
ABSTRACT
50% of Caucasians carry a Thr300Ala variant (T300A) in the protein encoded by the macroautophagy/autophagy gene ATG16L1. Here, we show that the T300A variant confers protection against urinary tract infections (UTIs), the most common infectious disease in women. Using knockin mice carrying the human T300A variant, we show that the variant limits the UTI-causing bacteria, uropathogenic Escherichia coli (UPEC), from establishing persistent intracellular reservoirs, which can seed UTI recurrence. This phenotype is recapitulated in mice lacking Atg16l1 or Atg7 exclusively in the urothelium. We further show that mice with the T300A variant exhibit urothelial cellular abnormalities, including vesicular congestion and aberrant accumulation of UPK (uroplakin) proteins. Importantly, presence of the T300A variant in humans is associated with similar urothelial architectural abnormalities, indicating an evolutionarily conserved impact. Mechanistically, we show that the reduced bacterial persistence is independent of basal autophagic flux or proinflammatory cytokine responses and does not involve Atg14 or Epg5. However, the T300A variant is associated with increased expression of the small GTPase Rab33b; RAB33B interacts with ATG16L1, as well as other secretory RABs, RAB27B and RAB11A, important for UPEC exocytosis from the urothelium. Finally, inhibition of secretory RABs in bladder epithelial cells increases intracellular UPEC load. Together, our results reveal that UPEC selectively utilize genes important for autophagosome formation to persist in the urothelium, and that the presence of the T300A variant in ATG16L1 is associated with changes in urothelial vesicle trafficking, which disrupts the ability of UPEC to persist, thereby limiting the risk of recurrent UTIs.
Abbreviations: 3-PEHPC: 3-pyridinyl ethylidene hydroxyl phosphonocarboxylate; ATG: autophagy; ATG16L1: autophagy related 16 like 1; BECs: bladder epithelial cells; dpi: days post infection; hpi: hours post infection; IF: immunofluorescence; IL1B: interleukin 1 beta; IL6: interleukin 6; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MVB: multivesicular bodies; T300A: Thr300Ala; TNF: tumor necrosis factor; QIR(s): quiescent intracellular reservoir(s); siRNA: short interfering RNA; UPEC: uropathogenic Escherichia coli; UTI(s): urinary tract infection(s); TEM: transmission electron microscopy; WT: wild type
KEYWORDS:
Introduction
Autophagy is a housekeeping process that maintains cellular homeostasis through recycling of nutrients and degradation of damaged or aged cytoplasmic contents. This process involves a series of dynamic membrane rearrangements that are mediated by a core set of autophagy-related (ATG) proteins. Autophagosome formation starts with the stepwise engulfment of cytoplasmic material by the phagophore, which later matures into a double-membrane-bound vesicle [Citation1–Citation5]. Vesicle nucleation requires the activation of the class III phosphatidylinositol 3-kinase complex, whose activation depends on the formation of a complex that includes ATG14. The vesicle elongation process involves 2 ubiquitin-like conjugation systems. The first involves the covalent conjugation of ATG12 to ATG5, which then interacts non-covalently with ATG16L1 (autophagy related 16 like 1). The second involves the conjugation of phosphatidylethanolamine to MAP1LC3B/LC3 (microtubule associated protein 1 light chain 3 beta) by the sequential action of ATG4, ATG7, and ATG3. Autophagosomes then undergo maturation by fusion with lysosomes to create autolysosomes, a process which requires various proteins including EPG5 [Citation6,Citation7].
Autophagy genes control bacterial infection and inflammation. Depletion or mutation of autophagy genes is associated with inflammatory diseases in humans. For example, a single nucleotide polymorphism (rs2241880, corresponding to the T300A variant) in ATG16L1 is present in 50% of Caucasians and is associated with a higher risk of developing Crohn disease, a form of inflammatory bowel disease [Citation8,Citation9]. The ATG16L1T300A variant does not affect basal autophagy but affects unconventional autophagy [Citation10] and xenophagy (a specific form of autophagy targeting pathogens [Citation11]) and the inflammatory response to certain infections or metabolic stress signals [Citation12–Citation17].
Autophagy is also involved in the pathogenesis of urinary tract infections (UTIs), one of the most common, recurrent infectious diseases worldwide. UTIs afflict 150 million women per year and recur in up to 40% of cases [Citation18]. The majority of infections are caused by uropathogenic Escherichia coli (UPEC), which invade into superficial bladder epithelial cells, replicate, and persist in a small number of urothelial cells as quiescent intracellular bacterial reservoirs (QIRs) [Citation19]. QIRs are refractory to antibiotic therapies and do not elicit an active immune response but can seed recurrent infections. UPEC are recognized by autophagy machinery and reside in autophagosomal and/or lysosomal compartments [Citation20,Citation21]; mice hypomorphic for ATG16L1 have reduced UPEC persistence [Citation21,Citation22]. Furthermore, UPEC co-opt ferritinophagy, a selective form of autophagy involved in iron release from ferritin stores, to survive and persist in bladder epithelial cells [Citation20]. However, whether ATG16L1 deficiency-mediated protection from UTIs depends on the canonical autophagy machinery or is specific to some other role for ATG16L1 is unclear. Additionally, whether other autophagy pathway genes play a role in orchestrating the response to UTIs remains to be elucidated. Further, given the important role of ATG16L1 in urothelial intracellular membrane trafficking and UPEC persistence, it remains unknown whether the T300A variant in ATG16L1 plays a role in pathogenesis of UTIs.
Superficial urothelial cells contain unique cytoplasmic organelles including fusiform vesicles, which deliver preassembled UPK (uroplakin) proteins to the apical cell surface during bladder filling. Recent studies have demonstrated that specific small RAB GTPases (RAB27B and RAB11A) are highly enriched in superficial urothelial cells and are important for vesicle trafficking, UPK recycling [Citation23–Citation28], and exosome-mediated expulsion of intracellular UPEC [Citation29–Citation31]. Interestingly, network mapping of autophagy pathways has identified a Golgi-resident small GTPase, RAB33B, which interacts directly with ATG16L1 and thereby modulates autophagosome formation [Citation32]. Whether the T300A variant is associated with changes to RAB33B, RAB27B or RAB11A, which are implicated in altered vesicular trafficking, remains to be determined.
Here, we report that UPEC selectively utilize autophagy proteins important for autophagosome formation to persist in the urothelium. T300A mice carrying the human mutant allele at the Atg16l1 gene locus exhibit altered urothelial architecture and harbor significantly fewer persistent bacterial reservoirs in the urothelium. Importantly, this feature is conserved in humans with the rs2241880 GG genotype (homozygous for the T300A variant in human ATG16L1). We demonstrate that these effects are independent of the canonical autophagy pathway and bladder mucosal immunity, but appear to be associated with increased expression of small RAB GTPases that are known to be important for UPK recycling and UPEC expulsion. Together, our findings suggest that the T300A variant protects the bladder mucosa from persistent UTIs.
Results
Presence of the ATG16L1T300A variant confers protection from UPEC persistence in urothelium
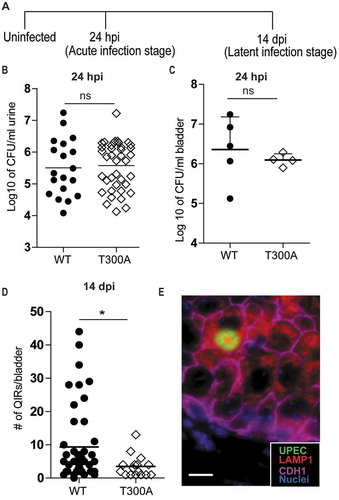
We first sought to determine whether the T300A variant of ATG16L1 was sufficient to confer protection from UPEC persistence in the bladder epithelium, a phenotype observed in mice with reduced Atg16l1 expression (Atg16l1HM mice) [Citation22]. We induced UTIs in adult female wild-type (WT) and mice carrying the human mutant allele at the Atg16l1 gene locus (hereafter referred to as T300A mice) and quantified the number of QIRs at 14 d post-infection (dpi) (). Urothelial cells of T300A mice harbored significantly fewer QIRs than did those of WT mice (). To confirm that the reduced QIR formation was not due to reduced UPEC invasion or defects in early colonization, we measured bacterial titers in the urine and bladder at 24 h post-infection (hpi) and found comparable bacterial titers between WT and T300A mice (). Thus, the T300A variant specifically confers increased resistance to UPEC persistence within the urothelium but does not affect UPEC invasion or early colonization, as was noted in Atg16l1HM mice [Citation22].
Figure 1. Mice carrying the ATG16L1T300A variant exhibit reduced UPEC persistence. (a) Schematic of the experimental setup. (b) Colony forming unit (CFU) counts of bacteriuria at 24 hpi plotted as mean of the Log10 value. n = 19 for WT; n = 45 for T300A; n = 6 experiments. ns, not significant by Mann-Whitney test. (c) CFU counts of the bladder plotted as mean ± SD of the Log10 value at 24 hpi. n = 5 for WT; n = 4 for T300A mice. ns, not significant by Mann-Whitney test. (d) Quantification of QIRs in WT and T300A bladders at 14 dpi. n = 8 sections/bladder, n = 17 mice for T300A, n = 45 mice for WT. *P < 0.05 by Mann-Whitney test. (e) IF image of a bacterial QIR. Bar: 10 µm.
The T300A variant is sufficient to induce cellular architectural alterations in superficial urothelial cells
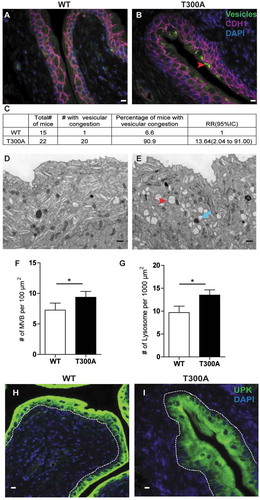
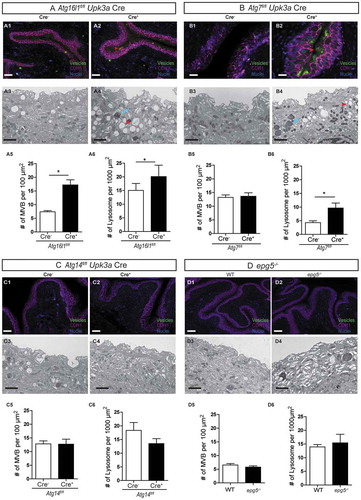
In a healthy bladder, the plasma membranes of superficial epithelial cells contain hexagonal arrays of UPK proteins, which are transported by discoidal storage vesicles and multivesicular bodies (MVBs) and are critical for normal epithelial flexibility and barrier function [Citation33,Citation34]. Previous work has demonstrated a role for ATG16L1 in superficial cell vesicular trafficking in mice globally hypomorphic for Atg16l1 [Citation22]. We next sought to determine whether the T300A variant was sufficient to confer the same vesicular phenotype associated with decreased UPEC persistence. Indeed, we found substantially more MVBs and lysosomes in T300A mice compared to those of WT mice (). Blinded analysis of sections revealed that 90% of T300A mice had vesicle congestion in superficial cells, whereas only 7% of WT mice had this phenotype (). Transmission electron microscopy (TEM) and immunofluorescence (IF) analysis confirmed that the density of MVBs, lysosomes, and UPK expression in urothelial cells was significantly higher in T300A mice than in WT mice (). These observations indicate that the human T300A variant is sufficient to disrupt urothelial cellular architecture in mice.
Figure 2. T300A mice exhibit superficial cell architectural changes. (a and b) IF imaging analysis of urothelium from WT (a) and T300A (b) mice. Green, vesicles; magenta, CDH1; blue, DAPI; red arrowhead, vesicular congestion. Bar: 10 µm. (c) Percentage of WT and T300A mice that showed vesicular congestion. (d and e) Representative TEM of superficial cell ultrastructure in WT (d) and T300A (e) mice. Red arrowhead, multi-vesicular bodies; blue arrow, lysosomes. Bar: 1 µm. (f and g) Quantification of MVBs (f) and lysosomes (g) in TEM images of urothelium from WT and T300A mice. Bars represent mean ± SEM. n = 3 mice/genotype.*P < 0.05 by unpaired two-tailed T test. (h and i) IF of UPK (green) in bladders of WT (g) and T300A (h) mice. Blue, DAPI. Bar: 10 µm.
UPEC hijack a specific set of autophagy genes important for normal tissue architecture to persist intracellularly in the urothelium
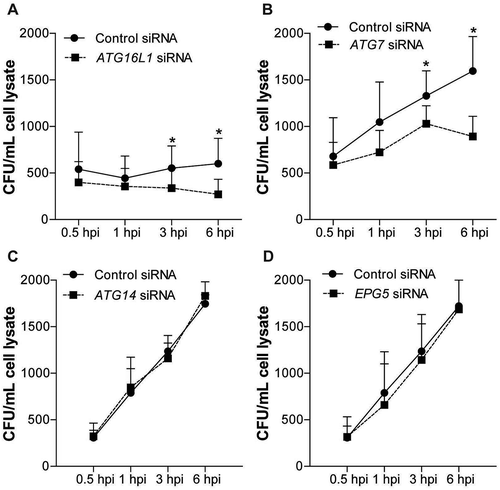
We next examined if the T300A variant-conferred restriction of UPEC persistence and urothelial architectural changes were unique to ATG16L1 or were a shared feature with other canonical autophagy related proteins. First, we depleted key genes for each step of the autophagy pathway via short interfering RNA (siRNA)-mediated gene-silencing in human bladder epithelial cells (BECs). RT-qPCR and immunoblotting validated the knockdown of mRNA levels of ATG16L1, ATG7, ATG14 and EPG5, and protein levels of ATG16L1 and ATG7 (Figure S1). We confirmed that silencing of ATG16L1 led to a significant reduction in the intracellular bacterial load in BECs at 3 and 6 hpi (). The effects were phenocopied in ATG7-silenced cells (). In contrast, knockdown of ATG14 or EPG5 did not decrease intracellular UPEC load (). We did not observe significant differences in bacterial load at earlier time points (0.5 and 1 hpi) in cells depleted of ATG16L1 or ATG7, indicating that these genes are not required for early invasion and colonization of BECs. Crystal violet assays did not reveal significant changes in cell growth in ATG16L1- or ATG7-silenced cells, indicating that the observed reduction in the intracellular load of UPEC was not likely due to decreased cell viability (Figure S2).
Figure 3. Loss of ATG16L1 or ATG7 but not ATG14 or EPG5 leads to decreased intracellular UPEC growth in human BECs. Quantification of intracellular UPEC CFU in human BECs treated with control siRNA and ATG16L1 siRNA (a), ATG7 siRNA (b), ATG14 siRNA (c) or EPG5 siRNA (d), Bars represent mean ± SD. *P < 0.05 by Mann-Whitney test, n = 3 replicates.
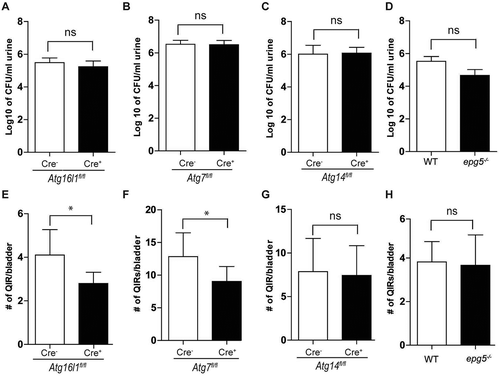
To extend the in vitro findings, we systematically examined whether loss of autophagy genes specifically in the urothelium affects UPEC persistence. To generate urothelial-cell-specific knockouts, we crossed Atg16l1fl/fl, Atg7fl/fl, and Atg14fl/fl mice to Upk3a-GFP-CreERT2 mice, which drive deletion exclusively in superficial urothelial cells [Citation35]. We also examined bladders of mice globally deficient in Epg5 (epg5−/-), a gene important for autophagosome-lysosome fusion. Next, we induced UTIs in Atg16l1fl/flUpk3a Cre+, Atg7fl/flUpk3a Cre+, Atg14fl/flUpk3a Cre+, and epg5−/- mice and examined urine bacterial loads in these mice at 24 hpi and quantified the number of bacterial QIRs at 14 dpi. We found that loss of each of the genes had no effect on bacteriuria at 24 hpi (). However, at 14 dpi, Atg16l1fl/flUpk3a Cre+ and Atg7fl/flUpk3a Cre+ mice harbored significantly fewer QIRs in the bladder than their Cre− littermates (), an effect not seen following loss of Atg14 in urothelial cells or global deletion of Epg5 (). Together, these data indicate that UPEC co-opts proteins of the elongation phase of the autophagy pathway important for autophagosome formation for persistence in QIRs but does not require proteins governing the initiation or autophagosome-lysosome fusion phases.
Figure 4. Loss of Atg16l1 or Atg7 in urothelium but not Atg14 or Epg5 leads to significant reduction in QIR formation. (a-d) CFU counts of bacteriuria at 24 hpi plotted as mean ± SEM of the Log10 value in Atg16l1fl/flUpk3a Cre− and Atg16l1fl/flUpk3a Cre+ mice (a), Atg7fl/flUpk3a Cre− and Atg7fl/flUpk3a Cre+ mice (b), Atg14fl/flUpk3a Cre− and Atg14fl/flUpk3a Cre+ mice (c) and WT and epg5−/- mice (d). ns, not significant by unpaired two-tailed T test. (e-h) Quantification of QIRs in Atg16l1fl/flUpk3a Cre− and Atg16l1fl/flUpk3a Cre+ bladders (e), Atg7fl/flUpk3a Cre− and Atg7fl/flUpk3a Cre+ bladders (f), Atg14fl/flUpk3a Cre− and Atg14fl/flUpk3a Cre+ bladders (g) and WT and epg5−/- bladders (h) at 14 dpi. n = 8 sections/bladder, n = 18–25 mice/genotype. Bar shows mean ± SD. *P < 0.05 by Mann-Whitney test; ns, not significant.
To determine if depletion of Atg16l1 specifically in the urothelium was sufficient to alter urothelial architecture, we examined bladders from Atg16l1fl/flUpk3a Cre+ mice by TEM and IF. These mice also display the vesicular congestion phenotype, characterized by accumulation of MVBs and lysosomes (), which has been documented in Atg16l1HM mice [Citation22] and in T300A mice, as shown above. Previously, it has been shown that loss of Atg16l1 in the myeloid compartment or in the adaptive immune compartment does not alter urothelial cellular architecture, supporting the idea that the structural changes were epithelial intrinsic [Citation22]. To further examine if other autophagy proteins were also involved, we examined urothelial architecture in Atg7fl/flUpk3a Cre+, Atg14fl/flUpk3a Cre+, and epg5−/- mice by IF and TEM. We found that only mice with urothelial cells lacking Atg7 showed similar vesicular congestion as seen with loss of Atg16l1 (). Blinded quantification revealed that whereas 34% of Atg16l1fl/flUpk3a Cre+ mice and 55% of Atg7fl/flUpk3a Cre+ mice had vesicle congestion in superficial cells, only 7% of Atg16l1fl/flUpk3a Cre− and 22% of Atg7fl/flUpk3a Cre− littermate controls showed this phenotype. Quantification of MVBs and lysosomes by TEM further corroborated these findings ( and S3). Vesicular congestion was not seen in superficial cells from Atg14fl/flUpk3a Cre+ or epg5−/- mice ( and S3). Together, our data reveal a role for ATG16L1 and ATG7 but not ATG14 or EPG5 in regulating both UPEC persistence in bladder epithelial cells as well as cellular membrane trafficking that is known to be important for bacterial persistence.
Figure 5. Loss of Atg16l1 or Atg7 but not Atg14 or Epg5 leads to urothelial architecture alterations. (a-d, 1 and 2) IF imaging analysis of urothelium from Atg16l1fl/flUpk3a Cre− and Atg16l1fl/flUpk3a Cre+ (A1-A2), Atg7fl/flUpk3a Cre− and Atg7fl/flUpk3a Cre+ (B1-B2), Atg14fl/flUpk3a Cre− and Atg14fl/flUpk3a Cre+ (C1-C2) and WT and epg5−/- mice (D1-D2). Green, vesicles; magenta, CDH1; blue, biz-benzimide DNA stain; red arrowhead, vesicular congestion. Bar: 10 µm. (A-D, 3 and 4) TEM of superficial cell ultrastructure in Atg16l1fl/flUpk3a Cre− and Atg16l1fl/flUpk3a Cre+ (A3-A4), Atg7fl/flUpk3a Cre− and Atg7fl/flUpk3a Cre+ (B3-B4), Atg14fl/flUpk3a Cre− and Atg14fl/flUpk3a Cre+ (C3-C4) and WT and epg5−/- mice (D3-D4). Red arrowhead, multivesicular bodies; blue arrow, lysosomes. n = 3 mice. Bar: 2 µm. (a-d, 5) Quantification of MVBs of Atg16l1fl/flUpk3a Cre− and Atg16l1fl/flUpk3a Cre+ mice (A5), Atg7fl/flUpk3a Cre− and Atg7fl/flUpk3a Cre+ mice (B5), Atg14fl/flUpk3a Cre− and Atg14fl/flUpk3a Cre+ mice (C5) and WT and epg5−/- mice (D5) urothelium. Bars represent mean ± SEM. (A-D, 6) Quantification of lysosomes of Atg16l1fl/flUpk3a Cre− and Atg16l1fl/flUpk3a Cre+ (A6), Atg7fl/flUpk3a Cre− and Atg7fl/flUpk3a Cre+ (B6), Atg14fl/flUpk3a Cre− and Atg14fl/flUpk3a Cre+ (C6) and WT and epg5−/- mice urothelium (D6). Bars represent mean ± SEM.
Decreased UPEC persistence in ATG16L1T300A urothelium is independent of innate immune responses in the bladder mucosa
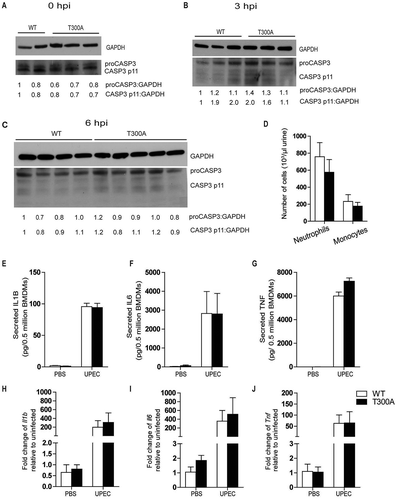
Recent studies in the intestinal mucosa have shown that the T300A variant sensitizes mice to cellular stress induced by intestinal infections with Salmonella enterica serovar Typhimurium and Yersinia enterocolitica, in large part due to increased ATG16L1 cleavage by the apoptotic proteases, CASP3 (caspase 3) and CASP7 [Citation13,Citation14]. Here, we found that levels of CASP3 were equivalent in urothelium of WT and T300A mice before infection () and at 3 and 6 hpi (). Next, we determined whether T300A variant-mediated resistance to UPEC persistence was associated with infiltration of innate immune cells into the bladder. We found that the urine of infected T300A mice harbored similar numbers of neutrophils and monocytes upon infection as did the urine of WT mice (). Previous studies have shown that depletion of ATG16L1 in macrophages amplifies inflammatory IL1B signaling in response to bacterial lipopolysaccharide or UPEC [Citation36,Citation37]. However, we found that bone-marrow-derived macrophages from T300A mice produced similar levels of IL1B, IL6, and TNF () in response to UPEC infection as those from WT mice. Further analysis of mRNA levels of Il1b, Il6, and Tnf from bladders at 24 hpi did not show a significant difference between WT and T300A mice (). Together, these data suggest that the T300A variant does not elicit an elevated inflammatory cellular influx or inflammatory cytokine response upon UPEC infection.
Figure 6. The ATG16L1T300A variant does not affect cytokine response to UPEC infection. (a-c) Western blot detection of CASP3 in whole bladder protein from WT and T300A mice before infection (a), 3 hpi (b), and 6 hpi (c). Numbers below the blots indicate densitometric quantification of the blots. (d) Neutrophil and monocyte counts in the urine at 24 hpi. n = 4–5 mice/genotype, n = 2 experiments. Bars represent mean ± SD. (e–g) Quantification of IL1B (e), IL6 (f), and TNF (g) levels produced by bone-marrow derived macrophages from WT and T300A mice at 24 hpi. (h–j) Quantification of Il1b (h), Il6 (i), and Tnf (j) mRNA levels in WT and T300A mouse bladders 24 hpi. Bars represent mean ± SD.
The ATG16L1T300A variant does not affect bulk autophagy in the bladder
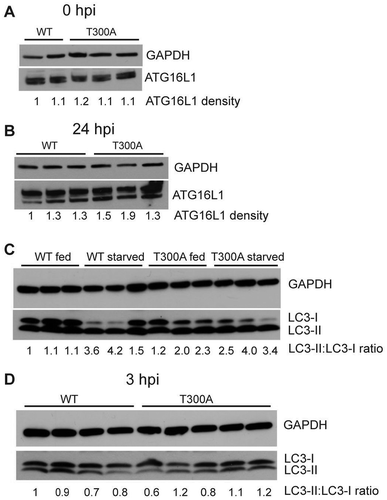
We next reasoned that, if ATG16L1 is critical for bacterial persistence in the bladder, then the T300A variant may cause decreased persistence by affecting ATG16L1 protein expression or autophagic activity in the bladder. However, we found that levels of ATG16L1 protein in WT and T300A mouse bladders before infection () or after infection (), were indistinguishable, indicating that the stability of ATG16L1 is not decreased in T300A mice. Next, to examine autophagic activity, we measured levels of LC3-II before and after starving WT and T300A mice for 24 h. Although the LC3-II:LC3-I ratio increased upon starvation in both WT and T300A mice, we detected no differences in LC3-II levels between the 2 genotypes (). To examine whether the T300A variant impaired autophagy during UTI, we measured LC3-II at 3 hpi and found equivalent levels of LC3-II between bladders from WT and T300A mice (). Thus, the T300A variant does not alter bulk autophagy activity in the bladder.
Figure 7. The ATG16L1T300A variant does not affect canonical autophagy response to UPEC infection. (a-b) Western blot detection of ATG16L1 and GAPDH (loading control) in whole bladder protein before infection (a) and 24 hpi (b). (c) Western blot detection of LC3 in whole bladder protein from fed and starved WT and T300A mice. (d) Western blot detection of LC3 in whole bladder protein from WT and T300A mice at 3 hpi. Numbers below the blots indicate densitometric quantification of the blots.
Presence of the T300A variant alters expression of RAB GTPases that are important for UPEC expulsion
Collectively, our findings suggest that the intersection of UPEC with the autophagy machinery occurs at the phagophore elongation phase wherein ATG16L1 plays a critical role as a central adaptor [Citation38,Citation39]. Given the important role of ATG16L1 in urothelial intracellular membrane trafficking and UPEC persistence, we sought to investigate how the T300A variant might alter the RAB GTPase network in the urothelium. Network mapping of autophagy pathways has identified proteins, notably, a Golgi-resident small GTPase, RAB33B, which interacts directly with ATG16L1 and modulates autophagosome formation [Citation32,Citation40,Citation41]. Thus, we hypothesized that RAB33B could be implicated in the altered UPEC pathogenesis in T300A mice. To test this, we examined expression levels of Rab33b in WT and T300A bladders before and after UPEC infection. Before infection, we found no significant change in expression levels of Rab33b. However, upon induction of a UTI, we noted a significant increase in Rab33b expression in T300A bladders (), in contrast to its downregulation in WT controls.
Figure 8. The presence of the T300A variant differentially regulates genes encoding RAB GTPases upon UPEC infection. (a) RT-qPCR analysis of mRNA levels of Rab27b, Rab11a, and Rab33b in bladders from WT and T300A mice before infection and 24 hpi. (b) Quantification of intracellular UPEC CFU in human BECs treated with vehicle and 3-PEHPC at the indicated time post infection. *P < 0.05; **P < 0.01 by Mann-Whitney test. (c) Quantification of intracellular UPEC CFU in human BECs treated with control siRNA and siRNAs directed against RAB33B, RAB27B, and RAB11A. *P < 0.05; **P < 0.01 by two-way ANOVA with Bonferroni post-test.
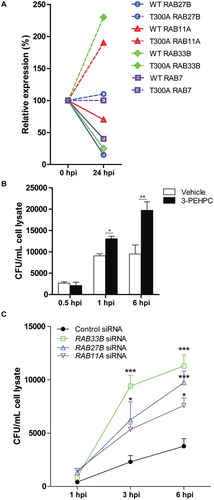
Recent studies have demonstrated that specific small secretory RAB GTPases, RAB27B and RAB11A, are highly enriched in superficial urothelial cells and are important for vesicle trafficking, UPK recycling, and expulsion of intracellular UPEC [Citation23–Citation31]. Examination of Rab27b and Rab11a mRNA levels in WT and T300A mice revealed that the T300A variant was again associated with increases in expression levels of both Rab27b and Rab11a upon induction of a UTI in contrast to decreases in expression in WT bladders (). Thus, secretory RABs associated with ATG16L1 and autophagosome formation and/or UPK recycling appear to be altered in T300A mice, a result consistent with a role for this variant in modulating urothelial architecture. Given the established role of RAB27B and RAB11A in urothelial stretch-induced trafficking [Citation25–Citation27] and in UPEC exocytosis from human BECs [Citation29,Citation30], we mechanistically determined whether inhibition of RAB activity would lead to increased bacterial load in BECs. We treated human BECs with 3-pyridinyl ethylidene hydroxyl phosphonocarboxylate (3-PEHPC), an inhibitor of RABGGT (Rab geranylgeranyl transferase) that inhibits all secretory RAB GTPase activity [Citation42–Citation44]. Pretreatment of human BECs with 3-PEHPC prior to infection with UPEC revealed a significant increase in intracellular bacteria load upon infection (). The highest load was noted at 6 hpi, which corresponds to the time point shown to be the peak of exocytosis of UPEC via RAB27B [Citation30]. Further, we depleted RAB33B, RAB27B and RAB11A via siRNA-mediated gene-silencing in human BECs. We found that silencing of RAB33B, RAB27B and RAB11A led to a significant increase in the intracellular bacterial load in human BECs at 3 and 6 hpi (), which is consistent with previous findings that RAB small GTPases mediate bacterial clearance from the urothelium [Citation29]. Taken together, our in vivo and in vitro data suggest that changes in RAB33B, RAB27B and RAB11A levels due to the presence of the T300A variant likely contribute to reduced UPEC persistence in urothelium.
Humans carrying the ATG16L1T300A variant display aberrant urothelial cell architecture in bladder tissue biopsies
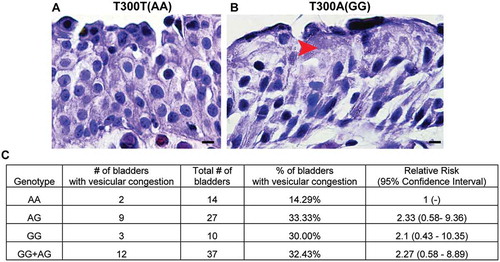
Given that UPEC persistence correlates with urothelial-specific aberrant vesicular trafficking (and not with canonical autophagy pathways or non-cell-autonomous immune cell effects) in mice carrying the human T300A variant, we next determined whether the incidence of the T300A variant in human populations would be correlated with similar urothelial architectural alterations. We obtained bladder tissues from patients undergoing transurethral resections for bladder cancer diagnosis and examined them in a blinded fashion for the presence/absence of vesicles. Remarkably, our analysis revealed that 33% of individuals with the GG (homozygous carriage of the T300A allele) or AG (heterozygous) genotypes displayed urothelial vesicular congestion, whereas only 14% of those with the AA (homozygous for the WT ATG16L1 allele) genotype had this phenotype (). Thus, the strong association between the presence of the T300A variant and increased urothelial vesicular abnormalities in mouse and human bladder urothelium indicates a key role for the T300A variant in the bladder mucosa with significant implications for UPEC pathogenesis.
Figure 9. The ATG16L1T300A variant leads to superficial cell architectural alterations in human bladders. (a and b) Hematoxylin and eosin staining of human bladder tissue biopsies from participants carrying ATG16L1 rs2241880 AA (a) and GG (b) alleles. Red arrowhead, vesicles. (c) Percentage of human bladders of the indicated genotype that showed a vesicular congestion phenotype.
Discussion
In this report, we demonstrated that the common ATG16L1T300A variant confers protection from UPEC persistence primarily due to an alteration in urothelial cell architecture and consequent depletion of UPEC-protective niches. We found that mice carrying the human ATG16L1T300A variant phenocopied urothelial abnormalities noted in mice carrying a hypomorphic allele of Atg16l1 with an 80% knockdown of the protein. Both Atg16l1HM and T300A mice harbored fewer persistent UPEC reservoirs than WT mice, underscoring a pro-pathogenic role for ATG16L1 in the bladder mucosa.
Interestingly, we observed substantial differences at the early stages of UPEC infection between Atg16l1HM and T300A mice. In T300A mice, as opposed to Atg16l1HM mice, we did not observe faster clearance of bacteria from the urine, increased neutrophil infiltration, or increased IL1B/IL-1β production by macrophages. These differences are likely due to the means of generation of these mice. Atg16l1HM mice were generated using gene-trap mutations to introduce a false splice acceptor in the intronic regions adjacent to exon 6 or 10, resulting in truncated core protein. Conversely, the T300A mutation occurs in the WD repeat C-terminal domain of ATG16L1 which is not associated with canonical autophagy [Citation10,Citation13,Citation14]. Our study demonstrated that T300A-associated urothelial alterations are independent of bulk autophagy activity, which is consistent with previous findings that the ATG16L1T300A variant does not reduce basal autophagy [Citation15–Citation17]. Boada-Romero et al., recently showed that the ATG16L1T300A variant also alters the ability of the WD repeat domain to interact with a transmembrane protein, TMEM59, and thereby affects TMEM59-mediated endosome trafficking. This impairment does not affect canonical autophagy and does not require caspase processing of ATG16L1 [Citation12]. Our findings are consistent with these observations because the presence of the T300A variant does not sensitize caspase-dependent cleavage of ATG16L1 upon UPEC infection. In other words, the level of ATG16L1 in the bladder of T300A mice is not affected by the mutation.
T300A mice exhibited alterations in the recycling of intracellular membranes in superficial urothelial cells, increased expression of UPKs, and accumulation of MVBs, which is consistent with an important role of ATG16L1 in governing the secretory pathway in epithelial cells [Citation13,Citation22,Citation45]. Because MVBs are required for degrading and recycling of UPKs [Citation46], we suggest that accumulation of MVBs in the urothelium may impair intracellular trafficking of UPKs and UPEC such that the bacteria are rerouted to exosomes for exocytosis [Citation30] rather than to phagophores where they can persist within the cell (i.e., inside autophagosomes). RAB27B is important for stretch-induced exocytosis in bladder umbrella cells [Citation26]. RAB11A, a protein that labels recycling endosomes, recruits WIPI2 for ATG16L1-mediated LC3 conjugation and autophagosome formation [Citation47] and thus may be involved in ATG16L1T300A-induced vesicle congestion in the urothelium. Recent work by Miao, et al., has demonstrated that concerted actions of the secretory RAB GTPases, RAB11A and RAB27B are involved in expulsion of UPEC from bladder epithelial cells [Citation29]. Indeed, our findings that the T300A variant was associated with increased expression of Rab33b, Rab27b, and Rab11a, may explain the reduced UPEC persistence in T300A bladders. This notion is further underscored by significantly higher intracellular UPEC loads in bladder epithelial cells treated with a pan-secretory RAB inhibitor or depleted of RAB33B, RAB27B, or RAB11A by siRNA. Further mechanistic studies are needed to understand how ATG16L1 deficiency or T300A polymorphism alters urothelial trafficking and how this may be mediated via RAB33B, RAB27B, or RAB11A. Nevertheless, it is clear that this variant is associated with decreased UPEC persistence, likely via disruption of UPEC trafficking to phagophores, allowing the bacteria to persist within autophagosomes.
Our findings are similar to a recent study demonstrating that the T300A variant is associated with structural defects in Paneth cells caused by defects in secretory autophagy due to the presence of this variant. Interestingly, the consequences of T300A variant-induced structural defects in Paneth cells and superficial bladder cells are quite the opposite. Bel et al., showed that disrupted secretory autophagy in Paneth cells in T300A mice is associated with increased Salmonella enterica serovar Typhimurium infection [Citation48]. However, in the bladder, T300A variant-induced structural changes are beneficial to the host as these alterations impair the ability of UPEC to persist in the urothelium. Thus, our data highlight a positive role for secretory autophagy in response to uropathogenic infection and point towards the tissue- and cell-type-specific roles of the T300A variant.
We demonstrate that UPEC adopt a unique strategy by selectively exploiting a specific set of autophagy proteins to establish latency, confirming that bacterial pathogens can utilize different host cell compartments and pathways during their intracellular cycle. Autophagy gene-dependent cellular functions in controlling intracellular pathogens have been reported in a variety of disease models. For example, Starr et al., showed selective subversion of autophagy initiation proteins by Brucella abortus [Citation49]. Huang et al., showed that IFNG/IFN-γ inhibits murine norovirus growth in macrophages in a manner dependent on Atg5, Atg7, and Atg16l1, but not on Atg14 [Citation50]. More recently, Park et al., showed that loss of autophagy genes including Atg5, Atg16l1, Atg7, and Atg14 but not Atg4b in myeloid cells results in inhibition of MHV68 reactivation from latently infected macrophages [Citation51]. Additionally, Kimmey et al., demonstrated a unique role for Atg5 but not for other autophagy genes in modulating Mycobacterium tuberculosis infection [Citation52].
The exact mechanism by which UPEC hijacks ATG16L1 and ATG7 and the bacterial proteins that are involved in mediating this process remain poorly described. We suggest that the absence of ATG16L1 or ATG7 leads to alteration of intracellular membrane trafficking, which in turn reroutes UPEC to compartments where they cannot persist. Urothelium-specific loss of these autophagy genes appears sufficient to induce urothelial vesicular congestion and confer resistance to UPEC persistence, further supporting our conclusion that regulation of QIR formation is not governed by loss of autophagy gene-mediated inflammatory responses. Loss of autophagy genes in myeloid cells or in the adaptive immune compartment does not affect intracellular membrane trafficking or bacterial persistence within the urothelium [Citation22]. Furthermore, we previously reported that Atg16l1HM macrophages had increased CASP1-dependent secretion of IL1B upon UPEC infection [Citation37]. Likewise, recent studies showed that the T300A polymorphism was associated with increased IL1B production in macrophages due to increased CASP3-mediated degradation of ATG16L1 in the context of metabolic stress or infection [Citation13,Citation14]. However, we did not find any differences in production of IL1B or other proinflammatory mediators between macrophages from ATG16L1T300A or WT mice before or in response to UPEC infection. Together, our findings suggest that the T300A variant specifically affects urothelial cells and indicate an epithelial-intrinsic role of autophagy genes in intracellular trafficking of UPEC inside the urothelium.
UTIs have been described as early as 1550 B.C., are the second most common infection in humans, and are the most common infection in women worldwide. It is remarkable that presence of the T300A variant in humans with no known bladder disease is sufficient to confer structural alterations in urothelial cells. This association is reminiscent of the observation that the harmful sickle cell trait is maintained in humans because it confers resistance to another common infection, malaria, by structurally altering red blood cells, thus limiting the ability of the parasite to invade [Citation53]. Together, our findings underscore a key role for ATG16L1 and its associated human polymorphism, T300A, in maintaining bladder epithelial architecture and altering the trafficking patterns of intracellular UPEC, thus having significant clinical implications for understanding the etiology of recurrent UTIs.
Materials and methods
Mice
Atg16l1fl/fl and Atg14fl/fl mice were kindly provided by Dr. Herbert Virgin (Washington University School of Medicine). Atg7fl/fl mice were a kind gift from Dr. Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science). Upk3a-GFP-Cre−ERT2 mice were purchased from Jackson laboratory. Atg16l1fl/fl, Atg7fl/fl or Atg14fl/fl mice were bred to Upk3a-GFP-Cre−ERT2 mice to generate Atg16l1fl/flUpk3a Cre−ERT2, Atg7fl/fl Upk3a Cre−ERT2 or Atg14fl/flUpk3a Cre−ERT2 mice. Atg16l1fl/flUpk3a Cre+, Atg7fl/flUpk3a Cre+ or Atg14fl/flUpk3a Cre+ mice and their Cre− counterparts were treated with tamoxifen (1 mg/20 g body weight; Sigma, T5648) intraperitoneally 3 times over the course of 1 wk. The mice were then infected 1 wk after the last tamoxifen treatment. epg5−/- mice were kindly provided by Dr. Hong Zhang (Chinese Academy of Sciences) [Citation54]. T300A mice were generated as described previously [Citation13]. Experiments were performed according to approved protocols. In all experiments, adult female mice, 8–10 wk of age were used. Mice were maintained under specified pathogen-free conditions in a barrier facility and under a strict 12-h light/dark cycle. All experimental procedures were approved by the animal studies committee of the Washington University School of Medicine (Animal Welfare Assurance #A-3381–01).
Bacterial strains, mouse inoculations, and urinalysis
UTI89, a clinical UPEC isolate from a patient with cystitis [Citation55] was grown statically for 17 h in Luria-Bertani broth (10 g/L tryptone, 5 g/L yeast extract, and 10g/L NaCl) at 37°C prior to infection in all studies [Citation22]. Mice were anesthetized and inoculated via transurethral catheterization with 107 colony forming units (CFUs) of UTI89 in phosphate-buffered saline (PBS; Sigma-Aldrich, D8537). Urine and bladders were collected at 24 hpi to measure bacterial titers. Bladders were collected before infection and at indicated time points (3–24 hpi for acute stage of infection and 14 dpi for persistent stage) following bacterial inoculation.
Quantification of QIRs
Mice were sacrificed at 14 dpi and the bladders were processed as described previously [Citation22]. Eight separate 5 μm step sections over a thickness of 300 μm were immunostained with antibodies against E. coli (United States Biological, E3500-26), LAMP1 (Developmental Studies Hybridoma Bank, 1D4B), and CDH1/E-cadherin (BD Transduction Laboratories, 610182). The total number of LAMP1-positive UPEC reservoirs were counted using a 63x oil objective on a Zeiss Apotome microscope and reported as number of QIRs/bladder.
Immunofluorescence and immunoblotting analysis
Mouse bladders were processed as described previously [Citation22]. The following primary antibodies were used: rabbit polyclonal antibodies to E. coli, mouse monoclonal antibody to CDH1/E-cadherin, rat monoclonal antibody to LAMP1 (Developmental Studies Hybridoma Bank, 1D4B), and mouse anti-UPK3/uroplakin III (Fitzgerald, 10R-U103A). Antigen-antibody complexes were detected with Alexa Fluor 488-, 594-, and 647-conjugated secondary antibodies (ThermoFisher, A32731, A-21209 and A32728). Images were obtained on a Zeiss Apotome microscope using 10-20x and 40-63x (oil) objectives.
Blots were probed with the following primary antibodies: rabbit polyclonal to ATG16L1 (Abgent, AP1817b), ATG7 (Sigma-Aldrich, A2856), LC3B (Novus Biologicals, NB600-1384), and CASP3/caspase3 (Cell Signaling Technology, 9664), and rabbit monoclonal to GAPDH (Cell Signaling Technology, 2118). Secondary antibodies used were goat anti-rabbit IgG-HRP (sc-2004) and donkey anti-goat IgG-HRP (sc-2020) from Santa Cruz Biotechnology. The protein bands on the developed film were quantified with ImageJ (NIH: http://rsb.info.nih.gov/ij/).
Transmission electron microscopy (TEM) and measurement of organelle density
Bladders were fixed with fixative containing 2% glutaraldehyde and 3% paraformaldehyde in 0.1 M sodium cacodylate. Samples were rinsed 3 times in sodium cacodylate buffer and post-fixed in 1% osmium tetroxide for 1 h, stained in 1 % uranyl acetate for a further hour, then rinsed, dehydrated, and dried using critical point drying. Samples were then gold-coated and viewed in an ULTRA 55 FEG (Zeiss). TEM images were taken from the superficial cells area at 2500X. The number of lysosomes and MVBs and the surface area of the superficial cells was counted using ImageJ. The densities of MVBs and lysosomes were reported as the number of MVBs and lysosomes per surface area examined (n = 200–300 TEM sections from 3 mice).
Quantitative PCR (qPCR)
Bladders from WT and T300A mice were collected before infection and 24 hpi. RNA was isolated from bladder tissue by using TRIzol (Invitrogen, 15596026) and treated with DNase I (Invitrogen, AM1907) to remove contaminating DNA. cDNAs were synthesized from 1 μg of total RNA using Superscript II reverse transcriptase (Invitrogen, 18064014). Gene expression was detected by real-time PCR using a CFX 96 Real-Time System (Bio-Rad, Hercules, CA, USA) and SYBR green PCR master mix (Bio-Rad, 172574). Each sample was measured in triplicate. Relative quantification was determined by using the comparative threshold cycle (CT) method with Rn18s/18S expression as a control.
Neutrophil and monocyte counts in urine
Urine samples were collected before infection and 24 hpi. HEMAVET® 950, Veterinary 5 part WBC Hematology System (Americas Drew Scientific Inc, Waterbury, CT, USA; Department of Comparative Medicine, WUSM) was used to determine neutrophil and monocyte counts.
Isolation, differentiation and challenge of bone marrow-derived macrophages (BMDMs)
Differentiation of BMDMs from WT and T300A mice was performed as described previously [Citation37]. Briefly, bone marrow was aseptically isolated and differentiated on non-tissue-culture-treated petri dishes for 7 d at 37°C with 5% ambient CO2 in DMEM (Gibco, 11330–032) with 15% FBS, 30% L929-conditioned medium (prepared as described previously [Citation56]), 1% Glutamax [Gibco, 35050–061], 1% Na-pyruvate, and 1% penicillin and streptomycin. On day 7 post-isolation, non-attached cells were aspirated, and the medium was changed to DMEM with 10% FBS, 1% Glutamax, and 1% Na-pyruvate. On day 8, cells were incubated with ice-cold DPBS (Gibco, 14190–136), removed with a cell scraper, counted, and plated at 5 × 105 cells/ml. On day 9, BMDMs were challenged with a multiplicity of infection of 0.1 of UTI89. BMDMs were incubated with UTI89 for 2 h, treated with medium containing 100 µg/ml gentamicin for 1 h, and then incubated with medium containing 10 µg/ml gentamicin for 21 h. Then, the supernatant was collected for ELISAs. DuoSet ELISA kits for IL1B (DY401), IL6 (DY406), and TNF/TNFα (R&D systems, DY 410) were used according to the manufacturer’s instructions.
Cell culture, bacterial infection, and gentamycin protection assays
Human bladder epithelial cell line 5637 (BECs; ATCC, HTB-9) were used for all experiments. BECs were seeded into 24-well plates and grown to confluency in RPMI 1640 medium (GIBCO, 11875-085) supplemented with 10% FBS, infected with UTI89 at a multiplicity of infection of 100 and incubated for 1 h at 37°C. Extracellular bacteria were then removed from the BECs by washing twice with PBS and incubating with medium containing 0.1 mg/mL gentamicin. At the indicated times, BECs were washed 3 times in PBS and lysed in 1 ml of PBS containing 0.1% Triton X-100 (Sigma, 9002-93-1). Lysates were plated on LB agar plates to determine numbers of surviving intracellular bacteria.
siRNA transfection
Human BECs were transfected using Trans-X2 (Mirus, MIR 6003) reagent with siRNA targeting ATG16L1 (IDs30071), ATG7 (IDs20651), ATG14 (ID22135), EPG5 (ID148797), RAB33B (ID120671), RAB27B (ID120374), RAB11A (ID120400) or non-targeting control siRNA (Life Technologies, 4390844).
RAB inhibitor studies
3-Pyridinyl ethylidene hydroxyl phosphonocarboxylate (3-PEHPC), an analog of the bisphosphonate drug risedronate was generated as previously described [Citation43,Citation44]. The 5637 cells were treated with 5 mM 3-PEHPC for 24 h before being infected with UPEC. The cells were collected at 0.5, 3 and 6 hpi for quantification of intracellular bacterial load using the gentamicin protection assay.
Tissue analysis of human bladder biopsy samples
Paraffin-embedded bladder biopsy samples from patients who underwent transurethral tissue resections were retrieved from the Pathology service of Barnes-Jewish Hospital, St. Louis, MO, USA. All samples were de-identified, and tissues with pathological evidence of malignancy were excluded as intact superficial urothelial cells were not evident. Tissues were sectioned and stained with hematoxylin & eosin and screened in a blinded manner for vesicular congestion by bright field differential interference contrast microscopy at 40X and 100X magnification. The presence/absence of vesicles was scored. Images were acquired with Olympus DP71 software.
rs2241880 genotyping
Genomic DNA was extracted from paraffin-embedded bladder sections using the QIAamp DNA FFPE Tissue Kit (Qiagen, 56404). Genotyping to detect the rs2241880 polymorphism in ATG16L1 was performed using the TaqMan 5´-exonuclease allelic discrimination assay, which contains sequence-specific forward and reverse primers to amplify the polymorphic sequences and 2 probes labeled with VIC and FAM dyes to detect both alleles of each polymorphism (Applied Biosystems, 4351379). PCR reactions were carried out with TaqMan universal PCR Master Mix (Applied Biosystems, 4304437) following the manufacturer’s instructions in a Step-One Plus Real-time PCR system.
Statistical analyses
To assess the significance of differences between mouse groups, unpaired t-test or non-parametric Mann-Whitney U test or two-way ANOVA with Bonferroni post-test was performed with Graph Prism software. P < 0.05 was considered significant.
Supplemental Material
Download Zip (6.3 MB)Acknowledgments
We thank Drs. Herbert Virgin (Washington University in St Louis School of Medicine) for Atg16l1fl/fl and Atg14fl/fl mice; Hong Zhang (Chinese Academy of Science) for epg5−/- mice; Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science) for Atg7fl/fl mice; Frank Ebetino (University of Rochester) for providing 3-PEHPC; Wandy Beatty for TEM assistance; Jason Mills, Deborah Frank, Brooke Liang for comments.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplementary data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Fullgrabe J, Klionsky DJ, Joseph B. The return of the nucleus: transcriptional and epigenetic control of autophagy. Nat Rev Mol Cell Biol. 2014 Jan;15(1):65–74. PubMed PMID: 24326622.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016 Jan 2;12(1):1–222. PubMed PMID: 26799652.
- Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011 Jan 20;469(7330):323–335. PubMed PMID: 21248839; PubMed Central PMCID: PMC3131688.
- Lassen KG, Xavier RJ. Mechanisms and function of autophagy in intestinal disease. Autophagy. 2017 Nov 13. PubMed PMID: 29130415. DOI:10.1080/15548627.2017.1389358
- Lassen KG, Xavier RJ. Genetic control of autophagy underlies pathogenesis of inflammatory bowel disease. Mucosal Immunol. 2017 May;10(3):589–597. PubMed PMID: 28327616.
- Zhao YG, Zhao H, Sun H, et al. Role of Epg5 in selective neurodegeneration and Vici syndrome. Autophagy. 2013 Aug;9(8):1258–1262. PubMed PMID: 23674064; PubMed Central PMCID: PMCPMC3748201.
- Lu Q, Yokoyama CC, Williams JW, et al. Homeostatic control of innate lung inflammation by Vici syndrome Gene Epg5 and additional autophagy genes promotes influenza pathogenesis. Cell Host Microbe. 2016 Jan 13;19(1):102–113. PubMed PMID: 26764600; PubMed Central PMCID: PMCPMC4714358.
- Franke A, McGovern DP, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010 Dec;42(12):1118–1125. PubMed PMID: 21102463; PubMed Central PMCID: PMCPMC3299551.
- Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007 Feb;39(2):207–211. PubMed PMID: 17200669.
- Fletcher K, Ulferts R, Jacquin E, et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. Embo J. 2018 Feb 15;37(4):e97840. PubMed PMID: 29317426; PubMed Central PMCID: PMCPMC5813257.
- Bauckman KA, Owusu-Boaitey N, Mysorekar IU. Selective autophagy: xenophagy. Methods. 2015 Mar;75:120–127. PubMed PMID: 25497060; PubMed Central PMCID: PMCPMC4355331.
- Boada-Romero E, Serramito-Gomez I, Sacristan MP, et al. The T300A Crohn’s disease risk polymorphism impairs function of the WD40 domain of ATG16L1. Nat Commun. 2016;7:11821. PubMed PMID: 27273576.
- Lassen KG, Kuballa P, Conway KL, et al. Atg16L1 T300A variant decreases selective autophagy resulting in altered cytokine signaling and decreased antibacterial defense. Proc Natl Acad Sci USA. 2014 May 12;111:7741–7746. PubMed PMID: 24821797.
- Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014 Feb 27;506(7489):456–462. PubMed PMID: 24553140.
- Fujita N, Saitoh T, Kageyama S, et al. Differential involvement of Atg16L1 in Crohn disease and canonical autophagy: analysis of the organization of the Atg16L1 complex in fibroblasts. J Biol Chem. 2009 Nov 20;284(47):32602–32609. PubMed PMID: 19783656; PubMed Central PMCID: PMC2781674.
- Kuballa P, Huett A, Rioux JD, et al. Impaired autophagy of an intracellular pathogen induced by a Crohn’s disease associated ATG16L1 variant. PloS one. 2008;3(10):e3391. PubMed PMID: 18852889; PubMed Central PMCID: PMC2566595.
- Messer JS, Murphy SF, Logsdon MF, et al. The Crohn’s disease: associated ATG16L1 variant and Salmonella invasion. BMJ open. 2013;3(6):e002790. PubMed PMID: 23794574; PubMed Central PMCID: PMC3686164.
- Foxman B. Urinary tract infection syndromes: occurrence, recurrence, bacteriology, risk factors, and disease burden. Infect Dis Clin North Am. 2014 Mar;28(1):1–13. PubMed PMID: 24484571.
- Mysorekar IU, Hultgren SJ. Mechanisms of uropathogenic Escherichia coli persistence and eradication from the urinary tract. Proc Natl Acad Sci USA. 2006 Sep 19;103(38):14170–14175. PubMed PMID: 16968784; PubMed Central PMCID: PMC1564066.
- Bauckman KA, Mysorekar IU. Ferritinophagy drives uropathogenic Escherichia coli persistence in bladder epithelial cells. Autophagy. 2016 Mar 22;12:850–863. PubMed PMID: 27002654.
- Wang C, Symington JW, Mysorekar IU. ATG16L1 and pathogenesis of urinary tract infections. Autophagy. 2012 Nov;8(11):1693–1694. PubMed PMID: 22874553; PubMed Central PMCID: PMCPMC3494604.
- Wang C, Mendonsa GR, Symington JW, et al. Atg16L1 deficiency confers protection from uropathogenic Escherichia coli infection in vivo. Proc Natl Acad Sci USA. 2012 Jul 3;109(27):11008–11013. PubMed PMID: 22715292; PubMed Central PMCID: PMCPMC3390880.
- Rodriguez-Boulan E, Kreitzer G, Musch A. Organization of vesicular trafficking in epithelia. Nat Rev Mol Cell Biol. 2005 Mar;6(3):233–247. PubMed PMID: 15738988.
- Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001 Feb;2(2):107–117. PubMed PMID: 11252952.
- Chen Y, Guo X, Deng FM, et al. Rab27b is associated with fusiform vesicles and may be involved in targeting uroplakins to urothelial apical membranes. Proc Natl Acad Sci USA. 2003 Nov 25;100(24):14012–14017. PubMed PMID: 14625374; PubMed Central PMCID: PMCPMC283537.
- Gallo LI, Dalghi MG, Clayton DR, et al. RAB27B requirement for stretch-induced exocytosis in bladder umbrella cells. Am J Physiol Cell Physiol. 2017 Nov 22. ajpcell 00218 2017. ajpcell 00218 2017. DOI:10.1152/ajpcell.00218.2017
- Wankel B, Ouyang J, Guo X, et al. Sequential and compartmentalized action of Rabs, SNAREs, and MAL in the apical delivery of fusiform vesicles in urothelial umbrella cells. Mol Biol Cell. 2016 May 15;27(10):1621–1634. PubMed PMID: 27009205; PubMed Central PMCID: PMCPMC4865319.
- Khandelwal P, Ruiz WG, Balestreire-Hawryluk E, et al. Rab11a-dependent exocytosis of discoidal/fusiform vesicles in bladder umbrella cells. Proc Natl Acad Sci USA. 2008 Oct 14;105(41):15773–15778. PubMed PMID: 18843107; PubMed Central PMCID: PMCPMC2572972.
- Miao Y, Bist P, Wu J, et al. Collaboration between distinct Rab small GTPase trafficking circuits mediates bacterial clearance from the bladder epithelium. Cell Host Microbe. 2017 Sep 13;22(3):330–342 e4. PubMed PMID: 28910634.
- Miao Y, Li G, Zhang X, et al. A TRP channel senses lysosome neutralization by pathogens to trigger their expulsion. Cell. 2015 Jun 4;161(6):1306–1319. PubMed PMID: 26027738; PubMed Central PMCID: PMCPMC4458218.
- Bishop BL, Duncan MJ, Song J, et al. Cyclic AMP-regulated exocytosis of Escherichia coli from infected bladder epithelial cells. Nat Med. 2007 May;13(5):625–630. PubMed PMID: 17417648.
- Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature. 2010 Jul 1;466(7302):68–76. PubMed PMID: 20562859; PubMed Central PMCID: PMCPMC2901998.
- Arrighi S. The urothelium: anatomy, review of the literature, perspectives for veterinary medicine. Ann Anat. 2015 Mar;198:73–82. PubMed PMID: 25533627.
- Hickling DR, Sun TT, Wu XR. Anatomy and physiology of the urinary tract: relation to host defense and microbial infection. Microbiol Spectr. 2015 Aug;3(4). PubMed PMID: 26350322; PubMed Central PMCID: PMCPMC4566164. DOI:10.1128/microbiolspec.UTI-0016-2012
- Gandhi D, Molotkov A, Batourina E, et al. Retinoid signaling in progenitors controls specification and regeneration of the urothelium. Dev Cell. 2013 Sep 16;26(5):469–482. PubMed PMID: 23993789; PubMed Central PMCID: PMCPMC4024836.
- Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008 Nov 13;456(7219):264–268. PubMed PMID: 18849965.
- Symington JW, Wang C, Twentyman J, et al. ATG16L1 deficiency in macrophages drives clearance of uropathogenic E. coli in an IL-1beta-dependent manner. Mucosal Immunol. 2015 Nov;8(6):1388–1399. PubMed PMID: 25669147; PubMed Central PMCID: PMCPMC4532666.
- Fujioka Y, Noda NN, Nakatogawa H, et al. Dimeric coiled-coil structure of Saccharomyces cerevisiae Atg16 and its functional significance in autophagy. J Biol Chem. 2010 Jan 8;285(2):1508–1515. PubMed PMID: 19889643; PubMed Central PMCID: PMCPMC2801276.
- Fujita N, Itoh T, Omori H, et al. The Atg16L complex specifies the site of LC3 lipidation for membrane biogenesis in autophagy. Mol Biol Cell. 2008 May;19(5):2092–2100. PubMed PMID: 18321988; PubMed Central PMCID: PMCPMC2366860.
- Fukuda M, Itoh T. Direct link between Atg protein and small GTPase Rab: Atg16L functions as a potential Rab33 effector in mammals. Autophagy. 2008 Aug;4(6):824–826. PubMed PMID: 18670194.
- Itoh T, Fujita N, Kanno E, et al. Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol Biol Cell. 2008 Jul;19(7):2916–2925. PubMed PMID: 18448665; PubMed Central PMCID: PMCPMC2441679.
- Baron RA, Tavare R, Figueiredo AC, et al. Phosphonocarboxylates inhibit the second geranylgeranyl addition by Rab geranylgeranyl transferase. J Biol Chem. 2009 Mar 13;284(11):6861–6868. PubMed PMID: 19074143; PubMed Central PMCID: PMCPMC2652301.
- Coxon FP, Ebetino FH, Mules EH, et al. Phosphonocarboxylate inhibitors of Rab geranylgeranyl transferase disrupt the prenylation and membrane localization of Rab proteins in osteoclasts in vitro and in vivo. Bone. 2005 Sep;37(3):349–358. PubMed PMID: 16006204.
- Tian X, Jin RU, Bredemeyer AJ, et al. RAB26 and RAB3D are direct transcriptional targets of MIST1 that regulate exocrine granule maturation. Mol Cell Biol. 2010 Mar;30(5):1269–1284. PubMed PMID: 20038531; PubMed Central PMCID: PMCPMC2820885.
- Cadwell K. Crohn’s disease susceptibility gene interactions, a NOD to the newcomer ATG16L1. Gastroenterology. 2010 Nov;139(5):1448–1450. PubMed PMID: 20875485.
- Guo X, Tu L, Gumper I, et al. Involvement of vps33a in the fusion of uroplakin-degrading multivesicular bodies with lysosomes. Traffic. 2009 Sep;10(9):1350–1361. PubMed PMID: 19566896; PubMed Central PMCID: PMCPMC4494113.
- Puri C, Vicinanza M, Ashkenazi A, et al. The RAB11A-positive compartment is a primary platform for autophagosome assembly mediated by WIPI2 recognition of PI3P-RAB11A. Dev Cell. 2018 Apr 9;45(1):114–131 e8. PubMed PMID: 29634932; PubMed Central PMCID: PMCPMC5896254.
- Bel S, Pendse M, Wang Y, et al. Paneth cells secrete lysozyme via secretory autophagy during bacterial infection of the intestine. Science. 2017 Sep 8;357(6355):1047–1052. PubMed PMID: 28751470; PubMed Central PMCID: PMCPMC5702267.
- Starr T, Child R, Wehrly TD, et al. Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host Microbe. 2012 Jan 19;11(1):33–45. PubMed PMID: 22264511; PubMed Central PMCID: PMC3266535.
- Hwang S, Maloney NS, Bruinsma MW, et al. Nondegradative role of Atg5-Atg12/Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe. 2012 Apr 19;11(4):397–409. PubMed PMID: 22520467; PubMed Central PMCID: PMC3348177.
- Park S, Buck MD, Desai C, et al. Autophagy genes enhance murine gammaherpesvirus 68 reactivation from latency by preventing virus-induced systemic inflammation. Cell Host Microbe. 2016 Jan 13;19(1):91–101. PubMed PMID: 26764599; PubMed Central PMCID: PMCPMC4714357.
- Kimmey JM, Huynh JP, Weiss LA, et al. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015 Dec 24;528(7583):565–569. PubMed PMID: 26649827; PubMed Central PMCID: PMCPMC4842313.
- Bunn HF. The triumph of good over evil: protection by the sickle gene against malaria. Blood. 2013 Jan 3;121(1):20–25. PubMed PMID: 23118217.
- Zhao H, Zhao YG, Wang X, et al. Mice deficient in Epg5 exhibit selective neuronal vulnerability to degeneration. J Cell Biol. 2013 Mar 18;200(6):731–741. PubMed PMID: 23479740; PubMed Central PMCID: PMCPMC3601354.
- Mulvey MA, Schilling JD, Hultgren SJ. Establishment of a persistent Escherichia coli reservoir during the acute phase of a bladder infection. Infection and Immunity. 2001 Jul;69(7):4572-9. doi: 10.1128/IAI.69.7.4572-4579.2001. PubMed PMID: 11402001; PubMed Central PMCID: PMC98534.
- Weischenfeldt J, Porse B. Bone Marrow-derived Macrophages (BMM): Isolation and applications. Csh Protoc. 2008 Dec 1;2008:pdbprot5080. doi:10.1101/pdb.prot5080. PubMed PMID: 21356739.