
ABSTRACT
Ketoconazole is a broad-spectrum antifungal agent, which has recently been characterized as a potential anticancer agent in several cancer types. However, the molecular mechanism underlying the anticancer effect of ketoconazole is not clearly defined. Our recent findings demonstrate that ketoconazole suppresses the growth of hepatocellular carcinoma (HCC) cells by exacerbating mitophagy in vitro and in vivo. Mechanistically, PINK1-PRKN-mediated mitophagy are led by ketoconazole-induced suppression of PTGS2 (prostaglandin-endoperoxide synthase 2), which in turn results in mitochondrial dysfunction and consequent apoptosis. These data link PTGS2 to mitophagy machinery and implicate ketoconazole as a potential therapeutic option for HCC treatment.
Ketoconazole is a broad-spectrum antifungal agent and is primarily used to treat fungal infections. The use of ketoconazole oral tablets for fungal infections has been limited due to potential risk of liver injury. However, growing evidence suggests that ketoconazole alone or in combination with other agents exhibits promising anticancer activity against multiple cancers including glioblastoma, prostate, breast, colon, and bladder cancers. Recently, we demonstrated that ketoconazole suppresses hepatocellular carcinoma (HCC) cell growth by eliciting apoptosis, with no obvious effect on human liver cells [Citation1]. The antitumor effect of ketoconazole in HCC was further confirmed in both a cell line-derived xenograft model and patient-derived xenograft model, supporting the idea of ketoconazole as a potential anticancer agent for HCC treatment.
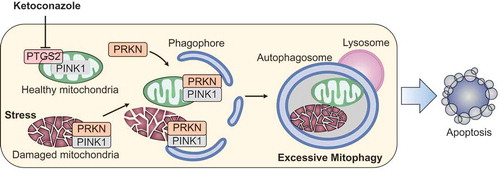
Figure 1. Schematic illustrating the mechanism by which ketoconazole exacerbates mitophagy to induce apoptosis in HCC. Ketoconazole-induced downregulation of PTGS2 activates PINK1-PRKN signaling, which initiates and exacerbates mitophagy of healthy mitochondria. The induction of excessive mitophagy leads to mitochondrial dysfunction and consequent apoptosis, resulting in growth suppression of HCC. HCC, hepatocellular carcinoma.
To ascertain the mechanism underlying the antitumor effect of ketoconazole, we examined whether ketoconazole induced mitochondrial dysfunction, which has been noted as a common consequence of azole antifungal drugs. Consistent with prior studies, massive mitochondrial fragmentation and loss are observed in HCC cells in response to ketoconazole treatment. Because mitophagy, a cargo-specific form of autophagy, has been recognized as a main form of mitochondrial quality control to remove dysfunctional mitochondria, we investigated whether ketoconazole induced mitophagy in HCC cells and confirmed the occurrence of mitophagy. Interestingly, inhibiting mitophagy suppresses ketoconazole-induced mitochondrial dysfunction and apoptotic cell death, which contradicts the paradigm that mitophagy maintains mitochondrial dynamics.
Mitophagy is critical for maintaining cellular homeostasis and is proposed to protect cells from accumulated mitochondrial damage. To date, the induction of mitophagy is mainly attributed to the PINK1-PRKN axis, both of which are involved in Parkinson disease. Mechanistically, PINK1 accumulates on the outer membrane of damaged mitochondria and recruits PRKN from the cytosol, which then ubiquitinates mitochondrial proteins to mark damaged mitochondria for disposal via the autophagy pathway. Indeed, we found that ketoconazole induces complete mitophagy in a PINK1-PRKN-dependent manner in HCC cells. However, a causal relationship between mitophagy and mitochondrial dysfunction is observed, indicating that mitophagy may determine cell fate in a context-dependent manner. Despite the initially recognized protective role for mitophagy in mitochondrial metabolism, several functionally distinct variants of mitophagy have been described recently; each may lead to cell death in an apoptosis-dependent or – independent manner. These previous studies confirm the lethal consequences of mitophagy, but the upstream mechanism leading to the variation of mitophagy, especially on the involved regulatory network in cancer cells, remains largely unknown. It is thus of interest to further investigate the mechanism by which mitophagy is induced and exacerbated in cancer cells.
In our study, ketoconazole-induced mitophagy in HCC cells is mediated by the downregulation of PTGS2/COX-2 (prostaglandin-endoperoxide synthase 2), a key enzyme catalyzing the conversion of arachidonic acid into prostaglandins. Enforced expression of PTGS2 prevents mitophagy-mediated mitochondrial dysfunction by impeding PINK1 accumulation and PRKN recruitment on mitochondrial membrane in ketoconazole-treated HCC cells, supporting the concept of PTGS2 as a negative regulator of mitophagy induction. These results, in conjunction with the causal role of ketoconazole-induced mitophagy in mitochondria loss, suggest that PTGS2-mediated mitophagy not only occurs in damaged mitochondria but also is incurred by healthy mitochondria. Together, these data revealed a novel role for PTGS2 in mitophagy induction, which may eventually result in mitochondria dysfunction via a vicious cascade including massive mitochondria loss and mitochondrial fragmentation in HCC cells. However, the molecular basis for PTGS2 interference with PINK1-PRKN-mediated mitophagy still requires further investigation.
Linking PTGS2 to mitophagy machinery is of particular interest considering the tissue-specific expression of PTGS2 and its localization to mitochondria in HCC cells. Because PTGS2 expression is inducible under pathological conditions, it is not detectable in human liver cells under normal circumstances. Notably, an association between ketoconazole efficacy and basal PTGS2 expression in multiple cell lines was convincingly demonstrated, which provides an explanation for the distinct response to ketoconazole between normal liver cells and HCC cells. As our data demonstrated that PTGS2-mediated mitophagy induction, rather than other potential effects of ketoconazole, contributes to HCC suppression, the basal PTGS2 level in HCC tissues may represent a potential indicator for identifying candidate patients suitable for ketoconazole treatment. Further studies with larger sample sizes will be needed to validate this clinical relevance.
Upregulated PTGS2 expression in cancer tissues correlates with aggressive phenotypes and poor prognosis. As such, the anticancer efficacy of PTGS2 inhibitors alone or in combination with other chemotherapeutic agents has been determined in multiple tumor models including HCC. In line with this evidence, we found that ketoconazole acts synergistically with sorafenib, a first-line chemotherapeutic agent for HCC, in the suppression of HCC growth in vitro and in vivo. Given the inhibitory effect of ketoconazole on PTGS2 expression, the exacerbated mitophagy mediated by PTGS2 inhibition may provide another explanation for the anticancer effect of PTGS2 inhibitors as well as their synergistic effects with other chemotherapeutic agents. However, the effect of PTGS2 inhibitors on mitophagy induction and the relevant consequences in different cancers still merits further investigation.
In summary, our study provides new evidence as to the mechanism by which ketoconazole reduces PTGS2 expression, exacerbates mitophagy, and induces mitochondrial dysfunction in HCC cells (). These data provide a novel role for PTGS2 in mitophagy machinery and implicate ketoconazole as a potential therapeutic agent for HCC treatment.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Reference
- Chen Y, Chen H-N, Wang K, et al. Ketoconazole exacerbates mitophagy to induce apoptosis by downregulating cyclooxygenase-2 in hepatocellular carcinoma. J Hepatol. 2018. DOI:10.1016/j.jhep.2018.09.022