
ABSTRACT
N6-methyladenosine (m6A) mRNA modifications play critical roles in various biological processes. However, no study addresses the role of m6A in macroautophagy/autophagy. Here, we show that m6A modifications are increased in H/R-treated cardiomyocytes and ischemia/reperfusion (I/R)-treated mice heart. We found that METTL3 (methyltransferase like 3) is the primary factor involved in aberrant m6A modification. Silencing METTL3 enhances autophagic flux and inhibits apoptosis in H/R-treated cardiomyocytes. However, overexpression of METTL3 or inhibition of the RNA demethylase ALKBH5 has an opposite effect, suggesting that METTL3 is a negative regulator of autophagy. Mechanistically, METTL3 methylates TFEB, a master regulator of lysosomal biogenesis and autophagy genes, at two m6A residues in the 3ʹ-UTR, which promotes the association of the RNA-binding protein HNRNPD with TFEB pre-mRNA and subsequently decreases the expression levels of TFEB. Further experiments show that autophagic flux enhanced by METTL3 deficiency is TFEB dependent. In turn, TFEB regulates the expression levels of METTL3 and ALKBH5 in opposite directions: it induces ALKBH5 and inhibits METTL3. TFEB binds to the ALKBH5 promoter and activates its transcription. In contrast, inhibition of METTL3 by TFEB does not involve transcriptional repression but rather downregulation of mRNA stability, thereby establishing a negative feedback loop. Together, our work uncovers a critical link between METTL3-ALKBH5 and autophagy, providing insight into the functional importance of the reversible mRNA m6A methylation and its modulators in ischemic heart disease.
Abbreviations
ACTB, actin beta; ALKBH5, alkB homolog 5, RNA demethylase; ANXA5, annexin A5; ATG, autophagy-related; BafA, bafilomycin A1; CASP3, caspase 3; ELAVL1, ELAV like RNA binding protein 1; FTO, FTO, alpha-ketoglutarate dependent dioxygenase; GFP, green fluorescent protein; GST, glutathione S-transferase; HNRNPD, heterogeneous nuclear ribonucleoprotein D; H/R, hypoxia/reoxygenation; I/R, ischemia/reperfusion; LAD, left anterior descending; m6A, N6-methyladenosine; MEFs, mouse embryo fibroblasts; Mer, mutated estrogen receptor domains; METTL3, methyltransferase like 3; METTL14, methyltransferase like 14; mRFP, monomeric red fluorescent protein; MTORC1, mechanistic target of rapamycin kinase complex 1; NMVCs, neonatal mouse ventricular cardiomyocytes; PCNA, proliferating cell nuclear antigen; PE, phosphatidylethanolamine; PI, propidium iodide; PTMs, post-translational modifications; PVDF, polyvinylidenedifluoride; RIP, RNA-immunoprecipitation; siRNA, small interfering RNA; SQSTM1, sequestosome 1; TFEB, transcription factor EB; TUBA: tublin alpha; WTAP, WT1 associated protein; YTHDF, YTH N6-methyladenosine RNA binding protein
Introduction
Autophagy is an evolutionarily conserved degradation pathway which is tightly regulated by autophagy-related (ATG) proteins and transcription factors [Citation1]. Given the wide variety of the cytoplasmic targets of autophagy, its dysregulation is associated with many diseases such as neurodegenerative syndromes, cardiovascular disease, and cancer [Citation2,Citation3]. During autophagy, cytoplasmic materials are sequestered within the autophagosome (a double-membraned structure) and transported to the lysosome for digestion [Citation4]. Although there are significant insights into each of these steps, the mechanisms of autophagosome-lysosome fusion are least understood. TFEB (the transcription factor EB) is a master gene for lysosomal biogenesis, and its function during the autophagic process is well documented [Citation5]. The previous study indicated that post-translational modifications (PTMs) such as mitogen-activated protein kinase-dependent phosphorylation regulated the nuclear localization and activity of TFEB [Citation6]. However, it is still unknown whether modification at the nucleotide level can control the activity and expression of TFEB and which enzyme mediates this modification. Answers to these questions will provide the opportunity to develop new therapeutic strategies by controlling the activity of autophagy.
N6-methyladenosine (m6A) modification is the most prevalent internal modification in eukaryotic mRNAs [Citation7]. Usually, m6A modification is embedded within the consensus sequence 5′-RRACU-3′ (R = A or G; methylated adenosine residue is underscored) and happens mostly at the beginning of the 3′-UTR near the translation termination codon [Citation8]. Although transcriptome-wide m6A profiling shows that m6A modification is present in thousands of RNA transcripts with unique distribution patterns [Citation9], the function of the majority of RNA modifications found in mRNAs and mechanisms remain unknown. Recently, the roles of m6A modifications in biological processes have just begun to be appreciated and become of great interest. In mammalian cells, the modification of m6A is catalyzed by a methyltransferase complex that contains METTL3 (methyltransferase like 3), METTL14 (methyltransferase like 14), and WTAP (WT1 associated protein) [Citation10]. In turn, ALKBH5 (alkB homolog 5, RNA demethylase) and FTO (FTO, alpha-ketoglutarate dependent dioxygenase) function as two mammalian RNA demethylases to reverse the m6A modifications [Citation11]. Variant m6A levels exert diverse biological functions in mammals, such as embryonic stem cell maintenance and differentiation, transcription splicing, nuclear RNA export, protein translation control, cell fate determination, circadian rhythm modification, heat shock response, meiotic progression, and neuronal function [Citation12–Citation14]. Research indicates that m6A modification has been most strongly linked to increased mRNA degradation [Citation15]. Therefore, perturbations of the regulatory machinery of m6A modification might be associated with human diseases. However, the status of m6A modification and the underlying regulatory mechanisms in human heart diseases remain incompletely understood.
This study explored the role of m6A modification in the autophagy of cardiomyocytes treated with hypoxia/reoxygenation (H/R) and addressed the mechanisms by which m6A participates in the ischemic heart disease.
Results
H/R induces METTL3 upregulation in cardiomyocytes, which accounts for the aberrant m6A modification
To determine whether m6A modification was involved in the H/R-induced cardiomyocytes injury, H9c2 cells and neonatal mouse ventricular cardiomyocytes (NMVCs) were exposed to H/R. The levels of m6A modification were measured using m6A mRNA dot blot. It was observed that the levels of m6A modification were significantly increased in H9c2 cells and NMVCs following H/R (,). To assess whether the studies of m6A modification in H9c2 cells and NMVCs were relevant to mice in vivo, we measured the m6A levels in mouse hearts after I/R. Consistent with the results in the cardiomyocytes after H/R, the levels of myocardial m6A modification were elevated after I/R () and Fig. S2A). These results indicate that m6A levels are dynamically regulated when cardiomyocytes are treated by H/R or when mice are submitted to I/R.
Figure 1. H/R and I/R induces elevated levels of m6A RNA modification in cardiomyocytes and mice heart respectively. RNA dot blot analysis (upper panel) of m6A levels in H/R-treated H9c2 cells (a), NMVCs (b), and focal I/R-treated mice heart tissues (c). Methylene blue staining served as a loading control (middle panel). And quantification of RNA dots are shown (lower panel). (mean ± SD; n = 3; **P < 0.01 vs. untreated/sham). (d) The levels of m6A modification–associated genes in 10 pairs of heart tissues from infarct patients and normal heart tissues (mean ± SD; n = 10; ***P < 0.001 vs. normal heart). (e) The mRNA and protein levels of METTL3 and METTL14 in H/R-treated H9c2 cells, NMVCs, and focal I/R-treated mice heart tissues (mean ± SD; n = 6; **P < 0.01 and ns: no significant difference vs. untreated/sham). (f) H9c2 cells and NMVCs with or without METTL3 knockdown were exposed to H/R, or WT and mice lacking cardiac METTL3 (Mettl3-KO) (WT: Mettl3flox/flox MerCreMer+/+ treated with oil; TAM: Mettl3+/+ MerCreMer+/+ treated with tamoxifen; KO: Mettl3flox/flox MerCreMer+/+ treated with tamoxifen) were exposed to focal I/R. Total RNA was extracted, and m6A content was determined (mean ± SEM; n = 6; **P < 0.01 vs. shCtrl/Sramble/WT Mettl3 mice). All above the heart tissue samples were collected from the injured region of I/R-treated mice. P values were calculated with student’s t-test.
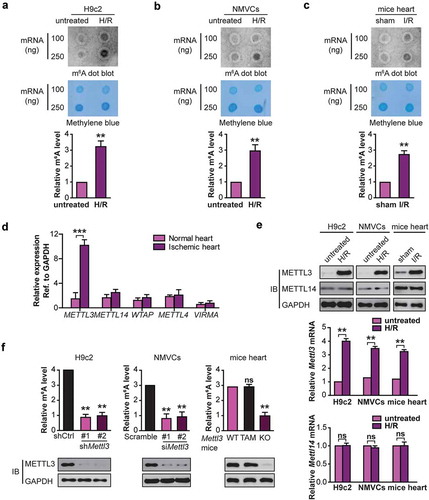
Given that the m6A modification is primarily catalyzed by an m6A methyltransferase complex containing METTL3, METTL14, WTAP, and VIRMA/KIAA1429 (vir like m6A methyltransferase associated) [Citation16], we hypothesized that the abnormal m6A modification in cardiomyocytes after H/R or I/R was caused by the dysregulation of the key m6A methyltransferase member. To investigate our assumption, we measured the expression levels of these genes in 10 pairs of heart tissues from infarct patients and normal tissues (Table S1). Interestingly, METTL3 was significantly upregulated in heart tissues from infarct patients compared with the control tissues ()). On the contrary, the expression of METTL14, WTAP, METTL4, and VIRMA in the heart tissues did not change significantly ()).
Consistent with the results in the infarct patients, the levels of METTL3 but not another methyltransferase METTL14 were significantly increased in H9c2 cells and NMVCs after H/R or in mouse myocardial tissues with I/R compared with their controls () and Fig. S2B). Our data suggest that METTL3 may be largely responsible for the elevated m6A modification of RNA in H/R-treated cardiomyocytes or I/R-treated mice heart.
To further confirm that increased m6A modification of RNA in H/R-treated cells was caused by the increased METTL3 expression, we used two different shRNAs or siRNA targeting Mettl3 (#1 and #2) to reduce the endogenous expression of METTL3 in H9c2 cells and NMVCs respectively. Compared with the control group, knockdown of METTL3 in H9c2 cells and NMVCs cells significantly blocked H/R-induced m6A levels in total RNA isolated from cardiomyocytes ()). To confirm the above findings in vivo, we generated cardiomyocyte-specific mettl3 knockout mice. The construction of this knockout strain and genotyping identification is shown in Fig. S1A and B. In this mouse model, Cre recombinase is fused to 2 mutated Esr (estrogen receptor) domains (Mer) [Citation17], and the cardiac-specific Myh6/α-myosin heavy chain promoter directs inducible Cre-mediated excision (MerCreMer) of the Mettl3 gene restricted in mouse cardiomyocyte. After 5 days of tamoxifen (TAM) treatment, mice homozygous for MerCreMer+/+ and Mettl3flox/flox alleles demonstrated excision of the floxed Mettl3 gene in the cardiac tissue only (Fig. S1C), accompanied by the reduced expression of METTL3 (Fig. S1D). Cardiac-specific Mettl3 deleted mice were viable and the baseline cardiac structure or functions was not affected. Body weight, heart/body weight (HW/BW), fractional shortenings (FS) and ejection fractions (EF) were significantly unchanged in sham mice after 5 days of TAM treatment relative to oil treatment (Fig. S1E and S1F). As expected, the similar data was observed when the cardiomyocyte-specific Mettl3 knockout mice were exposed to I/R () and Fig. S2C).
Taken together, the data in indicate that METTL3 expression mediates m6A methylation in total cellular RNA pools of H/R-dependent cardiomyocytes or I/R-treated mice heart tissues.
Upregulation of METTL3 impairs autophagic flux and enhances apoptosis in H/R-treated cardiomyocytes
Because we observed a change of METTL3 expression induced by H/R which is well-known to negatively regulate autophagy of cardiomyocyte [Citation18], we then assessed the effect of METTL3 on the autophagy marker LC3B in H/R-treated cardiomyocytes by immunoblotting. As shown in ), H/R induced LC3B-II in H9c2 cells with or without METTL3 knockdown. The increases in LC3B-II:LC3B-I ratio observed in our western blots can indicate an increase in lipidation or a defect in degradation via the lysosome [Citation19]. To distinguish between these two possibilities, we first used the lysosomal inhibitor bafilomycin A1 (BafA). We found that the BafA-induced accumulation of LC3B-II was significantly enhanced in Mettl3-silenced H9c2 cells compared with the control cells ()), indicative of an enhanced autophagic flux in Mettl3-depleted cells. Second, because SQSTM1 (sequestosome 1) was negatively correlated with autophagic flux [Citation20], we then assessed the effect of Mettl3 silencing on the SQSTM1 levels. We observed that H/R induced the increase in the levels of SQSTM1 and LC3B-II, suggesting that autophagic flux was impaired by METTL3 upregulation ()). And METTL3 knockdown significantly decreased the accumulation of SQSTM1 in the H/R-treated H9c2 cells, indicating to an increased autophagic flux ()). Similar data were observed when Mettl3 WT and KO mice were exposed to I/R () and Fig. S2D). Thirdly, tandem fluorescent mRFP-GFP-LC3 transfection was performed in NMVCs (Ad-LC3-NMVCs). The normal Ad-LC3-NMVCs had basal autophagy with few autolysosomes (red only puncta) and few autophagosomes (yellow puncta) ()). Once subjected to H/R, Ad-LC3-NMVCs without METTL3 knockdown had accumulated autophagosomes and few autolysosomes ()), suggesting that autophagosome clearance was inhibited and autophagic flux was blocked or impaired during myocardial H/R. However, in the siMettl3-treated group, Ad-LC3-NMVCs subjected to H/R had more autolysosomes and fewer autophagosomes than in the scramble-treated group ()), indicating that METTL3 knockdown induces the consumption of autophagosomes and enhanced autophagic flux. These data are consistent with the immunoblot analysis of LC3B and SQSTM1 protein levels.
Figure 2. Upregulation of METTL3 impairs autophagic flux and enhances apoptosis in H/R-treated cardiomyocytes. (a) Representative images of western blots in H/R-treated H9c2 cells along with or without BafA incubation. The LC3B-I, LC3B-II, and SQSTM1 levels were determined by western blotting. Quantification of LC3B-II protein levels are shown (mean ± SD; n = 3; *P < 0.05 and **P < 0.01). (b) Representative images of western blots of the LC3B-I, LC3B-II, and SQSTM1 of heart tissues of injured region from Mettl3 wild-type and knockout mouse with or without focal I/R. (WT: Mettl3flox/flox MerCreMer+/+ treated with oil; TAM: Mettl3+/+ MerCreMer+/+ treated with tamoxifen; KO: Mettl3flox/flox MerCreMer+/+ treated with tamoxifen). Quantification of SQSTM1 protein levels are shown (mean ± SD; n = 6; ***P < 0.001 and ns: no significant difference). (c) NMVCs were transfected with adenovirus harboring tandem fluorescent mRFP-GFP-LC3 (Ad-LC3-NMVCs) for 24 h and then subjected to different treatments. Representative pictures of immunofluorescent NMVCs expressing mRFP-GFP-LC3 are shown. GFP dots are green, and mRFP dots are red. Semi-quantitative analysis of autophagosomes (AP/autophagosome: yellow dots in merged images) and autolysosomes (AL/autolysosome: red-only dots in merged images). (mean ± SD; n ≥ 35; *P < 0.05 and ns: no significant difference). (d) NMVCs were infected with an adenovirus siRNA for Mettl3 (Ad-siMettl3) and then cultured under H/R conditions or normoxia. Apoptosis analysis was performed, and representative flow cytometry images are shown. The percentage of apoptosis was increased significantly after H/R whereas knockdown of METTL3 attenuated the effects of H/R on NMVCs. Quantifications of apoptotic cell percentage were shown (mean ± SD; n = 3; *P < 0.05 and **P < 0.01). P values were calculated with student’s t-test.
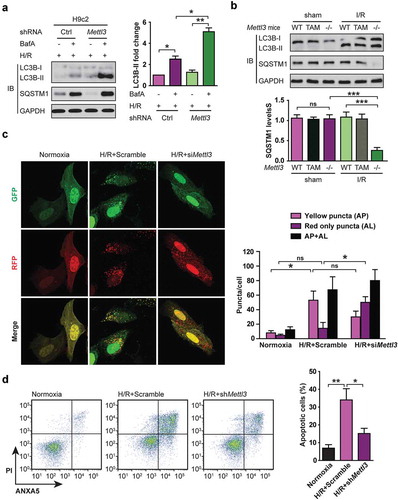
Since the inhibition of autophagy leads to apoptosis [Citation3], we then examined the effect of METTL3 silencing on the cardiomyocyte apoptosis. As seen in ), the amount of apoptosis in NMVCs under control conditions was unaffected by decreased expression of METTL3 (Ad-siMettl3, data not shown). However, METTL3 knockdown significantly reduced the percentage of apoptosis during H/R ()). Taken together, these results indicate that METTL3 upregulation inhibits cellular autophagic flux and promotes apoptosis in H/R-treated cardiomyocytes.
TFEB is required for shMettl3-enhanced autophagy flux in H/R-treated cardiomyocytes
Given the known role of TFEB in the regulation of a gene expression program of autophagy flux and lysosomal biogenesis [Citation6], we hypothesized that TFEB is required for shMettl3-enhanced autophagy flux and decreased apoptosis in H/R-treated cardiomyocytes. To test this hypothesis, we first prepared a stable knockdown of TFEB H9c2 cells with or without METTL3 deficiency and verified the efficiency of the silencing by western blotting. Then we submitted H9c2 cells to H/R treatment. As expected, knockdown of TFEB rescued shMettl3-mediated LC3B-II increase and the decrease of SQSTM1, cleaved CASP3 expression in H9c2 cells (,). Then we carried out the autophagic flux. The BafA-induced accumulation of LC3B-II and changes in the levels of SQSTM1 further confirmed the above findings () right panel). We also repeated the above experiments with NMVCs (,). Our findings show that autophagy induced by METTL3 deficiency is TFEB dependent.
Figure 3. TFEB is required for shMettl3-enhanced autophagy flux in H/R-treated cardiomyocytes. (a) H9c2 cells were transfected with the indicated treatment before cells were lysed and subjected to immunoblot analysis. (b) Immunoblot analysis of cleaved CASP3 expression in H9c2 cells with the indicated treatment. (c) NMVCs were transfected with indicated treatment before cells were lysed and subjected to immunoblot analysis. (d) Immunoblot analysis of cleaved CASP3 expression in NMVCs with the indicated treatment. (e) The Mettl3+/+ and mettl3−/- mice were subjected to focal I/R. Myocardial TFEB proteins and activities in these mice model were compared. Nuclear TFEB was isolated and indicated TFEB activity. Quantitative protein levels are shown on the right (mean ± SD; n = 6; **P < 0.01 and ***P < 0.001). (f) H9c2 cells with or without Mettl3 knockdown were transfected with or without siRNAs targeting Tfeb as indicated. After 72 h, these cells were fixed and then stained with LysoTracker Red and DAPI. Total lysosomal area per image was counted using an ImageJ script (mean ± SD; n = 3; **P < 0.01 and ***P < 0.001). P values were calculated with student’s t-test.
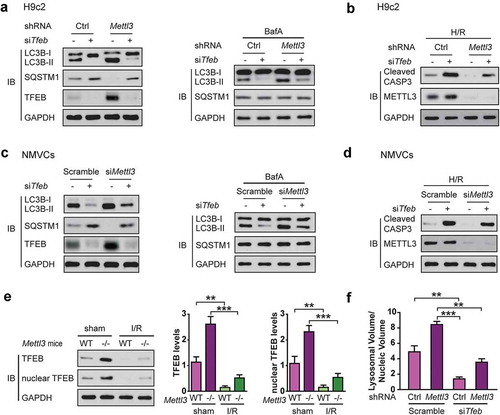
) shows a very dramatic increase in myocardial METTL3 protein levels in wild type mice after I/R. If the conclusions drawn from subsequent cell culture experiments hold true in animals, it is expected that myocardial TFEB protein level and activity would significantly decreased during I/R and that this decrease be significantly attenuated in mettl3−/- mice. To confirm this proposition, we examined myocardial TFEB protein levels and activities in the Mettl3+/+ and mettl3−/- mouse models subjected to I/R. As shown in ) and Fig. S2E, myocardial nuclear TFEB levels significantly decreased in wild type mice during I/R, which suggests a decline in its transcriptional activity [Citation6]. However, this decrease was significantly attenuated in mettl3−/- mice.
Since TFEB-mediated lysosomes are required for the completion of autophagy [Citation6], we hypothesized that TFEB might modulate autophagic flux by regulating the lysosome biogenesis. To test this, we again silenced Tfeb and Mettl3 using siRNA and shRNA respectively in H9c2 cells to quantitate the total lysosomal area with LysoTracker Red in H9c2 cells. As expected, METTL3 knockdown dramatically increased the number of lysosomes per cell while TFEB silencing reverted this increase in the lysosomal area ()), indicating that the shMettl3-mediated increase in lysosomal area was mediated through TFEB.
METTL3 decreases TFEB expression and inhibits its activity in H9c2 cells
To better understand how shMettl3 enhances autophagic flux and decreases apoptosis in H/R-treated cardiomyocytes via TFEB, we examined whether METTL3 regulates the expression and activity of TFEB. It was observed that shRNA-mediated knockdown of METTL3 in H9c2 cells significantly induced TFEB protein levels ()). Conversely, we observed a significant decrease in TFEB protein and mRNA levels in H9c2 cells in response to METTL3 overexpression ()). Since nuclear translocation of TFEB leads to increased expression of itself [Citation21], we asked whether METTL3 affects the nuclear translocation of TFEB. We transfected GFP-TFEB into H9c2 cells and observed that knockdown of METTL3 resulted in a significant increase in GFP-TFEB levels ()). Imaging analysis indicated that nuclear localization of GFP-TFEB was significantly increased upon knockdown of METTL3 as compared with the control cells ()). Western blotting assays of the nuclear and cytoplasmic fractions further confirmed that GFP-TFEB was enriched in the nucleus upon knockdown of METTL3 ()).
Figure 4. METTL3 knockdown stimulates TFEB expression and activity in H9c2 cells. (a) Western analysis of TFEB levels after knockdown of METTL3 in H9c2 cells. (b) H9c2 cells were transfected with a different dose of METTL3 expression vector and then subjected to immunoblot and qRT-PCR analysis. Quantification of relative Tfeb mRNA level from three independent experiments are shown on the right. (mean ± SD; n = 3; *P < 0.05, **P < 0.01 and ***P < 0.001, ns: no significant difference). (c) H9c2 cells were transfected with GFP-tagged TFEB, and shMettl3 or shcontrol and then the lysates were analyzed using an antibody to GFP. Quantification of GFP-TFEB levels after knockdown of METTL3 in H9c2 cells shown in bar graph (mean ± SD; n = 3; **P < 0.01 vs. shCtrl). (d) Representative image of the nuclear enrichment of GFP-TFEB after knockdown of METTL3 in H9c2 cells. And quantification of GFP-TFEB localization is shown in the bar graph (mean ± SD; n = 3; **P < 0.01 vs. shCtrl). (e) Immunoblot analysis indicates that silencing Mettl3 increases the nuclear levels of GFP-TFEB. (f) H9c2 cells were treated as indicated and subjected to qRT-PCR analysis to quantify the mRNAs levels of 6 established transcriptional targets of TFEB (mean ± SD; n = 3; *P < 0.05, **P < 0.01 and ***P < 0.001 vs. shCtrl). P values were calculated with student’s t-test.
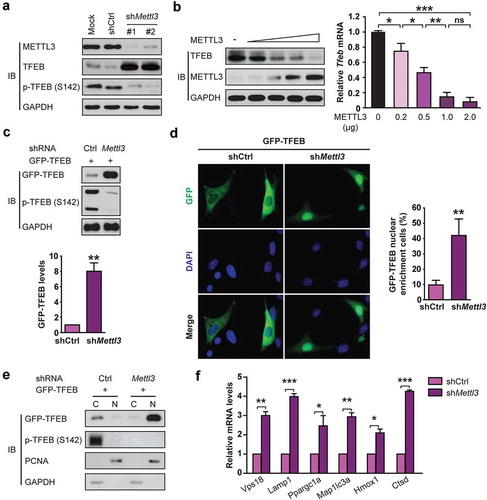
We then investigated whether METTL3 knockdown could increase the expression of endogenous TFEB target genes which have been well curated [Citation21]. As shown in ), silencing Mettl3 led to a significant increase in the mRNA levels of all 6 TFEB target genes (Vps18, Lamp1, Ppargc1a, Map1lc3a, Hmox1, and Ctsd) under H/R condition. Taken together, our data indicate that METTL3 regulates the expression and transcriptional activity of TFEB in H/R-treated cardiomyocytes.
METTL3 decreases Tfeb mRNA stability by catalyzing its m6A methylation in H/R-treated cardiomyocytes
We then set out to investigate the mechanism underlying how METTL3 regulates TFEB expression in H/R-treated cardiomyocytes. Given that METTL3 is a methyltransferase and selectively binds to m6A-containing RNA to regulate mRNA stability [Citation22], we wondered whether the altered TFEB expression could be a consequence of METTL3-mediated m6A methylation modification. To test this hypothesis, total cellular RNA was immunoprecipitated with an antibody that recognizes m6A, and the immunoprecipitated RNA was subjected to qRT-PCR to amplify the TFEB 3′-UTR. We found that H/R exposure led to the increased levels of Tfeb mRNA m6A methylation in H9c2 cells ()) and NMVCs ()). However, silencing Mettl3 led to the decreased levels of Tfeb m6A methylation under H/R conditions (,), suggesting that Tfeb mRNA may be subject to METTL3-dependent m6A methylation.
Figure 5. METTL3 decreases Tfeb mRNA stability by catalyzing its m6A methylation in H/R-treated cardiomyocytes. (a and b) H9c2 cells (a) and NMVCs (b) with or without METTL3 knockdown were exposed to H/R. m6A immunoprecipitation and qRT-PCR were performed to determine the change of Tfeb mRNA with m6A methylation. (c) Sequence analysis of TFEB 3′-UTR revealed two matches to the 5′-RRACU-3′ (methylated adenosine residue is underscored) m6A consensus sequence. (d and e) Luciferase assays were performed by transfecting H9c2 cells (d) and NMVCs (e) with a reporter plasmid containing luciferase coding sequences followed by the Tfeb-3′-UTR or empty vector only containing luciferase. The ratio indicating the luciferase activity in cells transfected with Tfeb 3′-UTR relative to empty vector was determined (relative luciferase activity: RLU). (f and g) Luciferase assays were performed in H9c2 cells (e) or NMVCs (f) transfected with WT or mutant (MUT) luciferase-Tfeb 3′-UTR reporter. The ratio of luciferase activity in cells transfected with Tfeb-3′-UTR relative to empty vector was determined. (mean ± SD; n = 3; *P < 0.05, **P < 0.01 and ***P < 0.001). P values were calculated with student’s t-test.
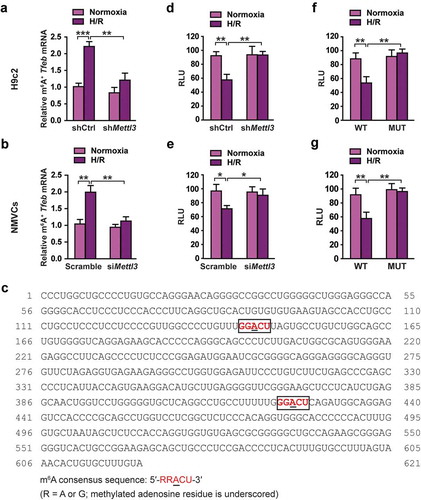
To further confirm that Tfeb mRNA is a direct target of METTL3-dependent m6A methylation, we searched sequence within the TFEB 3′-UTR. Sequence analysis revealed two matches to the 5′- RRACU-3′ (methylated adenosine residue is underscored) m6A consensus sequence within the TFEB 3′-UTR () and Fig. S3). Reporter plasmids that contained the entire Tfeb 3′-UTR were transiently transfected into H9c2 cells ()) and NMVCs cells ()), followed by incubation at H/R and measurement of luciferase activity. It was observed that H/R significantly decreased luciferase activity, whereas this effect was rescued in the H9c2 cells and NMVCs with METTL3 knockdown.
Next, the adenine residue embedded within the consensus sequence in the TFEB 3′-UTR was mutated (5′-GGACU-3′ to 5′-GGUCU-3′ in mRNA), and luciferase assays were performed comparing WT and mutant reporters. Mutation of the adenosine residue in both identified m6A consensus sequences resulted in constitutively increased luciferase activity in H9c2 cells ()) and NMVC ()), suggesting that the mutation prevented methylation and thereby increased the stability of the luciferase-Tfeb 3′-UTR fusion mRNA. These results suggest that the mutated adenosine residues were the targets for methylation. Together, the results presented in indicate that H/R-induced METTL3 expression increases TFEB mRNA methylation and decreases its stabilization and accumulation in cardiomyocytes. Supporting our findings, METTL3 knockdown might promote GFP-TFEB protein level via upregulating endogenous TFEB to affect the AMPK-MTOR pathway (Fig. S4).
METTL3 promotes HNRNPD association with Tfeb pre-mRNA in H/R-treated cardiomyocytes
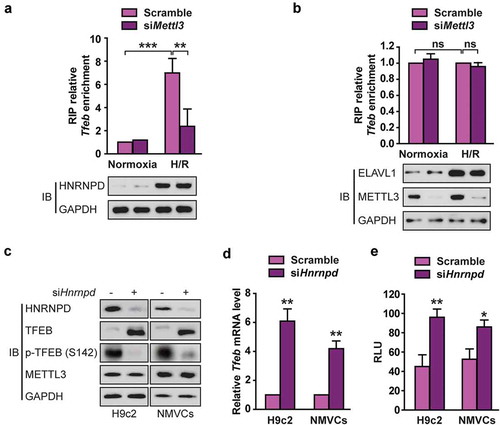
The RNA binding proteins heterogeneous nuclear ribonucleoprotein D0 (HNRNPD) and ELAVL1 (ELAV like RNA binding protein 1) levels are elevated in human heart failure patients [Citation23]. Importantly, HNRNPD and ELAVL1 reportedly regulates pre-mRNA expression and have been shown to bind to m6A-modified RNA [Citation24]. To further explain the negative correlation between m6A methylation modification and TFEB abundance, we investigated whether HNRNPD and ELAVL1 are involved in the METLL3-mediated stability of TFEB mRNA. We first tested whether HNRNPD or ELAVL1 interacted with TFEB nascent transcripts. RIP-qRT-PCR experiments revealed that both HNRNPD and ELAVL1 interacted with Tfeb pre-mRNA. H/R not only upregulated HNRNPD expression but also promoted its binding to Tfeb pre-mRNA and this interaction significantly decreased following METTL3 knockdown (,). In contrast, silencing Mettl3 did not affect the association of ELAVL1 with Tfeb pre-mRNA ()). Moreover, transient HNRNPD knockdown enhanced TFEB expression (,) and increased Tfeb 3′-UTR luciferase activity ()) in H9c2 cells. These data are consistent with the previous conclusion that the binding by HNRNPD usually lead to rapid degradation of the mRNA and reduced protein production [Citation25], suggesting that HNRNPD plays a vital role in the regulation of TFEB expression mediated by METTL3.
Figure 6. METTL3 promotes HNRNPD association with TFEB pre-mRNA in H/R-treated cardiomyocytes. (a and b) RIP analysis of the interaction of HNRNPD (A) or ELVAL1 (b) with Tfeb pre-mRNA using total cell lysates of H9c2 cells with or without METTL3 knockdown under normoxia or H/R condition. Enrichment of Tfeb pre-mRNA with an antibody against HNRNPD or ELVAL1 was measured by qRT-PCR and normalized to input. Western blotting was performed as indicated (mean ± SD; n = 3; **P < 0.01, ***P < 0.001 and ns: no significant difference). (c) Western blotting of TFEB expression in H9c2 cells and NMVCs with or without HNRNPD knockdown. (d) qRT-PCR analysis of Tfeb in H9c2 cells and NMVCs with or without HNRNPD knockdown (mean ± SD; n = 3; **P < 0.01 vs. Sramble in each cell line). (e) Relative activity of Tfeb 3ʹ-UTR firefly luciferase reporter in H9c2 cells and NMVCs with or without HNRNPD knockdown (relative luciferase activity: RLU). Mean ± SD; n = 3; *P < 0.05 and **P < 0.01 vs. Scramble in each cell line. P values were calculated with student’s t-test.
ALKBH5 not FTO reverses H/R-mediated m6A modification of Tfeb mRNA in cardiomyocytes
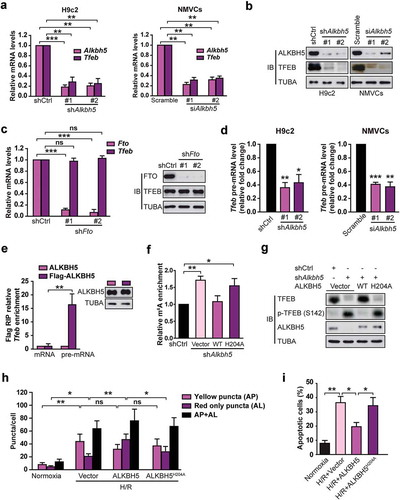
Modification of m6A is dynamic and reversible in mammalian cells [Citation26]. FTO and ALKBH5 have been reported to function as two mammalian RNA demethylases to reverse the m6A methylation [Citation11]. We then investigated whether FTO or ALKBH5 reverses the m6A modification of Tfeb in H/R-treated cardiomyocytes. We first measured the change in total mRNA levels of Tfeb upon ALKBH5 knockdown. Both H9c2 cells and NMVCs with ALKBH5 knockdown displayed approximately 75% lower expression of Tfeb mRNA ()). Consistent with the gene expression data, both transient and stable knockdown significantly decreased TFEB protein abundance ()). In contrast, stable FTO knockdown did not change TFEB expression, suggesting that TFEB may not be a direct target of FTO in H/R-treated cardiomyocytes ()). It was also observed that ALKBH5 knockdown strongly reduced the levels of Tfeb pre-mRNA ()). RNA RIP assay revealed that Tfeb pre-mRNA but not mRNA directly interacts with ALKBH5 ()). As expected, Tfeb pre-mRNA but not mRNA also directly interacts with METTL3 (Fig. S5). These data suggest that TFEB nascent transcript may be a substrate for ALKBH5 demethylase.
Figure 7. ALKBH5, but not FTO, reverses H/R-mediated m6A modification of TFEB mRNA in cardiomyocytes. (a) qRT-PCR analysis of Tfeb mRNA expression in H9c2 cells and NMVCs with or without ALKBH5 knockdown. Samples were normalized to Gapdh mRNA (mean ± SD; n = 3; **P < 0.01 and ***P < 0.001 vs. shCtrl). (b) Western blotting of TFEB in H9c2 cells and NMVCs with or without ALKBH5 knockdown (mean ± SD; n = 3; **P < 0.01 vs. Scramble). (c) qRT-PCR analysis of Tfeb mRNA expression and western blotting of TFEB in H9c2 cells treated with control or FTO shRNAs (mean ± SD; n = 3; ***P < 0.001 and ns: no significant difference vs. shCtrl). (d) qRT-PCR analysis of Tfeb pre-mRNA in H9c2 cells and NMVCs with or without ALKBH5 knockdown (mean ± SD; n = 3; *P < 0.05, **P < 0.01 ***P < 0.001 vs. shCtrl or Scramble). (e) RIP analysis of transcripts from the nuclear extracts of H9c2 cells expressing exogenous ALKBH5 with or without Flag tag. Enrichment of Tfeb mature and pre-mRNA with Flag was measured by qPCR and normalized to input. Western blotting of ALKBH5 showing equal expression of tagged or untagged proteins. (mean ± SD; n = 3; *P < 0.05). (f) Me-RIP-qPCR assay of m6A levels of Tfeb pre-mRNA in H9c2 cells with or without ALKBH5 knockdown and transfected with control, wild-type, or mutant ALKBH5 plasmid and treated with H/R (mean ± SD; n = 3; *P < 0.05 and **P < 0.01). (g) Western blotting of TFEB in H9c2 cells with or without ALKBH5 knockdown and transfected with control, wild type, or mutant ALKBH5 plasmid. (h) NMVCs were transfected with adenovirus harboring tandem fluorescent mRFP-GFP-LC3 for 24 h and then subjected to different treatments. And semi-quantitative analysis of autophagosomes/AP and autolysosomes/AL. (mean ± SD; n ≥ 35; *P < 0.05, **P < 0.01 and ns: no significant difference). (i) NMVCs were infected with an adenovirus ALKBH5 (Ad-ALKBH5) and then cultured under H/R conditions or normoxia. Apoptosis analysis was performed. The percentage of apoptosis was increased significantly after H/R whereas WT ALKBH5 but not its mutant significantly attenuated the effects of H/R on NMVCs. Quantifications of apoptotic cell percentage are shown (mean ± SD; n = 3; *P < 0.05 and **P < 0.01). P values were calculated with student’s t-test.
To examine this idea, we used methylated RNA immunoprecipitation (Me-RIP) from H/R-treated H9c2 cells combined with qRT-PCR to determine the levels of Tfeb m6A methylation following ALKBH5 knockdown. Our analysis confirmed that m6A methylation was readily detectable on Tfeb pre-mRNA under H/R condition ()). ALKBH5 knockdown further increased the m6A level of Tfeb pre-mRNA compared with the control ()). However, overexpression of the wild-type ALKBH5 but not the mutant H204A was able to restore the m6A level of Tfeb ()). The similar changes in TFEB protein expression were observed following ALKBH5 knockdown or reintroduction ()), indicating that ALKBH5 mainly affects TFEB expression through its demethylation activity.
Next, the effects of ALKBH5 on autophagy and apoptosis in H/R-treated cardiomyocytes were examined. As shown in ,i), the overexpression of WT ALKBH5 but not the mutant ALKBH5H204A enhanced autophagic flux and inhibited the apoptosis of H/R-treated cardiomyocytes.
TFEB induces ALKBH5 through direct binding and transcriptional activation of its promoter in cardiomyocytes
TFEB belongs to the MITF/TFE family of bHLH-leucine zipper transcription factors [Citation6]. We wondered whether TFEB is a novel regulator of ALKBH5 expression. We first made a luciferase reporter controlled by the human ALKBH5 promoter (−2230 bp upstream of the transcription start site, ALKBH5-Luc) and tested if overexpression of TFEB activates ALKBH5-Luc activity. We observed that TFEB increased ALKBH5-Luc activity in a dose-dependent manner ()). Moreover, qRT-PCR and immunoblot analysis of lysates from H9c2 cells indicated that overexpression of TFEB significantly increased endogenous Alkbh5 not Fto mRNA expression and protein content ()); TFEB knockdown had an opposite effect ()).
Figure 8. TFEB is identified as a novel activator of the human ALKBH5 promoter in cardiomyocytes. (a) Luciferase assays performed on cell extracts of HEK293T cells transfected with ALKBH5-Luc (−2230 bp) with increasing amounts of Flag-TFEB or empty vector. Luciferase activity was normalized to CMV-LacZ expression. GAPDH (glyceraldehyde 3-phosphate dehydrogenase) level acted as a loading control (RLU: relative luciferase activity) (mean ± SD; n = 3; *P < 0.05, **P < 0.01 and ***P < 0.001 vs. Vector). (b) H9c2 cells were transfected with indicated plasmids for 24 h. Immunoblotting with indicated antibodies and qRT-PCR were performed (mean ± SD; n = 3; *P < 0.05, **P < 0.01 and ns: no significant difference vs. Vector). (c) H9c2 cells were transfected with or without shRNA targeting TFEB (shTfeb, 100 nmol/L each) for 24 h. Immunoblotting with the indicated antibodies and qRT-PCR were performed (mean ± SD; n = 3; **P < 0.01, ***P < 0.001 and ns: no significant difference vs. shCtrl). (d) Schematic diagram of the human ALKBH5 promoter. Positions of conserved E-box motifs (CANNTG) in the ALKBH5 promoter relative to the transcription start are indicated. (e) HEK293T cells were transfected with a TFEB expression plasmid and the indicated ALKBH5 promoter constructs harboring E-box mutations. Luciferase assays were performed, and data are represented as mean ± SD (mean ± SD; n = 3; *P < 0.05, **P < 0.01 and ***P < 0.001 and ns: no significant difference). (f) ChIP assay was performed on chromatin from serum starvation-treated H9c2 cells using antibodies against TFEB. Primers flanking E-boxes 1, 2, 3, and 4 of the Alkbh5 promoter were used. (g) ChIP assay was performed on chromatin from H/R-treated and untreated NMVCs. Chromatin was immunoprecipitated with antibodies against TFEB. Antibodies against IgG were used as a control. (h) ChIP assay was performed on chromatin from doxorubicin- and DMSO-treated NMVCs. P values were calculated with student’s t-test.
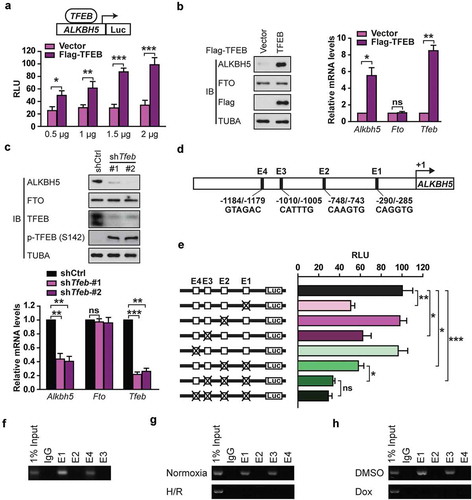
Because TFEB is well-known to bind to specific E-box motifs in the promoter of target genes [Citation5], we analyzed the sequence of the ALKBH5 promoter and found 4 E-box sites () and Fig. S6). To investigate the importance of these E-boxes for TFEB-induced ALKBH5 expression, site-directed mutagenesis was used to mutate these E-box motifs from CANNTG to ATNNTG in the ALKBH5-Luc construct ()). Mutation of E-box 1 and 3 was found to significantly decrease TFEB-induced ALKBH5 expression while mutation of E-box 2 and 4 had only minor effects ()), indicating that E-boxes 1 and 3 majorly mediate TFEB-induced ALKBH5 expression. ChIP experiments further confirmed direct binding of endogenous TFEB to E-boxes 1 and 3 of the endogenous Alkbh5 promoter in H9c2 cells with starvation treatment ()). Compared to the normoxia, H/R significantly decreased the binding of endogenous TFEB to the Alkbh5 promoter in NMVCs ()). Because doxorubicin is known to repress cellular TFEB activity and block NMVCs’ autophagic flux [Citation27], we tested if doxorubicin regulates TFEB binding to the Alkbh5 promoter in NMVCs. ChIP-PCR assays with doxorubicin–treated NMVCs showed that doxorubicin completely abolished TFEB binding to the E-boxes 1 and 3 in the endogenous Alkbh5 promoter ()).
Collectively, our data show that TFEB constitutively binds to conserved E-box elements in the Alkbh5 promoter in cardiomyocytes and that doxorubicin or H/R treatment strongly decreases the amount of TFEB bound to the ALKBH5 promoter. Thus, TFEB is a transcriptional activator of ALKBH5.
TFEB inhibits METTL3 expression by reducing the stability of its mRNA in cardiomyocytes
Increased METTL3 protein and mRNA expression upon TFEB knockdown indicate that, contrary to ALKBH5, METTL3 is under negative regulation by TFEB ()). Although most known TFEB targets are transcriptionally activated rather than inhibited [Citation5], we still proceeded to test the ability of TFEB to repress the human METTL3 promoter at the transcriptional level. As shown in ), overexpression of TFEB in H9c2 cells did not affect the basal transcriptional activity of a reporter containing a 1-kb fragment of the human METTL3 promoter harboring three E-box elements. We also found that neither transient nor stable knockdown of TFEB affected METTL3 promoter transcription ()).
Figure 9. TFEB inhibits METTL3 at the posttranscriptional level in cardiomyocytes. (a) H9c2 cells were transfected with or without shRNA targeting TFEB (shTfeb, 100 nmol/L each) for 24 h. Immunoblotting with the indicated antibodies and qRT-PCR were performed (mean ± SD; n = 3; **P < 0.01 vs. shCtrl). (b) TFEB overexpression does not affect the transcription of the human METTL3 promoter. H9c2 cells were transfected with 0.5 μg of the METTL3 promoter (−1.0 kb) along with 1 μg of indicated vectors. (mean ± SD; n = 3; ns: no significant difference vs. Vector). (c) Transient and stable knockdown of TFEB does not alter METTL3 transcription by using −1.0 kb METTL3 promoter luciferase reporters (mean ± SD; n = 3; ns: no significant difference vs. Scramble or shCtrl). (d) TFEB expression decreases Mettl3 mRNA stability. H9c2 cells transfected for 48 h with or without a shRNA against TFEB were incubated for the indicated periods with 10 μg of Act D before relative mRNA levels of Mettl3 and Actb were assessed by qRT-PCR (mean ± SD; n = 3; *P < 0.05, **P < 0.01 and ns: no significant difference vs. shCtrl Act D 6 h/24 h). (e) A proposed model for the critical link between METTL3, ALKBH5 and autophagy in H/R-treated cardiomyocytes. P values were calculated with student’s t-test.
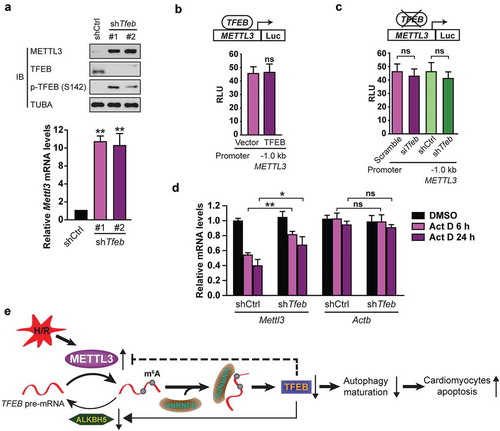
Although it cannot be ruled out that TFEB could transcriptionally repress METTL3 via binding to more upstream or downstream regulatory regions, we decided to explore the potential regulation of METTL3 by TFEB at an alternate level. We examined whether TFEB expression affects METTL3 mRNA stability. To this end, H9c2 cells transiently or stably knocked down for TFEB were treated with actinomycin D (Act D) to inhibit RNA elongation. Then Mettl3 mRNA levels were examined by qRT-PCR. It was observed that following treatment with Act D, Mettl3 mRNA was more stable in H9c2 cells with TFEB knockdown than in counterpart control cells ()). However, mRNA levels of the housekeeping gene Actb were not regulated by TFEB and unaffected by TFEB knockdown ()). Altogether, these results indicate that TFEB negatively controls METTL3 expression, at least in part, through regulation of its mRNA stability.
Discussion
Our results uncover an opposite role for METTL3 and ALKBH5 in regulating H/R-treated cardiomyocyte autophagy. This deduction is based on the following evidence. We first indicated that the level of m6A is increased in H/R-treated cardiomyocytes and I/R-treated mouse heart, and that METTL3, not METTL14, is the primary factor leading to aberrant m6A modification. Then we demonstrated that increased METTL3 inhibits autophagic flux and promotes apoptosis in H/R-treated cardiomyocytes, suggesting that METTL3 is a negative regulator of autophagy. Furthermore, we identified TFEB as a critical target gene downstream of METTL3. We found that METTL3 not only methylates TFEB at two m6A residues in the 3ʹ-UTR but also promotes the association of HNRNPD with TFEB pre-mRNA, thereby decreasing expression of TFEB mRNA. However, overexpression of TFEB or the RNA demethylase ALKBH5 reversed the effect of METTL3 on H/R-treated cardiomyocytes. Further experiments indicated that TFEB, in turn, regulates METTL3 and ALKBH5 in opposite directions: induces ALKBH5 and inhibits METTL3. TFEB binds to the ALKBH5 promoter and activates its transcription. In contrast, inhibition of METTL3 by TFEB does not involve transcriptional repression but rather a downregulation of mRNA stability. Based on the data presented here, for the first time, we address the regulation of TFEB activity by m6A modification and propose an important role of METTL3 and ALKBH5 in dictating the fate of H/R-treated cardiomyocytes. Hence, the discovery of this novel mechanism that controls the lysosomal-autophagic pathway suggests a new approach to ischemic diseases.
Despite the growing appreciation of the biological significance of METTL3-mediated m6A modification, the mechanisms on how m6A regulates gene expression remain poorly understood. Emerging models imply that the so-called reader proteins, such as the YTH domain-containing proteins (YTHDF1, 2, and 3), selectively bind m6A-modified RNAs [Citation28]. In the case of YTHDF2, binding to m6A-modified RNAs leads to their transport to processing bodies in the cell cytoplasm and subsequent degradation [Citation29]. Conversely, YTHDF1 has been implicated in the control of translation through an interaction with the eIF3 initiation factor [Citation28]. Recent studies report an important role of METTL3 in promoting translation of a specific subset of target mRNAs independent of its methyltransferase activity, binding-partners METTL14 and WTAP, or downstream m6A reader proteins including YTHDF1 and YTHDF2 [Citation30]. For example, METTL3 promotes EGFR and TAZ protein expression in a YTHDF1-independent manner [Citation30]. However, in our current report, we propose a novel model in which METTL3 methylates TFEB at two m6A residues in the 3ʹ-UTR, thereby promoting the association of RNA-binding protein HNRNPD with TFEB pre-mRNA and decreasing the mRNA level of TFEB. Therefore, our results provide an alternative model and independent pathway for the role of METTL3 in mRNA control. Supporting our findings, the previous study reported that m6A modification could alter RNA conformation to influence interaction with RNA-binding proteins [Citation31].
Notably, based on our data, METTL3 suppresses autophagy likely by destabilizing TFEB at the transcript level. Therefore, Mettl3 cannot directly target the GFP-TFEB plasmid. However, very surprisingly, we found that Mettl3 knockdown significantly enhanced GFP-TFEB protein level. We reasoned that Mettl3 knockdown might promote GFP-TFEB protein level via upregulating endogenous TFEB to affect the AMPK-MTOR pathway. Our reasons are as follows. First, TFEB has been shown to activate AMPK and inhibit mTOR activity [Citation32]. Secondly, Inhibition of mTOR activity has been reported to upregulate the GFP-TFEB protein level [Citation33]. Supporting this explanation, we do find that Mettl3 knockdown leads to increased p-AMPK-T172 and decreased p-RPS6KB1/S6K1-S389. However, without endogenous TFEB, no significant changes in GFP-TFEB, p-AMPK-T172, and p-RPS6KB1-S389 levels were observed even though Mettl3 was knocked down. Thus, our evidence indicates the effect of METTL3 knockdown on the counter-regulation of AMPK and MTOR phosphorylation, which depends on endogenous TFEB.
Although we reported METTL3 methylating TFEB at two m6A residues in the 3ʹ-UTR, two papers most recently reported that dynamic m6A modification in the 5′-UTR of certain mRNAs promotes stress-induced translation [Citation34], suggesting that METTL3 might display selective binding and translational regulation in a specific subset of mRNAs. Although there are multiple METTL3 binding sites located in 5′-UTR and CDS regions of mRNA, METTL3 does not regulate the translation of all mRNAs containing m6A peaks. Thus, it may be interesting to investigate the global effects of METTL3 on transcription and translation of other autophagy-related genes beyond TFEB and to determine the molecular determinants conferring mRNA specificity. It also may be interesting to examine whether autophagy, in turn, can regulate METTL3 or other METTL3-complex components. Future experiments will provide more insight into the selectivity of METTL3 in regulating autophagy. Considering our data demonstrating that METTL3 depletion enhances H/R-treated cardiomyocytes’ viability, the future efforts aimed at inhibiting this target could lead to the development of new therapies against ischemic diseases.
It is well-known that TFEB binds to selected E-box or E-box–like sequence (CANNTG) in the promoters of its targets [Citation6]. Unsurprisingly, TFEB activates ALKBH5 expression by direct binding to the E-box of its promoter. However, although we identified three optimal E-box elements in the first 1.0 kb of the human TFEB promoter, all of them failed to recruit TFEB, potentially suggesting that the binding of TFEB to its target genes might also be modulated by the nucleotides surrounding the core E-box or other coactivators such as EP300 (E1A binding protein p300) [Citation35]. Interestingly, the regulation of METTL3 by TFEB in cardiomyocytes occurs at the posttranscriptional level because knockdown of TFEB reduced the effect of Act D over Mettl3 mRNA, indicating that TFEB inhibits METTL3 expression at least partially by reducing the stability of its mRNA. Since several RNA-binding proteins are known to protect or degrade mRNA [Citation25], it remains to be elucidated whether the inhibition of METTL3 expression by TFEB involves RNA-binding proteins. Together, our findings suggest that TFEB may not only act as a transcriptional factor but also regulate gene expression through different mechanisms.
Finally, although we reported that treatment with 5 days of tamoxifen at 40mg/kg did not significantly affect the structure and function of cardiac myocyte from mettl3 null mice, the findings are in stark contrast to what has been noted in prior publication where depressed ejection performance was seen with a lower dose of 20 mg/kg [Citation17]. Another group also reported a similar observation wherein a dose of 40 mg/kg for 5 days was used, similar to the protocol in the current study [Citation36]. The reasons for the discrepancy with our findings are not clear. It is possible that the differences are because of the mice used. Based on our experiences, different mouse backgrounds might affect the toxicity of tamoxifen. In addition, the sensitivity of mice to tamoxifen from various vendors is significantly different. Supporting this possibility, one group treated 8-week-old male cardiomyocyte-specific lepr (leptin receptor) knockout mice (Leprflox/flox MerCreMer+/+) with tamoxifen (20 mg/kg intraperitoneal [ip]) daily for 7 days to induce cardiac-specific Lepr excision [Citation37]. They observed that the excision of the Lepr gene induced by tamoxifen does not affect the myocardial structure or contractile performance during a short (4 week) period of observation [Citation37]. Together, the toxicity of tamoxifen to the mouse heart might be context-dependent.
In summary, we have uncovered a novel link between autophagy and m6A modification ()). By expanding the mechanisms by which METTL3 and ALKBH5 regulate autophagy and apoptosis of H/R-treated cardiomyocytes, the results presented here reinforce their roles as potential therapeutic targets in ischemic diseases.
Materials and methods
Cell lines and culture conditions
H9c2 cardiomyocytes derived from rat myocardium and HEK293T were obtained (ATCC, CRL-1446, CRL-3216) and cultured in DMEM. Primary neonatal mouse ventricular cardiomyocytes (NMVCs) were isolated from mice with 1- to 3-day-old using a Pierce Primary Cardiomyocyte Isolation Kit (Thermo Fisher Scientific, 88281) according to the manufacturer’s instructions. Myocytes were seeded into each well of 6-well plates at a concentration of 2 × 106 cells per plate and cultured in DMEM with 5% fetal bovine serum (Sigma-Aldrich, F7524) and penicillin-streptomycin (100 μg/mL, Thermo Fisher Scientific, 15140163). After 24 h, the complete medium was replaced with fresh DMEM containing cardiomyocyte growth supplement at 37°C and 5% CO2. H9c2 cells and NMVCs were treated with 4 h of hypoxia and then followed by 3 h of reoxygenation as described previously [Citation3]. An incubator with O2 control, 5% CO2, and balanced with N2 (NBS Galaxy 48R) was used to culture the cells under hypoxic condition (1% O2). The medium was replaced daily. In a separate group of experiments, bafilomycin A1 (Sigma-Aldrich, SML1661) was added to the incubation medium (final concentration of 50 nM) at the time of reoxygenation to neutralize lysosome pH as well as to block lysosome-autophagosome fusion.
Chemical reagents and antibodies
Reagents used were as follows: Transfection was done with Oligofectamine (Invitrogen Life Technologies, 12252–011) as recommended by the manufacturer. The following antibodies were used: anti-m6A antibody (Synaptic Systems, 202003), METTL3 (Proteintech, 15073–1-AP), METTL14 (Sigma-Aldrich, HPA038002), anti-GAPDH (Bosterbio, 0411), anti-FLAG (Sigma-Aldrich, F3165), anti-SQSTM1 (Sigma-Aldrich, P0068), rabbit polyclonal anti-LC3B antibody (Sigma-Aldrich, L7543), TFEB (Bethyl Laboratories, A303-673A), cleaved CASP3/caspase-3 (Abcam, Ab2302), PCNA (Abcam, ab18197), GFP (Abcam, ab13970), HNRNPD (Abcam, ab61193), ELAVL1 (Abcam, ab54987), TFEB-phospho-Ser142 (MilliporeSigma, ABE1971), phospho-AMPK-Thr172 (Cell Signaling Technology, 2535S), phospho-RPS6KB/p70 S6 Kinase-Thr389 (Cell Signaling Technology, 9205), ALKBH5 (Abcam, ab69325), FTO (Abcam, ab92821), and TUBA (Abcam, ab6046).
Quantification of the m6A modification
Total RNA was isolated using TRIzol (Invitrogen, 15596018) according to the manufacturer’s instructions and treated with deoxyribonuclease I (Sigma-Aldrich, 04716728001). RNA quality was analyzed using a NanoDrop. The change of global m6A levels in mRNA was measured by EpiQuik m6A RNA Methylation Quantification Kit (colorimetric; Epigentek, P-9005–48) following the manufacturer’s protocol. Poly-A-purified RNA (200 ng) was used for each sample analysis. Briefly, 200 ng RNAs were coated on assay wells. Capture antibody solution and detection antibody solution were then added to assay wells separately in a suitable diluted concentration. The m6A levels colorimetrically quantified by reading the absorbance at a wavelength of 450 nm, and then calculations were performed based on the standard curve.
Apoptosis analysis
Cardiomyocytes were treated as indicated, harvested, washed with phosphate-buffered saline (PBS; Sigma-Aldrich, P4417). Cell death assay was performed using ANXA5 (BD, 556419). Briefly, cells were suspended in ANXA5 binding buffer (10 mM HEPES, pH 7.4, 2.5 mM CaCl2, 140 mM NaCl) and stained with ANXA5. Samples were analyzed on the FACSCantoTM II Flow Cytometer (BD Biosciences, USA). FlowJo V7 software (Tree Star Inc., USA) was used to calculate the percentage of cells positive for ANXA5 and propidium iodide (PI). Both ANXA5- and PI-negative staining represents viable cells, early apoptotic cells were positive for ANXA5 staining, both ANXA5 and PI-positive staining means late apoptosis, but necrotic cells were positive for PI staining.
Myocardial ischemia/reperfusion (I/R) mouse model
C57BL6/J mouse hearts were subjected to ischemia/reperfusion (I/R) in vivo as described previously [Citation3,Citation38]. This animal study was approved by the Institutional Animal Care Committee. Briefly, mice were anesthetized with pentobarbital sodium (50 mg/kg; Sigma-Aldrich, P3761) through intraperitoneal injection. Then the left anterior descending (LAD) coronary artery was ligated after performing a left thoracotomy using a 6–0 silk suture with a slip knot. To cause I/R injury, mice were subjected to 30 min of LAD ischemia followed by 60 min of reperfusion. The sham group was treated with a suture passed under the LAD or not. Finally, mice were euthanized with pentobarbital sodium (200 mg/kg) by an intraperitoneal injection, and the whole heart tissue was collected for further assays. The Langendorff-perfused isolated mouse heart model of I/R was performed as described previously. In brief, hearts were isolated from C57Bl/6J male mice and cannulated via the aorta and perfused with Krebs-Henseleit buffer (118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.25 mM CaCl2, 1.2 mM KH2PO4, pH 7.4, 25 mM NaHCO3, 11 mM glucose) in a retrograde fashion. The hearts were perfused to achieve stabilization for 20 min, and then paced at 420 beats/min to make baseline functional measurements. The global ischemia was achieved by closing an in-line stopcock to stop perfusion for 20 min, which was followed by 45 min of reperfusion by re-opening the stopcock. The whole heart tissue was collected for further assays. The tissue samples from ischemic area were used.
RNA immunoprecipitation
RNA immunoprecipitation (RIP) was performed as previously described with minor modifications [Citation39]. According to the manufacturer’s instructions, RNA immunoprecipitation was performed with the Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit (Millipore Sigma, 17–700). Indicated cells were transfected with the same amount of either Flag-tagged or untagged plasmid expressing the same gene (METTL3, or HNRNPD, or ELAVL1). Then all cells were subjected to the same treatment. Indicated cells’ nuclear pellet was resuspended and homogenized in 1 ml RIP buffer (150 mM KCl, 0.5 mM DTT, 25 mM Tris, pH 7.4, 0.5% NP40 [Thermo Fisher Scientific, 28324], 1 mM PMSF [Sigma-Aldrich, 10837091001] and protease inhibitor [Roche, 4693116001]). RNA enriched by Flag RIP of cells expressing untagged protein was served as negative control. Briefly, magnetic beads (Millipore Sigma, 17–700) coated with 5 μg of normal antibodies against mouse immunoglobulin G (Millipore Sigma, 12–371), HNRNPD or ELAVL1, or Flag were incubated with pre-frozen indicated cell lysates or nuclear extracts overnight at 4°C. Associated RNA-protein complexes were collected. After being washed 6 times, the samples were subjected to proteinase K (VWR, 97062–670) digestion and RNA extraction by TRIzol. The relative interaction between protein and RNA was determined by qPCR. And the data were normalized to input.
Immunoblotting
Indicated cells were lysed using a low-stringency lysis buffer (50 mM Tris-HCl, pH 7.5, 120 mM NaCl, 0.5 mM EDTA, 0.5% NP40 and 10% glycerol). The cell suspension was incubation and centrifuged at 14,000 × g for 10 min at 4°C. The protein supernatant was collected, and concentration was measured by BCA assay (Pierce, 23225). Approximately 0.5 mg of whole-cell extract was used for each immunoprecipitation. Indicated antibodies or control rabbit IgGs were used to carry out the immunoprecipitatio ns overnight. Immunocomplexes were collected with a mixture of protein A and protein G-Sepharose (Abcam, ab193262) and washed three times in washing buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 0.1% Triton X-100 [Sigma-Aldrich, X100-100ML], 5% glycerol [MilliporeSigma, G9012], 0.5% dithiothreitol). Immunocomplexes were eluted in loading buffer and loaded onto a 7.5% SDS-PAGE gel and then transferred to polyvinylidene difluoride membranes (Millipore Sigma, IPVH00010). TBST (0.1% Tween 20 [Sigma-Aldrich, P9416] plus TBS [50 mM Tris-Cl, pH 7.5, 150 mM NaCl]) buffer including 1% non-fat milk was used to block the membranes and then was incubated overnight at 4°C using the indicated antibodies. After being washed three times with TBST, membranes were incubated with an indicated concentration of HRP-conjugated secondary antibodies (Thermo Fisher Scientific, 32230, 32460) for 1 h in the blocking solution at room temperature. Then the membranes were washed with TBST five times. Protein signals were visualized using ECL solution (Millipore Sigma, WBKL S0500) and detected with an UVitec Alliance mini-chemiluminescence device (UVitec, U.K.). The intensity of the western blotting bands was quantified using NIH ImageJ software.
Luciferase reporter assay
The indicated cells were seeded in 12-well plates and transfected with the pMIR-REPORT luciferase vector (Thermo Fisher Scientific, AM5795) fused with or without the wild-type or mutated TFEB-3′-UTR or METTL3 or ALKBH5. The firefly luciferase and Renilla luciferase activities in each well were calculated by a dual-luciferase reporter assay system (Promega Corporation, E1910). The ratios between the 3′-UTR reporter of the indicated gene and Renilla control were determined 48 h after siRNA treatment. The relative luciferase activity was further normalized to that in cells transfected with the firefly luciferase vector control under the same treating conditions. The promoter activity of ALKBH5 and METTL3 was also normalized by co-transfection with the ACTB-Renilla luciferase reporter.
RNA m6A dot blots
Dot blots were performed as follows. Total RNA was isolated. The RNAs (100 and 250 ng respectively) were double diluted and spotted onto a nylon membrane (Sigma-Aldrich, GERPN1210B). Then the membranes were ultraviolet (UV) crosslinked and blocked in blocking buffer (5% milk in phosphate-buffered saline with 0.1% Tween 20) for 1 h. Rabbit anti-m6A antibody (Synaptic Systems, 202003) was diluted 1:1,000 and incubated with the membranes overnight (4°C). After washing twice with 0.1% phosphate-buffered saline-Tween 20, horseradish peroxidase conjugated anti-rabbit immunoglobulin G (Cell Signaling Technology, 7074S) was diluted 1:5,000 and incubated with the membranes for 1 h at room temperature. After extensive washing, membranes were detected with a 3,3′-diaminobenzidine peroxidase substrate kit (Yeasen Biotechnology, 36302ES01). The same amount of poly(A)+ RNAs were spotted on the membrane, stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2) for 2 h, and washed with ribonuclease-free water for 5 h.
Quantitative real-time PCR (qRT-PCR)
Total RNA from cells or tissues was extracted using Trizol (Invitrogen, 15596018) following the manufacturer’s instructions. The cDNA was generated using SuperScript III Reverse Transcriptase (Thermo Fisher Scientific, 18080093) for reverse transcription. qRT-PCR was performed using SYBR Green PCR Master Mix (Thermo Fisher Scientific, 4309155) on a 7500 Fast Real-time PCR System (Applied Biosystems, USA). ACTB, or GAPDH were used as internal controls for the normalization. For RNA stability assay, cells were plated in a poly-lysine coated 6-cm dish and incubated with actinomycin D (Santa Cruz Biotechnology, CAS 50–76-0) at 5 μg/ml for the indicated time. Total RNA was isolated for qRT-PCR analysis. All primers used in this study are as follows: METTL3-F 5′-CAAGCTGCACTTCAGACGAA-3′ and METTL3-R 5′-GCTTGGCGTGTGGTCTTT-3′, GAPDH-F 5′-AAATCCCATCACCATCTTCCAG-3′ and GAPDH-R 5′-AGGGGCCATCCACAGTCTTCT-3′, METTL14-F 5ʹ-AGAAACTTGCAGGGCTTCCT-3ʹ and METTL14-R 5ʹ-TCTTCTTCATATGGCAAATTTTCTT-3ʹ, METTL4-F 5ʹ- TATCCCTCTTGGTCTGTGGAG-3ʹ and METTL4-R 5ʹ- ACCTTCGTAGGGCTTTTTGTG −3ʹ, Tfeb-F 5ʹ- GGTGCAGTCCTACCTGGAGA-3ʹ and Tfeb-R 5ʹ- GTGGGCAGCAAACTTGTTCC-3ʹ, Alkbh5-F 5ʹ-CCCGAGGGCTTCGTCAACA-3ʹ and Alkbh5-R 5ʹ-CGACACCCGAATAGGCTTGA-3ʹ, Fto-F 5ʹ-TGGGTTCATCCTACAACGG-3ʹ and Fto-R 5ʹ-CCTCTTCAGGGCCTTCAC-3ʹ. WTAP-F 5ʹ-GGCGAAGTGTCGAATGCT-3ʹ and WTAP-R 5ʹ-CCAACTGCTGGCGTGTCT-3ʹ. Actb-F 5′-TTCTACAATGAGCTGCGTGTG-3′ and Actb-R 5′-GGGGTGTTGAAGGTCTCAAA-3′, Map1lc3a-F 5′-CGTCCTGGACAAGACCAAGT-3′ and Map1lc3a-R 5′-CTCGTCTTTCTCCTGCTCGT-3′; Lamp1-F 5′-GGACAACACGACGGTGACAAG-3′ and Lamp1-R 5′-GAACTTGCATTCATCCCGAACTGGA-3′; Ctsd-F 5′-GTGTAAACCGAGCCCTGATGACTT-3′ and Ctsd-R 5′-CAGCAGCAGGGAGGGGGCAGCACT-3′; Ppargc1a-F 5′-GGTATTAGGGTTGGAATTTAATGT-3′ and Ppargc1a-R 5′- ATCTCCAAATAAACTCAAACTCAATT-3′; Vps18-F 5′-TTGTCGTCTCCAGCAATCAG-3′ and Vps18-R 5′-CCTTGCCCAAGTCAATGC-3′. Hmox1-F 5′-GAGACGGCTTCAAGCTGGTGAT-3′ and Hmox1-R 5′-CCGTACCAGAAGGCCAGGTC-3′.
Plasmids, transfection and RNA knockdown
Human METTL3 cDNA was generated by PCR and cloned into NotI and BglII sites of the pFlag-CMV2 expression plasmid (provided by Dr. XH Hong from Xiamen University). The pcDNA3.1-DYK-ALKBH5, HNRNPD and ELAVL1 were generated by GenScript. Wild-type and a H204 mutant of ALKBH5 were cloned into pLVX (Clontech, 632164) for stable expression. TFEB–3× Flag plasmid was a gift from Dr. BB Wang (Rutgers University). Transfection of plasmids was performed using Lipofectamine 2000 (Thermo Fisher Scientific, 11668027) according to the manufacturer’s instructions. Transient knockdown of target genes by siRNA was performed with Lipofectamine RNAi Max (Thermo Fisher Scientific, 13778030). AAV-r-METTL3-shRNA (shAAV-294678) was purchased from VECTOR BIOLABS. The pLKO.1 lentiviral shRNA [Citation40] constructs targeting the human METTL3 (shMETTL3-1: RHS3979-201764032, shMETTL3-2: RHS3979-201764033) were purchased from Dharmacon. The previously confirmed siRNA targeting sequences used in this study are: siMettl3-#1: CTGCAAGTATGTTCACTATGA, siMettl3-#2: UCAUAGUGAACAUACUUGCAG. Lentiviral vectors expressing non-targeting pLKO.1 control shRNA (SCH002), and two shRNA constructs targeting Alkbh5-shRNA1 (TRCN0000064783) and -shRNA2 (TRCN0000064787) were obtained from Sigma-Aldrich. ShRNA for Hnrnpd was generated according to the pLKO.1 protocol from Addgene. To establish stable cell lines, H9c2 cells were transduced with the indicated plasmids. After 72 h of transduction, cells were selected with 2 μg/ml puromycin. For the ALKBH5 rescue experiment, shRNA targeting the 3′-UTR of Alkbh5 (shRNA1) was used for knockdown. ShRNAs against Fto (Sigma-Aldrich, SHCLND-NM_011936) and Tfeb (Sigma-Aldrich, SHCLNV-NM_011549) were used.
m6A MeRIP-qRT-PCR
MeRIP assay was adapted from a reported protocol [Citation41]. Briefly, mRNA was first purified from total RNA using PolyATtract mRNA Isolation Systems (Promega Corporation, Z5210). Then, mRNA was denatured to 70°C for 10 min, fragmented and immunoprecipitated with anti-m6A antibody in 1 ml buffer containing RNasin Plus RNase inhibitor (400 U; Promega Corporation, N2611), 50 mM Tris-HCl, pH 7.4, 750 mM NaCl, 0.5% (vol:vol) Igepal CA-630 (Sigma-Aldrich, I8896) for 2 h at 4°C. Dynabeads Protein G (Thermo Fisher Scientific, 10003D) were washed, added to the mixture and incubated for 2 h at 4°C with rotation. m6A RNA was eluted twice with 6.7 mM N6-methyladenosine 5ʹ-monophosphate sodium salt at 4°C for 1 h and precipitated with 5 μg glycogen, one-tenth volume of 3 M sodium acetate in 2.5 volumes of 100% ethanol at −80°C overnight. The m6A enrichment was determined by qRT-PCR analysis. Fragmented mRNA was directly incubated with m6A antibody containing buffer and treated similarly.
Chromatin immunoprecipitation (ChIP)
As described previously [Citation39], cells were fixed for 8 min at room temperature by adding 37% formaldehyde to a final concentration of 1%. Cross-linking was stopped by adding glycine to a final concentration of 0.125 M for 10 min at room temperature. The indicated cells were collected and washed three times with cold PBS containing proteinase inhibitors (Roche, 11836153001). After centrifugation at 4,000 x g for 5 min at 4°C, pellets were suspended in SDS ChIP lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1). Cell lysates were sonicated at 4°C to obtain DNA fragments of clarified chromatin. Following centrifugation at 13,000 x g for 10 min at 4°C, 50 μl of clarified chromatin was used as input control and 100 μl for immunoprecipitation. Clarified chromatin was diluted 1:10 with ChIP RIPA buffer containing 0.1% SDS (RPI Corp, 151–21-3). Chromatin was immunoprecipitated with antibodies against TFEB (Abcam, ab2636) and immunoglobulin G (Sigma-Aldrich, I5006), and anti-Flag or Ni2+-NTA agarose (Thermo Fisher Scientific, R90101) for 2 h at 4°C. Coupled protein-DNA complexes were washed twice in low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), twice in high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl) and twice in TE-buffer (10 mM Tris HCl, pH 8.8, 1 mM EDTA). Elution was performed by using 55 μl of TE-buffer with 1% SDS at 65°C and shaking at 1200 rpm for 10 min. Eluate (50 µl) or input were supplemented with 150 μl TE-buffer, 5 μl 10% SDS and 1 μl RNAse (Fermentas, EN0541) and incubated for 30 min at 37°C. Afterwards, proteins were digested with proteinase K (20 μg/μl; VWR, 97062–670) and lysates were incubated for 6 h at 37°C and 6 h at 65°C. DNA was isolated using DNA purification columns (Macherey-Nagel, 740609; NT buffer, Macherey-Nagel, 740595) as described by the manufacturer. Input DNA was eluted in 200 μl H2O and immunoprecipitated DNA was eluted in 50 μl H2O. Real-time PCRs were performed with QuantiFast SYBR green PCR master mix (Qiagen, 204054). Primers used were 5′-AGGCCAGGAGGTGAGACACA-3′ and 5′-CTTCTGGCCTTGGCTCCTCC-3′ for ALKBH5 E-box 1, 5′-GCAGGGGAACTTTATCCTGGG-3′ and 5′-ACAAAAAGGCGTAGCCGTGC-3′ for ALKBH5 E-box 2, 5′-AGAGATGGGGTTGCCATGTTGG-3′ and 5′-GGGGCCTCACGCCTGTAATC-3′ for ALKBH5 E-box 3, 5′-TCGCGGGTTTCAGAAGTTTCCT-3′ and 5′-CGAGCAGCGAGGACCCCTAA-3′ for ALKBH5 E-box 4. PCR products are ~110 bp. The ChIP assay was performed in triplicate and four times.
Immunofluorescence microscopy
Briefly, the indicated cells were fixed with 4% paraformaldehyde in PBS for 10 min. Cells were permeabilized with 0.25% Triton X-100 in PBS for 10 min and stained with indicated antibody at 4°C overnight. Nuclei were stained with DAPI for 1 min. The fluorescence images were acquired using a confocal laser-scanning microscope.
Lysosomal biogenesis quantification assay
Briefly, H9c2 cells were transfected as indicated. Then these cells were seeded on coverslips in charcoal-stripped media for 72 h before treatment. Next, 1 μl/ml of LysoTracker® Red (Life Technologies, L7528) was added to each well and incubate for 30 min at 37°C. Then the cells were fixed, stained with DAPI, and mounted onto slides for imaging. At least 10 randomly selected images per coverslip were taken. The NIH ImageJ software was used to analyze the images. Each coverslip was repeated in triplicate.
Cardiomyocyte-specific Mettl3 knock-out mouse
All animal use conformed to the Guide for the Institutional Animal Care, and Use Committee at the University Hospital approved this project. Gene targeting strategy was utilized to insert loxP sites into mouse Mettl3 gene introns 2 and 3. Cre-mediated recombination would cause deletion of exon 3 and result in inactivation of Mettl3 gene. To specifically disrupt Mettl3 gene in the cardiomyocyte, Mettl3flox/flox mice were subsequently bred with mice homozygous for the Myh6 promoter-driven MerCreMer transgene (Jackson Laboratories, USA, B6129SF1/J background, 005650). Subsequent inbreeding generated double homozygous (+/+) mice for the MerCreMer transgene and the Mettl3 floxed allele. To induce cardiac-specific METTL3 excision, 8-week-old male Mettl3flox/flox MerCreMer+/+ transgenic mice were treated with tamoxifen (tam; 40 mg/kg intraperitoneal [ip]) daily for 5 days, whereas vehicle (oil)-treated control mice received an equivalent volume of 98% peanut oil:2% ethanol. One week after oil or tam treatment, mice were subjected to open thoracotomy, and a suture was put around the left anterior descending coronary artery and left loose or tightly tied.
Patients
This study needs myocardial specimens from patients with ischemic heart and controls for RT-PCR experiment. The research contents and research programs were reviewed and approved by the Ethics Committee of the affiliated Hospital of Guilin Medical University. IRB Number is GLMU1A201705202. In the course of implementation, the Ethics Committee will supervise in accordance with the relevant national laws and regulations. Tissue samples used for this study were procured in accordance under an approved university IRB protocol, which uses written informed consent or a waiver of consent for discarded tissues. The university IRB complies with all regulatory requirements related to the protection of human research participants, and complies with the Guidelines of the International Conference on Harmonization (ICH). No HIPAA information was provided with any of the samples used in this study. Heart tissue samples were collected from 10 patients who died within 7 h of myocardial infarction and from 10 age- and sex-matched control cases. Heart tissue samples were obtained from forensic autopsies performed at the University Hospital between 2010 and 2014. The tissue samples from ischemic area were used. Control cases were selected by the following criteria: no pathological features of myocarditis, myocardial infarction, cardiomyopathy, congenital heart abnormalities, or coronary atherosclerosis. Our project was approved by the Ethics Committee of Medicine at University.
Statistics
Data are presented as mean ± SEM or mean ± SD. Differences between two groups or multiple groups were analyzed by Student’s t test and ANOVA, respectively. p values were calculated with student’s t-test using GraphPad Prism software (Version 5.0). P values <0.05 were considered significant, plus **P < 0.01 and ***P < 0.001. ns means no statistically significant.
Supplemental Material
Download Zip (6.2 MB)Acknowledgments
We thank Dr. Michelle Chadwick, Louis Osorio, Yanira Gonzalez from Rutgers University for critical reading and editing of the manuscript.
The authors declare that they have no competing financial interests.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material:
Supplementary material for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12(1):1–222.
- Yang ZJ, Chee CE, Huang S, et al. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10(9):1533–1541.
- Song H, Pu J, Wang L, et al. ATG16L1 phosphorylation is oppositely regulated by CSNK2/casein kinase 2 and PPP1/protein phosphatase 1 which determines the fate of cardiomyocytes during hypoxia/reoxygenation. Autophagy. 2015;11(8):1308–1325.
- Santarelli R, Granato M, Pentassuglia G, et al. KSHV reduces autophagy in THP-1 cells and in differentiating monocytes by decreasing CAST/calpastatin and ATG5 expression. Autophagy. 2016;12(12):2311–2325.
- Pastore N, Brady OA, Diab HI, et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy. 2016;12(8):1240–1258.
- Zhao E, Czaja MJ. Transcription factor EB: A central regulator of both the autophagosome and lysosome. Hepatology. 2012;55(5):1632–1634.
- Parashar NC, Parashar G, Nayyar H, et al. N 6-adenine DNA methylation demystified in eukaryotic genome: from biology to pathology. Biochimie. 2018;144:56–62.
- Brocard M, Ruggieri A, Locker N. m6A RNA methylation, a new hallmark in virus-host interactions. J Gen Virol. 2017;98(9):2207–2214.
- Patil DP, Pickering BF, Jaffrey SR. Reading m 6 A in the transcriptome: m 6 A-binding proteins. Trends Cell Biol. 2018;28(2):113–127.
- Wang X, Huang J, Zou T, et al. Human m6A writers: two subunits, 2 roles. RNA Biol. 2017;14(3):300–304.
- Meyer K, Jaffrey SR. Rethinking m6A readers, writers, and erasers. Annu Rev Cell Dev Biol. 2017;33:319–342.
- Roignant J-Y, Soller M. m 6 A in mRNA: an ancient mechanism for fine-tuning gene expression. Trends Genet. 2017;33(6):380–390.
- Guo M, Liu X, Zheng X, et al. m6A RNA modification determines cell fate by regulating mRNA degradation. Cell Reprogram. 2017;19(4):225–231.
- Wei W, Ji X, Guo X, et al. Regulatory role of N6‐methyladenosine (m6A) methylation in RNA processing and human diseases. J Cell Biochem. 2017;118(9):2534–2543.
- Niu Y, Zhao X, Wu Y-S, et al. N 6-methyl-adenosine (m 6 A) in RNA: an old modification with a novel epigenetic function. Genomics Proteomics Bioinformatics. 2013;11(1):8–17.
- Schwartz S, Mumbach MR, Jovanovic M, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8(1):284–296.
- Lexow J, Poggioli T, Sarathchandra P, et al. Cardiac fibrosis in mice expressing an inducible myocardial-specific Cre driver. Dis Model Mech. 2013;6(6):1470–1476.
- Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8(4):445–544.
- Hale CM, Cheng Q, Ortuno D, et al. Identification of modulators of autophagic flux in an image-based high content siRNA screen. Autophagy. 2016;12(4):713–726.
- Jeong K, Kwon HY, Jeong MS, et al. CNOT2 promotes degradation of p62/SQSTM1 as a negative regulator in ATG5 dependent autophagy. Oncotarget. 2017;8(28):46034.
- Chua JP, Reddy SL, Merry DE, et al. Transcriptional activation of TFEB/ZKSCAN3 target genes underlies enhanced autophagy in spinobulbar muscular atrophy. Hum Mol Genet. 2013;23(5):1376–1386.
- Kudou K, Komatsu T, Nogami J, et al. The requirement of Mettl3-promoted MyoD mRNA maintenance in proliferative myoblasts for skeletal muscle differentiation. Open Biol. 2017;7(9):170119.
- Misquitta CM, Iyer VR, Werstiuk ES, et al. The role of 3′-untranslated region (3′-UTR) mediated mRNA stability in cardiovascular pathophysiology. Mol Cell Biochem. 2001;224(1–2):53–67.
- Zhang S, Zhao BS, Zhou A, et al. m 6 A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem-like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell. 2017;31(4):591–606. e6.
- Gratacós FM, Brewer G. The role of AUF1 in regulated mRNA decay. Wiley Interdiscip Rev RNA. 2010;1(3):457–473.
- Wang X, He C. Dynamic RNA modifications in posttranscriptional regulation. Mol Cell. 2014;56(1):5–12.
- Bartlett JJ, Trivedi PC, Yeung P, et al. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem J. 2016;473(21):3769–3789.
- Wang X, Zhao BS, Roundtree IA, et al. N 6-methyladenosine modulates messenger RNA translation efficiency. Cell. 2015;161(6):1388–1399.
- Fu Y, Dominissini D, Rechavi G, et al. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet. 2014;15(5):293–306.
- Lin S, Choe J, Du P, et al. The m 6 A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell. 2016;62(3):335–345.
- Liu N, Zhou KI, Parisien M, et al. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucleic Acids Res. 2017;45(10):6051–6063.
- Fan Y, Lu H, Liang W, et al. Endothelial TFEB (Transcription Factor EB) positively regulates postischemic angiogenesis. Circ Res. 2018;122(7):945–957.
- Ugolino J, Ji YJ, Conchina K, et al. Loss of C9orf72 enhances autophagic activity via deregulated mTOR and TFEB signaling. PLoS Genet. 2016;12(11):e1006443.
- Zhou J, Wan J, Gao X, et al. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature. 2015;526(7574):591–594.
- Val P, Lefrançois-Martinez A-M, Veyssière G, et al. SF-1 a key player in the development and differentiation of steroidogenic tissues. Nucl Recept. 2003;1(1):8.
- Hall ME, Smith G, Hall JE, et al. Systolic dysfunction in cardiac-specific ligand-inducible MerCreMer transgenic mice. Am J Physiol Heart Circ Physiol. 2011;301(1):H253–H260.
- McGaffin KR, Witham WG, Yester KA, et al. Cardiac-specific leptin receptor deletion exacerbates ischaemic heart failure in mice. Cardiovasc Res. 2010;89(1):60–71.
- Pu J, Yuan A, Shan P, et al. Cardiomyocyte-expressed farnesoid-X-receptor is a novel apoptosis mediator and contributes to myocardial ischaemia/reperfusion injury. Eur Heart J. 2012;34(24):1834–1845.
- Hong X, Song R, Song H, et al. PTEN antagonises Tcl1/hnRNPK-mediated G6PD pre-mRNA splicing which contributes to hepatocarcinogenesis. Gut. 2014;63(10):1635–16347.
- Moffat J, Grueneberg DA, Yang X, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124(6):1283–1298.
- Dominissini D, Moshitch-Moshkovitz S, Salmon-Divon M, et al. Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nat Protoc. 2013;8(1):176–189.