
ABSTRACT
Receptor-driven selective macroautophagy/autophagy delivers ubiquitinated targets for autophagosomal clearance to maintain metabolic homeostasis and orchestrate reparative inflammatory responses. Deregulated autophagy is linked to tumor progression, but the exact mechanisms of selective autophagy in the spatiotemporal control of cell polarity signaling components during cancer metastasis remain ill-defined. Our recent study has demonstrated that TRIM59, an E3 ligase upregulated in metastatic breast cancer, is required for cancer cell survival and metastasis. Genetic depletion of TRIM59 suppresses cancer metastasis by promoting RNFT1-induced K63 polyubiquitination and SQSTM1-directed autophagic degradation of PDCD10, thereby boosting ROCK (Rho associated coiled-coil containing protein kinase)-induced actomyosin contractility and enhancing CDH1-mediated adhesion formation.
During metastasis, cancer cells adopt 3 major types of motility modes: (i) a mesenchymal type with elongated morphology directed by the RAC signals; (ii) a blebbing and amoeboid mode with small F-actin-rich protrusions regulated by RHO-ROCK signaling; and (iii) collective invasion of tumor cell clusters, which requires the spatiotemporal control of the turnover or stabilization of cell polarity signaling components. The biological functions of the tripartite motif (TRIM) family of E3 ligases in cancer mobility and metastasis remain largely an uncharted territory. We aim to bridge this gap by first analyzing the expression profiles of 68 TRIM genes using the Cancer Genome Atlas (TCGA) database and immunohistochemistry (IHC) staining on primary and metastatic human breast tumors. We detected augmented TRIM59 expression in metastatic breast cancer, a feature that is strongly correlated with advanced clinical stages and shortened patient survival [Citation1]. We, therefore, hypothesized a positive correlation between TRIM59 expression and breast cancer metastasis. To test this hypothesis, we first sought to modulate the expression level of TRIM59 in luciferase-expressing MCF7 and MDA-MB-231 xenografts, and monitor cancer cell growth and metastasis via in vivo imaging. We discovered that TRIM59 ablation suppresses tumor growth and cancer cell metastasis, which could be reversed by ectopic expression of TRIM59. TRIM59 overexpression promotes the survival of breast cancer cells, which may to some extent support the metastasis of cancer cells under adverse environmental stresses. Surprisingly, no significant changes in the TP53 and mitogen-activated protein kinase (MAPK) signaling pathways, 2 well-studied pathways in the regulation of tumor growth, are observed. This prompted us to explore unconventional molecular mechanisms underlying TRIM59-mediated tumor growth and metastasis.
Mesenchymal-to-amoeboid transition (MAT) is mainly controlled by RHO-ROCK-induced actomyosin contractility, as reflected in the elevated phosphorylation (p-) of MYL2/MLC2 (myosin light chain 2) and EZR/ezrin-RDX/radixin-MSN/moesin (p-ERM). Upon TRIM59 depletion, we found that cancer cells undergo MAT with strong focal adhesion formation (indicated by F-actin and CDH1 staining) and membrane blebbing. At the molecular level, we further validated a negative correlation between TRIM59 expression and p-ERM based on bioinformatic information mined from the Clinical Proteomic Tumor Analysis Consortium (CPTAC). EZR is required for ROCK-mediated CDH1-dependent focal adhesion formation. The concomitant upregulation of p-ERM and accumulation of CDH1 in TRIM59-depleted cells indicates that TRIM59 might inhibit MAT and cell adhesion, so as to curtail cancer metastasis by suppressing RHO-ROCK activation.
After confirming the phenotypic changes associated with TRIM59 depletion, we next moved on to identify direct targets of TRIM59 by yeast two-hybrid screening. PDCD10, the inactivation of which often drives the development of vascular lesions in the brain or cerebral cavernous malformation, stands out as the strongest hit. PDCD10 expression level was found to negatively correlate with RHO-ROCK-mediated actomyosin contractility and breast cancer patient survival. We reasoned that TRIM59 as a RING domain-containing E3 ligase might promote the ubiquitination and degradation of PDCD10. Unexpectedly, we found that TRIM59 stabilizes PDCD10 and further modulates the expression of PDCD10-associated downstream targets. Furthermore, PDCD10 overexpression or administration of a ROCK inhibitor, Y-27632, not only reverses TRIM59 loss-induced contractile phenotypes but also accelerates cancer cell growth, migration and invasion, suggesting a pro-oncogenic role of the TRIM59-PDCD10-ROCK-ERM axis. Our findings argue against the idea of using ROCK inhibitors to suppress tumor metastasis, which is still debated in the metastasis field.
Capitalizing on a high-throughput reverse phase protein array (RPPA) assay, we performed unbiased quantitative functional proteomics studies to unveil the differential expression or phosphorylation of additional key cancer-associated signaling proteins upon TRIM59 deletion. For instance, pro-oncogenic factors, such as MYC, IGF1R, and KDR/VEGFR2, are downregulated. By contrast, CDH1 and the tumor suppressor INPP4B are upregulated. Moreover, we revealed a close involvement of WNT signaling components (upregulation of CTNNB1 and AXIN2) during metastasis.
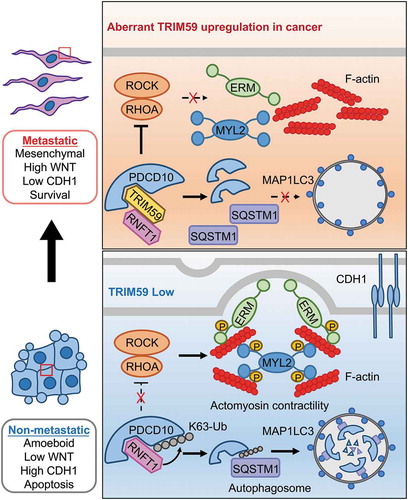
Protein degradation constantly involves the proteasome and autophagosomes. We discovered that PDCD10 destabilization arising from TRIM59 deletion can be restored by autophagy or autolysosome inhibitors, but not proteasome inhibitors. TRIM59 does not seem to affect the basal autophagosome formation as reflected by unaltered levels of the autophagosomal marker GFP-MAP1LC3. It has been established that different cargo receptors such as SQSTM1 can direct ubiquitinated proteins for autophagic degradation by interacting with autophagosome-localized MAP1LC3. We discovered that RNFT1-induced K63 polyubiquitination of PDCD10 acts as a ‘death’ signal to engage SQSTM1 for selective autophagic degradation. In this case, TRIM59 plays a non-proteolytic role in the blockade of the interaction between RNFT1 and PDCD10, causing cytosolic accumulation of PDCD10 to suppress RHO-ROCK-induced actomyosin contractility (). The exact molecular determinants governing the TRIM59-PDCD10-RNFT1 interplay are yet to be worked out in follow-up studies.
Figure 1. A working model depicting the TRIM59-PDCD10 interplay during cancer metastasis. TRIM59 is essential for breast cancer cell mesenchymal movement and cell survival by maintaining low cell adhesion and high WNT signaling. TRIM59 stabilizes PDCD10 by inhibiting RNFT1-induced K63 ubiquitination and subsequent SQSTM1-selective autophagic degradation. TRIM59 ablation removes the stabilizing effect on PDCD10 to cause autophagic degradation of PDCD10, and further activates the downstream RHOA-ROCK signaling to promote the phosphorylation of MYL2/MLC2 and ERM. These changes contribute to the amoeboid phenotype and focal adhesion formation, ultimately curtailing tumor formation and metastasis.
PDCD10 works with Ste20-like kinases (STKs) to regulate cell survival and cell junctions. Under oxidative stress, PDCD10 interacts with STKs to activate ERM. Moreover, Pdcd10-STKs negatively regulate Rhoa activation by activating Msn in endothelial cells derived from zebrafish. These findings hint that PDCD10-STKs promotes ERM activation with RHOA acting as the downstream but not upstream target of ERM. It remains to be clarified whether cancer cells have hijacked a pathway (such as the TRIM59-PDCD10-RHOA-ERM axis reported herein) other than the PDCD10-STK-ERM axis. Further genetic studies are also needed to resolve whether STKs participate in TRIM59-PDCD10 signaling to modulate the activities of RHOA-ROCK and ERM. Given the physiological importance of TRIM59 and PDCD10 in metastatic breast cancer and vascular integrity, the TRIM59-PDCD10 interplay promises to be a viable target for developing new therapies against cancer and cerebral cavernous malformation.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
Related Research Data
Reference
- Tan P, Ye Y, He L, et al. TRIM59 promotes breast cancer motility by suppressing p62-selective autophagic degradation of PDCD10. PLoS Biol. 2018;16(11):e3000051.