damage response, as well as protein quality and degradation control mechanisms [Citation13]. In this context, VCP is involved in the unfolded protein response (UPR) of the endoplasmic reticulum (ER) that is activated by an accumulation of unfolded or misfolded proteins in the ER lumen and subsequently ER stress [Citation14]. This leads to the cytosolic proteasomal degradation of misfolded proteins, a process called ERAD (endoplasmic reticulum-associated degradation) [Citation14]. Recent evidence suggests that during ER stress and UPR activation macroautophagy is the predominant degradation pathway whereas the UPS seems to play only a minor role [Citation15,Citation16].
Defective proteolytic mechanisms are often associated with pathological protein aggregation disorders such as IBMPFD. This multisystem disease is included in the group of myofibrillar myopathies (MFM) since muscle biopsies from affected patients exhibit DES (desmin)-positive protein aggregates, rimmed vacuoles and myofibrillar degeneration. MFMs are severe striated muscle disorders often affecting both, heart and skeletal muscle cells [Citation17,Citation18]. The function of VCP critically depends on its association with specific cofactors such as UFD1 (ubiquitin recognition factor in ER associated degradation 1), NPLOC4/NPL4 (NPL4 homolog, ubiquitin recognition factor), PSMF1 (proteasome inhibitor subunit 1) or WASHC5/Strumpellin (WASH complex subunit 5) [Citation19,Citation20]. Recently, Washc4/Swip (WASH complex subunit 4), a member of the WASH (Wiskott-Aldrich syndrome protein and scar homolog) complex that contributes to actin polymerization and endosomal transport processes, has been identified as a novel VCP interactor in Dictyostelium discoideum [Citation20]. Furthermore, a Washc4 point-mutation has been shown to result in autosomal recessive intellectual disability [Citation21].
To elucidate the in vivo role of the novel Vcp interactor Washc4 in the vertebrate heart and skeletal muscle and to define its impact on protein homeostasis, we generated Washc4-deficient zebrafish. We found that, similar to the situation in Vcp-depleted zebrafish, Washc4 deficiency resulted in severe myopathy and heart failure due to pronounced ultrastructural heart and skeletal muscle pathologies. Notably, whereas Vcp ablation led to impaired function of the proteasomal system and the autophagy/lysosome machinery, Washc4 deficiency induced ER stress and solely interfered with autophagy-mediated degradation in vivo, implying distinct roles of Washc4 and Vcp during proteostasis in striated muscle cells.
Results
Inactivation of vcp leads to heart failure and myopathy in zebrafish embryos
MFMs are sporadic or hereditary diseases of the striated muscle, which lead to severe physical disability and premature death [Citation22]. Point mutations in the VCP gene cause, beside neurodegenerative diseases, MFM [Citation22], but the molecular pathomechanisms that provoke muscle degeneration in these patients are still poorly understood. Here, to investigate the in vivo role of Vcp in the vertebrate heart and skeletal muscle, we inactivated zebrafish vcp using morpholino-modified antisense oligonucleotide (MO)- and Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR associated protein 9 (CRISPR/Cas9)-mediated reverse genetics as well as pharmacological approaches.
We first microinjected 2 independent MOs either directed against the translational start site (MO1-vcp) or the splice-donor site of exon 2 (MO2-vcp) into fertilized wild-type zebrafish oocytes at the 1-cell-stage. When injected with 9.1 ng of MO1-vcp or 8.4 ng of MO2-vcp, 81.38% ran: [78.57% to 84.29%] and 79.10% ran: [76.36% to 83.33%] of the injected embryos (n = 242, N = 4 and n = 212, N = 3, respectively) displayed a severe myopathic phenotype and heart failure at 60 h post fertilization (hpf) ( and (e); Fig. S1B). Injection of the same amount of the corresponding 5 base pair mismatch morpholino (MO1-control/MO2-control) as control did not interfere with regular cardiac and skeletal muscle structure or function (n = 259, N = 4 and n = 161, N = 3, respectively) (); Fig. S1A). To ensure that both used MOs are effectively knocking down zebrafish Vcp in vivo, we performed western blot analyses and reverse transcription-polymerase chain reaction (RT-PCR)-based messengerRNA (mRNA) splicing assays in MO1-vcp- and MO2-vcp-injected embryos, respectively. We found that Vcp protein levels were reduced in MO1-vcp-injected embryos compared to MO1-control-injected embryos ()), demonstrating that MO1-vcp effectively blocks Vcp translation in vivo. Additionally, to demonstrate the effectiveness of MO2-vcp, we analyzed vcp mRNA splicing by RT-PCR and found that MO2-vcp led to an aberrant vcp splicing product (skipping of exon 2), which led to premature termination of translation of zebrafish Vcp ()).
Figure 1. Phenotypic analysis of zebrafish after morpholino-mediated knockdown of vcp or ML240 treatment. (a to d) Lateral view of embryos at 60 hpf. Zebrafish embryos were either injected with 5 base pair (bp) mismatch vcp start morpholino (MO1-control; a) and vcp start MO (MO1-vcp; b) or treated with DMSO (c) and VCP-Inhibitor ML240 (VCP-I; d). scale bar: 500 μm (a, c); scale bar: 650 μm (b, d). (e) Statistical analysis of affected embryos after MO1/2-control or MO1/2-vcp injection and DMSO or ML240 treatment. N = 3/4 Experiments with in total n = 259 (MO1-control), n = 161 (MO2-control), n = 242 (MO1-vcp), n = 212 (MO2-vcp), n = 152 (DMSO), n = 148 (VCP-I); N two-sided Fisher exact tests combined with a modified Fisher method. Ratios of N experiments are displayed. MO1-vcp-injected embryos reveal reduced levels of Vcp by western blot analysis. Actb1/β-actin was used as loading control. RT-PCR after MO2-vcp injection shows specific effect on mRNA splicing. (f) Ventricular fractional shortening (FS) measurements of MO1-control and MO1-vcp-injected embryos at 48, 72 and 96 hpf, n = 18 to 30 (MO1-control), n = 19 to 32 (MO1-vcp). The individual measurements are shown (two-sided Wilcoxon rank-sum test). (g) Heart beat per minute at 48, 72 and 96 hpf of vcp morphants and control embryos. n = 30 (MO1-control), n = 29 or 30 (MO1-vcp). The individual measurements are shown (two-sided unpaired Wilcoxon rank-sum test. (h and i) False-colored superimposed overviews of spontaneous movement assay with MO1-control- (h) and MO1-vcp-injected (i) embryos at 24 hpf; red pictures = 0 s; green pictures = 10 s; yellow = merge. scale bar: 1 mm. (j) Quantification of spontaneous movement assay; N = 3 experiments with n = 30 (MO1-control), n = 29 or 30 (MO1-vcp) per experiment (N two-sided Fisher exact tests combined with modified Fisher method). The ratios of N experiments are shown. (k) Statistical analysis of touch-evoke flight response of control and vcp morphants at 72 hpf. n = 30, N = 3 (MO1-control), n = 29 or 30, N = 3 (MO1-vcp); data represent means.
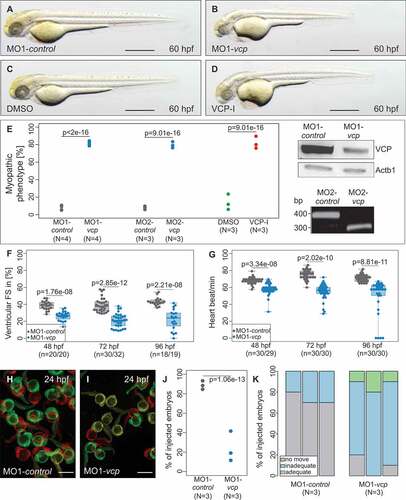
To further prove the specificity of the Vcp knockdown, we pharmacologically inactivated Vcp function by incubating wild-type zebrafish embryos with ML240, a selective VCP inhibitor [Citation23]. After incubation of 10 hpf wild-type zebrafish embryos for 38 h in 15 µM ML240, 80% ran: [76% to 89.58] (n = 148, N = 3) of these embryos exhibited, identical to Vcp morphants, myopathy and heart failure (). Additionally, we depleted Vcp by injecting a CRISPR/Cas9 ribonucleoprotein (RNP) complex that enables fast and efficient disruption of the targeted gene [Citation24]. Injection of vcp-specific RNP complexes into one-cell stage zebrafish embryos resulted in CRISPR/Cas9-induced mutants (crispants) that perfectly resemble the vcp morpholino knockdown as well as the VCP-inhibitor phenotype as demonstrated by functional and structural analyses (Fig. S2A,B,D,E,F,H). Remarkably, vcp morphants, crispants and VCP-inhibitor-treated zebrafish embryos die 120 hpf due to their severe cardiac and skeletal muscle defects (Table S1). In summary, these findings indicate that Vcp plays an essential role in the regulation of cardiac and skeletal muscle function in vivo.
Next, to describe the cardiac and skeletal muscle pathologies in Vcp-deficient embryos in more detail, we measured cardiac fractional shortening and heart rate as well as motility. At 24 hpf, Vcp-deficient zebrafish embryos showed strong peristaltic contractions that were indistinguishable from wild-type contractions. By contrast, starting at 48 hpf, contractile force of the ventricular chamber started to decline in Vcp morphants (Fractional shortening (FS): 26.3% interquartile range (IQR): [23.7% to 29.53%]; n = 20) compared to MO-control-injected embryos (FS: 38.6% IQR: [35.04% to 42.16%]; n = 20) ()). At 72 and 96 hpf, cardiac contractile performance in Vcp-deficient embryos further worsened compared to control embryos (vcp morphants at 72 hpf: Median (MED) = 20.96% IQR: [15.01% to 25.92%]; n = 32; at 96 hpf: 21.97% IQR: [14.6% to 29.68]; n = 19; control embryos at 72 hpf: MED = 37.62% IQR: [32.83% to 43.02%]; n = 30; at 96 hpf: 42.5% IQR: [40.85% to 44.77%]; n = 18) ()). This led to blood congestion at the cardiac inflow tract and diminished blood circulation through the vasculature. Additionally, Vcp-deficient zebrafish embryos displayed significant bradycardia with a heart rate of 144 IQR: [138 to 150] beats per minute (bpm) (n = 29) compared to 169 IQR: [168 to 174] bpm in control embryos (n = 30) at 48 hpf. During further development the heart rate of Vcp-deficient embryos further decreased slightly at 72 hpf to 138.8 IQR: [132 to 150] bpm (n = 30) compared to 188.1 IQR: [180 to 198] bpm in control embryos (n = 30) and to 135 IQR: [126 to 156] bpm (n = 30) compared to 178.9 IQR: [168 to 186] bpm in controls (n = 30) at 96 hpf ()). To evaluate how Vcp deficiency leads to heart failure in zebrafish, we next analyzed cardiac structure in Vcp morphants. Histological analysis of Vcp-deficient hearts revealed defined cardiac chambers with atrium and ventricle separated by the atrio-ventricular ring. Endocardial and myocardial layers of both heart chambers were developed properly (Fig. S1C and D). Whole-mount antisense RNA in situ hybridizations against myh6/amhc (myosin heavy chain 6, cardiac muscle, alpha) and myh7/vmhc (myosin heavy chain 7) at 48 hpf showed that both cardiac chambers were regularly differentiated (Fig. S1E to H).
Already at 24 hpf, Vcp-deficient embryos exhibited compromised spontaneous movements (moving embryos: 19.05% ran: [11.54% to 41.67%]; n = 71, N = 3) compared to control embryos (moving embryos: 88.46% ran: [84.62% to 93.33%]; n = 82, N = 3) ()). At 48 hpf and after touch-evoked stimulation, Vcp morphants showed severely impaired motility (inadequate: 80% ran: [70.00% to 80.00%]/no movement ran: 10% [10.00% to 20.00%]; n = 40, N = 3), whereas only a small fraction of the control embryos showed reduced motility after tactile stimulation (inadequate: 30% ran: [20.00% to 30.00%]/no movement 0% ran: [0.00% to 0.00%]; n = 40, N = 3) ()). As the expression of the essential myogenic markers myod1 and myog (myogenin) was unaffected at the 16 to 18 somite stage (Fig. S1I to L), impaired motility in VCP-deficient embryos cannot be attributed to defective skeletal muscle differentiation.
Vcp deficiency results in ultrastructural striated muscle pathologies
To elucidate whether defective heart and skeletal muscle function of Vcp-deficient zebrafish embryos were caused by structural striated muscle alterations, we first assessed the muscle architecture of Vcp-deficient embryos by a non-invasive birefringence assay. Due to the high structural organization of striated muscle tissue light polarizes after illumination resulting in a strong birefringence signal [Citation25]. We found that the birefringence signal intensity was severely reduced in Vcp-deficient zebrafish embryos 47.93 IQR: [39.23 to 64.53] (n = 23) at 48 hpf compared to control embryos 122.46 IQR: [98.21 to 134.23] (n = 25) (), suggesting that Vcp deficiency leads to disruption or disorganization of muscle tissue. To substantiate this finding, we next evaluated the muscle structure by histological analyses. At 72 hpf, histological sectioning of Vcp-deficient zebrafish embryos revealed unorganized and vacuolated skeletal muscle tissue () and (e)), implying that impaired skeletal muscle function is due to defective myofibrillar organization.
Figure 2. Vcp deficiency results in a severe skeletal muscle myopathy. (a and b) Lateral view of control (a) and vcp morphants (b) showing birefringence at 48 hpf. scale bar: 450 μm. (c) Densitometric analysis of birefringence (n = 23/25). Individual samples are shown (two-sided Wilcoxon rank-sum test). (d and e) Skeletal muscle H&E-stained sagittal histological sections of MO1-control- (d) and MO1-vcp-injected embryos (e) at 72 hpf; scale bar: 100 μm. (f to g``) Transmission electron microscopy in control embryos (f to f``) and vcp morphants (g to g``) at 48 hpf. (f to f``) MO1-control-injected embryos show organized myofibrils and sarcomeric units with highly structured thick and thin filaments (f`) and mitochondria with many cristae (f``). (g to g``) VCP-deficient muscle cells present disorganized myofibrils with Z-disc alterations (arrow, g`), dysmorphic mitochondria (g``) and an accumulation of numerous vesicular bodies (double arrowheads, g); scale bar: 2.5 μm (f, g); scale bar: 1 μm (f` and f``, g` and g``).
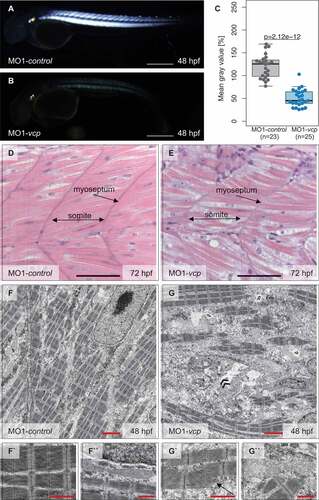
Additionally, to assess whether impaired striated muscle function is due to ultrastructural alterations, we conducted transmission electron microscopy studies of Vcp-deficient muscle tissue at 48 hpf. As shown in ), skeletal muscle cells of MO1-control-injected embryos appear unaffected. These cells were densely packed with sarcomeres that are assembled by highly structured thick and thin filaments and flanked by Z-disks () and (f’)). Furthermore, mitochondria were composed of numerous cristae and muscle cell nuclei appeared elongated in control embryos (``)). In contrast, Vcp-deficient muscle cells exhibited reduced numbers of properly organized myofibrils and dysmorphic mitochondria with massively reduced cristae numbers (’’)). Additionally, Vcp deficiency resulted in the accumulation of numerous vesicular bodies ()), suggesting that the pathologically altered striated muscle ultrastructure accounts for the observed skeletal muscle failure in Vcp-deficient zebrafish embryos.
Loss of vcp interferes with UPS- and autophagy-mediated protein degradation
VCP is critically involved in the degradation of poly-ubiquitinated substrates via the proteasome [Citation7]. To assess whether loss of Vcp function in zebrafish also interferes with protein degradation via the UPS as observed in other model systems [Citation19], we performed western blot analyses using ubiquitin-specific antibodies on protein lysates derived from control- and MO1-vcp-injected zebrafish embryos. Here, we demonstrated that levels of ubiquitinated proteins were markedly increased in Vcp-deficient zebrafish embryos compared to controls ()), implying that loss of Vcp in the zebrafish model indeed leads to compromised UPS function in vivo. To finally prove that Vcp deficiency in zebrafish interferes with proteasomal function, we inactivated Vcp and subsequently incubated these zebrafish embryos with either dimethyl sulfoxide (DMSO) as control or the UPS inhibitor MG132. We found that MG132 treatment did not result in a further increase of the levels of ubiquitinated proteins, demonstrating that loss of Vcp completely blocked UPS function in zebrafish in vivo ()).
Figure 3. Inactivation of Vcp leads to an accumulation of ubiquitinated proteins and Sqstm1. (a) Western blot analysis using an anti-K48-linkage specific ubiquitin antibody after MO1-control and MO1-vcp injection and DMSO or MG132 treatment (n = 4). (b) Vcp morphants show a significant increase of Sqstm1 by western blot analysis (n = 3); quantification of gray values. Actb1/β-actin was used as loading control. Data represent means ± SD, unpaired Student t test, *P value< 0.03 (c) Western blot analysis of Lc3 after MO1-control and MO1-vcp injection and DMSO or Ammonium chloride (NH4Cl) treatment (top) and quantification of gray values (bottom). NH4Cl treated control embryos reveal significant increase of Lc3-II levels, compared to DMSO treated embryos (n = 5). Actb1/β-actin was used as loading control. The individual samples are displayed (two-sided Wilcoxon rank-sum test).
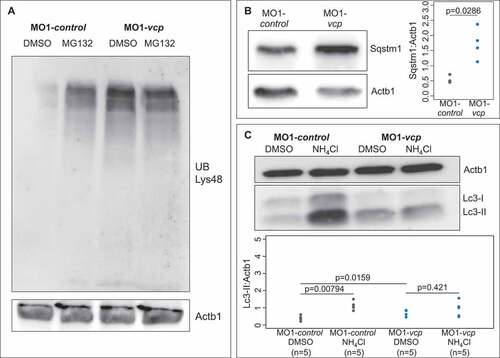
Recently, VCP was demonstrated to be also involved in autophagic degradation of ubiquitinated substrates [Citation26,Citation27]. To evaluate whether autophagy-mediated protein degradation, similar to UPS-mediated degradation, is impaired by the loss of Vcp in vivo, we assessed protein levels of Sqstm1/p62 (sequestosome 1) and Lc3A/B-II, the lipidated and autophagosome-associated form of Lc3A/B or Map1lc3A/B (microtubule-associated protein 1 light chain 3) (hereafter referred to as Lc3), 2 established markers of autophagy function [Citation28]. These studies revealed significantly increased Sqstm1 protein levels in Vcp-deficient zebrafish embryos compared to control embryos ()). To assess whether increased Sqstm1 protein levels are due to elevated transcription of the sqstm1 gene, we performed quantitative real-time PCR analyses and found no increase in sqstm1 transcript levels in Vcp-deficient zebrafish embryos (Fig. S3A). These findings imply that the loss of Vcp function also directly has an impact on the autophagy system.
Similar to increased Sqstm1 protein levels, Lc3-II levels were also significantly increased in Vcp-deficient embryos ()). Additionally, incubation with the lysosome-autophagosome fusion inhibitor ammonium chloride (NH4Cl) revealed no significant changes in Lc3-II levels in Vcp-deficient embryos compared to DMSO-treated Vcp morphants (DMSO: 0.78 IQR: [0.62 to 0.86] and after NH4Cl treatment: 0.96 IQR: [0.54 to 1.06]; n = 5), consistent with a strongly diminished autophagic flux ()). These results indicated that Vcp deficiency in zebrafish embryos at 72 hpf did interfere with the function of the autophagy machinery in vivo.
To further substantiate this finding, we next assessed the impact of Vcp deficiency on the function of the autophagy system using the novel striated muscle-specific transgenic autophagy-reporter zebrafish line Tg(unc45b:mCherry-EGFP-LC3B). Here, assessment of autophagic function is based on the pH sensitivity of green fluorescent protein (GFP), with the GFP signal being quenched when the tandem-tagged LC3 protein is delivered to the acidic lysosome, while the red fluorescent mCherry signal remains stable. Thus, autophagic flux can be quantified by calculating the ratio between green:yellow (autophagosomes) and red-only (autolysosomes) fluorescence. We injected 9.1 ng of MO1-vcp into Tg(unc45b:mCherry-EGFP-LC3B) autophagy-reporter embryos at the one-cell-stage, evaluated mCherry and GFP fluorescence in striated muscle cells by confocal microcopy and calculated GFP:mCherry signal ratios (KCl+ DMSO: 1.15 IQR: [0.0 to 5.01], n = 43; MO1-vcp+ DMSO: 17.02 IQR: [10.07 to 20.65], n = 24; MO1-vcp+ NH4Cl: 20.82 IQR: [12.32 to 28.57], n = 20) (Fig. S3B to E). We found that there was a significant increase of the GFP:mCherry ratio in Vcp morphant skeletal muscle cells (Fig. S3B to E), further substantiating that autophagic flux was impaired by the loss of VCP function in zebrafish in vivo.
In summary, these findings demonstrate that Vcp deficiency in the zebrafish embryo leads to severe myopathy, defective striated muscle architecture and both, impaired UPS and autophagy-mediated protein degradation.
VCP and WASHC4 colocalize at the endoplasmic reticulum
To identify and define novel and pathology-related VCP interactors, we previously identified by coimmunoprecipitation several novel proteins that physically associate with VCP such as WASHC5, PSMF1 or WASHC4 [Citation20]. Whereas the functional roles of WASHC5 and PSMF1 are partially characterized [Citation19,Citation20], the in vivo function of WASHC4 and in particular its role in striated muscle cells is still enigmatic. WASHC4 is an evolutionarily highly conserved protein that showed high amino acid identity between human, mouse and zebrafish orthologs (Figure S4). Zebrafish Washc4 displays 86% amino acid identity to human WASHC4 and 84% identity to the murine ortholog, suggesting conserved biological function across species. To test whether zebrafish (Danio rerio) Vcp (DrVcp) and zebrafish Washc4 (DrWashc4) interact directly, we performed affinity isolation experiments using glutathione S-transferase (GST)-tagged DrVcp and His-tagged DrWashc4. We found that, similar to the situation of the human and D. discoideum orthologs, zebrafish Vcp and Washc4 physically associated ()). Furthermore, we were able to demonstrate that DrVcp carrying the arginine to histidine mutation at position 155 (R155H), the most common human IBMPFD mutation, was still able to bind to DrWashc4 ()), implying that loss of their association might not be the molecular cause for the IBMPFD pathologies observed in human VCP mutation carriers.
Figure 4. VCP and its interactor WASHC4 colocalize in HEK293T cells and mouse FDB fibers. (a) Western blot analysis of a representative Washc4 pull-down experiment using anti-His (upper panel) and anti-GST (middle and lower panels) antibodies. Extracts of E. coli cells expressing recombinant His-tagged zebrafish (Danio rerio, Dr) Washc4 (His-DrWashc4) were incubated with either no beads (ctrl) or glutathione beads carrying GST (GST), GST–DrVcp wt or GST–DrVcpR155H. Coomassie stained SDS–PAGE gel of a Washc4 affinity isolation experiment (lower panel). ctrl = beads incubated with PBS. (b to b``) Transfection of HEK293T cells using a VCP-eGFP expression construct (b`) and staining with an anti-WASHC4 antibody (b) show cytoplasmic colocalization of both proteins. (c to c``) HEK293T cell immunostaining of CANX (c`) and WASHC4 (c) display colocalization at the endoplasmic reticulum. Scale bar = 5 µm. (d to d``) FDB muscle fiber of a male CD1 mouse transfected with WASHC4-eGFP (d) and VCP-mCherry (d`) expressing constructs demonstrated colocalization of both proteins. (e to e``) Close-ups of the squared area of the FDB muscle fiber of D``. (e```) Line-scan profiles demonstrated overlapping fluorescence intensities. Cell nuclei were counterstained with DAPI.
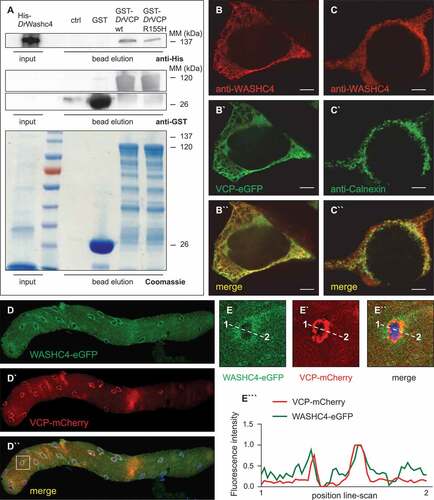
To assess whether Washc4 colocalizes with Vcp in HEK293T cells, we transfected cells with a construct for the expression of eGFP-tagged Vcp and performed coimmunostaining with a WASHC4-specific antibody. We found cytoplasmic staining of WASHC4 which mostly colocalized with Vcp-eGFP (”)), demonstrating that VCP and WASHC4 not only interact but also colocalize in HEK293T cells. To further investigate the subcellular distribution of WASHC4, we coimmunostained HEK293T cells using a WASHC4-specific antibody and an antibody against CANX (calnexin), an established marker of the endoplasmic reticulum (ER) [Citation20]. We found that WASHC4 is present at the endoplasmic reticulum in HEK293T cells (-c”)). To test whether WASHC4 and VCP also colocalize in striated muscle cells, we performed in vivo electroporation-mediated transfection of constructs for the expression of WASHC4-eGFP and VCP-mCherry in mouse flexor digitorum brevis (FDB) muscle. Remarkably, we found colocalization of WASHC4 and VCP in FDB muscle fibers at the ER (-d”)). Representative line-scan profiles derived from these fluorescence images demonstrated overlapping fluorescence intensities for WASHC4-eGFP and VCP-mCherry (-e”’)), substantiating their colocalization at the ER and further implying an in vivo role of WASHC4 in striated muscle cells.
Knockdown of washc4 leads to striated muscle dysfunction in vivo
To investigate the in vivo role of Washc4 in the vertebrate, zebrafish model, we inactivated zebrafish Washc4 by injecting morpholino-modified antisense oligonucleotides directed against either the translational start site (MO1-washc4) or the splice-donor site of exon 5 (MO2-washc4) into zebrafish embryos at the one-cell stage. As shown in ) and (d), 67% ran: [66.67% to 76.92%] of MO1-washc4 (n = 226, N = 3; control embryos: 7.23% [3.75% to 11.02%]; n = 290, N = 3) and 79.5% ran: [72.41% to 83.54%] of MO2-washc4 (n = 259, N = 4; control embryos: 5.47% ran: [4.76% to 14.61%]; n = 238, N = 4) injected zebrafish embryos developed a severe myopathic phenotype (). Hence, to investigate the efficacy of MO1-washc4, we assessed Washc4 protein levels in morphant embryos and found severely reduced proteins levels of Washc4 ()). Additionally, isolation of mRNA from MO2-washc4-injected embryos and subsequent analysis by RT-PCR confirmed the predicted effect on pre-mRNA splicing resulting in skipping of exon 5, leading to a premature stop codon in MO2-washc4 morphant embryos ()). To analyze whether the observed myopathic phenotype of Washc4-deficient embryos is specific, we performed rescue experiments by coinjecting myc-tagged washc4 mRNA together with MO2-washc4. We found that ectopic overexpression of washc4 mRNA reversed the striated muscle pathology in MO2-washc4 morphants (Fig. S5B). Only 12.04% ran: [7.69% to 15.38%] of coinjected embryos developed a myopathic phenotype (n = 216; N = 4) (); Fig. S5A and B), demonstrating that the effects on striated muscle function induced by the 2 independent MOs targeting Washc4 were specific. Furthermore, we injected washc4-specific CRISPR/Cas9 ribonucleoproteins (RNP) into wild-type zebrafish embryos at the one-cell stage to ablate Washc4 function and were able to phenocopy the heart and skeletal muscle defects observed in washc4 morphant embryos (Fig. S2A,C,D,E,G,H).
Figure 5. Phenotypic analysis washc4 morpholino-mediated knockdown in zebrafish embryos. (a to d) Lateral view of embryos at 48-hpf injected with washc4 5 bp mismatch start or splice morpholino (MO1/2-control; a, c) or washc4 start or splice MO (MO1/2-washc4; b, d). MO1/2- washc4 injection leads to a myopathic phenotype (b, d). scale bar: 450 μm. (e) Quantification of myopathic embryos after MO1/2-control or MO1/2- washc4 injection. N = 3/4 Experiments with in total n = 290 (MO1-control), n = 238 (MO2-control) n = 226 (MO1- washc4), n = 259 (MO2- washc4). The data were analyzed via N two-sided Fisher exact tests combined with a modified Fisher combination. The ratios of N experiments are shown. Western blot analysis shows reduction of Washc4 protein level in MO1-washc4-injected embryos. Actb1/β-actin was used as loading control. MO2-washc4 injection effects on mRNA splicing shown by RT-PCR. (f) Ventricular fractional shortening (FS) measurements at 48, 72 and 96 hpf of control and washc4 morphants. n = 30 (MO1-control), n = 30 (MO1-washc4); The individual measurements are shown (two-sided Wilcoxon rank-sum test). (g) Heart beat per minute of MO1-control- or MO1-washc4-injected embryos at 48, 72 and 96 hpf. n = 30 (MO1-control), n = 30 (MO1- washc4). The individual measurements are shown (two-sided Wilcoxon rank-sum test). (h, i) Spontaneous movement assay shown by false-colored superimposed pictures of control embryos (h) or Washc4 morphants (i) at 24 hpf; red pictures = 0 s; green pictures = 10 s; yellow = merge. scale bar: 1 mm. (j) Statistical analysis of spontaneous movement assay; N = 3 experiments with in total n = 56 (MO1-control), n = 60 (MO1-washc4) were performed (N two-sided Fisher exact tests combined with a modified Fisher method). The ratios of N experiments are shown. (k) Quantification of touch-evoke flight response assay of MO1-control- or MO1-washc4-injected embryos at 60 hpf. n = 40, N = 3 (MO1-control), n = 40, N = 3 (MO1-washc4); data represent means.
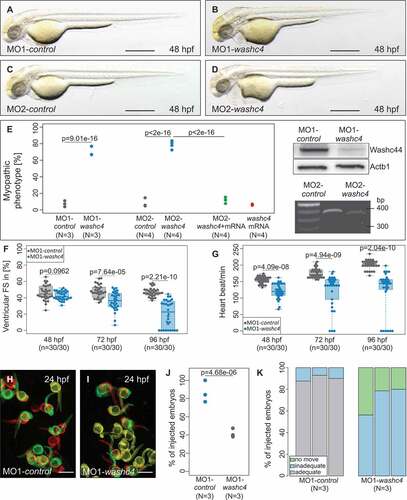
In Washc4 morphants, FS was reduced to 35.95% IQR: [29.25% to 44.82%] (n = 30) compared to control morphants (FS: 47% IQR: [41.38% to 52.81%]; n = 30) at 72 hpf and further decreased to 20.38% IQR: [0% to 35.06%] compared to control (FS: 45.86% IQR: [43.76% to 49.77%]; n = 30) at 96 hpf ()). At 48 hpf, although slightly reduced, FS of Washc4 morphants was not significantly impaired compared to control-injected embryos (FS: 41.42% IQR: [38.25% to 47.83%] (morphants) and FS: 44.6% IQR: [40.93% to 53.22%] (controls); n = 30) ()). By contrast, heart beat frequency in Washc4-deficient embryos was significantly reduced as early as 48 hpf (120.6 IQR: [108 to 144] bpm; n = 30) compared to controls (155.3 IQR: [150 to 162] bpm; n = 30) and remained reduced during development ()). Similar to the situation in Vcp-deficient zebrafish embryos, Washc4-deficient hearts displayed defined cardiac chambers with atrium and ventricle separated by the atrio-ventricular ring and properly developed endocardial and myocardial layers of both heart chambers (Fig. S5G and H). Additionally, expression analyses of myh6 and myh7 at 48 hpf revealed no alterations (Figure S5 C to F), demonstrating that both cardiac chambers were regularly differentiated in Washc4-deficient hearts.
Inactivation of Washc4 in zebrafish embryos resulted in reduced motility as shown by voluntary movement measurements at 24 hpf (-j)). 84.21% ran: [76.47% to 100%] of control embryos moved spontaneously (n = 56, N = 3), whereas only 40% ran: [38.1% to 47.37%] of Washc4-deficient embryos exhibited spontaneously movements at 24 hpf (n = 60, N = 3) (-j)). To further analyze the progression of skeletal muscle dysfunction, we assessed the flight response after touch-evoked stimulation at 72 hpf. Control embryos reacted rapidly and showed a powerful escape response after tactile stimulation ()). By contrast, Washc4-deficient embryos showed severely impaired motility at 72 hpf (inadequate: 78.57 ran: [56.25% to 80.00%]/no movement 21.43 ran: [20.00% to 43.75%; n = 40, N = 3) ()). Myod1 and myog expression was, similar to the situation in Vcp morphants, unaffected at the 16 to 18 somite stage (Fig. S6A to D), demonstrating that defective skeletal muscle differentiation does not account for impaired motility in Washc4-deficient embryos.
Washc4 deficiency disrupts muscle ultrastructure without affecting motor neuron development
To investigate whether defective motility in Washc4-deficient zebrafish embryos is due to structural muscle alterations, we first assessed myofibrillar organization of Washc4 morphants by birefringence assays. In fact, birefringence signal intensity from the skeletal muscle was severely diminished in Washc4-deficient embryos (75.34 IQR: [64.9 to 90.8]; n = 30) compared to control-injected embryos at 72 hpf (171.36 IQR: [159.91 to 185.3]; n = 30) (). Next, we performed histological analyses of the skeletal muscle and found indeed severely disorganized myofibrils and even vacuolization of skeletal muscle tissue (). Both assays imply that loss of Washc4 might interfere with regular myofibrillar organization.
Figure 6. Inactivation of Washc4 in zebrafish embryos results in skeletal muscle ultrastructural alterations. (a and b) Lateral view of MO1-control- (a) and MO1-washc4-injected (b) embryos showing birefringence at 72 hpf. scale bar: 400 μm. (c) Statistical analysis of birefringence signal. n = 30 (MO1-control), n = 30 (MO1-washc4); two-sided Wilcoxon rank-sum tests. (d and e) H&E-stained sagittal histological sections of skeletal muscle of control embryos (d) and Washc4 morphants (e) at 72 hpf; scale bar = 100 μm. (f to g```) Transmission electron microscopy in MO1-control (f to f``) and MO1-washc4 (g to g```) skeletal muscle at 72 hpf. (f to f``) Control muscle cells exhibit highly organized myofibrils (f``) and normal mitochondria, next to the sarcomeric Z-discs (f`). (g to g``) Washc4 morphant muscle cells show less myofibrils, dysmorphic mitochondria (g``) and an accumulation of various vesicular structures; AV = autophagic vesicle, M = mitochondria, MeB = membranous bodies; scale bar: 2 μm (f, g); scale bar = 1 μm (f` and f``, g` to g```). (j and k) Immunostaining using an anti-acetylated tubulin (acTub) antibody of MO1-control- (j) or MO1-washc4-injected (k) embryos at 72 hpf. scale bar: 200 μm.
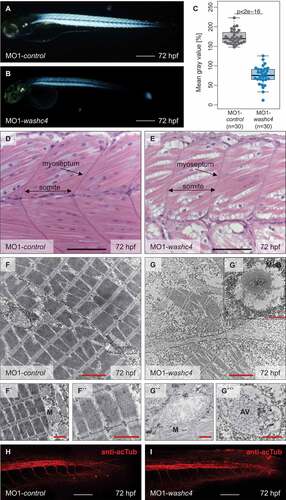
Transmission electron microscopy analyses of control muscles showed densely packed sarcomeres composed of precisely aligned thick and thin filaments and flanking Z-disks as well as elongated muscle cell nuclei (-f”)). Similar to the situation in Vcp-deficient zebrafish embryos, Washc4 morphant muscle cells exhibited reduced numbers of myofibrils and disorganized sarcomeres as well as dysmorphic mitochondria with diminished cristae numbers (-g””)). Notably, Washc4 deficiency leads to the accumulation of numerous membranous vesicular structures (autophagic vesicles) (`) and (g”’)) often linked to the rough endoplasmic reticulum.
To investigate whether loss of Washc4, similar to the situation in Washc5-deficient zebrafish embryos [Citation20], leads to neuronal defects, we conducted immunostaining of Washc4-deficient embryos using antibodies against acetylated tubulin and against the primary motor neuronal marker Syt2b (synaptotagmin IIb). In contrast to Washc5 morphants, Washc4 morphants showed no gross alterations in the development of the caudal neural tube or the ventral and caudal motoneurons compared to control-injected embryos () and (i) and Fig. S6E and F), suggesting that loss of Washc4 predominantly and directly affects striated muscles.
Loss of washc4 leads to ER stress and interferes with autophagy-mediated protein degradation
To assess whether Washc4, similar to its interactor Vcp, functions in the regulation of protein degradation via the 26S proteasome, we first evaluated the levels of poly-ubiquitinated proteins in Washc4-deficient embryos compared to control-injected embryos by western blot analyses. In contrast to Vcp deficiency, loss of Washc4 in zebrafish embryos did not result in elevated levels of poly-ubiquitinated proteins compared to control-injected embryos ()), suggesting no primary role of Washc4 in the regulation of UPS-mediated protein degradation.
Figure 7. Loss of Washc4 leads to an upregulation of ER stress markers and to an impaired autophagy. (a) Western blot analysis of ubiquitinated proteins after MO1-control and MO1-washc4 injection. (b) qRT-PCR of ER stress markers (hspa5, atf4 and ppp1r15a) of control embryos or washc4 morphants at 3 dpf. The individual measurements are shown (log10). The 3 markers were analyzed via Wilcoxon rank-sum tests. A combined P value (comb. p) was determined via a modified Fisher method. slc25a5 was used as housekeeping gene. (c) Western blot analysis using anti-Sqstm1 antibody after MO1-control and MO1-washc4 injection (upper panel); quantification of gray values (lower panel; n = 6). (d) Representative western blotting of Lc3 of control embryos or washc4 morphants (upper panel); quantification of gray values (lower panel; n = 6). Individual measurements are shown (Wilcoxon rank-sum test). (e) MO1-control-or MO1-washc4-injected embryos treated with DMSO or ammonium chloride (NH4Cl). Control embryos show a significant increase of Lc3-II levels after NH4Cl treatment in comparison to DMSO controls. Ammonium chloride treatment of Washc4 morphants did not significantly enhance the Lc3-II level in comparison to DMSO treated embryos (n = 6). Actb1/β-actin was used as loading control. The individual measurements are shown (Wilcoxon rank-sum test).
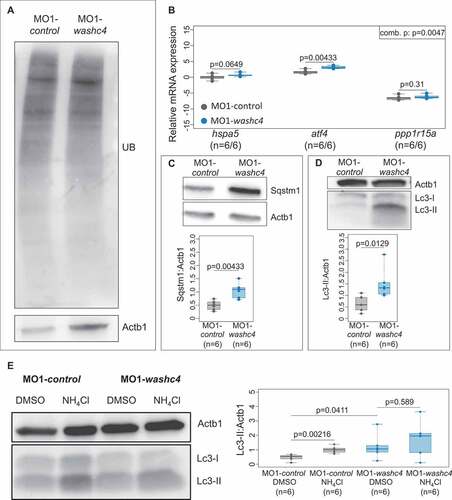
As shown before, WASHC4 colocalizes with VCP at the endoplasmic reticulum in striated muscle cells. To define whether loss of WASHC4 leads to ER stress and UPR in vivo, we investigated transcript levels of the established ER stress markers hspa5/BiP (heat shock protein 5), atf4 (activating transcription factor 4) and ppp1r15a/gadd34 (protein phosphatase 1 regulatory subunit 15A) in Washc4-deficient zebrafish embryos. We found all 3 markers upregulated in Washc4 morphants (1.55 IQR: [1.46 to 1.59] hspa5; 8.38 IQR: [6.92 to 10.19] atf4; 0.01 IQR: [0.01 to 0.02] ppp1r15a; n = 5) ()), implying that Washc4 deficiency leads to ER stress in vivo.
ER stress and subsequently the unfolded protein response (UPR) were recently shown to trigger predominantly the autophagy-lysosome degradation machinery [Citation14]. To test whether autophagic degradation is activated in Washc4-deficient embryos, we first assessed protein levels of Sqstm1 and Lc3-II and found that both markers were significantly upregulated in Washc4 morphant embryos (Sqstm1: 1.08 IQR: [0.84 to 1.16]; Lc3-II: 1.41 IQR: [1.12 to 1.5]; n = 6) compared to controls (Sqstm1: 0.5 IQR: [0.37 to 0.6]; Lc3-II: 0.61 IQR: [0.42 to 0.83]; n = 6) () and (d)), indicating that autophagy might be indeed induced by the loss of Washc4. Next, we performed measurements of the autophagic flux by treating Washc4-deficient and control embryos with the lysosome-autophagosome fusion inhibitor ammonium chloride (NH4Cl) followed by western blot determination of Lc3-II levels. In contrast to control embryos, we found in Washc4 morphants in response to NH4Cl treatment no significant increase in Lc3-II protein levels (DMSO: 1.13 IQR: [0.88 to 1.25] and after NH4Cl treatment: 1.86 IQR: [1.09 to 2.09]; n = 6) ()), suggesting that Washc4 deficiency interferes with the autophagic flux, possibly autophagosome and lysosome fusion, and thereby the function of the autophagy machinery in vivo.
In summary, our findings suggest a role of the novel Vcp-interactor Washc4 in autophagy-mediated proteostasis in striated muscle cells and when defective leads to severe myopathy in zebrafish in vivo.
Discussion
The UPS and autophagy are both essential machineries for the control of protein quality and quantity in vivo. Here, VCP has emerged as a crucial element of protein degradation via both degradation machineries [Citation7]. Autosomal dominant mutations in the human VCP gene cause the multisystemic degenerative disease IBMPFD where disease symptoms usually manifest late in life [Citation18]. Mice carrying the most frequent human VCP mutation, arginine to histidine at position 155 (R155H), develop normally but exhibit severe degenerative pathologies during adulthood [Citation29–Citation31]. Interestingly, nullizygous vcp mice are not viable and die at a peri-implantation stage, demonstrating that VCP is fundamentally important for embryogenesis in mice [Citation32]. Additionally, knockdown of Vcp in zebrafish using morpholino-modified antisense-oligonucleotides leads to severe defects in neuronal outgrowth and neurodegeneration [Citation33], whereas the impact of Vcp inactivation on striated muscles has not been investigated in this study.
Here, we characterized the in vivo role of Vcp in zebrafish striated muscle and found that Vcp-deficient zebrafish embryos display severe muscle pathologies but survive up to 5 days post fertilization allowing the dissection of the underlying molecular mechanisms. Genetic and pharmacological inhibition of Vcp led to severely impaired cardiac and skeletal muscle function without interfering with early differentiation of these organ systems. Ultrastructural analyses in Vcp-deficient zebrafish muscles revealed severely impaired myofibrillar organization, dysmorphic mitochondria and numerous vesicular bodies. This is consistent with previous findings in muscle tissue from mice carrying the human mutations VCPR155H and VCPA232E which also display pronounced vacuolization, degenerated mitochondria and loss of myofilament organization [Citation29,Citation31]. VCP plays a key role in protein degradation via the UPS, and haploinsufficient mice display significantly elevated levels of poly-ubiquitinated proteins due to severely impaired proteasomal function [Citation19]. Additionally, VCP ablation in HeLa cells also leads to the accumulation of poly-ubiquitinated proteins [Citation34], substantiating a fundamental role of VCP in UPS-mediated proteostasis in mammals.
We also found severely increased levels of poly-ubiquitinated proteins in Vcp-deficient zebrafish, implying conserved biological function in the control of UPS-mediated protein degradation in vivo. In addition to these loss-of-function models, VCPR155H transgenic knock-in mice display autophagy pathologies resulting in increased Sqstm1 and LC3-II protein levels, both sensitive markers for autophagy function in vitro and in vivo [Citation28]. We found significantly increased levels of Sqstm1 in Vcp-deficient zebrafish, suggesting that loss of Vcp effects autophagy in zebrafish in vivo. To further investigate a possible impairment of the autophagy machinery, we measured changes in the autophagic flux in Vcp-deficient and control zebrafish embryos after the incubation with the lysosome-autophagosome fusion inhibitor ammonium chloride (NH4Cl). In a control situation, ammonium chloride treatment leads to the accumulation of Lc3-II proteins due to the blockade of the lysosomal degradation of Lc3-II. Interestingly, we did not find significantly elevated Lc3-II levels in ammonium chloride treated MO-vcp-injected zebrafish embryos compared to DMSO-treated Vcp morphants, clearly demonstrating that, similar to the situation in HeLa cells and mice devoid of functional VCP [Citation27], autophagic flux and thereby autophagic function is also impaired in Vcp-deficient zebrafish embryos. To further substantiate this finding, we assessed the impact of Vcp deficiency on the function of the autophagy system using a therefore generated novel striated muscle-specific transgenic autophagy-reporter zebrafish line Tg(unc45b:mCherry-EGFP-LC3B) and found, similar to the Lc3-II western blot analyses, impaired autophagic flux in Vcp-deficient zebrafish muscle in vivo.
In summary, both, autophagic proteolysis and the ubiquitin-proteasome system are recognized to play important roles in muscle protein homeostasis, and Vcp crucially regulates the function of both degradation machineries thereby regulating striated muscle structure and function and when mutated leads to myopathy [Citation19,Citation35,Citation36].
VCP functions are orchestrated by many cofactors, especially several UPS substrates need VCP-cofactor interaction in order to be properly processed [Citation14]. Recently, we have identified PSMF1 as a specific VCP interactor and find that this crucial UPS regulator controls cellular proteostasis via proteasome-mediated protein degradation in an antagonistic manner [Citation19]. Additionally, WASHC5 is defined as a novel VCP interactor that is involved in the pathogenesis of protein aggregate diseases impairing striated muscles and the central nervous system (CNS) [Citation20]. In this context, Washc4/Swip (WASH complex subunit 4), known to interact with WASHC5, is also identified as a novel VCP binding partner in vitro (HEK293 cells) and in vivo (D. discoideum) [Citation20]. WASHC4 is described as a component of the WASH complex, which plays a role in actin polymerization and endosomal transport processes [Citation21]. A WASHC4 variant (WASHC4P1019R) results in autosomal recessive intellectual disability. Expression analyses and studies using cells derived from patients carrying the WASHC4P1019R variant show that this mutation destabilizes the protein thereby interfering with the stability of the entire WASH complex [Citation21]. Furthermore, WASHC4 SNPs (single-nucleotide polymorphisms) are associated with Alzheimer disease (AD) in an African American cohort [Citation37]. Nevertheless, the in vivo role of WASHC4 and the role of its association with VCP in striated muscle cells were completely unknown.
We found here that WASHC4 colocalizes with VCP mainly at the ER in HEK293 but also in murine mouse flexor digitorum brevis muscle cells, suggesting an important role of WASHC4 in striated muscles. In fact, Washc4-deficient zebrafish embryos displayed severely diminished heart and skeletal muscle function and structural as well as ultrastructural abnormalities, mimicking the phenotypic characteristics observed in Vcp-depleted fish. Transmission electron microscopy analyses in Washc4-deficient zebrafish muscles revealed myofibrillar disorganization, degenerated mitochondria and vesicular bodies that were often associated with the ER, again suggesting an important role of Washc4 at the ER. ER-associated VCP is involved in the UPR, a process that is activated by ER stress leading to the proteasomal degradation of misfolded ER proteins, called ERAD (endoplasmic reticulum-associated degradation) [Citation38]. Interestingly, recent data imply that although UPS-mediated protein degradation is involved, autophagy seems to be the predominant degradation pathway during ER stress and UPR activation [Citation15,Citation16]. We found here that Washc4 deficiency resulted in ER stress in vivo without impairing the proteasomal protein degradation machinery. By contrast, protein levels of the established autophagy markers Sqstm1 and Lc3-II were significantly upregulated in Washc4-deficient zebrafish embryos and the autophagic flux was impaired in Washc4 morphants, consistent with impaired autophagic function. As a result of compromised autophagy, severely increased numbers of autophagic vesicles are present in Washc4-deficient embryos. In summary, these findings suggest that Vcp and Washc4, even though their interaction and similar myopathic pathologies after inactivation, might regulate proteostasis in striated muscle cells by at least partly distinct molecular degradation mechanisms.
The pathological relevance of impaired proteostasis, as observed in Vcp-deficient zebrafish, mice or humans as well as in Washc4 morphant zebrafish embryos, is emphasized by several studies. These link defective proteasomal and autophagy-mediated protein degradation to a wide variety of human diseases including neurodegenerative disorders, immune defects, metabolic diseases, cancer but also striated muscle disease, such as muscular dystrophies, myofibrillar myopathies or cardiomyopathies [Citation39,Citation40]. Whether mutations in WASHC4 might contribute to the pathogenesis of these severe disorders is currently not known. However, based on our findings in the vertebrate model system zebrafish, careful analysis of WASHC4 as a novel candidate gene might help to further dissect the molecular mechanisms that drive the pathology in human diseases such as neurodegenerative disorders or striated muscle diseases.
Materials and methods
Zebrafish strains
All procedures and experiments in this study were carried out after appropriate institutional approvals (Tierforschungszentrum (TFZ) Ulm University, No. 0183), which conform to the EU Directive 2010/63/EU. Care and breeding of zebrafish Danio rerio was carried out as described [Citation41]. Pictures and movies were recorded at 16 to 18 somite stage and 24, 48, 60, 72 and 96 h post fertilization. For documentation, zebrafish embryos were treated with 0.003% 1-phenyl-2-thiourea (Sigma-Aldrich, P7629) to inhibit pigmentation.
Transgenesis
To generate a striated muscle-specific transgenic autophagy reporter zebrafish line, pDestTol2pA2 was used as backbone for multisite gateway cloning reaction. The final vector construct for microinjection into zebrafish oocytes and the generation of germline transgenic lines includes the zebrafish striated muscle-specific minimal unc45b promoter and mCherry-EGFP-LC3B (rat LC3B) sequences to visualize autophagy function in striated muscle cells in vivo [Citation42].
Injection procedures, ML240, ammonium chloride and MG132 treatment
Morpholino-modified antisense oligonucleotides (MOs) (Gene Tools, LLC) and mRNA were injected into one-cell stage zebrafish embryos. The sense-capped mRNA of Myc-tagged zebrafish washc4 was synthesized using the mMESSAGE mMASCHINE kit (Thermo Fisher Scientific, AM1340) and used for rescue experiments.
To knock down zebrafish vcp, MOs targeting the translational start site of zebrafish vcp (MO1-vcp: 5´-AAGCCATATTGCTTCTCCCGCAAAA-3´) or the splice-donor site of exon 2 (MO2-vcp: 5´-TTGACATCCAAAACTGTACCTGAGA-3´) with the corresponding 5 base pair mismatch MOs (MO1-control: 5´-AACCAATATTGATTCTACCGAAAAA-3´ and MO2-control: 5´-TTCACATCAAAAACTCTACCTCACA −3´) were injected into fertilized oozytes at the one-cell stage. MOs were injected with 9.1 ng MO1-vcp, MO1-control and 8.4 ng MO2-vcp, MO2-control in 0.2 M potassium chloride (KCl; Sigma-Aldrich, P9541).
To knock down zebrafish washc4, MOs directed against the translational start site of zebrafish washc4 (MO1-washc4: 5´-CAGGAGATAATGAATCCACAGCCAT-3´) or the splice-donor site of exon 5 (MO2-washc4: 5´-AAACAGCAATTATTACCTCCTTCCC-3´) with the appropriate 5 base pair mismatch MOs (MO1-control: 5´-CAGGACATAATCAATACACACCAAT-3´ and MO2-control: 5´-AAAAAGAAATTATTAACTCATTCAC-3´) were injected. MOs were injected with 6.5 ng MO1-washc4, MO2-washc4, MO1-control and MO2-control in 0.2 M potassium chloride. At 10 hpf wild-type zebrafish embryos were treated with 1% DMSO (Sigma-Aldrich, D8418) or 15 µM ML240 (Sigma-Aldrich, SML1071) in E3 (4 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4) for 38 h. At 48 hpf, control zebrafish embryos or vcp and washc4 morphants were treated with DMSO or ammonium chloride (100 mM NH4Cl; Sigma-Aldrich, A9434) in E3 for 4 h or MG132 (100 µM) (Tocris Bioscience, 1748) [Citation28].
For CRISPR/Cas9 injections 400 ng/µl recombinant Cas9-RFP protein (Eupheria Biotech GmbH, CAS9-NLS-taqRFP, conc. 2 µg/µl) were mixed with a synthetic trans-activating crRNA (tracrRNA) (100 ng/µl; AAACAGCAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUGGCACCGAGUCGGUGCU) and a gene-specific crRNA (Eurofins Genomics) against vcp (GAAGGUUCGCAUGAACAGGGGUUUUAGAGCUAUGCUG UUUUG) or washc4 (GCUGAAACCAAGUUCUACAAGUUUUAGAGCUAUGCUG UUUUG), respectively, at 50 ng/µl in 300 µM KCl injection buffer. A non-targeting CRISPR RNA (crRNA) was used as negative control. The Cas9-crRNA mix was injected into one-cell stage zebrafish embryos. Gene disruption of the target DNA locus was confirmed via PCR.
Coimmunoprecipitation experiments, zebrafish protein lysate extraction, Coomassie gels and western blot analysis
Affinity-isolation experiments were performed as described previously [Citation43]. Briefly, GST-tagged fusion proteins were directly immobilized on glutathione-Sepharose beads (GE Healthcare, GE17–0756–01) from E. coli extracts and incubated with purified proteins for 1 h at 4°C in PBS under rotation. Afterwards, the beads were washed 3 times with phosphate-buffered saline (PBS; Gibco, 10,010,023) and eluted with 20 mM reduced glutathione (GE Healthcare, GE17–5132–01) in 50 mM Tris (Sigma Aldrich, T1503). Bead elutions were analyzed by Coomassie Brilliant Blue (SERVA Electrophoresis GmbH, 17,521) staining and anti-His western blotting. His6-tagged proteins were purified by affinity capture (HisTrap; GE Healthcare, GE17–5247–01).
Protein extraction of embryonic zebrafish and immunoblotting was carried out as described previously [Citation44]. SDS gels were stained with Coomassie Brilliant Blue or further used for western blot analysis. For immunoblotting the following primary antibodies were used: rabbit anti-VCP (1:5000; Novus Biologicals, NB100–1558) [Citation20], rabbit anti-K48-linkage-specific polyubiquitin (1:1000; Cell Signaling Technology, 4289), mouse anti-ubiquitin (P4D1, 1:1000; Cell Signaling Technology, 3936), rabbit anti-SQSTM1 (1:2000; Abcam, ab109012), rabbit anti-LC3A (1:2000; Novus Biologicals, LLC, NB100–2331) (this antibody detects both LC3A and LC3B), mouse anti-His (1:10,000; Sigma-Aldrich, H1029), rabbit anti-GST (1:10,000; Cell Signaling Technology, 2622), rabbit anti-WASHC4/SWIP (1:2000; Abcam, ab111109), mouse anti-Syt2b (1:100; Developmental Studies Hybridoma Bank (DSHB), Clone znp-1) and mouse anti-MYC (1:10,000; Cell Signaling Technology, 2276). For loading, control mouse anti-ACTB/β-actin (1:2000; Sigma-Aldrich, A5441) was used. Signals were detected by chemiluminescence (anti-mouse IgG HRP-linked, anti-rabbit IgG HRP-linked; Cell Signaling Technology, 7076, 7074) using a Luminescent image analyzer (Image Quant LAS4000mini, Freiburg, Germany). Protein levels were compared by measuring mean gray values.
Cell culture, transfection and immunostaining
HEK293T human embryonic kidney cells were cultured in Dulbecco Modified Eagle Medium (Gibco, 31,966–021) with 10% fetal calf serum (Sigma-Aldrich, F2442) and 1% penicillin-streptomycin mixture (Gibco, 15,140–122) at 37°C and 5% CO2. Cells were grown on glass coverslips in 4/8-well culture plates and transiently transfected with a VCP-eGFP-expressing construct using the Lipofectamine 2000 method (Invitrogen, 11,668,027). After 24 to 48 h of transfection, cells were washed with 1x PBS and fixed with 4% formalin (Sigma-Aldrich, HT501128) for 20 min at room temperature or methanol (Sigma-Aldrich, 32,213) for 10 min at 4°C. Cells were permeabilized with 0.2% Triton X-100 (Sigma-Aldrich, T8787) in 1x phosphate-buffered saline (PBST) for 5 min. For indirect immunostaining, cells were further blocked with 1% BSA (Sigma-Aldrich, A8531) in 1x PBS for 60 min. As primary antibody, rabbit anti-WASHC4/SWIP (1:100; Abcam, ab111109) was used and incubated overnight at 4°C. Secondary Alexa Fluor 555-labelled goat anti-rabbit antibody (1:1000; Invitrogen, A-21,429) was used for 40 min. For endoplasmic reticulum staining, cells were incubated with mouse monoclonal anti-CANX (calnexin)-Alexa Fluor 488 (1:200) (Abcam, ab202574) for 40 min.
Animals, in vivo transfection experiments and single-fiber isolation
Animals (adult, 2-month-old male CD1 mice) were handled by specialized personnel under the control of inspectors of the Veterinary Service of the Local Sanitary Service (ASL 16-Padova), the local officers of the Ministry of Health. All procedures are specified in the projects approved by the Italian Ministero della Salute, Ufficio VI (authorization numbers C65). In vivo transfection was performed by intramuscular injection of expression plasmids (WASHC4-eGFP and VCP-mCherry) in flexor digitorum brevis (FDB) muscle followed by electroporation as described [Citation45]. Flexor digitorum brevis muscle muscles were digested in type I collagenase (Worthington Biochemical Corp., LS004194) at 37°C for 1 h, dissociated into single fibers, plated on glass coverslips coated with laminin (Sigma-Aldrich, L2020) and cultured as described [Citation46].
RNA isolation, reverse-transcriptase (RT)-PCR and quantitative real-time PCR
RNA extraction was carried out by using either Qiazol Lysis Reagent (Qiagen, 79,306) or by RNeasy Plus Mini Kit (Qiagen, 74,104), according to the manufacturer’s instructions. cDNA synthesis was performed by using 1 µg or 700 ng total RNA, oligo(dT) primer and SuperScript III Reverse Transcriptase (Invitrogen, 18,080,044). RT-PCR was carried out according to standard protocols using vcp- or washc4-specific primers. Quantitative real-time PCR was performed according to standard protocols with the usage of SYBRGreen master mix (Roche Diagnostics, 04887352001) and a Roche LightCycler 480 II (Roche Diagnostics Deutschland GmbH, Mannheim, Germany) The housekeeping genes slc25a5 and 18s ribosomal RNA (18S) were used for normalization.
Spontaneous movement, touch-evoke escape response assay and birefringence
False-colored superimposed overviews of 24-hpf embryos were analyzed for the spontaneous movement assay. The touch-evoke escape response assay and non-invasive birefringence was performed and analyzed as described previously [Citation25]. Motility after tactile stimulation was classified in 3 groups: adequate, inadequate, and no movement.
Histology and transmission electron microscopy
For histology, fixed embryos were embedded in JB-4 (Polysciences, Inc., 00226–1) and 5-µm sections were cut, dried and stained with hematoxylin & eosin [Citation47]. For transmission electron microscopy analyses, zebrafish embryos were fixed in 2.5% glutaraldehyde and 1% paraformaldehyde (Sigma-Aldrich, P6148) in 0.1 M phosphate buffer (pH 7.3). Afterwards, samples were post-fixed in 1% osmium tetroxide (dehydrated). Embryos were embedded in Epon (Sigma-Aldrich, 45,359) and then sections and ultrathin sections (50 nm) were cut. The ultrathin sections were contrasted using uranyl acetate and lead citrate and subsequently imaged on an electron microscope JEM-1400 (JEOL USA, Inc., Peabody, MA, USA) [Citation48].
Functional assessment and statistical analysis
Zebrafish images were taken with an Olympus SZX 16 microscope (Olympus Europa SE & Co. KG, Hamburg, Germany) and movies were recorded with a Leica DM IL LED microscope (Leica Mikrosysteme Vertrieb GmbH, Wetzlar, Germany). Fractional shortening was measured with the help of the zebraFS software [Citation49]. HEK293T cell, transgenic fish and FDB muscle fiber images were taken with the Zeiss LSM710 (Carl Zeiss Microscopy GmbH, Jena, Germany) and Leica SP8 microscopes (Leica Mikrosysteme Vertrieb GmbH, Wetzlar, Germany).
Statistical analyses and graphs were done in R (www.r-project.org). Generally, the nonparametric two-sided Wilcoxon test was used as indicated. For count data Fisher exact test was used. Test combinations were done with a modified Fisher method [Citation50]. Location and variation data are either given as median and interquartile range (IQR) (n > 4) or median (MED) and range (ran) (n < 5). A detailed description of animal numbers and number of independent experiments for the statistical analyses is provided in Table S2.
Abbreviations
| ACTB | = | actin beta |
| Atf4 | = | activating transcription factor 4 |
| Hspa5/BiP | = | heat shock protein 5 |
| bpm | = | beats per minute |
| CANX | = | calnexin |
| CRISPR/Cas9 | = | clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9 |
| crRNA | = | CRISPR RNA |
| DMSO | = | dimethyl sulfoxide |
| Dr | = | Danio rerio |
| EGFP | = | enhanced green fluorescent protein |
| ER | = | endoplasmatic reticulum |
| FDB | = | flexor digitorum brevis |
| FS | = | fractional shortening |
| GFP | = | green fluorescent protein |
| GST | = | glutathione S-transferase |
| h | = | hours |
| HEK | = | human embryonic kidney |
| hpf | = | hours post fertilization |
| IBMPFD | = | inclusion body myopathy with Paget disease of the bone and frontotemporal dementia |
| IQR | = | interquartile range |
| KCl | = | potassium chloride |
| Map1lc3A/B | = | microtubule-associated protein 1 light chain 3 |
| MED | = | median |
| MFM | = | myofibrillar myopathy |
| MO | = | morpholino-modified antisense oligonucleotides |
| mRNA | = | messengerRNA |
| Myh6/Amhc | = | myosin, heavy chain 6, cardiac muscle, alpha |
| Myh7/Vmhc | = | myosin heavy chain 7 |
| Myod1 | = | myogenic differentiation 1 |
| Myog | = | myogenin |
| ng | = | nanogram |
| NH4Cl | = | ammonium chloride |
| Ppp1r15a/Gadd34 | = | protein phosphatase 1, regulatory subunit 15A |
| PSMF1 | = | proteasome inhibitor subunit 1 |
| ran | = | range |
| RNP | = | ribonucleoprotein |
| RT-PCR | = | reverse transcription-polymerase chain reaction |
| SQSTM1/p62 | = | sequestosome 1 |
| Syt2b | = | synaptotagmin IIb |
| tracrRNA | = | trans-activating crRNA |
| unc45b | = | unc-45 myosin chaperone B |
| UPR | = | unfolded protein response |
| UPS | = | ubiquitin-proteasome system |
| VCP | = | valosin containing protein |
| WASH | = | Wiskott-Aldrich syndrome protein and scar homolog |
| WASHC4 | = | WASH complex subunit 4 |
| WASHC5 | = | WASH complex subunit 5 |
| µM | = | micromolar |
Supplemental Material
Download Zip (7.7 MB)Acknowledgments
We thank Regine Baur, Renate Durst, Karin Strele, Katrin Vogt, Sabrina Diebold, Jessica Rudloff, Anaïs Franco Romero and Carolin Berwanger for their excellent technical assistance. This work was supported by the Deutsche Forschungsgemeinschaft (DFG) RO2173/4–1 (WR), RO2173/4–2 (WR), JU2859/1–2 (SJ) and the German Federal Ministry of Education and Research (BMBF) (e:Med-SYMBOL-HF grant #01ZX1407A and #01ZX1708C) (SJ & HAK) and the Ministerium für Wissenschaft, Forschung und Kunst Baden-Württemberg (MWK) Juniorprofessurenprogramm (SJ).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary data
Supplementary data can be accessed here
Additional information
Funding
Related Research Data
References
- Wang X, Robbins J. Proteasomal and lysosomal protein degradation and heart disease. J Mol Cell Cardiol. 2014 Jun;71:16–24. PubMed PMID: 24239609; PubMed Central PMCID: PMC4011941.
- Jung T, Catalgol B, Grune T. The proteasomal system. Mol Aspects Med. 2009 Aug;30(4):191–296. . PubMed PMID: 19371762.
- Sorokin AV, Kim ER, Ovchinnikov LP. Proteasome system of protein degradation and processing. Biochem Biokhimiia. 2009 Dec;74(13):1411–1442. PubMed PMID: 20210701.
- Bohnert KR, McMillan JD, Kumar A. Emerging Roles of ER Stress and Unfolded Protein Response Pathways in Skeletal Muscle Health and Disease. J Cell Physiol. 2017 Feb 08. PubMed PMID: 28177127. DOI:10.1002/jcp.25852
- Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu Rev Biochem. 2015;84:435–464. PubMed PMID: 25784053; PubMed Central PMCID: PMC4539002.
- Rinaldi C, Mager I, Wood MJ. Proteostasis and Diseases of the Motor Unit. Front Mol Neurosci. 2016;9:164. PubMed PMID: 28082869; PubMed Central PMCID: PMC5187379.
- Meyer H, Weihl CC. The VCP/p97 system at a glance: connecting cellular function to disease pathogenesis. J Cell Sci. 2014 Sep 15;127(Pt 18):3877–3883. . PubMed PMID: 25146396; PubMed Central PMCID: PMC4163641.
- Tang WK, Xia D. Mutations in the Human AAA+ Chaperone p97 and Related Diseases. Front mol biosci. 2016;3:79. PubMed PMID: 27990419; PubMed Central PMCID: PMC5131264.
- Abramzon Y, Johnson JO, Scholz SW, et al. Valosin-containing protein (VCP) mutations in sporadic amyotrophic lateral sclerosis. Neurobiol Aging. 2012 Sep;33(9):2231 e1–2231 e6. PubMed PMID: 22572540; PubMed Central PMCID: PMC3391327.
- de Bot ST, Schelhaas HJ, Kamsteeg EJ, et al. Hereditary spastic paraplegia caused by a mutation in the VCP gene. Brain: J neurology. 2012 Dec;135(Pt 12):e223; author reply e224. PubMed PMID: 22991237.
- Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004 Apr;36(4):377–381. PubMed PMID: 15034582.
- Figueroa-Bonaparte S, Hudson J, Barresi R, et al. Mutational spectrum and phenotypic variability of VCP-related neurological disease in the UK. J Neurol Neurosurg Psychiatry. 2016 Jun;87(6):680–681. PubMed PMID: 26105173; PubMed Central PMCID: PMC4893144.
- Baek GH, Cheng H, Choe V, et al. Cdc48: a swiss army knife of cell biology. J Amino Acids. 2013;2013:183421. PubMed PMID: 24167726; PubMed Central PMCID: PMC3791797.
- Zhang T, Ye Y. The final moments of misfolded proteins en route to the proteasome. DNA Cell Biol. 2014 Aug;33(8):477–483. . PubMed PMID: 24833120; PubMed Central PMCID: PMC4117260.
- Nijholt DA, de Graaf TR, van Haastert ES, et al. Endoplasmic reticulum stress activates autophagy but not the proteasome in neuronal cells: implications for Alzheimer’s disease. Cell Death Differ. 2011 Jun;18(6):1071–1081. PubMed PMID: 21252911; PubMed Central PMCID: PMC3131935.
- Scheper W, Nijholt DA, Hoozemans JJ. The unfolded protein response and proteostasis in Alzheimer disease: preferential activation of autophagy by endoplasmic reticulum stress. Autophagy. 2011 Aug;7(8):910–911. PubMed PMID: 21494086; PubMed Central PMCID: PMC3149694.
- Weihl CC. Valosin containing protein associated fronto-temporal lobar degeneration: clinical presentation, pathologic features and pathogenesis. Curr Alzheimer Res. 2011 May;8(3):252–260. PubMed PMID: 21222596; PubMed Central PMCID: PMC3246398.
- Hubbers CU, Clemen CS, Kesper K, et al. Pathological consequences of VCP mutations on human striated muscle. Brain: J neurology. 2007 Feb;130(Pt 2):381–393. PubMed PMID: 16984901.
- Clemen CS, Marko M, Strucksberg KH, et al. VCP and PSMF1: antagonistic regulators of proteasome activity. Biochem Biophys Res Commun. 2015 Aug 07;463(4):1210–1217. PubMed PMID: 26086101.
- Clemen CS, Tangavelou K, Strucksberg KH, et al. Strumpellin is a novel valosin-containing protein binding partner linking hereditary spastic paraplegia to protein aggregation diseases. Brain: J neurology. 2010 Oct;133(10):2920–2941. PubMed PMID: 20833645.
- Ropers F, Derivery E, Hu H, et al. Identification of a novel candidate gene for non-syndromic autosomal recessive intellectual disability: the WASH complex member SWIP. Hum Mol Genet. 2011 Jul 01;20(13):2585–2590. PubMed PMID: 21498477.
- Schroder R, Schoser B. Myofibrillar myopathies: a clinical and myopathological guide. Brain pathology. 2009 Jul;19(3):483–492. . PubMed PMID: 19563540.
- Chou TF, Li K, Frankowski KJ, et al. Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. Chem Med Chem. 2013 Feb;8(2):297–312. PubMed PMID: 23316025; PubMed Central PMCID: PMC3662613.
- Kotani H, Taimatsu K, Ohga R, et al. Efficient Multiple Genome Modifications Induced by the crRNAs, tracrRNA and Cas9 Protein Complex in Zebrafish. PLoS One. 2015;10(5):e0128319. PubMed PMID: 26010089; PubMed Central PMCID: PMC4444095.
- Buhrdel JB, Hirth S, Kessler M, et al. In vivo characterization of human myofibrillar myopathy genes in zebrafish. Biochem Biophys Res Commun. 2015 May 29;461(2):217–223. PubMed PMID: 25866181.
- Bug M, Meyer H. Expanding into new markets–VCP/p97 in endocytosis and autophagy. J Struct Biol. 2012 Aug;179(2):78–82. . PubMed PMID: 22450227.
- Ju JS, Fuentealba RA, Miller SE, et al. Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J Cell Biol. 2009 Dec 14;187(6):875–888. PubMed PMID: 20008565; PubMed Central PMCID: PMC2806317.
- Buhler A, Kustermann M, Bummer T, et al. Atrogin-1 Deficiency Leads to Myopathy and Heart Failure in Zebrafish. Int J Mol Sci. 2016 Jan 30;17(2). PubMed PMID: 26840306; PubMed Central PMCID: PMC4783921.
- Badadani M, Nalbandian A, Watts GD, et al. VCP associated inclusion body myopathy and paget disease of bone knock-in mouse model exhibits tissue pathology typical of human disease. PLoS One. 2010 Oct 05;5(10). PubMed PMID: 20957154; PubMed Central PMCID: PMC2950155.
- Nalbandian A, Llewellyn KJ, Kitazawa M, et al. The homozygote VCP(R(1)(5)(5)H/R(1)(5)(5)H) mouse model exhibits accelerated human VCP-associated disease pathology. PLoS One. 2012;7(9):e46308. PubMed PMID: 23029473; PubMed Central PMCID: PMC3460820.
- Custer SK, Neumann M, Lu H, et al. Transgenic mice expressing mutant forms VCP/p97 recapitulate the full spectrum of IBMPFD including degeneration in muscle, brain and bone. Hum Mol Genet. 2010 May 01;19(9):1741–1755. PubMed PMID: 20147319.
- Muller JM, Deinhardt K, Rosewell I, et al. Targeted deletion of p97 (VCP/CDC48) in mouse results in early embryonic lethality. Biochem Biophys Res Commun. 2007 Mar 09;354(2):459–465. PubMed PMID: 17239345.
- Imamura S, Yabu T, Yamashita M. Protective role of cell division cycle 48 (CDC48) protein against neurodegeneration via ubiquitin-proteasome system dysfunction during zebrafish development. J Biol Chem. 2012 Jun 29;287(27):23047–23056. . PubMed PMID: 22549779; PubMed Central PMCID: PMC3391108.
- Wojcik C, Yano M, deMartino GN. RNA interference of valosin-containing protein (VCP/p97) reveals multiple cellular roles linked to ubiquitin/proteasome-dependent proteolysis. J Cell Sci. 2004 Jan 15;117(Pt 2):281–292. . PubMed PMID: 14657277.
- Masiero E, Sandri M. Autophagy inhibition induces atrophy and myopathy in adult skeletal muscles. Autophagy. 2010 Feb;6(2):307–309. PubMed PMID: 20104028.
- Weihl CC, Pestronk A, Kimonis VE. Valosin-containing protein disease: inclusion body myopathy with Paget’s disease of the bone and fronto-temporal dementia. Neuromuscular disorders: NMD. 2009 May;19(5):308–315. . PubMed PMID: 19380227; PubMed Central PMCID: PMC2859037.
- Vardarajan BN, Bruesegem SY, Harbour ME, et al. Identification of Alzheimer disease-associated variants in genes that regulate retromer function. Neurobiol Aging. 2012 Sep;33(9):2231 e15–2231 e30. PubMed PMID: 22673115; PubMed Central PMCID: PMC3391348.
- Wolf DH, Stolz A. The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim Biophys Acta. 2012 Jan;1823(1):117–124. . PubMed PMID: 21945179.
- Gomes AV. Genetics of proteasome diseases. Scientifica (Cairo). 2013;2013:637629. PubMed PMID: 24490108; PubMed Central PMCID: PMC3892944.
- Jankowska E, Stoj J, Karpowicz P, et al. The proteasome in health and disease. Curr Pharm Des. 2013;19(6):1010–1028. PubMed PMID: 23016682.
- Just S, Hirth S, Berger IM, et al. The mediator complex subunit Med10 regulates heart valve formation in zebrafish by controlling Tbx2b-mediated Has2 expression and cardiac jelly formation. Biochem Biophys Res Commun. 2016 Sep 02;477(4):581–588. PubMed PMID: 27343557.
- Rudeck S, Etard C, Khan MM, et al. A compact unc45b-promoter drives muscle-specific expression in zebrafish and mouse. Genesis. 2016 Aug;54(8):431–438. PubMed PMID: 27295336; PubMed Central PMCID: PMC5113797.
- Neller J, Dunkler A, Rosler R, et al. A protein complex containing Epo1p anchors the cortical endoplasmic reticulum to the yeast bud tip. J Cell Biol. 2015 Jan 05;208(1):71–87. PubMed PMID: 25547157; PubMed Central PMCID: PMC4284228.
- Hirth S, Buhler A, Buhrdel JB, et al. Paxillin and Focal Adhesion Kinase (FAK) Regulate Cardiac Contractility in the Zebrafish Heart. PLoS One. 2016;11(3):e0150323. PubMed PMID: 26954676; PubMed Central PMCID: PMC4782988.
- Sandri M, Sandri C, Gilbert A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004 Apr 30;117(3):399–412. PubMed PMID: 15109499; PubMed Central PMCID: PMC3619734.
- Mammucari C, Milan G, Romanello V, et al. FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab. 2007 Dec;6(6):458–471. PubMed PMID: 18054315.
- Just S, Raphel L, Berger IM, et al. Tbx20 Is an Essential Regulator of Embryonic Heart Growth in Zebrafish. PLoS One. 2016;11(12):e0167306. PubMed PMID: 27907103; PubMed Central PMCID: PMC5132222.
- Kessler M, Berger IM, Just S, et al. Loss of dihydrolipoyl succinyltransferase (DLST) leads to reduced resting heart rate in the zebrafish. Basic Res Cardiol. 2015 Mar;110(2):14. PubMed PMID: 25697682; PubMed Central PMCID: PMC4335124.
- Meder B, Just S, Vogel B, et al. JunB-CBFbeta signaling is essential to maintain sarcomeric Z-disc structure and when defective leads to heart failure. J Cell Sci. 2010 Aug 01;123(Pt 15):2613–2620. PubMed PMID: 20605922.
- Dai H, Leeder JS, Cui Y. A modified generalized Fisher method for combining probabilities from dependent tests. Front Genet. 2014;5:32. PubMed PMID: 24600471; PubMed Central PMCID: PMC3929847.