
ABSTRACT
Several studies have shown that dysfunction of macroautophagy/autophagy is associated with many human diseases, including neurodegenerative disease and cancer. To explore the molecular mechanisms of autophagy, we performed a cell-based functional screening with SH-SY5Y cells stably expressing GFP-LC3, using an siRNA library and identified TMED10 (transmembrane p24 trafficking protein 10), previously known as the γ-secretase-modulating protein, as a novel regulator of autophagy. Further investigations revealed that depletion of TMED10 induced the activation of autophagy. Interestingly, protein-protein interaction assays showed that TMED10 directly binds to ATG4B (autophagy related gene 4B cysteine peptidase), and the interaction is diminished under autophagy activation conditions such as rapamycin treatment and serum deprivation. In addition, inhibition of TMED10 significantly enhanced the proteolytic activity of ATG4B for LC3 cleavage. Importantly, the expression of TMED10 in AD (Alzheimer disease) patients was considerably decreased, and downregulation of TMED10 increased amyloid-β (Aβ) production. Treatment with Aβ increased ATG4B proteolytic activity as well as dissociation of TMED10 and ATG4B. Taken together, our results suggest that the AD-associated protein TMED10 negatively regulates autophagy by inhibiting ATG4B activity.Abbreviations: Aβ: amyloid-β; AD: Alzheimer disease; ATG: autophagy related; BECN1: beclin 1; BiFC: bimolecular fluorescence complementation; CD: cytosolic domain; GFP: green fluorescent protein; GLUC: Gaussia luciferase; IP: immunoprecipitation; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; LD: luminal domain; PD: Parkinson disease; ROS: reactive oxygen species; siRNA: small interfering RNA; SNP: single-nucleotide polymorphisms; TD: transmembrane domain; TMED10: transmembrane p24 trafficking protein 10; VC: C terminus of Venus fluorescent protein; VN: N terminus of Venus fluorescent protein.
Introduction
Autophagy is a highly conserved, lysosome-dependent degradation process involved in the removal of cytoplasmic components, and maintenance of cellular homeostasis during stress [Citation1,Citation2]. Several studies have shown that defects in autophagy are linked to neurodegenerative diseases, such as Alzheimer disease (AD) and Parkinson disease (PD), which are characterized by the accumulation of toxic protein aggregates [Citation3,Citation4]. Since autophagy has been implicated in the pathogenesis of many diseases, its modulation might provide therapeutic strategies for combating autophagy-associated diseases.
Autophagy is regulated by multiple signaling pathways, and requires proteins encoded by the autophagy-related (ATG) genes [Citation5,Citation6]. Autophagy is initiated with the formation of a phagophore mediated by ULK1 complex, and two ubiquitin-like systems mediate the elongation and maturation of autophagosomes [Citation7]. MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) is cleaved by the cysteine protease ATG4B, leading to the generation of LC3-I [Citation8]. The ATG12–ATG5 complex assists the conjugation of the LC3-I to phosphatidylethanolamine (PE), forming lipidated LC3-II on phagophore membranes [Citation9]. The resulting autophagosomes then fuse with lysosomes to degrade engulfed contents [Citation6]. As LC3-II remains associated with the autophagosomes until degradation, green fluorescent protein (GFP)-fused LC3 is considered an effective marker protein for monitoring the activation of autophagy in cells [Citation10]. Recently, genetic and epigenetic mechanisms for the regulation of autophagy-related proteins have been reported [Citation11,Citation12]. Several transcription factors, such as NR1H4/FXR (nuclear receptor subfamily 1 group H member 4) and TFEB (transcription factor EB), coordinately regulate the expression of ATG genes [Citation11]. Posttranslational modifications, including phosphorylation of ATG proteins, are also involved in the regulation of autophagy [Citation13]. Previously, several groups have performed genome-wide screening to identify novel autophagy regulators [Citation14,Citation15]. To date, several hundred proteins have been suggested to be involved in autophagy regulation. More than 40 specific ATG genes have been identified in yeast, of which 16 have mammalian homologs [Citation16]; however, the molecular mechanisms of autophagy regulation are still not fully understood. To identify novel autophagy regulators, we performed an image-based high-content screening using an siRNA library. Using this approach, we identified TMED10 (transmembrane p24 trafficking protein 10) as a novel autophagy regulator.
TMED10/TMP21, is a member of the p24 cargo receptor family that is involved in vesicular protein trafficking in the early secretory pathway [Citation17,Citation18]. TMED10 is transcriptionally regulated by NFAT, and its degradation is mediated by the ubiquitin-proteasome pathway [Citation19,Citation20]. TMED10 is expressed at high levels in the heart, lung, liver, adrenal gland, and kidney [Citation21]. Interestingly, TMED10 is a member of the γ-secretase complex and decreased in Alzheimer disease (AD) patients [Citation22]. TMED10 negatively regulates the production of the amyloid-β (Aβ) peptides, Aβ40 and Aβ42, in AD models [Citation22]. However, the molecular mechanisms underlying the role of TMED10 in autophagy are largely unknown. In this study, we found that TMED10 suppression increases autophagy activation and TMED10 binds to ATG4B. However, the interaction was diminished during autophagy activation and the reduction in TMED10 enhanced ATG4B protease activity. In addition, we found that TMED10 decreased in AD patient brain tissues induces Aβ production, which promotes autophagy by activating ATG4B.
Results
Downregulation of TMED10 activates autophagy in SH-SY5Y cells
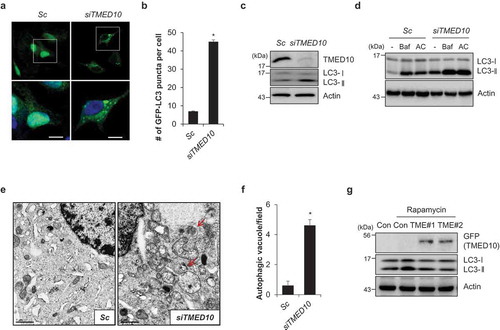
Although abnormal regulation of autophagy has been implicated in various pathophysiological conditions, the precise molecular mechanisms are not fully understood. To elucidate the mechanisms of autophagy, we developed a cell-based functional screening system using GFP-LC3, a widely used maker for monitoring autophagy, in SH-SY5Y neuroblastoma cells (SY5Y/GFP-LC3). Then, we performed screening using a siRNA library consisting of approximately 2,000 genes, including AD-related proteins, mitochondrial proteins, and the human kinome. From this screen, we identified TMED10 as a novel, potent regulator of autophagy. We confirmed the screening results by knockdown of TMED10 in SY5Y/GFP-LC3 cells. In accordance with the screening results, downregulation of TMED10 induced the formation of punctate structures by the GFP-LC3 proteins (). In addition, knockdown of TMED10 induced the accumulation of LC3-II (). The levels of LC3-II were higher in the cells co-treated with the lysosome fusion inhibitors, bafilomycin A1 or ammonium chloride than in the control cells (), suggesting that the knockdown of TMED10 increases autophagy flux in SH-SY5Y cells. Furthermore, an electron microscopic analysis showed that downregulation of TMED10 increased the accumulation of autophagosomes, confirming our previous observations (). To evaluate the effects of TMED10 on autophagy, we generated a TMED10-overexpressing SH-SY5Y cell line, and treated the cells with rapamycin to induce the conversion of LC3-I to LC3-II. Reciprocally, overexpression of TMED10 slightly suppressed the rapamycin-induced conversion of LC3-I to LC3-II, compared to that in the control SH-SY5Y cells (). As ATG5 plays a critical role in the formation of autophagosomes, we further investigated its effect on TMED10-mediated autophagy inhibition. Consistent with previous reports, knockdown of ATG5 suppressed autophagy induced by suppression of TMED10 (Figure S1). Taken together, these results suggest that downregulation of TMED10 activates autophagy by canonical pathway.
Figure 1. Downregulation of TMED10 induces autophagy activation in SH-SY5Y cells. (A and B) SH-SY5Y cells stably expressing GFP-LC3 (SY5Y/GFP-LC3) were transiently transfected with either scrambled negative control siRNA (Sc) or TMED10 siRNA (siTMED10). The cells were stained with DRAQ5 dye and imaged (A), and counted using a fluorescence microscopy (B). Data are presented as mean ± SEM (n = 3, * P < 0.05, SEM of # of GFP puncta structure per cell with approximately 100 cells). (C) SH-SY5Y cells were transiently transfected with TMED10 siRNA (#1 and #2). After 3 d, the cells were analyzed by western blotting with the indicated antibodies. (D) SH-SY5Y cells were transfected with Sc or siTMED10. After 2 d, the cells were further treated with bafilomycin A1 (Baf, 10 nM) or ammonium chloride (AC, NH4Cl 10 mM) for 24 h, and analyzed by western blotting (upper panel); the ratio of LC3-II:LC3-I was determined by densitometer analysis. (E and F) SH-SY5Y cells, transfected with Sc or siTMED10, were analyzed using electron microscopy. Red arrows indicate autophagosomes (E) and the number of autophagosomes in the field was counted (F). Scale bar: 1 μm. (G) SH-SY5Y cells stably expressing pEGFP (Cont) or pEGFP-TMED10 (TME #1, #2) were treated with rapamycin (5 μM) for 3 h, and analyzed by western blotting using anti-GFP and anti-LC3 antibodies. Actin was used as an internal loading control.
TMED10 interacts with ATG4B in SH-SY5Y cells
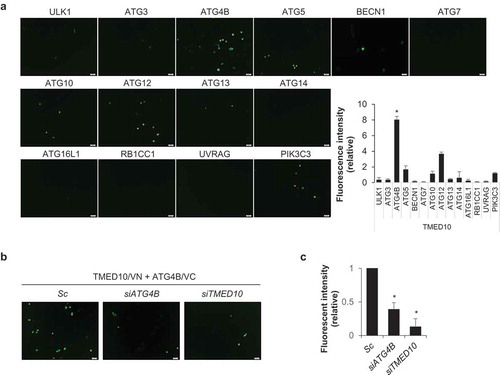
Because downregulation of TMED10 induced autophagy, we investigated its effect on the levels of other autophagy-regulating proteins, including ULK1, ATG3, ATG4B, BECN1/Beclin 1, ATG13, ATG16L1, ATG101, RB1CC1, ZBTB24, UVRAG, and WIPI2. The expression levels of these proteins were not notably changed in the TMED10-knockdown cells, except for a slight enhancement in the levels of ULK1 as compared to that in the control cells (Figure S2). To delineate the molecular mechanisms of TMED10-mediated autophagy inhibition, we employed a bimolecular fluorescence complementation (BiFC) assay, which enables the visualization of protein-protein interactions in living cells [Citation23]. In this assay, two proteins of interest, A and B, are fused to the N- and C-terminal non-fluorescent fragments of the Venus fluorescent protein (VN173 and VC155), respectively. If A and B interact, the VN173 and VC155 fragments are brought into proximity and produce a fluorescent signal. For the BiFC assay, we generated a TMED10-fused N-terminal Venus BiFC vector (TMED10-VN), and various ATG-fused C-terminal Venus BiFC vectors (ATG-VC) (Figure S3). To identify TMED10-binding proteins, SH-SY5Y cells were cotransfected with paired combinations of TMED10-VN with the various ATG-VC plasmids. As compared to the control cells, the intensity of the BiFC signal was remarkably increased in cells expressing TMED10-VN and ATG4B-VC, and moderately enhanced in cells expressing TMED10-VN and ATG12-VC (). However, in cells co-expressing TMED10-VN with the ULK1-, ATG3-, ATG5-, BECN1-, ATG7-, ATG10-, ATG13-, ATG14-, ATG16L1-, RB1CC1-, UVRAG-, or PIK3C3-VC protein, the BiFC signal was not sufficiently changed as compared to that of the control. These results suggest that TMED10 may interact with ATG4B in SH-SY5Y cells (). Then, we verified the BiFC signal by TMED10-VN and ATG4B-VC with ATG4B- or TMED10-knockdown conditions. In the TMED10- or ATG4B-depleted cells, the BiFC fluorescence intensity was significantly diminished as compared with that of the control cells (), indicating that TMED10 may bind to ATG4B in SH-SY5Y cells.
Figure 2. BiFC analysis of the interaction between TMED10 and ATG proteins. (A) SH-SY5Y cells were transiently cotransfected with the BiFC plasmids, pFlag/TMED10-VN173 and pHA/ATG-VC155 (ATGx-VC). After 48 h, the cells were imaged by fluorescence microscopy, and the relative fluorescence intensity of Venus was measured using a fluorescence microplate reader. Scale bar: 20 μm. Data are presented as mean ± SEM (n = 3, * P < 0.05). (B and C) SH-SY5Y cells transiently transfected with BiFC assay plasmids (pVC155-ATG4B and pVN173-TMED10) were additionally transfected with siRNA against either ATG4B or TMED10. Then the fluorescent cells were imaged (B) andthe fluorescence intensity was measured with a microplate reader (C). Scale bar: 20 μm. Data are presented as mean ± SEM (n = 3, * P < 0.05).
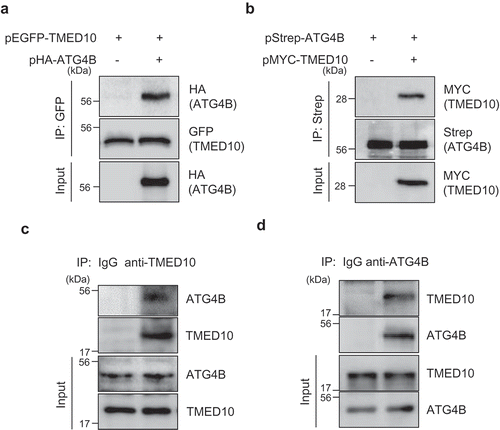
We further confirmed the interaction of TMED10 with ATG4B using immunoprecipitation (IP) assays. SH-SY5Y cells overexpressing pEGFP-TMED10 and pHA-ATG4B, or pStrep-ATG4B and pMyc-TMED10 were prepared, and IP assays were performed. Consistent with the results of the BiFC assay, we found that TMED10 interacted with ATG4B (Figure 3(A, B)). In addition to overexpression experiments, we confirmed the interaction between TMED10 and ATG4B at the endogenous level in SH-SY5Y cells. As shown in , IP assays with endogenous TMED10 and ATG4B further indicated that TMED10 interacts with ATG4B in neuroblastoma cells.
Figure 3. TMED10 interacts with ATG4B in SH-SY5Y cells. (A) SH-SY5Y cells overexpressing GFP-fused TMED10 and HA-tagged ATG4B were immunoprecipitated with anti-GFP antibody conjugated to agarose beads, then the immunoprecipitates were further analyzed by Western blotting with anti-HA and anti-GFP antibodies. (B) SH-SY5Y cells overexpressed pStrep-ATG4B and pMyc-TMED10 were immunoprecipitated with antibody conjugated Strep bead, then the immune-complex were further analyzed by western blotting with anti-Strep and anti-HA antibodies. (C and D) SH-SY5Y cell lysates were immunoprecipitated with anti- TMED10 (C) or anti-ATG4B antibody (D). Subsequently, the immune-complexes were analyzed by western blotting using anti-ATG4B (C) and anti-TMED10 antibody (D).
To map the binding region of TMED10, deletion mutants of TMED10 were examined for their abilities to bind to ATG4B protein. IP assay with TMED10 deletion mutants revealed that C-terminal deletion mutants, which contain the lumenal domain- transmembrane domain (LC-TD) and transmembrane domain-cytosolic domain (TC-CD) weakly interacted with ATG4B compared with that of wild-type TMED10. whereas, the LD-only mutant of TMED10 did not bind to ATG4B (Figure S4A,B) suggesting that the C-terminal region of TMED10 is involved in the interaction with ATG4B. Reciprocal analysis with ATG4 deletion mutants suggested that both the N-terminal and C-terminal parts of ATG4B might participate in the interaction with TMED10 (Figure S4C,D). Taken together, these results indicate that TMED10 binds to ATG4B in SH-SY5Y cells.
Autophagy activation diminishes the TMED10 and ATG4B interaction
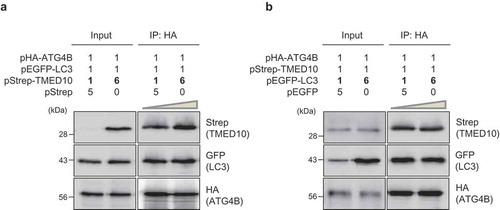
The LC3 protein is a well-known substrate for ATG4B, and its binding can induce conformational changes in ATG4B. As we found that TMED10 interacts with ATG4B, we investigated whether TMED10 and LC3 compete with each other for binding to ATG4B. Three different proteins, HA-tagged ATG4B, GFP-fused LC3, and Strep-tagged TMED10 were overexpressed in SH-SY5Y cells, and IP assays were performed. As shown in , the IP assays showed that increasing levels of TMED10 (up to a 1:6 molar ratio of LC3 to TMED10) did not affect the extent of interaction between LC3 and ATG4B. In reciprocal IP experiments, the interaction between TMED10 and ATG4B was not influenced by an increase in the levels of LC3 (), suggesting that TMED10 and LC3 may not compete with each other for binding to ATG4B in SH-SY5Y cells.
Figure 4. TMED10 does not compete with LC3 for ATG4B binding. (A) SH-SY5Y cells were transiently transfected with 3 plasmids: pHA-ATG4B, pEGFP-LC3, and different amounts of pStrep-TMED10 (up to a 1:6 molar ratio of LC3 to TMED10). After 2 d, the cells were lysed and immunoprecipitated using anti-HA antibody conjugated to agarose beads. Subsequently, the proteins were analyzed by western blotting using anti-Strep and anti-GFP antibodies. (B) SH-SY5Y cells overexpressing HA-tagged ATG4B, Strep-tagged TMED10, and GFP-tagged-LC3 (up to a 1:6 molar ratio of TMED10 to LC3) were assessed by immunoprecipitation using anti-HA antibody conjugated to agarose beads. The immunoprecipitated proteins were analyzed by western blotting using anti-Strep and anti-GFP antibody.
To investigate the functional role of the TMED10-ATG4B interaction, we assessed their binding under autophagy-inducing conditions. To induce autophagy, SH-SY5Y cells overexpressing Strep-tagged TMED10 and HA-tagged ATG4B were treated with rapamycin. Remarkably, the interaction between TMED10 and ATG4B was markedly decreased in rapamycin-treated cells (). Furthermore, the interaction between TMED10 and ATG4B was also decreased under serum-free conditions (), suggesting that TMED10 may dissociate from ATG4B during autophagy activation. We next examined the TMED10-ATG4B interaction under these conditions using the BiFC assay. SH-SY5Y cells overexpressing TMED10-VN and ATG4B-VC were treated with rapamycin or were exposed to serum-free conditions, and the fluorescence intensity was measured. In accordance with the IP assays, the BiFC assay also showed a decrease in binding affinity between TMED10 and ATG4B upon both rapamycin treatment and under serum starvation (). Taken together, these results indicate that the activation of autophagy may diminish the interaction between TMED10 and ATG4B.
Figure 5. TMED10-ATG4B interaction is diminished upon activation of autophagy. (A) SH-SY5Y cells transfected with pStrep- TMED10 and pHA-ATG4B were additionally treated with rapamycin (5 μM) for 3 h. The cells were harvested and subjected to immunoprecipitation using the indicated antibodies conjugated to Strep beads. The samples were analyzed by western blotting with the indicated antibodies. (B) SH-SY5Y cells overexpressing Strep-tagged TMED10 and HA-tagged ATG4B were incubated under serum-free conditions (–) for the indicated times. Then, the cells were subjected to immunoprecipitation using antibodies conjugated to Strep beads, and analyzed by western blotting with the indicated antibodies. (C) SH-SY5Y cells were transfected with the BiFC plasmids (pVC155-ATG4B and pVN173-TMED10) and treated with 5 μM rapamycin for 3 h, or incubated under serum-starved conditions for 24 h. Then, the fluorescence signals were imaged (C). (D-F) SH-SY5Y cells were transfected with pVC155-ATG4B (ATG4/VC), pVC155-ATG4BS 383,392A (S383,392A/VC) and pVC155-ATG4B S383,392E (S383,392E/VC) with pVN173-TMED10 (TMED10/VN). The fluorescent cells were imaged (D) and measured the fluorescence intensity with a microplate reader (E). Then, the cells were analyzed by Western blotting using GFP antibody (F). Scale bar: 20 μm. Data are presented as mean ± SEM (n = 3, ** P < 0.01).
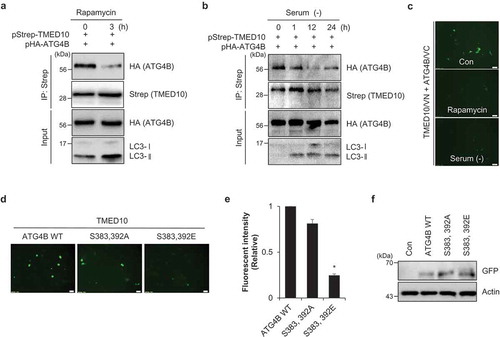
Recently, it was reported that phosphorylation of ATG4B at Ser383 and Ser392 regulates ATG4B activity and increases autophagic flux [Citation24,Citation25]. To investigate the consequence of Ser383 and Ser392 phosphorylation of ATG4B, we generated non-phosphorylated mutant (S383,392A) or phospho-mimetic mutant (S383,392E). Interestingly, the interaction of TMED10 and ATG4B was reduced in the phospho-mimetic mutant (). These results suggest that the phosphorylation of ATG4B promotes dissociation of ATG4B from TMED10.
Downregulation of TMED10 increases ATG4B protease activity
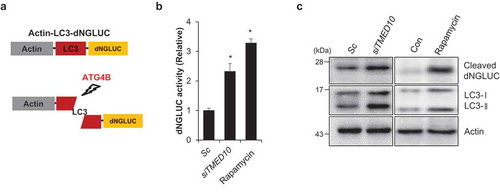
Because phosphorylation of ATG4B stimulates its activity, we elucidated the effect of TMED10 knockdown on ATG4B activity. Using a cell-based assay system wherein the proteolytic activity of ATG4B can be monitored by measuring Gaussia luciferase (GLUC) activity (Figure 6(A)) [Citation26]. SH-SY5Y cells were generated overexpressing pEAK12-Actin-LC3-dNGLUC, which is secreted after cleavage by ATG4B [Citation26]. Then autophagy activation was induced in these cells by either TMED10 knockdown or rapamycin treatment followed by the measurement of luciferase activity. Notably, the luciferase activity was significantly increased in both TMED10-depleted and rapamycin-treated cells compared to that of the control cells, suggesting that reduction of TMED10 enhances the cleavage of GLUC-linked LC3 by activating ATG4B in the SH-SY5Y cells (). Furthermore, we confirmed the cleavage of Actin-LC3-dNGLUC in these cells by examining the cleaved luciferase using western blotting. Consistent with the luciferase assay, the luciferase fragment released upon cleavage by ATG4B was also markedly increased in TMED10-knockdown cells (). These results suggest that downregulation of TMED10 increases autophagy by enhancing the proteolytic activity of ATG4B.
Figure 6. Downregulation of TMED10 increases the proteolytic activity of ATG4B. (A) Schematic diagram of the quantification of ATG4B activity using an assay based on a luciferase-release system. (B and C) SH-SY5Y cells transfected with pEAK12-Actin-LC3-dNGLUC were transfected with scrambled siRNA (Sc) or TMED10 siRNA (siTMED10), or treated with rapamycin (5 μM). The supernatants were collected, and the relative luciferase activity was measured (B). The collected supernatants were further analyzed by western blotting using an anti-luciferase antibody (C). Data are presented as mean ± SEM (n = 3). * P < 0.05.
Increased Aβ by downregulation of TMED10 enhances dissociation of TMED10 and ATG4B
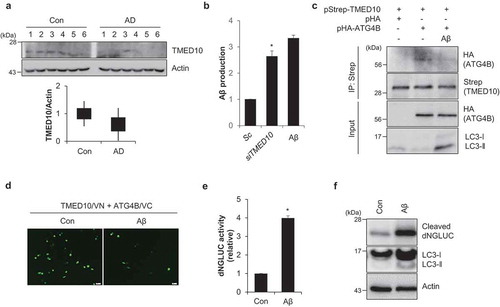
Previously it was reported that TMED10 expression was decreased in the brain tissue of AD patients and the decreased TMED10 enhanced Aβ production [Citation22,Citation27]. In accordance, we confirmed the reduced expression of TMED10 in brain tissue samples from AD patients () and increased Aβ production in TMED10-depleted cells (). Since treatment of Aβ activates autophagy, we further investigated the effect of Aβ on the interaction of TMED10 and ATG4B. Interestingly, the interaction between TMED10 and ATG4B was reduced in Aβ-treated cells (). We further examined the interaction using the BiFC assay. In accordance with the IP assays, the fluorescence intensity in the Aβ-treated cells was significantly diminished compared with that of the control cells, indicating that Aβ induces dissociation of TMED10 and ATG4B (). Finally, we measured ATG4B activity in Aβ-treated SY5Y cells. Consistently, we found that luciferase activity was significantly increased in Aβ-treated cells (). Taken together, these results indicate that Aβ regulates ATG4B activity in AD models.
Figure 7. Aβ1-42 regulates ATG4B activity. (A) The expression level of TMED10 was examined in brain tissues from normal (Cont.) and age-matched Alzheimer disease (AD) patients. The tissue samples were assessed by western blotting with TMED10 antibodies. Actin was used as an internal loading control. The relative expression value was analyzed using densitometry. (B) SH-SY5Y cells were transfected with scrambled siRNA (Sc) or TMED10 siRNA (siTMED10), or treated with Aβ (10 μM). Then Aβ ELISA was measured. (C) SH-SY5Y cells transfected with pStrep-TMED10 and pHA-ATG4B were treated with Aβ (10 μM) for 24 h. The cells were harvested and subjected to immunoprecipitation using antibody conjugated to Strep beads. The samples were analyzed by western blotting with the indicated antibodies. (D) SH-SY5Y cells were transfected with the BiFC plasmids (pVC155-ATG4B and pVN173-TMED10) and treated with Aβ (10 μM) for 24 h. Then, the fluorescence signals were imaged. (E and F) SH-SY5Y cells transfected with pEAK12-Actin-LC3-dNGLUC were treated with Aβ (10 μM). The supernatants were collected, and the relative luciferase activity was measured (E) and further analyzed by western blotting using an anti-luciferase antibody (F). Scale bar: 10 μm. Data are presented as mean ± SEM (n = 3, * P < 0.05).
Discussion
Autophagy plays a fundamental role in maintaining cellular homeostasis under various stress conditions. In this study, we constructed an siRNA library, and performed a cell-based functional screening of neuroblastoma SH-SY5Y cells to identify novel modulators of autophagy. We identified various autophagy regulators (Figure S5A), including already known autophagy regulators, such as PHB2 (prohibitin 2), INPP5E (inositol polyphosphate-5-phosphatase E), RAB10 (RAB10, member RAS oncogene family), and NDUFA13 (NADH:ubiquinone oxidoreductase subunit A13), which validate our screening approach (Figure S5B) [Citation28–Citation31]. Based on the screening results, we further explored the role of TMED10 as a potent autophagy regulator.
TMED10 interacts with PRKCD (protein kinase C delta), which is involved in the activation of autophagy and regulation of cell death in cancer cells [Citation32]. In addition, TMED10 modulates cell growth in papillary thyroid cancer cells. Depletion of TMED10 increases the level of AMPK phosphorylation, but decreases level of mTOR phosphorylation [Citation33], indicating that TMED10 plays a role in the regulation of autophagy. In this study, we demonstrated that TMED10 interacts with ATG4B, and this interaction negatively regulates autophagy. Consistent with previous notion, downregulation of TMED10 remarkably increased the activation of autophagy (). Molecular genetic studies in yeast have identified more than 40 ATG proteins. Among the various ATG proteins, ATG4 is the only known cysteine protease, of which there are 4 different isoforms in mammals (ATG4A, 4B, 4C, and 4D) [Citation34]. Although ATG4 is an essential protein in the regulation of autophagy, little is known about its underlying molecular mechanisms. During autophagy, ATG4B not only cleaves the newly generated LC3 (pro-LC3) protein before its conjugation to lipids in the phagophore, but also deconjugates PE from LC3-II to produce LC3-I for recycling [Citation35,Citation36]. Interestingly, our data suggest that TMED10 directly interacts with ATG4B without competition with LC3. TMED10 is a member of the p24 family of proteins, which can bind to STX17 (syntaxin 17) to cycle between the endoplasmic reticulum (ER) and ER-Golgi intermediate compartment through trafficking pathways involving coat protein complex vesicles [Citation17,Citation18,Citation37]. STX17 recruits ATG14 to the ER-mitochondria contact site and localizes to the outer membrane of completed autophagosomes, and is required for their fusion with endosomes or lysosomes [Citation38,Citation39], indicating that TMED10 may play a role at organelles membrane to modulate autophagy. Although we did not validate the autophagosomal localization of TMED10, it may be possible that TMED10 suppresses ATG4B activity by recruiting ATG4B to membrane. Our deletion mapping analysis also indicated that the C terminus of TMED10 containing the transmembrane and cytosolic domains is involved in the interaction with ATG4B. In addition, Blum et al. demonstrated that the C terminus of TMED10 contributes to its cellular localization [Citation40]. Thus, further investigation of localization of TMED10 and ATG4B during autophagy will help to elucidate the regulatory mechanism of ATG4B by TMED10.
Recently, we and others reported that posttranslational modifications, such as O-GlcNAcylation and phosphorylation, target on ATG4B, and positively regulate its hydrolase activity and autophagic flux [Citation25,Citation41]. Rapamycin treatment induces phosphorylation at the C terminus of ATG4B, which enhances its catalytic activity and autophagy [Citation24,Citation25]. Although O-GlcNAcylation and phosphorylation share target residues, the interplay of these modulations is not yet elucidated. We found that Ser383 and Ser392 phosphorylation of ATG4B regulates the interaction with TMED10 and ATG4B (); however, we did not evaluate whether TMED10 modulates phosphorylation of ATG4B by STK26/MST4 kinase. Nonetheless, we observed that both suppression of TMED10 expression and rapamycin treatment potentiated the peptidase activity of ATG4B (). The dissociation of ATG4B from TMED10 resulted in an increase in ATG4B activity during autophagy. Therefore, further investigation of the interaction of TMED10 with post-translationally modified ATG4B is needed to delineate the mechanisms of regulation of ATG4B under starvation conditions.
The primary role of autophagy is a protective response under various stress conditions by degradation of abnormal harmful proteins and damaged organelles, thus dysregulation of autophagy results in the accumulation of toxic aggregates, which are commonly identified in neurodegenerative disease [Citation42]. The major hallmarks of AD are the accumulation of Aβ and hyper-phosphorylated tau proteins [Citation43,Citation44]. Interestingly, TMED10 was originally identified through its interaction with the γ-secretase complex; the suppression of TMED10 increased Aβ peptide production by enhancing the secretase activity in AD models [Citation22]. In this study, we also identified TMED10 as an autophagy regulator from a siRNA library consisting of AD-related genes. Aβ may activates autophagy by increasing reactive oxygen species (ROS) production with mitochondrial damage or suppressing MTOR signaling pathway [Citation45,Citation46]. Although proper autophagic flux involves the execution of clearance for target molecules by lysosomal degradation, impairment of the autophagy-lysosomal pathway has been implicated in neurodegenerative disorders, including AD [Citation42]. For example, BECN1 expression is significantly decreased in the early stages of AD, and overexpression of BECN1 reduces Aβ pathology in AD mice model [Citation47]. In addition, lysosomal acidification is impaired in AD patients [Citation48]. Lysosomal malfunction accompanies accumulation of autophagic cargos as well as autophasomes [Citation49]. Autosomal dominant mutations of PSEN1/PS1 (presenilin 1) and PSEN2 cause an early onset form of AD. Loss of PSEN1 functions compromise proton-pumping activity of the vacuolar-type H+-ATPase on lysosomes, which induces defective lysosomal acidification and marked impairment of autophagy [Citation48,Citation50]. Despite its potential relevance, the molecular mechanisms underlying the interplay between autophagy and AD are not fully explored. Meaningfully, downregulation of TMED10 causes increased Aβ production, and AD patients have low TMED10 expression () [Citation22,Citation27]. However, the role of TMED10-mediated inhibition of autophagy in AD is still largely unknown. To identify novel genetic variants of TMED10, which may be associated with AD pathogenesis, we performed a genome-wide exome-sequencing analysis of Korean AD patients. Unfortunately, we could not identify any significant genetic variations (single-nucleotide polymorphisms [SNPs]) from our genetic screening. However, Xiaojie et al. recently reported that a novel SNP in intron 4 of TMED10 is genetically associated with AD [Citation51]. Since, we found that Aβ regulates ATG4B activity through the interaction of TMED10 and ATG4B (), further studies of autophagy regulation by TMED10 will be helpful to understand pathogenesis of Alzheimer disease.
In conclusion, we identified TMED10 as a noble autophagy modulator. Although, it is possible that TMED10 may affect autophagy via multiple mechanisms, we showed that TMED10 directly interacts with ATG4B and negatively regulates ATG4B’s activity during autophagy.
Materials and methods
Cell culture
SH-SY5Y cells were obtained from the American Type Culture Collection (ATCC® CRL-2266™). Cells were maintained in Dulbecco’s modified Eagle’s medium (Hyclone, SH30243.01) supplemented with 10% fetal bovine serum (Hyclone, SH30084.03) and 1% penicillin-streptomycin (Invitrogen, 15140122) and were maintained with 5% CO₂at 37°C. To generate a stable cell line (SY5Y/GFP-LC3), SH-SY5Y cells were transfected with pEGFP-LC3 using Lipofectamine 2000 (Invitrogen, 11668019), and the transfectants were collected following 7 days selection with G418 (Invitrogen, 10131035).
Reagents and plasmids
Bafilomycin A1 (B1793), NH4 Cl (A0171) and rapamycin (R0395) were purchased from Sigma-Aldrich. Amyloid β (Aβ1–42, 4014447) was purchased from American Peptide Co. The short interfering RNA (siRNA) targeting ATG5 (#1: 5-CAGGUAAGUCAAGCCUACAUU-3) and negative scrambled siRNA (5-CCUACGCCACCAAUUUCGU-3) were synthesized from Genolution. The siGENOME SMART pool of TMED10 siRNA (#1: 5-UCACAAGGACCUGCUAGUG-3; #2: 5- GCGGAUACCUGACCAACUC-3) were purchased from Dharmacon. ATG4B siRNA (sc-72584) was purchased from Santa Cruz Biotechnology. The expression plasmid for green fluorescent protein fused tomicrotubule-associated protein 1 light chain 3 (pEGFP-LC3) was kindly provided by Noboru Mizushima (Tokyo University, Japan). pcDNA4/MYC-TMED10 plasmid was a gift from Weihong Song (University of British Columbia, Canada) [Citation20]. GFP-fused TMED10 and its mutant plasmids were kindly provided from Alexandra Lepier [Citation40]. Wild-type TMED10 cDNA and its deletion mutants were subcloned into expression vectors, including pEGFP-N1, pcDNA3/Myc, pEXPR-IBA105/Strep and pBiFC-VN 173 vector (addgene, 22010). TMED10 mutant plasmids were cloned into pcDNA3/Myc. ULK1, ATG3, ATG4B, ATG5, BECN1, ATG7, ATG9, ATG10, ATG12, ATG13, ATG14, ATG16L1, RB1CC1, UVRAG, and PIK3C3 were cloned into a pBiFC-VC 155 vector (addgene, 22011). ATG4B was cloned into pcDNA3/HA and pEXPR-IBA 105/Strep. To create ATG4BS383,392A, and ATG4BS383,392E mutants, site directed mutagenesis was performed following Muta- Direct Site Directed Mutagenesis Kit (Intron Biotechnology, 15071) and specificity of mutagenesis was confirmed by direct sequencing. ATG4B mutants were subcloned into pBiFC-VC 155 and pcDNA3/HA vectors. The Actin-LC 3-dNGLUC construct in a pEAK12 vector was kindly provided by Dr. Brian Seed (Massachusetts General Hospital, Boston, MA) [Citation26].
SiRNA library screening
An arrayed library (Dharmacon, Thermo-Fisher Scientific, Rockford, IL, USA) consisting with AD-related proteins, mitochondrial proteins, and the human kinome was used for a cell based screening. Each pool consisted of 4 oligonucleotides targeting a different region of the same gene, and each assay plate included non-targeting siRNA as a control. SY5Y/GFP-LC3 cells were seeded into 96-well plates, transiently transfected with 50 pmol siRNA using Lipofectamine 2000 (Invitrogen, 11668019) in Opti-MEM (Thermo Fisher Scientific, 31985–070) for 4 h, and then incubated under standard culture conditions for 72 h. For the screening, fluorescence images of GFP-LC3 protein in the cells were obtained from the Operetta High Content Imaging System (Perkin-Elmer, Waltham, MA, USA) and analyzed using the Harmony Analysis Software (Perkin-Elmer)
Transmission electron microscopy analysis
SH-SY5Y cells stably expressing GFP-LC3 transfected with scrambled siRNA or TMED10 siRNA for 3 days were fixed in 4% glutaraldehyde containing 2% paraformaldehyde in 30 mM phosphate buffer, pH 7.4. After dehydration with ethanol, ultrathin sections were prepared using a Sorvall MT5000 microtome (DuPont Instruments, MT5000, Liverpool, NY, USA) and collected on 150 mesh copper grids. The sections were stained with 2% uranyl acetate and/or Reynolds’ lead citrate, and 6 images of each sample were obtained with a JEOL 100CX transmission electron microscope at 50 kV (JEOL, Japan).
Western blotting
All cells lysates were prepared with 2× Laemmli sample buffer [(62.5 mM Tris–HCl, pH 6.8, 2% sodium dodecyl sulfate (SDS)], 25% glycerol, 5% β-mercaptoethanol, 0.01% bromophenol blue) (Bio-Rad, 1610737). Proteins (approximately 50 μg) were quantified by using Bradford solution (Bio-Rad, 5000001). Then the samples were resolved by SDS-10% polyacrylamide gel electrophoresis, and transferred to polyvinylidene difluoride (PVDF) membrane for western blot analysis. After blocking, the membranes were incubated with specific primary antibodies at 4°C overnight. Anti-LC3 (NB100-2220; 1:10,000), anti-ATG5 (NB110-53818; 1:3000) were purchased from NOVUS Biologicals; anti-ULK1 (ab128859; 1:2000), anti-ATG4B (ab69937; 1:3000), anti-ATG13 (ab105392; 1:2000), anti-ATG16L1 (ab47946; 1:2000), anti-ATG 101 (ab105387; 1:2000), anti-RB 1CC1 (ab176816; 1:2000), anti-ZBTB 24 (ab111404; 1:2000), anti-UVRAG (ab174550; 1:2000), anti-WIPI 2 (ab131271; 1:2000) were purchased from Abcam; HA (sc-7392; 1:4000), MYC (sc-40; 1:2000) and GFP (sc-8334; 1:5000) antibody were purchased from Santa Cruz Biotechnology; anti-TMED 10 (2076S; 1:1000), anti-SQSTM 1/p62 (5114S; 1:3000), anti-ATG 3 (3415S; 1:2000), anti-BECN 1 (3738S; 1:3000) antibodies were purchased from Cell Signaling Technology; anti-GLUC (E8023S; 1:1000) antibody was purchased from New England BioLabs; anti-Strep (2–1507; 1:3000) antibody was purchased from IBA-Solutions For Life Sciences; anti-actin (MAB1501; 1:10,000) antibody was purchased from Millipore. For protein detection, the membranes were incubated with secondary antibodies conjugated to horseradish peroxidase (Thermo Fisher Scientific, 34095).
Immunoprecipitation
For the immunoprecipitation assay, cells were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.5, 150 mM sodium chloride, 0.5% sodium deoxycholate, 1% Triton X-100, 0.1% SDS, 2 mM EDTA) (Intron biotechnology, IBS-BR002) containing protease inhibitors (GenDEPOT, P3100) for 1 hr at 4°C. The supernatant was immunoprecipitated with anti-GFP (Santa Cruz Biotechnology, sc-8334-AC), anti-HA (Santa Cruz Biotechnology, sc-7392 AC), anti-Strep (IBA Lifesciences, 2–1201)-conjugated agarose, anti-TMED10 (Santa Cruz Biotechnology, sc-68398), or anti-ATG 4B (Cell Signaling Technology, 13507S) added to the lysate for 24 h at 4°C. Then protein G plus-protein A agarose (CalbioChem, IP05) was added and the samples were incubated for 2 ~ 4 h at 4°C. All cells lysates were prepared with 2× Laemmli sample buffer (Bio-Rad, 1610737). Then all samples were analyzed by western blotting.
Bimolecular fluorescence complementation (BiFC) assay
TMED10 cDNA was subcloned into a Flag-tagged N-terminal Venus plasmid (pBiFC/Flag-VN173) and cDNAs coding for autophagy regulatory proteins were subcloned into a HA-tagged C-terminal Venus vector (pBiFC/HA-VC155). A BiFC assay was performed as previously described [Citation34]. SH-SY5Y cells were co-transfected with TMED10 fused to the N-terminus (TMED10-VN) and each ATG protein fused to the C-terminus (ATGs-VC) fragment of Venus by using Lipofectamine 2000 (Invitrogen, 11668019) according to the manufacturer’s protocol. After 48 h, TMED10-VN and ATGs-VC fusion proteins were imaged using a fluorescence microscope (Olympus, IX71) or measured using a fluorescence microplate reader (PerkinElmer, Victor X3).
ATG4 activity assay (luciferase activity assay)
As previously reported by Ketteler et al, cellular ATG4B activity was analyzed using an N-terminal deleted form of Gaussia luciferase (dNGLUC) [Citation26,Citation41]. Briefly, SH-SY5Y cells transfected with siRNAs were transiently transfected with pEAK12-Actin-LC3-dNGLUC. When DNGLUC is fused to the C-terminus of β-actin, dNGLUC activity was not detected in supernatant (SN). However, when LC3 is cleaved by ATG4B, dNGLUC is increased in SN in the protein expressing cells. After 48 h of transfection, the assays were performed using the dual luciferase reporter assay system (Promega Corporation, E2810) according to the manufacturer’s instructions.
Human Alzheimer disease (AD) samples
Inferior parietal lobule and cerebellum specimens from the brains of 6 patients with AD and 6 control subjects that had been enrolled in the University of Kentucky Alzheimer Disease Center Autopsy Program were used for this study. All patients with AD met both clinical diagnostic criteria and neuropathological diagnostic criteria of AD [Citation52]. The control subjects had no history or neuropathological signs of a brain disorder. At autopsy, tissue specimens were rapidly removed and frozen, and were stored at −80°C [Citation53].
Aβ ELISA assay
Aβ production levels were analyzed using the Human β-Amyloid (1–42) ELISA Kit (FUJIFILM Wako Chemicals, 298–62401). SH-SY5Y cells plated in 96-well plate were transfected with scrambled siRNA or TMED10 siRNA. After 72 h, the cells were analyzed using the Human β-Amyloid (1-42) ELISA kit according to the manufacturer’s instructions and were measured using a fluorescence micro plate reader (wavelength 450) (Perkin Elmer, Victor X3).
Statistical analysis
All results are expressed as the mean ± standard error. The statistical analysis was performed using multiple comparisons tests and SPSS17.0 software (SPSS Inc., Chicago, IL, USA). A P-value < 0.05 or <0.01 was considered significant.
Supplemental Material
Download Zip (7.1 MB)Disclosure statement
No potential conflict of interest was reported by the authors.
Supplemental material
Supplemental data for this article can be accessed here.
Additional information
Funding
Related Research Data
References
- Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12:823–830. PMID: 20811354.
- Mizushima N. Autophagy in protein and organelle turnover. Cold Spring Harb Symp Quant Biol. 2011;76:397–402. PMID:21813637.
- Li Q, Liu Y, Sun M. Autophagy and Alzheimer’s Disease. Cell Mol Neurobiol. 2016;37:377–388. PMID: 27260250.
- Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69–79. PMID:24323045.
- Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat Rev Mol Cell Biol. 2007;8:931–937. PMID:17712358.
- Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Patho. 2010;221:3–12. PMID:20225336.
- Itakura E, Mizushima N. Characterization of autophagosome formation site by a hierarchical analysis of mammalian Atg proteins. Autophagy. 2010;6:764–776. PMID: 20639694.
- Kabeya Y, Mizushima N, Ueno T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. Embo J. 2000;19:5720–5728. PMID:11060023.
- Mizushima N, Yoshimori T, Ohsumi Y. Role of the Apg12 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2003;35:553–561. PMID:12672448.
- Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. PMID:26799652.
- Shin HJ, Kim H, Oh S, et al. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature. 2016;534:553–557. PMID:27309807.
- Füllgrabe J, Ghislat G, Cho DH, et al. Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci. 2016;16:3059–3066. PMID:27528206.
- Xie Y, Kang R, Sun X, et al. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy. 2015;11:28–45. PMID:25484070.
- Lipinski MM, Hoffman G, Ng A, et al. A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell. 2010;18:1041–1052. PMID:20627085.
- Orvedahl A, Sumpter R Jr, Xiao G, et al. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature. 2011;480:113–117. PMID:22020285.
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9:1102–1109. PMID:17909521.
- Bremser M, Nickel W, Schweikert M, et al. Coupling of coat assembly and vesicle budding to packaging of putative cargo receptors. Cell. 1999;96:495–506. PMID:10052452.
- Gommel DU, Memon AR, Heiss A, et al. Recruitment to Golgi membranes of ADP-ribosylation factor 1 is mediated by the cytoplasmic domain of p23. Embo J. 2001;20:6751–6760. PMID:11726511.
- Liu S, Zhang S, Bromley-Brits K, et al. Transcriptional regulation of TMP21 by NFAT. Mol Neurodegener. 2011;6:21–32. PMID:21375783.
- Liu S, Bromley-Brits K, Xia K, et al. TMP21 degradation is mediated by the ubiquitin-proteasome pathway. Eur J Neurosci. 2008;28:1980–1988. PMID:19046380.
- Xie J, Yang Y, Li J, et al. Expression of tmp21 in normal adult human tissues. Int J Clin Exp Med. 2014;7:2976–2983. PMID:25356171.
- Chen F, Hasegawa H, Schmitt-Ulms G, et al. TMP21 is a presenilin complex component that modulates gamma-secretase but not epsilon-secretase activity. Nature. 2006;440:1208–1212. PMID:16641999.
- Hu CD, Grinberg AV, Kerppola TK. Visualization of protein interactions in living cells using bimolecular fluorescence complementation (BiFC) analysis. Curr Protoc Protein Sci. 2005. Chapter19, Unit19.10. PMID:18429278. DOI: 10.1002/0471140864.ps1910s41
- Huang T, Kim CK, Alvarea AA, et al. MST4 phosprylation of ATG4B regulates autophagy acitivity, tumorigenecity, and radirsistance in glioblastoma. Cancer Cell. 2017;32:840–855. PMID: 29232556.
- Yang Z, Wilkie-Grantham RP, Yanagi T, et al. ATG4B (autophagin-1) phosphorylation modulates autophagy. J Biol Chem. 2015;290:26549–26561. PMID: 26378241.
- Ketteler R, Seed B. Quantitation of autophagy by luciferase release assay. Autophagy. 2008;4:801–806. PMID:18641457.
- Vetrivel KS, Kodam A, Gong P, et al. Localization and regional distribution of p23/TMP21 in the brain. Neurobiol Dis. 2008;32:37–49. PMID: 18652896.
- Kathiria AS, Butcher LD, Feagins LA, et al. Prohibitin 1 modulates mitochondrial stress-related autophagy in human colonic epithelial cells. PLoS One. 2012;7:e31231. PMID:22363587.
- Hasegawa J, Iwamoto R, Otomo T, et al. Autophagosome-lysosome fusion in neurons requires INPP5E, a protein associated with Joubert syndrome. Embo J. 2016;35:1853–1867. PMID:27340123.
- Li Z, Schulze RJ, Weller SG, et al. A novel Rab10-EHBP1-EHD2 complex essential for the autophagic engulfment of lipid droplets. Sci Adv. 2016;2:e1601470. PMID: 28028537.
- Yue X, Zhao P, Wu K, et al. GRIM-19 inhibition induced autophagy through activation of ERK and HIF-1α not STAT3 in Hela cells. Tumour Biol. 2016;37:9789–9796. PMID:26810068.
- Wang H, Xiao L, Kazanietz MG. p23/Tmp21 associates with protein kinase Cdelta (PKCdelta) and modulates its apoptotic function. J Biol Chem. 2011;286:15821–15831. PMID:21454541.
- Xu X, Gao H, Qin J, et al. TMP21 modulates cell growth in papillary thyroid cancer cells by inducing autophagy through activation of the AMPK/mTOR pathway. Int J Clin Exp Pathol. 2015;8:10824–10831. PMID:26617795.
- Mariño G, Uría JA, Puente XS, et al. Human autophagins, a family of cysteine proteinases potentially implicated in cell degradation by autophagy. J Biol Chem. 2003;278:3671–3678. PMID:12446702.
- Satoo K, Noda NN, Kumeta H, et al. The structure of Atg4B-LC3 complex reveals the mechanism of LC3 processing and delipidation during autophagy. Embo J. 2009;28:1341–1350. PMID:19322194.
- Tanida I, Sou YS, Ezaki J, et al. HsAtg4B/HsApg4B/autophagin-1 cleaves the carboxyl termini of three human Atg8 homologues and delipidates microtubule-associated protein light chain 3- and GABAA receptor-associated protein-phospholipid conjugates. J Biol Chem. 2004;279:36268–36276. PMID:15187094.
- Muppirala M, Gupta V, Syntaxin SG. 17 cycles between the ER and ERGIC and is required to maintain the architecture of ERGIC and Golgi. Biol Cell. 2011;103:333–350. PMID:21545355.
- Diao J, Liu R, Rong Y, et al. ATG14 promotes membrane tethering and fusion of autophagosomes to endolysosomes. Nature. 2015;7548:563–566. PMID:25686604.
- Itakura E, Kishi-Itakura C, Mizushima N. The hairpin-type tail-anchored SNARE syntaxin 17 targets to autophagosomes for fusion with endosomes/lysosomes. Cell. 2012;151:1256–1269. PMID:23217709.
- Blum R, Lepier A. The luminal domain of p23 (Tmp21) plays a critical role in p23 cell surface trafficking. Traffic. 2008;9:1530–1550. PMID:18627576.
- Jo YK, Park NY, Park SJ, et al. O-GlcNAcylation of ATG4B positively regulates autophagy by increasing its hydroxylase activity. Oncotarget. 2016;7:57186–57196. PMID:27527864.
- Nixon RA. The role of autophagy in neurodegenerative disease. Nat Med. 2013;19:983–997. PMID:23921753.
- Glenner GG, Wong CW. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. PMID:6375662.
- Grundke-Iqbal I, Iqbal K, Tung YC, et al. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. PMID:3088567.
- Lipinski MM, Zheng B, Lu T, et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2010;107:14164–14169. PMID: 20660724.
- Zheng X, Xie Z, Zhu Z, et al. Methyllycaconitine alleviates amyloid-β peptides-induced cytotoxicity in SH-SY5Y cells. PLoS One. 2014;9:e111536. PMID: 25360664.
- Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest. 2008;118:2190–2199. PMID:18497889.
- Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–1158. PMID: 20541250.
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007;120:4081–4091. PMID: 18032783.
- Colacurcio DJ, Pensalfini A, Jiang Y, et al. Dysfunction of autophagy and endosomal-lysosomal pathways: roles in pathogenesis of down syndrome and Alzheimer’s disease. Free Radic Biol Med. 2018;114:40–51. PMID: 28988799.
- Zhang X, Wu Y, Cai F, et al. A novel Alzheimer-associated SNP in Tmp21 increases amyloidogenesis. Mol Neurobiol. 2018;55:1862–1870. PMID: 28233271.
- The National Institute on Aging and Reagan Institute Working Groupon Diagnostic Criteria for the Neuropathological Assessmen to fAlzheimer’s Disease, NIARI. Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. Neurobiol Aging. 1997;18:S1–S2.
- Jo DG, Arumugam TV, Woo HN, et al. Evidence that gamma-secretase mediates oxidative stress-induced beta-secretase expression in Alzheimer’s disease. Neurobiol Aging. 2010;31:917–925. PMID: 18687504.