
ABSTRACT
Accumulating evidence suggests that misfolded MAPT (microtubule associated protein tau), the main component of neurofibrillary tangles in tauopathies, is subject to degradation by the autophagy-lysosomal pathway. Selective autophagy is a subtype of macroautophagy that requires cargo receptors, such as OPTN (optineurin) or SQSTM1, to recognize specific targets for their sequestration within the autophagosome and their eventual degradation by the lysosome, although their roles in targeting distinct MAPT species have not been fully investigated. Using cargo receptor knockout cell lines and a seeding-based cellular assay in which neurofibrillary tangle pathology can be modeled in vitro, we reveal that while OPTN primarily targets soluble MAPT expressed in physiological conditions, SQSTM1 predominantly degrades insoluble but not soluble mutant MAPT. Endogenous SQSTM1 colocalizes with misfolded and aggregated MAPT species in vitro and in vivo, and both this colocalization and its function in MAPT clearance require both the LC3-interacting region (LIR) motif and also the PB1 self-polymerization domain of SQSTM1. Further, pathogenic MAPT accumulation reduces basal macroautophagy/autophagy in vitro and is associated with a compensatory upregulation of the lysosomal pathway in vivo. Finally, increased expression of SQSTM1 in MAPT transgenic mouse brains ameliorates MAPT pathology and prion-like spreading. Our results uncover distinct properties of selective autophagy receptors in targeting different MAPT species, implicate compromised autophagy as a potential underlying factor in mutant MAPT deposition, and demonstrate a potent and specific role of SQSTM1 in targeted clearance of pathogenic MAPT, through which it blocks neurofibrillary tangle accumulation and pathological spreading.
Abbreviations: AAV: adeno-associated virus; AD: Alzheimer disease; ALP: autophagy-lysosomal pathway; ALS: amyotrophic lateral sclerosis; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; FTD: frontotemporal dementias; HD: Huntington disease; HTT: huntingtin; LIR: LC3-interacting region; NBR1: autophagy cargo receptor; NFE2L2/Nrf2: nuclear factor, erythroid derived 2, like 2; NFTs: neurofibrillary tangles; MAPT: microtubule associated protein tau; OPTN: optineurin; p-MAPT: hyperphosphorylated MAPT; PFA: paraformaldehyde; TARDBP/TDP-43: TAR DNA binding protein; TAX1BP1 Tax1: binding protein 1; ThioS: thioflavin-S; UBA: ubiquitin-associated
Introduction
Tauopathies, consisting of a group of diseases including frontotemporal dementias (FTDs) and most commonly Alzheimer disease (AD), are characterized by the accumulation of intracellular neurofibrillary tangles (NFTs) mainly composed of aggregates of misfolded MAPT/Tau protein, and extensive neurodegeneration. In normal conditions, MAPT is an axonal protein that binds to and stabilizes microtubules. Aberrant MAPT hyperphosphorylation or other alterations lead to its misfolding and dissociation from microtubules, followed by redistribution and oligomerization in the cell bodies and dendrites, leading to NFT formation [Citation1]. Mutations in the MAPT gene are causal for a subtype of FTD, demonstrating its essential role in disease pathogenesis [Citation2,Citation3]. Although AD does not typically have mutations in MAPT, it is widely recognized that the NFT pathology correlates more closely with cognitive decline than β-amyloid plaques [Citation4]. As such, targeted removal of pathogenic MAPT species represents an attractive therapeutic avenue against tauopathies including AD.
The autophagy-lysosomal pathway (ALP) is a highly conserved degradation pathway that acts by engulfing cellular substrates within MAP1LC3B/LC3B (microtubule-associated protein 1 light chain 3 beta)-positive double-membrane autophagosomes before delivering them to the lysosome for eventual degradation [Citation5,Citation6]. Although mostly studied as a bulk degradation process induced by nutrient or energy depletion, increasing evidence supports the idea that a significant subset of autophagy can be selective and is starvation independent [Citation5,Citation7,Citation8]. This process, termed selective autophagy, is mediated by a family of cargo receptors, including SQSTM1 [Citation9], OPTN (optineurin) [Citation10], NBR1 (NBR1, autophagy cargo receptor) [Citation11], CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2) [Citation12], and TAX1BP1 (TAX1 binding protein 1) [Citation13] among others. These receptors mostly contain an LC3-interacting region (LIR) and a cargo binding motif such as ubiquitin-associated (UBA) domain, which binds to LC3 and ubiquitinated substrates, respectively, thereby allowing the recruitment of these substrates to phagophores, the precursors to autophagosomes [Citation13].
The cargo receptors appear to exhibit both overlapping and distinct properties in selective autophagy. In particular, Lazarou et al. reported that CALCOCO2 and OPTN, but not SQSTM1, engage PINK1 (PTEN induced kinase 1)-dependent mitophagy associated with Parkinson disease (PD) [Citation14]. NFE2L2/Nrf2 (nuclear factor, erythroid derived 2, like 2) modulates MAPT turnover through its downstream target CALCOCO2 instead of SQSTM1 [Citation15]. Importantly, mutations in multiple selective autophagy receptor genes have been linked to neurodegenerative diseases [Citation16–Citation20], highlighting a critical role of selective autophagy in intracellular quality control and neurodegeneration. Of note, absence of SQSTM1 results in childhood-onset neurodegeneration, and mutations in SQSTM1 and OPTN are genetically associated with FTD-amyotrophic lateral sclerosis (ALS) [Citation16–Citation20]. In mice, loss of Sqstm1 leads to the accumulation of hyperphosphorylated MAPT (p-MAPT) and neurodegeneration [Citation21]. Conversely, overexpression of SQSTM1 has been reported to remove pathological aggregates, such as Aβ [Citation22], AR (androgen receptor) [Citation23] and TARDBP/TDP-43 (TAR DNA binding protein) [Citation24].
Here we tested the role of multiple cargo receptors, in particular, OPTN and SQSTM1, in MAPT regulation. We found that whereas OPTN recognizes normal and soluble MAPT, SQSTM1 specifically targets pathological mutant MAPT species and suppresses insoluble mutant MAPT accumulation. Overexpression of SQSTM1 significantly reduced NFT pathology and its spreading in MAPT mouse models.
Results
OPTN and SQSTM1 target distinct MAPT species
To ascertain whether MAPT is a substrate for selective autophagy, we examined endogenous MAPT protein levels in HeLa cell lines lacking SQSTM1, OPTN, or 5 of the known selective autophagy receptors: SQSTM1, OPTN, NBR1, CALCOCO2, and TAX1BP1 (herein referred to as penta KO) [Citation14], using parental HeLa cells (WT) and ATG5 KO cells as controls (Figure S1(a)). We were particularly interested in SQSTM1 and OPTN due to their genetic association with FTD-ALS. Although total MAPT was readily detectable ()), we were not able to detect p-MAPT, probed using both AT8 and PHF1 antibodies that recognize pathological MAPT with phosphorylation at Ser202/Thr205 and Ser396/Ser404, respectively, in any of the cell lines (data not shown), suggesting MAPT mainly exists in its normal conformation in HeLa cells under physiological conditions. Consistent with previous reports that the MAPT protein is subject to ALP degradation [Citation25], the level of total MAPT was highly elevated in ATG5 KO cells ()). Compared with the parental HeLa cells (WT), we observed a similar mild but significant increases in MAPT protein levels in OPTN KO and penta KO cells, but not in SQSTM1 KO cells () and quantified in 1(b)), suggesting that a selective autophagy pathway contributes to physiological MAPT clearance and this process is OPTN dependent but SQSTM1 independent.
Figure 1. OPTN targets soluble MAPT species in vitro. (a) Representative western blot image of total MAPT levels in wild-type (WT), SQSTM1 KO, OPTN KO, penta KO, and ATG5 KO HeLa cells. (b) Quantitative analysis of the sum of all MAPT bands in (a). (c) Representative western blot image of total MAPT levels in penta KO HeLa cells transfected with empty vector (CMV), or plasmids encoding SQSTM1, or OPTN. SQSTM1 and OPTN overexpression was confirmed using their respective antibodies. (d) Quantitative analyses of the sum of all MAPT bands in (c) showing rescue of increased total MAPT in penta KO cells by OPTN but not SQSTM1. (e) Representative western blot image of total MAPT levels in OPTN KO HeLa cells transfected with empty vector (CMV) or a plasmid encoding OPTN. Western blots images show that total MAPT level is rescued by re-introducing OPTN in OPTN KO cells. (f) Quantification of the sum of all MAPT bands in (e) showing the rescue of MAPT clearance by re-introducing OPTN in penta KO cells. HeLa cells transfected with empty vector was used as a control in transfection studies. TUBG was used as a loading control in all western blot analyses. The total MAPT antibody was from DAKO. Data are presented as relative levels of protein:TUBG and expressed as mean ± SEM (n = 3 of 3 experiments). NS, non-significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
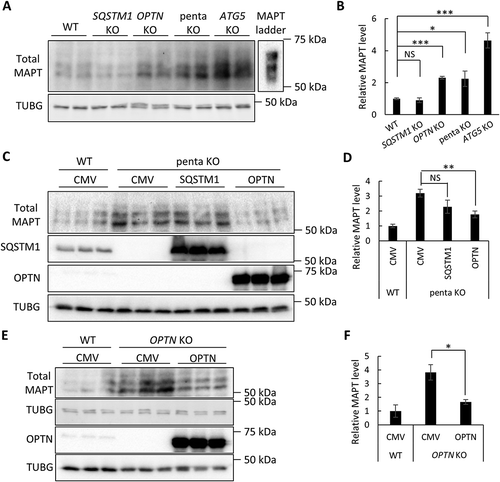
To further test the functional role of SQSTM1 and OPTN in MAPT degradation, we re-introduced SQSTM1 or OPTN to penta KO cells ()). Compared with CMV (vector-transfected controls), transfection of OPTN led to significant reduction of MAPT whereas expression of SQSTM1 had no significant effect ()). Likewise, re-introduction of OPTN to OPTN KO cells normalized the MAPT to control (WT) levels (,)). Hybridization using the Tau1 antibody that recognizes unphosphorylated MAPT revealed similar results as the total MAPT antibody (Figure S1(b)–S1(h)). Together, these results strengthen the notion that normal MAPT undergoes selective autophagy degradation pathway through OPTN, but not SQSTM1.
We next tested the activity of SQSTM1 and OPTN in handling pathological mutant MAPT using our established seeding-based cellular assay ()) [Citation26]. In this assay, when full-length MAPT-PL-V5 (human MAPT with the P301L mutation and V5 tag) is expressed in HEK293 cells, a significant amount of PHF1-positive p-MAPT accumulates but remains soluble. When inoculated in the culture medium, brain lysate from aged rTg4510 MAPT transgenic mice, but not that from wild-type controls, acts as an extracellular seed and effectively converts the soluble mutant MAPT to the insoluble form (see [Citation26] for details). When tested in this assay, OPTN expression eliminated PHF1 and V5 (total MAPT) in both soluble and insoluble fractions ()). This is consistent with its effect on endogenous MAPT, and the decreased insoluble MAPT is likely secondary to the depletion of soluble MAPT. Distinct from OPTN, SQSTM1 expression only mildly affected soluble MAPT () and quantified in )) but robustly reduced PHF1- and V5-positive MAPT in insoluble fractions. Co-immunoprecipitation revealed positive interaction of SQSTM1 with MAPT (Figure S2(a). Contrary to SQSTM1 overexpression, siRNA knockdown of SQSTM1 caused a significant increase in insoluble MAPT () and quantified in 2(e)). Importantly, in ATG5 KO HeLa cells, the levels of insoluble MAPT were no longer responsive to SQSTM1 overexpression () and quantified in 2(g)), suggesting that the effect of SQSTM1 on insoluble MAPT is mediated through autophagy.
Figure 2. SQSTM1 targets insoluble mutant MAPT species in vitro. (a) Experimental scheme of seeding-based cellular assay. Brain lysate from rTg4510 mice was added into the medium of HEK293 cells expressing MAPT-PL-V5, which was followed by ultracentrifugation of cell lysate into soluble and insoluble fractions. (b) Representative western blot image of PHF1 and V5 mutant MAPT in soluble and insoluble fractions harvested from the HEK293 cells transfected with empty vector (CMV), or a plasmid encoding SQSTM1 or OPTN. SQSTM1 and OPTN overexpression was confirmed using their respective antibodies. (c) Quantification of (b) demonstrating potent inhibition of both soluble and insoluble mutant MAPT by OPTN while significant reduction of only insoluble mutant MAPT was seen in SQSTM1 transfected cells. (d) Representative western blot image of PHF1 MAPT by siRNA knockdown of SQSTM1 (siSQSTM1) in the MAPT-PL-V5 expressing HEK293 cells. SQSTM1 reduction was confirmed using the anti-SQSTM1 antibody. (e) Quantitative analysis of (d) showing increases of insoluble mutant MAPT. HEK293 cells transfected with non-targeting siRNA was used as a control in transfection studies. (f) Representative western blot image of PHF1-positive MAPT in soluble and insoluble fractions harvested from the WT and ATG5 KO HeLa cells. (g) Quantitative analysis of (f) showing an autophagy-dependent function of SQSTM1. (h) Representative images of immunostaining showing that SQSTM1 decreases insoluble mutant MAPT (MC1) in seeded primary rTg4510 mouse neurons. Images are shown in pseudocolor: Gray, DAPI; red, MC1; green, SQSTM1. Scale Bar: 20 μm. (I) Quantitative analysis of (h). TUBG was used as a loading control in all western blot analyses. Data are presented as relative levels of protein:TUBG and expressed as mean ± SEM (n = 3 of 3 experiments). NS, non-significant; **P ≤ 0.01; ***P ≤ 0.001.
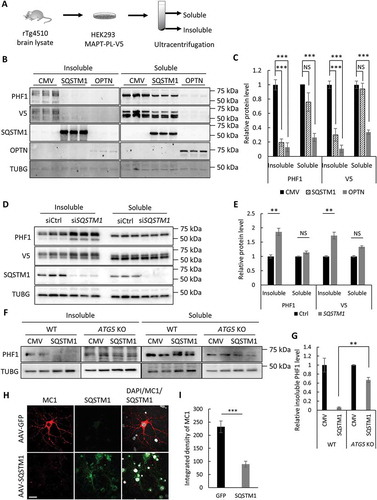
To determine if SQSTM1 has a similar effect on insoluble MAPT in neuronal cells, we next established a seeding-based assay using primary cultured mouse neurons from rTg4510 mice. Similar to that of HEK293 cells, brain lysate from rTg4510, but not from control wild-type mice, induced robust formation of insoluble mutant MAPT deposit in primary neurons from rTg4510 mice, as revealed by MC1 antibody staining (Figure S2(b)). Adeno-associated virus (AAV)-mediated expression of SQSTM1, but not GFP, significantly reduced the formation of such MC1-positive insoluble MAPT species () and quantified in 2(i)). Collectively, these results demonstrate that SQSTM1 predominantly targets insoluble MAPT species or potentially their intermediate precursors such as oligomeric misfolded MAPT for degradation in both neuronal and non-neuronal cells.
PB1 and LIR domains are essential for SQSTM1’s function
The preferential targeting of soluble and insoluble mutant MAPT by OPTN and SQSTM1, respectively, coincided with their intrinsic solubility evidenced by western blotting of fractionated cell lysates ()). The fact that OPTN is predominately soluble and SQSTM1 mainly resides in the insoluble fraction is likely attributed to differences in their biophysical properties. The autophagy function of SQSTM1 is largely mediated by its 3 protein-interacting motifs: the C terminus UBA domain that recognizes ubiquitinated substrates, a neighboring LIR motif that interacts with LC3 and phagophores, and the N terminus PB1 domain that mediates its self-polymerization that is important for its effectiveness in selective autophagy [Citation27]. To probe mechanistically how SQSTM1 promotes the clearance of insoluble MAPT, we therefore tested the involvement of these 3 SQSTM1 domains. To test the PB1 domain, which is absent in OPTN, we constructed FLAG-tagged SQSTM1 mutants either deleting PB1 (SQSTM1-ΔPB1) or containing only the PB1 domain (SQSTM1-PB1) and evaluated their effect on SQSTM1 solubility and mutant MAPT degradation in the seeding assay ()). Western blot analysis of fractionated cell lysates using anti-FLAG and PHF1 antibodies to detect SQSTM1 and p-MAPT, respectively, showed that, in contrast to full-length SQSTM1 (SQSTM1-FL), SQSTM1-ΔPB1 largely resided in the soluble fraction and it failed to promote insoluble MAPT degradation () and quantified in 3C). On the contrary, the SQSTM1-PB1 was exclusively present in the insoluble pool (), FLAG). SQSTM1-PB1 expression led to increased insoluble mutant MAPT similar to SQSTM1 knockdown () and quantified in 3E). Thus, expression of the PB1 domain likely exerted a dominant-negative effect by disrupting the function of endogenous SQSTM1. This result is consistent with the shift of endogenous SQSTM1 from soluble to insoluble forms by PB1 ()), presumably due to the formation of an inactive PB1-SQSTM1 complex and depletion of the functional SQSTM1.
Figure 3. Structure-function analysis of SQSTM1 in seeding assay. (a) Schematic representation of domain structures in FL (full-length) SQSTM1, the SQSTM1-ΔPB1 (SQSTM1 deleting the PB1 domain), SQSTM1-PB1 (PB1 domain alone), SQSTM1-LIR mut (LIR mutant 335-DDDW-338 to 335-AAAA-338) and SQSTM1-ΔUBA (deleting the UBA domain). All constructs were FLAG-tagged at the C terminus. (b) Representative western blot image of PHF1 mutant MAPT in soluble and insoluble fractions from the MAPT-PL-V5 expressing HEK293 cells transfected with empty vector (CMV), SQSTM1-FL, or ΔPB1 mutant. SQSTM1 overexpression was confirmed using the anti-FLAG antibody. (c) Quantitative analyses of (b) showing that SQSTM1-ΔPB1 failed to promote PHF1 mutant MAPT degradation. (d) Representative western blot image of PHF1 mutant MAPT in soluble and insoluble fractions from the MAPT-PL-V5-expressing HEK293 cells transfected with empty vector (CMV) or a plasmid encoding SQSTM1-PB1. Endogenous SQSTM1 was detected using an anti-SQSTM1 antibody (SQSTM1) while SQSTM1-PB1 expression was visualized using the anti-FLAG antibody (FLAG). (e) Quantification of (d) showing increased insoluble PHF1 mutant MAPT by SQSTM1-PB1 expression. (f) Representative western blot image of PHF1 mutant MAPT in soluble and insoluble fractions from the MAPT-PL-V5-expressing HEK293 cells transfected with empty vector (CMV), or a plasmid encoding SQSTM1-FL, or the SQSTM1-LIR mutant. SQSTM1 overexpression was confirmed using the anti-FLAG antibody. (j) Quantitative analyses of (f) showing that the SQSTM1-LIR mut failed to promote PHF1 mutant MAPT degradation. (h) Representative western blot image of PHF1 mutant MAPT in soluble and insoluble fractions from the MAPT-PL-V5-expressing HEK293 cells transfected with empty vector (CMV), or a plasmid encoding the SQSTM1-ΔUBA mutant. SQSTM1 overexpression was confirmed using the anti-FLAG antibody. (g) Quantitative analyses of (i) showing SQSTM1-ΔUBA was still functional in PHF1 mutant MAPT degradation. HEK293 cells transfected with empty vector was used as a control in transfection studies. TUBG was used as a loading control in all western blot analyses. Data are presented as relative levels of protein:TUBG and expressed as mean ± SEM (n = 3 of 3 experiments). NS, non-significant; *P ≤ 0.05; ***P ≤ 0.001.
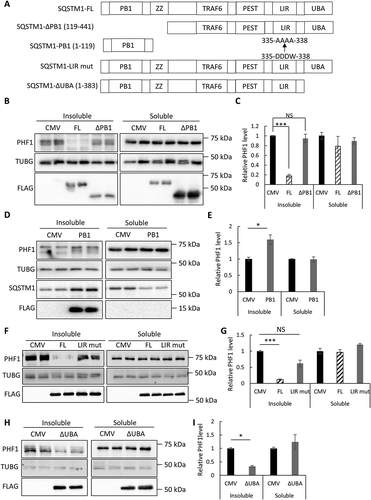
To test the requirement of LIR motif in SQSTM1-mediated insoluble MAPT clearance, we constructed a SQSTM1 mutant with alanine replacement of the essential amino acids within the LIR motif (DDDW-AAAA) [Citation9]. This SQSTM1 mutant completely lost its ability in promoting insoluble MAPT clearance () and quantified in 3(g)), supporting an essential role of LC3 binding and autophagy in SQSTM1’s function. Interesting, an SQSTM1 mutant lacking the UBA domain (SQSTM1-ΔUBA) was still largely effective in reducing the accumulation of insoluble MAPT () and quantified in 3(i)), implicating a ubiquitin-independent mechanism of SQSTM1 in mutant MAPT degradation.
SQSTM1 colocalizes with misfolded MAPT
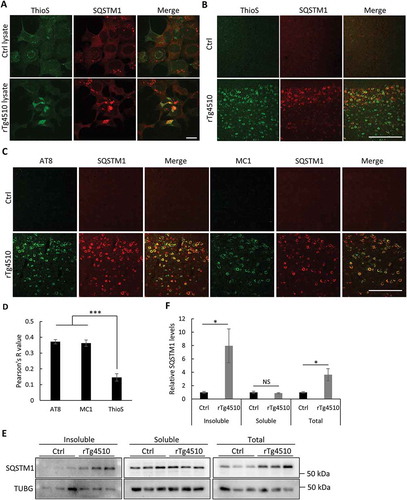
To further test the association of SQSTM1 and MAPT, we performed co-staining of SQSTM1 with thioflavin-S (ThioS), a dye that binds to β sheet structures including NFTs, in our seeding cultures. We found that SQSTM1 strongly colocalized with ThioS in HEK293 cells seeded with rTg4510 lysate but not control (Ctrl) lysate ()). A similar result was obtained when rTg4510 mouse brains were costained for ThioS and SQSTM1 ()) and was further validated by double immunostaining of SQSTM1 with AT8 and MC1 antibodies ()). Interestingly, there was a significantly higher degree of colocation of SQSTM1 with AT8-positive p-MAPT and MC1-marked misfolded MAPT than with ThioS ()), indicating that SQSTM1 targets mutant MAPT both as mature tangles and in pathological and misfolded forms preceding tangle formation. Consistent with its targeting of soluble MAPT, no OPTN was found to colocalize with ThioS in vitro and in vivo (Figure S3(a,b)). Western blotting of fractionated cell lysates from control and rTg4510 mouse brains revealed that the increased SQSTM1 can only be observed in insoluble but not soluble fractions, leading to overall higher SQSTM1 levels in rTg4510 brains () and quantified in 4(f)). As expected, the SQSTM1 immunoreactivity colocalized with the neuronal marker RBFOX3/NeuN but not the astrocyte marker GFAP (Figure S3(c)).
Figure 4. Mutant MAPT aggregates associate with SQSTM1. (a) Representative fluorescence images of ThioS, SQSTM1 or combined (Merge) of MAPT-PL-V5-expressing HEK293 cells seeded with control (Ctrl) or MAPT (rTg4510) lysates, showing a high degree of colocalization between SQSTM1 and ThioS. (b) Representative costaining images in the cortex of rTg4510 mice showing similar colocalization of SQSTM1 with ThioS. (c) Representative costaining images in the cortex of rTg4510 mice showing similar colocalization of SQSTM1 with AT8 and MC1. (d) Quantification of colocalization between SQSTM1 and ThioS, AT8, and MC1 in (b) and (c). (e) Representative western blot image of SQSTM1 in control (Ctrl) or rTg4510 in insoluble or soluble fractions or total brain lysates (Total). (f) Quantification of relative levels of SQSTM1:TUBG (n = 3). Scale Bar: 20 μm in (a); 200 μm in (b) and (c). All data are expressed as mean ± SEM. NS, non-significant; *P ≤ 0.05; ***P ≤ 0.001.
Mutant MAPT aggregates reduce basal autophagy
Accumulation of SQSTM1 has often been used as a marker for autophagy blockade. To assess the effect of mutant MAPT aggregates on autophagy, we analyzed the autophagosome marker MAP1LC3B. Using the seeding-based cell assay, we performed costaining of ThioS and MAP1LC3B and quantified the steady-state levels of MAP1LC3B puncta in ThioS-positive vs. -negative cells (), DMSO). The intensity of MAP1LC3B puncta was indistinguishable regardless of whether the cells were ThioS positive or negative ()). However, upon treatment with bafilomycin A1 ()), a specific inhibitor that blocks the fusion of autophagosomes with lysosomes, we observed a robust increase of MAP1LC3B puncta in ThioS-negative cells, indicating a strong autophagy flux (), compare DMSO vs. bafilomycin A1, ThioS−). In contrast, the degree of the increase was significantly decreased in cells positive for ThioS (), ThioS− vs. ThioS+, bafilomycin A1). Together, the data suggest an overall lower level of basal autophagy in cells containing mutant MAPT aggregates, which might contribute to the accumulation of SQSTM1.
Figure 5. Mutant MAPT aggregates reduce autophagy. (a) Representative fluorescence images of ThioS and MAP1LC3B in MAPT-seeded MAPT-PL-V5-expressing HEK293 cells treated with DMSO or bafilomycin A1 (200 nM, 16 h). Individual ThioS-positive cells are highlighted by dotted lines, ThioS-negative cells are indicated by white asterisks. (b) Quantitative analysis of MAP1LC3B puncta areas in ThioS-positive (ThioS+) or -negative (ThioS−) cells under control (DMSO) or bafilomycin A1 treatment (200 nM, 16 h) (n ≥ 60 cells each). (c) Representative fluorescence images of ThioS and p-ULK1 (Ser555) in MAPT-seeded MAPT-PL-V5-expressing HEK293 cells. Individual ThioS-positive cells are marked by a dotted line, and ThioS-negative cells are indicated by a white asterisk. (d) Quantitative analysis of p-ULK1 (Ser555) intensity in ThioS-positive (ThioS+) vs. -negative (ThioS−) cells (n ≥ 40 cells each). (e) Representative fluorescence images of ThioS and ULK1 in MAPT-seeded MAPT-PL-V5-expressing HEK293 cells. Individual ThioS-positive cells are marked by dotted lines, and a ThioS-negative cell is indicated by a white asterisk. (f) Quantitative analysis of ULK1 intensity in ThioS-positive (ThioS+) vs. -negative (ThioS−) cells (n = 38 cells each). Scale Bar: 20 μm. All data are expressed as mean ± SEM. NS, non-significant; *P ≤ 0.05.
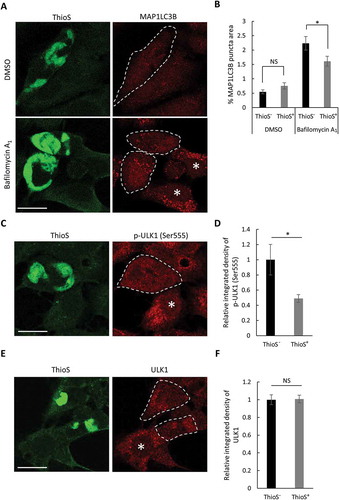
To further confirm this observation and to explore the potential mechanism, we next examined the level of ULK1 (unc-51 like autophagy activating kinase 1) phosphorylation at serine 555. ULK1 is a serine/threonine kinase essential for autophagosome biogenesis and its phosphorylation at serine 555 is required for its activity [Citation28,Citation29]. Immunostaining analysis validated the specificity of a p-ULK1 (Ser555) antibody (Figure S4). Costaining of ThioS with the anti-p-ULK1 (Ser555) antibody ()) and subsequent quantification ()) revealed that the level of p-ULK1 (Ser555) fluorescence intensity was significantly lower in ThioS-positive cells than that in ThioS-negative cells. However, the levels of total ULK1 were indistinguishable between ThioS-positive and -negative cells (,)). Combined, these results suggest that mutant MAPT accumulation attenuates autophagosome formation through affecting p-ULK1 (Ser555) activation.
Overexpression of SQSTM1 reduces MAPT pathology
The above in vitro data support the idea that mutant MAPT accumulation affects early steps of the autophagy pathway. To explore whether lysosomal profiles were altered as a function of mutant MAPT pathology in vivo, we performed gene set enrichment analysis of microarray data derived from 4-months-old wild-type and rTg4510 mutant MAPT transgenic mouse brains [Citation25]. Unexpectedly, we found a highly significant enrichment of lysosome genes in rTg4510 mice as compared to wild-type controls (Figure S5(a), Enrichment score = 0.74), indicating that the lysosomal pathway is induced by MAPT pathology. Upregulation of lysosomal genes was confirmed by qPCR analysis (Figure S5(b)).
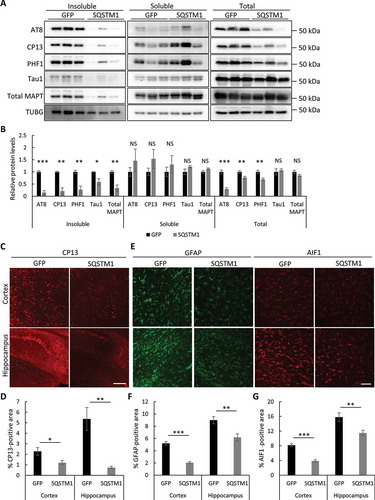
Considering the strong effect of SQSTM1 overexpression on insoluble mutant MAPT clearance in vitro (), and the compensatory activation of lysosomal gene expression in rTg4510 mutant MAPT mice, we hypothesize that enhancing SQSTM1-mediated selective autophagy may create a synergetic effect to promote the degradation of pathological mutant MAPT. We thus used an AAV-mediated gene delivery system to test the therapeutic effect of SQSTM1 in tauopathy mouse models. AAV-SQSTM1 or AAV-GFP driven by the chicken ACTB promoter was injected into the lateral ventricles of both hemispheres at postnatal day 0 of rTg4510 mice and their littermates (Figure S6(a,b)). Four months after injection, we performed biochemical and immunofluorescence analyses of mutant MAPT levels and associated pathology. Remarkably, and consistent with our in vitro results, SQSTM1 expression significantly reduced all forms of mutant MAPT in insoluble fractions when compared to GFP-injected controls, while soluble MAPT species were not significantly changed () and quantified in 6B). This is associated with reduced p-MAPT detected by AT8, CP13, and PHF1 antibodies in total lysates, while Tau1 (unphosphorylated MAPT) and total MAPT levels were not significantly changed () and quantified in 6(b)). Further, consistent with the western blot data, immunostaining using CP13 ()) and MC1 antibodies (Figure S6(c)) showed that the pathological forms of mutant MAPT and the NFT-like pathology were drastically reduced in both the cortex and hippocampus in the AAV-SQSTM1 injected cohort ()). This is accompanied by dampened reactive astrogliosis and microgliosis marked by GFAP and AIF1/Iba1 staining, respectively (–)). Together, these results demonstrate a primary and potent effect of SQSTM1 in targeting insoluble pathological mutant MAPT species, leading to the overall reduction of p-MAPT in total brain lysates and ameliorated brain pathology.
Figure 6. Overexpression of SQSTM1 reduces mutant MAPT pathology. (a) Representative western blots of AT8, CP13, and PHF1 (p-MAPT), Tau1 (unphosphorylated MAPT), and total MAPT in insoluble, soluble and total lysates of rTg4510 brains injected with AAV-GFP or AAV-SQSTM1. TUBG was used as a loading control. (b) Quantification of relative levels of mutant MAPT proteins:TUBG. n = 5/group for fractionated samples and n = 8/group for total lysates. (c) Representative immunofluorescence images of the cortex and hippocampus using the CP13 antibody. (d) Quantification of CP13-positive areas in (c). n = 4/group. (e) Representative GFAP and AIF1 immunofluorescence images of rTg4510 mice injected with AAV-GFP or AAV-SQSTM1. (f and g) Quantification of GFAP and AIF1/Iba1 immunoreactivity in (e). n = 4/group. Scale Bar: 500 μm in (c); 200 μm in (e). All data are expressed as mean ± SEM. NS, non-significant; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Overexpression of SQSTM1 reduces MAPT spreading
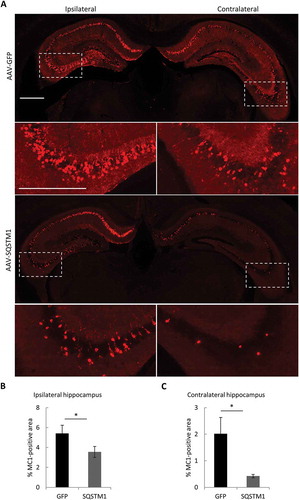
Numerous studies have demonstrated that pathological MAPT undergoes prion-like spreading. Specific to AD, the development of NFTs pathology follows defined network connections from the entorhinal cortex to the hippocampus, and then to other cortical areas [Citation30,Citation31]. Such a cell-to-cell transfer of MAPT pathology can be induced by inoculating MAPT fibrils to MAPT transgenic mouse brains [Citation32–Citation35]. Having documented a potent role of SQSTM1 in removing MAPT pathology, we wondered whether SQSTM1 expression can also restrict pathological MAPT spreading. We thus tested the effect of SQSTM1 in a MAPT spreading model in which PS19 mice expressing human MAPT with the P301S mutation that has been used as recipient mice for MAPT spreading [Citation32,Citation33] were injected with AAV-GFP or AAV-SQSTM1 at postnatal day 0 (P0), followed by stereotaxic injection of rTg4510 brain lysates into one hippocampal hemisphere at 3 months-of-age and analysis of mutant MAPT pathology 2 months later (). Immunofluorescence staining using MC1 ()) and AT8 (Figure S7) detected positive staining in both ipsilateral and contralateral hippocampus in GFP-injected controls. In SQSTM1-overexpression mice, misfolded mutant MAPT levels were significantly decreased in both ipsilateral and contralateral hippocampus compared to AAV-GFP injected controls ()). These data demonstrate that SQSTM1 functions as a barrier to prevent pathological MAPT spreading in vivo.
Figure 7. Overexpression of SQSTM1 attenuates pathological MAPT spreading. PS19 P0 pups were injected with AAV-GFP or AAV-SQSTM1, and at 2–3 months of age, 2 μl of MAPT brain lysate were stereotaxically injected into one hippocampal hemisphere (bregma, −2.5 mm; lateral, −2 mm, and depth, −1.8 mm). The mice were euthanized 2 months later for mutant MAPT spreading analysis. (a) Representative MC1-positive immunofluorescence images of ipsilateral and contralateral of hippocampus of PS19 mice injected with AAV-GFP or AAV-SQSTM1 and inoculated with MAPT lysates. The higher magnification views of bracketed areas are shown underneath each panel. (b and c) Quantitative analysis of MC1-positive areas in ipsilateral (b) or contralateral (c) hippocampus of AAV-GFP- or AAV-SQSTM1-treated PS19 mice. n = 9/group. Scale Bar: 1000 μm. Data are expressed as mean ± SEM. *P ≤ 0.05.
Discussion
In light of the pivotal role of MAPT and NFTs in AD and other tauopathies, identifying pathways that promote pathological MAPT and NFT clearance represent an appealing therapeutic strategy. It is known that MAPT is subject to a myriad of post-translational modifications and can exist in soluble monomeric, soluble/insoluble oligomeric forms as well as insoluble NFTs [Citation1,Citation2,Citation36]. Both the ubiquitin-proteasome system and autophagy-lysosomal pathway have been implicated in the degradation of these diverse MAPT species [Citation15,Citation37–Citation40]. We and others have shown that activation of ALP by inhibiting MTOR or upregulating TFEB are effective in MAPT clearance [Citation25,Citation41,Citation42]. However, these master regulators do not confer substrate specificity and may lead to adverse effects associated with systemic activation. In this regard, selective autophagy is advantageous as it targets selective substrates through discrete cargo receptors, and their activation is often MTOR independent. The distinct roles of cargo receptors were elegantly demonstrated by Lazarou et al., who revealed that PINK1-dependent mitophagy requires OPTN but not SQSTM1 in cultured cells [Citation14]. However, the potential differential roles of cargo receptors in MAPT clearance have not been carefully examined. Here we show that OPTN and SQSTM1 exhibit differential activities in MAPT handling. Whereas OPTN facilitates normal soluble MAPT degradation, SQSTM1 primarily targets misfolded and insoluble mutant MAPT both in vitro and in vivo, through which it effectively inhibits p-MAPT accumulation and pathological spreading.
We document that OPTN is for the most part soluble, but SQSTM1 is enriched in both soluble and insoluble fractions, consistent with earlier observations [Citation9,Citation43]. This intrinsic difference in solubility coincides with the differential targeting of soluble and insoluble MAPT species, respectively. The PB1 domain of SQSTM1, which is absent in OPTN, mediates its self-oligomerization [Citation27]. However, solubility alone is not sufficient to confer specificity or activity. A recent study demonstrated that SQSTM1 is effective in preventing the accumulation of amyloid β in plaque mouse model, and the LIR motif in SQSTM1, which mediates its association with the phagophore through LC3, is particularly important for such a beneficial effect [Citation22]. Consistently, we showed that LC3 binding is similarly important for SQSTM1-mediated clearance of insoluble mutant MAPT. However, an SQSTM1 mutant lacking PB1, which still contains intact LIR and UBA domains, shifts SQSTM1 from an insoluble to a soluble form but results in its loss of function instead of soluble MAPT targeting like OPTN; although this could be simply due to the general defect in SQSTM1 activity considering the observation that the polymerization of SQSTM1 through its PB1 domain is important for its effectiveness in selective autophagy [Citation9,Citation43,Citation44].
Interestingly, we found that SQSTM1-mediated clearance of insoluble mutant MAPT is likely to be ubiquitin independent, as deletion of the UBA domain does not affect the overall effectiveness of SQSTM1 on MAPT clearance. Notably, similar ubiquitin-independent mechanisms were also observed both for SQSTM1- and OPTN-mediated autophagic clearance of mutant SOD1 linked to ALS [Citation45–Citation47] and SQSTM1-mediated autophagic clearance of β-amyloid [Citation22]. A shared feature of these proteins, including mutant p-MAPT, SOD1, β-amyloid and Huntington disease (HD) protein HTT (huntingtin), is that they are all misfolding prone as mutants and easily polymerize. These scenarios bear much resemblance to the cytoplasm-to-vacuole targeting (Cvt) process, a well-studied selective autophagy-like pathway in yeast. The Cvt pathway is also independent of ubiquitination but requires the polymerization of cargo proteins prApe1 and Ams1 before their recognition by the cargo receptor Atg19 and subsequent recruitment into phagophores and delivery to the ALP pathway [Citation48]. Similarly, in protein misfolding diseases, aggregate formation is likely a dynamic and gradual process, involving many intermediate species with different conformation and oligomerization states and their associated solubility, before their eventual accumulation and maturation into large aggregates. The facts that SQSTM1 colocalizes with both ThioS- and MC1-positive MAPT and it is more effective in eliminating the p-MAPT in the insoluble pool support the notion that SQSTM1 recognizes multiple forms of mutant MAPT that are mainly insoluble. These include the less aggregated oligomeric species that may be more neurotoxic. Such an interpretation is consistent with the observation that SQSTM1 not only reduces insoluble mutant MAPT accumulation but also exerts a protective effect in vivo in suppressing mutant MAPT toxicity. Thus, the level of insoluble mutant MAPT might just be the end readout of in vivo SQSTM1 activity, instead of being the direct substrate of SQSTM1-mediated selective autophagy. This might also explain the existence of large mutant MAPT aggregates that are SQSTM1 positive, which could reflect the overburdened or overall inefficient ALP activities of the vulnerable cells. Although the precise structure-function relationships remain to be deciphered, we propose that the distinct physical properties of the cargo receptors are one contributing factor that dictates their substrate selectivity, with SQSTM1 harboring the ability to recognize and process certain oligomeric misfolding and aggregated mutant MAPT species that are pathogenic and being intermediates of eventual large insoluble mutant MAPT aggregates. Consistent with this notion, an earlier study showed that CALCOCO2, which is another selective receptor proposed to modulate ubiquitin-independent p-MAPT turnover, displays an inverse relationship with sarkosyl-insoluble MAPT opposite to SQSTM1 in human AD brain samples [Citation15].
A similar principle might also apply to other autophagy receptors as well as the group of misfolding-prone proteins that are known to be recognized by these receptors, including mutant SOD1, TARDBP, HTT, and SNCA/α-synuclein that are associated with various neurodegenerative diseases from ALS to HD and PD. Thus, although SQSTM1, OPTN and CALCOCO2 all were shown to promote MAPT degradation, each receptor may recognize different MAPT species in a way that is analogous to their distinct functions in mitophagy [Citation14]. Similarly, other misfolding proteins might also form varying aggregate species of different sizes and biochemical and toxic properties, which are differentially recognized by each of these autophagy receptors with differing affinity and specificity. Such a hypothesis would suggest that autophagy receptors are not functionally redundant, but might have both overlapping and distinct substrate preferences, and process different misfolding proteins and their aggregating derivatives with differential specificity and efficiency. Such a possibility, together with other factors such as the varying expression patterns and expression levels of these autophagy receptors and their substrate proteins in different tissues and brain cell types, might help explain why overexpressed SQSTM1 is effective in suppressing MAPT pathology in vivo, while mutations of SQSTM1 are linked to ALS with TARDBP pathology but not tauopathy or PD. It is possible that SQSTM1 can facilitate the autophagic clearance of both misfolding MAPT and TARDBP or other disease proteins, but in the brain, loss of SQSTM1 or OPTN can be sufficiently compensated by the presence of other autophagy receptors and/or protective mechanisms with an overlapping role in mutant MAPT clearance. However, in spinal cord, in the absence of SQSTM1 (or OPTN), the remaining capacity of the cellular clearance machineries are insufficient in processing the ALS-linked pathogens such as misfolding TARDBP, either because of its higher levels of expression in this tissue or the absence of other protective factors with redundant activity such as SQSTM1 in TARDBP clearance, leading to their accumulation and ALS pathology. Additionally, different neurons likely have different proteostasis environments, and are thus prone to develop a certain type of proteinopathy, such as MAPT in the neocortex in AD and FTD, and SNCA/α-synuclein in the substantia nigra in PD. Moreover, different neuronal types might have different susceptibility to different misfolding proteins and aggregates. These factors together might lead to the relatively distinct neuronal vulnerability in different protein misfolding diseases, a question that remains a substantial challenge to the neurodegeneration field.
Notably, although HeLa and HEK293 cells used in our in vitro studies are not of neuronal origin, our results provide support that such systems are informative in understanding the conservative mechanisms governing MAPT homeostasis. In particular, seeding-induced insoluble mutant MAPT formation, as well as the role of SQSTM1-mediated clearance of insoluble mutant MAPT species and its cellular protective effect, all can be recapitulated in vivo. Thus, although these non-neuronal systems might bear significant physiological differences from neuronal cells, they share conservative mechanisms in regulating MAPT misfolding, accumulation and clearance, and can be effectively employed in complementation with neuronal assays for detailed mechanistic studies. Another limitation of our study lies in the fact that mutant MAPT is used to examine the SQSTM1 effect. Since no MAPT mutations are identified in AD, whether SQSTM1 plays a similar role for wild-type MAPT is a question that requires further investigation.
The abnormal SQSTM1 accumulation has been observed in various neurodegenerative conditions including AD and FTD [Citation49,Citation50]. This feature could be used to indicate autophagy reduction [Citation51], although at which step this effect occurs is not understood. Our in vitro studies show that mutant MAPT aggregation is inversely correlated with the basal autophagic activity of the cell, as evidenced by a significant reduction of MAP1LC3-positive autophagosomes under bafilomycin A1 treatment condition as well as the reduced levels of active p-ULK1 (Ser555), the essential kinase for autophagosome biogenesis, in ThioS-positive cells. One possibility would be that MAPT aggregation could sequester toxic forms of MAPT to attenuate cellular stress and the need for autophagy response. However, it is difficult to probe the molecular pathways using the current system due to a lack of effective tools to distinguish various MAPT species. How mutant MAPT accumulation leads to changes of the ULK1 regulatory pathway remains an open question that warrants further investigation.
As discussed earlier, such mutant MAPT accumulation could simply reflect the overall reduced proteostasis capability of the cell, either because of their intrinsic lower activities or weakened effectiveness of the cellular proteostasis machineries such as chaperones and ALP pathways in the face of continuing insult from misfolding MAPT; It also raises a scenario that compromised cellular autophagy results in further abnormal MAPT accumulation, leading to a vicious feedback cycle of higher levels of MAPT and lower autophagy capacity that eventually overwhelms the cellular homeostasis mechanism, causing neuronal degeneration. Interestingly, we and others reported that the HD protein HTT functions as a scaffold in selective autophagy [Citation52,Citation53]. Of particular relevance, HTT interacts with both SQSTM1 and ULK1 to promote the autophagic clearance of a C-terminal truncated and aggregation-prone form of MAPT. Indeed, multiple lines of evidence have documented the existence of MAPT pathology in HD mouse models and HD patients [Citation54–Citation57], providing a tantalizing link between the 2 neurodegenerative conditions through SQSTM1-mediated selective autophagy.
Notably, ULK1 phosphorylates SQSTM1 and facilitates its function in substrate recognition and recruitment of autophagy machinery components [Citation58], which may be affected by MAPT accumulation. Our finding that AAV-mediated SQSTM1 expression leads to a robust reduction of p-MAPT and misfolded mutant MAPT pathology and spreading supports the idea that in mutant MAPT-expressing cells, especially at early stages, cellular autophagy pathways are still functional and higher levels of SQSTM1 are sufficient to overcome the weakened autophagy, and, in concert with the activated lysosomal pathways, potently direct the targeted clearance of pathogenic MAPT species. Indeed, consistent with our results from the in vitro seeding assay, SQSTM1 primarily reduces hyperphosphorylated and misfolded mutant MAPT in the insoluble fraction but with minimum effect on the soluble fraction in rTg4510 mice, leading to the overall reduction of p-MAPT in total brain lysates. The potent effect of SQSTM1 in mutant MAPT clearance supports the idea that enhanced lysosomal gene expression in MAPT transgenic mice serves as an adaptive mechanism and may cooperate with augmented autophagy by exogenous SQSTM1 expression. Similar upregulation of lysosomal genes was also observed in human AD and FTD [Citation50,Citation59], implicating a conserved lysosomal sensing mechanism in response to cellular stress such as protein aggregates. Overall, our findings provide mechanistic insight and functional support for harnessing the SQSTM1-mediated selective autophagy for diseases of tauopathy including Alzheimer disease.
Materials and methods
Mice
The rTg4510 [Citation60], PS19 [Citation61] MAPT transgenic mouse lines, and mouse mapt knockout and human MAPT transgenic mouse line [Citation62] were obtained from the Jackson Laboratories. Littermate wild-type mice were used as controls. Both males and females were used in the study and they were randomly assigned to treatment groups. Investigators were blinded to study group identity during experiments, data collection, and analysis. The sample size was determined based on experience with similar studies [Citation25] and power analysis described in ‘Statistics’. All procedures were performed according to the protocols approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine.
Reagents
CP13, PHF1, and MC1 antibodies were kind gifts from Peter Davies (Albert Einstein College of Medicine). The rest of the antibodies and key reagents were from: AT8 (Invitrogen, MN1020), total MAPT (DAKO, A0024), Tau1 (EMD Millipore, MAB3420), RBFOX3 (Chemicon, MAB377), GFAP (EMD Millipore, MAB360), AIF1 (Waco, 019-19741), MAP1LC3B (Sigma-Aldrich, L7543), SQSTM1 (Sigma-Aldrich, P0067), OPTN (Proteintech, 10837-1-AP), NBR1 (Abnova, H00004077-M01), CALCOCO2 (Cell Signaling Technology, 9036S), TAX1BP1 (Cell Signaling Technology, 5105S), p-ULK1 (Ser555; Cell Signaling Technology, 5869s), ULK1 (Cell Signaling Technology, 8054S), ATG5 (Cell Signaling Technology, 2630s), V5 (Thermo Fisher Scientific, R960-25), GFP (Santa Cruz Biotechnology, sc-8334), TUBG/γ-tubulin (Sigma-Aldrich, T6557), HRP-conjugated secondary antibodies (EMD Millipore, AP100P, AP307P), IRDye secondary antibodies (LI-COR, 926-32211, 926-32212, 926-68070, 926-68073), Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific, A21202, A21422, A21206, A31572), bafilomycin A1 (Cayman Chemical, 11038), ThioS (Sigma-Aldrich, T1892), Ctrl-siRNA and SQSTM1-siRNA (Santa Cruz Biotechnology, sc-37007, sc-29679), Lipofectamine 3000 (Thermo Fisher Scientific, L3000015), protease and phosphatase inhibitor cocktails (Roche, 04693116001, 04906845001), ECL (Pierce Protein Biology, 32106, 34096). The SQSTM1 expression vector was constructed previously [Citation63] and the OPTN vector was a gift from Beatrice Yue (Addgene, 27052) [Citation64]. The SQSTM1 mutant sequences with one FLAG tag in the C terminus were cloned into pCDNA3.1+ (Invitrogen, V79020), using the restriction enzymes HindIII and EcoRI. ULK1-WT and ULK1-4SA were gifts from Noboru Mizushima (Addgene, 24301) [Citation65] and Reuben Shaw (Addgene, 27631) [Citation29], respectively.
Cellular assays
All cells were maintained in DMEM supplemented with 10% FBS at 37°C with 5% CO2. HeLa cell lines including wild-type (WT), SQSTM1 knockout, OPTN knockout, SQSTM1, OPTN, NBR1, CALCOCO2, and TAX1BP1 penta knockout, and ATG5 knockout were kind gifts from Richard Youle (National Institutes of Health) [Citation14]. The MAPT seeding assay was carried out in HEK293 cells and primary neurons using a previously described protocol [Citation26]. Primary neurons were prepared from P0 pups using a previously described protocol [Citation66]. Primary neurons were infected with AAV at day 7 after culturing. The brain lysate from rTg4510 mice was added to the culture medium at day 5 after AAV infection. After 4 days incubation, cells were fixed for immunostaining. The transfections were performed according to the product’s manual. Briefly, plasmid, transfection agents P3000TM (Thermo Fisher Scientific, L3000015) and Lipofectamine 3000TM were diluted and mixed with 200 μl OPTI-MEM (Thermo Fisher Scientific, 31985088) with the ratio of 2 μg:2 μl:2 μl (for knockdown assay, the ratio was plasmid 1 μg, siRNA 0.1 nmol, P3000TM 2 μl and Lipofectamine 3000TM 2 μl) and the mixture was added to the cell culture medium after incubating at room temperature for 15 min. Cells were collected in ice-cold Tris-buffered saline (TBS; 50 mM Tris-HCl, pH 7.4, 150 mM NaCl) 2 days after transfection. All in vitro experiments were performed at least 3 times each in triplets.
In vivo gene delivery and MAPT spreading assay
The AAV 2/9-GFP or -SQSTM1 driven by the chicken ACTB promoter were produced by the Gene Vector Core of Baylor College of Medicine. Intraventricular injections were performed in P0 pups at 2.5 × 1010 viral particles per hemisphere. Injected rTg4510 mice were euthanized for biochemical and immunofluorescence analysis at 4 months of age. For the MAPT spreading assay, P0 AAV-GFP- or -SQSTM1-injected PS19 mice were stereotaxically injected with 2 μl of MAPT brain lysate at 2–3 months of age into one hippocampal hemisphere (bregma, −2.5 mm; lateral, −2 mm, and depth, −1.8 mm). The mice were euthanized 2 months later for MAPT spreading analysis.
Protein fractionation and western blotting
The cells or forebrain tissues were lysed in buffer (TBS with 1% NP-40 [Thermo Fisher Scientific, 85124], 1% sodium deoxycholic acid [EMD Millipore, 264101], 0.1% sodium dodecylsulfate [EMD Millipore, 428029], and protease and phosphatase inhibitor cocktails [Roche, 04693132001 and 4906845001]). Lysates were then sonicated and centrifuged at 20,000 × g for 15 min. Supernatants were boiled with loading buffer, used for SDS-PAGE and transferred to PVDF membranes. After incubation with primary and secondary antibodies, the signals were detected with a Chemidoc image system (Bio-Rad, Hercules, CA, USA) using ECL or Odyssey image system (LI-COR, Lincoln, NE, USA). The signal intensity was quantified using ImageJ (National Institutes of Health). The biological replicates are shown in the western blots figures.
For protein fractionation, cells were lysed in TBS with 1% Triton X-100 (Sigma-Aldrich, X100). The lysates were sonicated, followed by ultracentrifugation at 100,000 × g for 30 min. The supernatants were collected as soluble fraction. Pellets were resuspended in lysis buffer and subjected to ultracentrifugation again. Pellets were resuspended in 200 μl TBS with 1% SDS and sonicated, then labeled as the insoluble fraction. For brain tissues, hemispheres of forebrain were weighed, homogenized and centrifuged at 800 × g for 5 min. The supernatants were subjected to ultracentrifugation at 100,000 × g for 1 h. The supernatants were collected as the TBS soluble pool. The pellets were resuspended 1% sarkosyl (Teknova, S3376) in TBS with protease and phosphatase inhibitor cocktails, sonicated, and followed by ultracentrifugation as above. The supernatants were collected as the sarkosyl soluble pool, and the pellets were resuspended and sonicated in 200 μl of TBS with 1% SDS as the sarkosyl-insoluble pool.
Immunofluorescence staining
For brain samples, tissues were collected after saline perfusion, followed by overnight fixation in 4% paraformaldehyde (PFA) in TBS. The PFA was replaced with 30% sucrose (Acros Organics, 177142500) in TBS. After dehydration, brain tissues were cut into 30-μm frozen sections for staining. For in vitro cultures, cells were fixed with 4% PFA at room temperature for 20 min, followed by TBS wash. Tissue sections or cells were incubated with primary antibody in TBS with 0.4% Triton X-100 and 2% donkey serum (EMD Millipore, S30-100ML) overnight, followed by washing and incubation with secondary antibodies. For ThioS staining, cells were incubated with 0.002% ThioS (Sigma-Aldrich, T1892) in TBS, followed by rinsing with 50% ethanol twice, then 5 min in TBS before imaging. Cells were imaged by confocal microscopy (Leica SPE, Wetzlar, Germany). For quantification, the Analyze Particles model from ImageJ was used to calculate the percentage of the area or integrated density. Fields of view were randomly selected at 64X magnification for at least 20 images per group.
Statistics
All data are presented as mean ± SEM. Power analysis was performed using a confidence interval of α = 0.05. Pairwise comparisons were analyzed using a two-tailed Student’s t-test. P-values less than or equal to 0.05 were considered statistically significant. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Supplemental Material
Download MS Power Point (9 MB)Acknowledgments
We are grateful to R. Youle (NIH) for the gift SQSTM1 KO, OPTN KO, ATG5 KO and penta KO Hela cells and P. Davies (Feinstein Institute for Medical Research) for providing PHF1, CP13, and MC1 antibodies. We thank A. Cole and N. Aithmitti for expert technical assistance, M. Sardiello for GESA analysis, D. Swartzlander for critical reading of the manuscript, and members of the Zheng laboratory for insightful discussions. This project was supported by the Gene Vector Core of Baylor College of Medicine and by grants from the NIH (R01 NS093652, R01 AG020670, and RF1 AG054111 to HZ and R01 AG057509 to HZ and SZ).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol. 2016;12(1):15–27.
- Mandelkow EM, Mandelkow E. Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med. 2012 Jul;2(7):a006247.
- Gendron TF, Petrucelli L. The role of tau in neurodegeneration. Mol Neurodegener. 2009;4:13.
- Giannakopoulos P, Herrmann FR, Bussiere T, et al. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology. 2003;60(9):1495–1500.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd ed.). Autophagy. 2016;12(1):1–222.
- Martini-Stoica H, Xu Y, Ballabio A, et al. The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 2016 Apr;39(4):221–234.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741.
- Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014;16(6):495–501.
- Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282(33):24131–24145.
- Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014;111(42):E4439–4448.
- Lamark T, Kirkin V, Dikic I, et al. NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle. 2009;8(13):1986–1990.
- Thurston TL, Ryzhakov G, Bloor S, et al. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat Immunol. 2009;10(11):1215–1221.
- Deng Z, Purtell K, Lachance V, et al. Autophagy receptors and neurodegenerative diseases. Trends Cell Biol. 2017;27(7):491–504.
- Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524(7565):309–314.
- Jo C, Gundemir S, Pritchard S, et al. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun. 2014;5:3496.
- Cirulli ET, Lasseigne BN, Petrovski S, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436–1441.
- Teyssou E, Takeda T, Lebon V, et al. Mutations in SQSTM1 encoding p62 in amyotrophic lateral sclerosis: genetics and neuropathology. Acta Neuropathol. 2013;125(4):511–522.
- Pottier C, Bieniek KF, Finch N, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015;130(1):77–92.
- Haack TB, Ignatius E, Calvo-Garrido J, et al. Absence of the autophagy adaptor SQSTM1/p62 causes childhood-onset neurodegeneration with Ataxia, Dystonia, and Gaze Palsy. Am J Hum Genet. 2016;99(3):735–743.
- Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010;465(7295):223–226.
- Ramesh Babu J, Seibenhener Lamar M, Peng J, et al. Genetic inactivation of p62 leads to accumulation of hyperphosphorylated tau and neurodegeneration. J Neurochem. 2008;106(1):107–120.s.
- Caccamo A, Ferreira E, Branca C, et al. p62 improves AD-like pathology by increasing autophagy. Mol Psychiatry. 2017;22(6):865–873.
- Doi H, Adachi H, Katsuno M, et al. p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J Neurosci. 2013;33(18):7710–7727.
- Brady OA, Meng P, Zheng Y, et al. Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J Neurochem. 2011;116(2):248–259.
- Polito VA, Li H, Martini-Stoica H, et al. Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med. 2014;6(9):1142–1160.
- Xu Y, Martini-Stoica H, Zheng H. A seeding based cellular assay of tauopathy. Mol Neurodegener. 2016;11:32.
- Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004.
- Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141.
- Egan DF, Shackelford DB, Mihaylova MM, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331(6016):456–461.
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82(4):239–259.
- Goedert M. Alzheimer’s and Parkinson’s diseases: the prion concept in relation to assembled Abeta, tau, and alpha-synuclein. Science. 2015;349(6248):1255555.
- Iba M, Guo JL, McBride JD, et al. Synthetic tau fibrils mediate transmission of neurofibrillary tangles in a transgenic mouse model of Alzheimer’s-like tauopathy. J Neurosci. 2013;33(3):1024–1037.
- Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82(6):1271–1288.
- Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–913.
- de Calignon A, Polydoro M, Suarez-Calvet M, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73(4):685–697.
- Morris M, Maeda S, Vossel K, et al. The many faces of tau. Neuron. 2011;70(3):410–426.
- Chesser AS, Pritchard SM, Johnson GV. Tau clearance mechanisms and their possible role in the pathogenesis of Alzheimer disease. Front Neurol. 2013;4:122.
- Myeku N, Clelland CL, Emrani S, et al. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat Med. 2016;22(1):46–53.
- Dickey CA, Koren J, Zhang YJ, et al. Akt and CHIP coregulate tau degradation through coordinated interactions. Proc Natl Acad Sci U S A. 2008;105(9):3622–3627.
- Schaeffer V, Lavenir I, Ozcelik S, et al. Stimulation of autophagy reduces neurodegeneration in a mouse model of human tauopathy. Brain. 2012;135(Pt 7):2169–2177.
- Ozcelik S, Fraser G, Castets P, et al. Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One. 2013;8(5):e62459.
- Caccamo A, Magri A, Medina DX, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell. 2013;12(3):370–380.
- Bjorkoy G, Lamark T, Brech A, et al. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171(4):603–614.
- Ichimura Y, Kumanomidou T, Sou YS, et al. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008;283(33):22847–22857.
- Korac J, Schaeffer V, Kovacevic I, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013;126(Pt 2):580–592.
- Gal J, Ström AL, Kwinter DM, et al. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem. 2009;111(4):1062–1073.
- Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci. 2011;124(Pt 16):2692–2701.
- Lynch-Day MA, Klionsky DJ. The Cvt pathway as a model for selective autophagy. FEBS Lett. 2010;584(7):1359–1366.
- Kuusisto E, Salminen A, Alafuzoff I. Early accumulation of p62 in neurofibrillary tangles in Alzheimer’s disease: possible role in tangle formation. Neuropathol Appl Neurobiol. 2002;28(3):228–237.
- Piras A, Collin L, Gruninger F, et al. Autophagic and lysosomal defects in human tauopathies: analysis of post-mortem brain from patients with familial Alzheimer disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol Commun. 2016;4:22.
- Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140(3):313–326.
- Rui YN, Xu Z, Patel B, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17(3):262–275.
- Ochaba J, Lukacsovich T, Csikos G, et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A. 2014;111(47):16889–16894.
- Fernandez-Nogales M, Cabrera JR, Santos-Galindo M, et al. Huntington’s disease is a four-repeat tauopathy with tau nuclear rods. Nat Med. 2014;20(8):881–885.
- Jellinger KA. Alzheimer-type lesions in Huntington’s disease. J Neural Transm (Vienna). 1998;105(8–9):787–799.
- Vuono R, Winder-Rhodes S, de Silva R, et al. The role of tau in the pathological process and clinical expression of Huntington’s disease. Brain. 2015;138(Pt 7):1907–1918.
- Gratuze M, Cisbani G, Cicchetti F, et al. Is Huntington’s disease a tauopathy? Brain. 2016;139(Pt 4):1014–1025.
- Lim J, Lachenmayer ML, Wu S, et al. Proteotoxic stress induces phosphorylation of p62/SQSTM1 by ULK1 to regulate selective autophagic clearance of protein aggregates. PLoS Genet. 2015;11(2):e1004987.
- Bordi M, Berg MJ, Mohan PS, et al. Autophagy flux in CA1 neurons of Alzheimer hippocampus: increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy. 2016;12(12):2467–2483.
- Santacruz K, Lewis J, Spires T, et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309(5733):476–481.
- Yoshiyama Y, Higuchi M, Zhang B, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351.
- Andorfer C, Kress Y, Espinoza M, et al. Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms. J Neurochem. 2003;86(3):582–590.
- Xu Y, Zhang J, Tian C, et al. Overexpression of p62/SQSTM1 promotes the degradations of abnormally accumulated PrP mutants in cytoplasm and relieves the associated cytotoxicities via autophagy-lysosome-dependent way. Med Microbiol Immunol. 2014;203(2):73–84.
- Park BC, Shen X, Samaraweera M, et al. Studies of optineurin, a glaucoma gene: golgi fragmentation and cell death from overexpression of wild-type and mutant optineurin in two ocular cell types. Am J Pathol. 2006;169(6):1976–1989.
- Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991.
- Lian H, Yang L, Cole A, et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron. 2015;85(1):101–115.